

Key Points
Overexpression of Hhex transcription factor blocks myeloid differentiation at the promyelocyte stage.
Hhex cooperates with growth factor independence to elicit rapid promyelocytic leukemia in mice.
Abstract
The hematopoietically expressed homeobox (Hhex) transcription factor is overexpressed in human myeloid leukemias. Conditional knockout models of murine acute myeloid leukemia indicate that Hhex maintains leukemia stem cell self-renewal by enabling Polycomb-mediated epigenetic repression of the Cdkn2a tumor suppressor locus, encoding p16Ink4a and p19Arf. However, whether Hhex overexpression also affects hematopoietic differentiation is unknown. To study this, we retrovirally overexpressed Hhex in hematopoietic progenitors. This enabled serial replating of myeloid progenitors, leading to the rapid establishment of interleukin-3 (IL-3)–dependent promyelocytic cell lines. Use of a Hhex-ERT2 fusion protein demonstrated that continuous nuclear Hhex is required for transformation, and structure function analysis demonstrated a requirement of the DNA-binding and N-terminal–repressive domains of Hhex for promyelocytic transformation. This included the N-terminal promyelocytic leukemia protein (Pml) interaction domain, although deletion of Pml failed to prevent Hhex-induced promyelocyte transformation, implying other critical partners. Furthermore, deletion of p16Ink4a or p19Arf did not promote promyelocyte transformation, indicating that repression of distinct Hhex target genes is required for this process. Indeed, transcriptome analysis showed that Hhex overexpression resulted in repression of several myeloid developmental genes. To test the potential for Hhex overexpression to contribute to leukemic transformation, Hhex-transformed promyelocyte lines were rendered growth factor–independent using a constitutively active IL-3 receptor common β subunit (βcV449E). The resultant cell lines resulted in a rapid promyelocytic leukemia in vivo. Thus, Hhex overexpression can contribute to myeloid leukemia via multiple mechanisms including differentiation blockade and enabling epigenetic repression of the Cdkn2a locus.
Visual Abstract
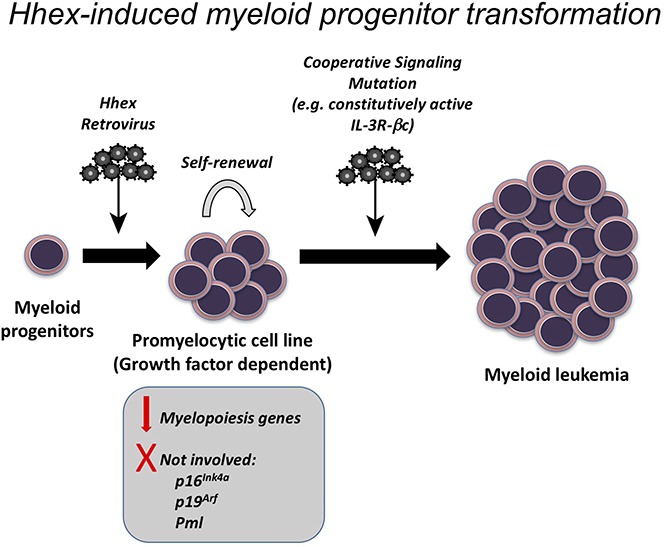
Introduction
The hematopoietically expressed homeobox gene (Hhex) is an oligomeric, nonclustered homeobox transcription factor that acts primarily as a transcriptional repressor.1,2 Hhex was first cloned via its expression in hematopoietic tissues, where it is highly expressed in hematopoietic stem cells (HSCs) and progenitors but then undergoes transcriptional downregulation during differentiation, particularly in the T-cell lineage.3-5 Consistent with the function of members of the homeobox gene family, Hhex has been shown to have important developmental roles during embryogenesis. Consequently, Hhex-knockout mice die during midgestation with perturbed mesodermal tissue development, resulting in defects in liver, thyroid, and cardiac formation.6-8
To analyze the role of Hhex in adult hematopoiesis, we and others have studied mice harboring a conditional knockout allele of Hhex.9,10 This showed that although Hhex is dispensable for the maintenance of HSCs and myeloid progenitors, it is required for HSC self-renewal during serial transplantation and myeloid progenitor expansion in response to hematopoietic stress.11 In addition, Hhex plays a critical physiologic role regulating commitment of common lymphoid precursors to diverse lymphoid lineages including B cells, T cells, natural killer cells, and dendritic cells.9 In contrast, overexpression of Hhex in T-cell progenitors results in aberrant thymocyte self-renewal and T-cell leukemia in mouse models.12,13 These studies establish Hhex as a dynamic regulator of lymphoid development that acts during early lymphopoiesis to promote differentiation of multiple lymphoid lineages, whereas downregulation during T-cell development is important to prevent the development of T-cell leukemia.
In addition to its key role in lymphoid development, Hhex is the target of a rare chromosomal translocation [t(10;11)(q23;p15)] in acute myeloid leukemia (AML), which results in the generation of a NUP98-HHEX fusion oncogene.14 In addition, we have recently found that Hhex is overexpressed in a variety of AML subtypes, including t(8;21) (RUNX1-RUNX1T1), t15;17 (promyelocytic leukemia–retinoic acid receptor α [PML-RARA]), inv(16)/t(16;16) (CBFB-MYH11), and t(11q23) (mixed-lineage leukemia [MLL]).15 Using Hhex conditional knockout mice, we found that Hhex plays a critical role in both initiation and maintenance of leukemic stem cell self-renewal in mouse models of AML induced by MLL-ENL or Hoxa9/Meis1 oncogenes.15 In these models, Hhex binds to members of the Polycomb-repressive complex 2 (PRC2) and promotes epigenetic repression of the Cdkn2a tumor suppressor locus (encoding p16Ink4a and p19Arf) by enabling PRC2-mediated silencing.15 In the absence of Hhex, PRC2-mediated repression is lost, leading to reactivation of Cdkn2a and consequent senescence of AML stem cells. AML stem cells were uniquely sensitive to upregulation of Cdkn2a following loss of Hhex, presumably due to alternative mechanisms to silence Cdkn2a in normal HSCs.
In mouse models of MLL-ENL–induced AML, deletion of Cdkn2a enabled AML maintenance in the absence of Hhex, indicating that loss of Hhex eliminates leukemia in this model solely via Cdkn2a activation and that Hhex is not required for MLL-ENL to block myeloid differentiation.15 However, in this model, Hhex expression was only modestly increased compared with normal HSCs, in contrast to human AML in which Hhex expression is increased up to 10-fold.15 It remains unknown whether this Hhex overexpression, which is observed in various subtypes of human AML, can also affect myeloid differentiation and cooperate in leukemogenesis. Here, we have studied the effects of Hhex overexpression on myeloid progenitors. We find that this results in a differentiation block at the promyelocyte stage, leading to the development of interleukin-3 (IL-3)–dependent promyelocyte cell lines that are dependent on DNA binding and N-terminal–repressive domains of Hhex. Upon abrogation of growth factor dependence, transplantation of Hhex-overexpressing cell lines results in a rapid leukemia in vivo. Thus, Hhex can promote myeloid leukemia via multiple mechanisms including inhibition of differentiation as well as enabling Polycomb-mediated silencing of tumor-suppressor pathways.
Materials and methods
Mice and transplantation experiments
All mice used were on a C57BL/6 background. Pml,16 p19Arf,17 and Cdkn2a (p16Ink4a/p19Arf)18 knockout mice have been published previously. CD45.1+ C57BL/6.Ly5.1 mice (Walter and Eliza Hall Institute [WEHI]) were used as recipient mice in chimeric transplant experiments, where they received a 650 rad (137Cs source) sublethal dose of irradiation immediately prior to IV injection of 106 cells. Blood was analyzed using an ADVIA 2120 hematology analyzer (Siemens Healthcare) device. All experiments were approved by the animal ethics committees of WEHI or the Alfred Medical Research and Education Precinct (AMREP).
Cell culture
Retrovirally transduced Lin−Sca-1+Kit+ (LSK) cells were cultured in Iscove modified Dulbecco medium (IMDM; WEHI) supplemented with 10% fetal bovine serum, in the presence of murine IL-3 (mIL-3) (10 ng/mL; WEHI), human erythropoietin (hEPO) (3 U/mL, EPREX; Janssen-Cilag), and murine stem cell factor (mSCF) (50 ng/mL; WEHI). For viable cell counts, cells were mixed with Accucheck beads (Invitrogen), stained with propidium iodide (1 μg/mL; Sigma-Aldrich), and enumerated by flow cytometry.
For colony assays, cells were suspended in mouse methylcellulose base media (R&D Systems) in the presence of the growth factors mentioned in the previous paragraph and colony numbers scored after 7 days. Cells were then resuspended and reseeded at 2000 cells per plate.
MSCV-IRES-GFP (MIG)-Hhex–transformed promyelocytic cell lines were maintained in complete IMDM containing 10 ng/mL mIL-3. MIG-Hhex-ERT2 cell lines were maintained in the same medium plus tamoxifen (1 μM; Sigma-Aldrich).
Retroviral transduction
Retroviruses were produced by calcium-phosphate transfection of HEK-293T cells using the pMD2-Gag-Pol and pCAG4-Eco packaging vectors. LSK cells were retrovirally transduced using Retronectin (Takara) precoated (1.5 mg/mL), non-tissue-culture–treated plates (Thermo Scientific). Transduced cells were cultured in StemPro-34 media (Gibco) supplemented with mIL-3 (10 ng/mL), mIL-6 (10 ng/mL), mSCF (50 ng/mL), and mFlt3 ligand (50 ng/mL). Two days posttransduction, cells were sorted for green fluorescent protein (GFP) expression and used in cell culture assays.
Molecular biology
The MIG-Hhex retroviral vector was obtained from David Izon (St. Vincent’s Institute, Melbourne, VIC, Australia). We used the MIG retroviral vector to directionally clone mutant forms of Hhex using 5′-XhoI and 3′-EcoRI restriction enzyme sites. To create MIG-Hhex-ERT2, the MSCV-Cre-ERT2-IRES-GFP vector was used to clone Hhex as a fusion protein with ERT2 at the 3′ end. The primer strategies and sequences used are shown in supplemental Table 1.
Flow cytometry
Antibodies used for lineage marker depletion included rat anti-mouse B220 (RA3-6B2), CD3 (KT3-1-1), CD4 (GK1.5-7), CD8 (53.6.7), CD19 (1D3), Gr-1 (RB6-8C5), Mac-1 (M1/70), and TER119 (TER119) all from the WEHI Monoclonal Antibody Facility. The exclusion of hematopoietic lineage cells from murine bone marrow (BM) was performed using anti-rat antibodies in combination with sheep anti-rat immunoglobulin G–coated immunomagnetic beads (Invitrogen). Antibodies used for flow cytometry included goat anti-rat immunoglobulin G–Alexa 680 (A21096; Invitrogen) and the following rat anti-mouse antibodies: c-Kit–allophycocyanin (ACK-2; WEHI), CD45.1–fluorescein isothiocyanate (A20.1; WEHI), CD45.1-BV650 (A20.1; Biolegend), CD45.2–peridinin-chlorophyll-protein-cyanine5.5 (A20.1; eBioscience), CD45.2–fluorescein isothiocyanate (S450-015-2; WEHI), CD45.2–Alexa 700 (S450-015-2; WEHI), Flt-3–phycoerythrin (A2F10; Biolegend), Gr-1–Alexa 700 (RB6-8C5; WEHI), Mac-1–peridinin-chlorophyll-protein-cyanine5.5 (M1/70; BD), and Sca-1–phycoerythrin-cyanine7 (D7; eBioscience). An Fc receptor blocking step was performed prior to staining using rat γ globulin (10 mg/mL; Jackson ImmunoResearch) at a 1/10 dilution for 10 minutes at 4°C. Cellular viability was determined by staining with 8.3 mg/mL FluoroGold (Sigma-Aldrich). Data were acquired on LSR Fortessa or FACSCanto II flow cytometers (BD Biosciences) and analyzed using Flowjo software (version 9.4.4; Tree Star). Cell sorting was done using either an Aria device (BD) or a MoFlo device (Beckman Coulter).
Histology
For cytocentrifuge preparations, 50 000 cells were centrifuged onto a microscope slide and stained with May-Grünwald-Giemsa (Sigma-Aldrich). Hematoxylin-and-eosin stains of mouse organs were performed in-house by WEHI Histology. Cell phenotypic enumeration was performed by visual assessment using a light microscope, scoring at least 200 cells per sample.
Next-generation sequencing
RNA was extracted from GFP+-sorted LSK cells 2 days postinfection using the RNeasy Plus Minikit (Qiagen). RNA quality and quantity were evaluated with a 2100 Bioanalyzer (Agilent). Sequencing was performed on an Illumina Hi-Sequation 2000, producing at least 12.4 million 100-bp paired-end (n = 2 per vector) or 100-bp single-end (n = 1 per vector) reads per sample. Reads were mapped to the mm10 mouse genome (Genome Reference Consortium GRCm38, December 2011) using the Subjunc aligner in the Subread package.19 Read counts were summarized at the gene level by featureCounts20 using National Center for Biotechnology Information RefSeq gene annotation. Differential expression analysis used the edgeR21 and limma22 software packages. Genes were filtered as not expressed if they failed to achieve at least 0.5 counts per million reads in at least half the samples. All Entrez gene identifiers without an official symbol were removed from further analysis leaving 12 493 genes for downstream analysis. Library sizes were normalized using the trimmed mean of M values method.23 The voom function of the limma package was used to convert read counts to log2 counts per million with associated precision weights.24 Differential expression was assessed using empirical Bayes moderated t statistics.25 Metascape analysis26 was performed using the express analysis conversion annotation membership enrichment workflow (www.metascape.org).
Results
Overexpression of Hhex leads to promyelocyte self-renewal and immortalization in vitro
We have previously shown that Hhex is overexpressed in human AML and promotes self-renewal of leukemic stem cells in a mouse model of MLL-ENL–induced AML.15 To study the effect of Hhex overexpression on myeloid differentiation, immature LSK hematopoietic progenitors were sorted and transduced with the MIG retroviral vector overexpressing Hhex (MIG-Hhex) or an empty-vector control (MIG). Retrovirally transduced (GFP+) cells were sorted and serially replated in methylcellulose culture in vitro in the presence of IL-3, SCF, and EPO. Although nontransduced LSK cells, and those transduced with the MIG empty vector, failed to form colonies beyond the third round of plating, transduction with MIG-Hhex allowed serial replating of colonies with a highly diffuse appearance (Figure 1A-C). Microscopic examination of cytocentrifuge preparations showed that progenitors in control cultures had undergone differentiation to granulocytes, macrophages, and mast cells within 14 days (Figure 1D). In contrast, Hhex overexpression resulted in a developmental block and subsequent expansion of promyelocytic myeloid progenitors (Figure 1D), which, upon transfer to liquid culture in IL-3 after 4 weeks, gave rise to immortalized cell lines that were dependent on IL-3 or granulocyte-macrophage colony-stimulating factor for growth (supplemental Figure 1). These MIG-Hhex–induced lines (hereafter termed MH lines) retained a promyelocytic appearance, expressed high levels of GFP, and were positive for the myeloid marker Mac-1 but negative for Kit and Gr-1 as expected for promyelocytes (Figure 1E-F). Thus, Hhex overexpression results in a myeloid differentiation block at the promyelocyte stage and rapid establishment of factor-dependent promyelocytic cell lines.
Figure 1.

Hhex induces self-renewal of promyelocytes. (A) Wild-type LSK cells were retrovirally transduced with empty (MIG) or Hhex-encoding retroviruses, cultured in methylcellulose in the presence of IL-3, SCF, and EPO, and colony counts determined weekly. (B) Cumulative cell counts of methylcellulose cultures as in panel A. (C) Representative fields from week 4 cultures of MIG and MIG-Hhex–transduced LSK cells (original magnification ×400). (D) Cells harvested from methylcellulose cultures at the indicated round of plating were cytocentrifuged, stained with May-Grünwald-Giemsa, and the proportion of each cell type was determined by morphological examination. Data are representative of 3 separate experiments. (E) May-Grünwald-Giemsa–stained cytocentrifuge preparation of week 4 Hhex-transformed promyelocytes (original magnification ×400). (F) Fluorescence-activated cell sorter (FACS) plots of phenotypic markers (Mac-1, Kit, and Gr-1) and GFP expression in a representative Hhex-induced promyelocyte cell line. E, erythroblast; G, neutrophilic granulocyte; M, monocyte/macrophage; pro, promyelocyte.
Promyelocyte transformation requires nuclear localization of Hhex
As Hhex is a transcription factor, we hypothesized that nuclear localization should be required for promyelocyte transformation. To test this, we constructed the MIG-HhexERT2 retrovirus, which expresses the full-length Hhex complementary DNA fused in-frame with the tamoxifen-controllable ERT2 domain,27 which confers cytoplasmic localization in the absence of tamoxifen but nuclear localization in its presence. This was used to infect mouse Lin− BM cells, which were then placed in liquid culture in the presence or absence of tamoxifen. As expected, overexpression of Hhex in the absence of the ERT2 fusion domain led to continuous growth of Mac1+GFP+ myeloid progenitors and the establishment of promyelocyte lines both in the presence and absence of tamoxifen (Figure 2). In contrast, the Hhex-ERT2 fusion protein could only promote myeloid growth and immortalization in the presence of tamoxifen, indicating that Hhex requires nuclear localization to transform myeloid progenitors (Figure 2).
Figure 2.

Promyelocyte self-renewal requires nuclear localized Hhex. (A) Growth curves showing expansion of virally transduced (GFP+) lineage marker–negative BM cells following infection with control virus (MIG) or viruses encoding Hhex, the ERT2 domain, or a Hhex-ERT2 fusion cultured in the presence of IL-3 with or without tamoxifen (Tam) to induce nuclear localization of ERT2. Data are representative of 3 separate experiments. (B) FACS analysis showing expression of Mac-1 and GFP of liquid cultures as in panel A at day 21 of culture.
To determine whether sustained Hhex localization is required for maintenance of Hhex-induced cell lines, Hhex-ERT2–transformed lines were washed and cultured in the absence of tamoxifen. This caused a growth arrest from day 2 of culture (Figure 3A), accompanied by a loss of cell viability (Figure 3B) and morphological granulocyte-macrophage maturation accompanied by an increase in the myeloid differentiation marker Gr-1 (Figure 3C-D). Thus, Hhex-transformed cell lines require continuous Hhex function in the nucleus to enable sustained growth and blocked differentiation.
Figure 3.

Promyelocyte self-renewal requires continuous nuclear Hhex expression. (A) Growth curves showing expansion of promyelocytic cell lines expressing Hhex or HhexERT2 fusion protein in the presence or absence of tamoxifen (Tam). (B) Viability of HhexERT2 cell lines cultured as in panel A, as assessed by exclusion of Fluoro-Gold. (C) Induction of the myeloid differentiation marker Gr-1 on a HhexERT2 line (as in panel A) upon culture without tamoxifen (-Tam). (D) Morphology of HhexERT2-transformed promyelocytic cell line cultured in the presence or absence of tamoxifen for 7 days. Cells were stained with Wright-Giemsa (original magnification ×400). Data are representative of 2 separate experiments. Max, maximum.
Structure-function analysis defines multiple domains required for Hhex-induced myeloid differentiation block
To determine the functional domains of Hhex required for myeloid transformation, we generated retroviral constructs containing mutant forms of Hhex (Figure 4A). These included several truncations of the N-terminal–repressive domain including regions critical for interaction with eukaryotic initiation factor 4E (eIF-4E) (amino acids 18-24)28 and the promyelocytic leukemia protein (Pml) (amino acids 50-115),29 a point mutation that abolishes interaction with the corepressor protein Groucho/TLE (F32E),30,31 and constructs that lack the homeodomain (ΔHD), abolish DNA binding (N194A),32 or lack the C-terminal activation region required for transcriptional activation by Hhex (ΔCT)33 (Figure 4A).
Figure 4.

Structure-function analysis reveals the importance of DNA binding and an N-terminal–repressive domain in promyelocytic transformation. (A) Design of Hhex mutants tested in differentiation assay and summary results. (B) LSK cells were infected with retroviruses expressing the indicated Hhex mutants (as in panel A) and cultured in myeloid growth conditions (IMDM with 10% fetal bovine serum [FBS], 3 U/mL EPO, 10 ng/mL IL-3, and 25 ng/mL SCF). Cytocentrifuge preparations were taken at weekly intervals and the percentage of blast/promyelocyte (Blast/pro), neutrophil (G), monocyte (M), basophil (Ba), and erythroid (E) cells was determined following Wright-Giemsa staining and microscopic examination. Data are representative of 3 separate experiments. (C) Pml is not required for myeloid differentiation block by Hhex. Wild-type and Pml-knockout LSK cells were retrovirally transduced with MIG or Hhex and cultured in myeloid growth conditions as in panel B. Cytocentrifuge preparations were taken at weekly intervals and the percentage of cell types determined by microscopic analysis as in panel B. CT, C-terminus; Gro, groucho; HD, homeodomain; NT, N-terminus; TLE, transducin-like enhancer.
To test the ability of mutant Hhex proteins to block myeloid differentiation, the retroviral constructs described in the previous paragraph were used to infect LSK cells that were then re-sorted and cultured in vitro in the presence of IL-3, SCF, and EPO. This showed that the N’-terminal 32 amino acids, the TLE/Gro-interaction residue F32, and the C-terminal acidic activation region were dispensable for Hhex transformation (Figure 4A-B). However, the region between residues 24 and 115, which encompasses the Pml interaction domain, the homeodomain, and the critical DNA-interacting residue N194 were essential for myeloid transformation. Thus, Hhex’s ability to block myeloid differentiation requires the ability to bind DNA as well as the homeodomain, and a critical N-terminal region encompassed by amino acids 24-115.
Pml and Cdkn2a are dispensable for Hhex-induced promyelocyte transformation
The structure-function studies described in the previous section identified an important role for an N-terminal region encompassing the Pml-interacting region (residues 24-115) in the Hhex-mediated myeloid differentiation block. As disruption of Pml nuclear bodies has been shown to be important for the pathogenesis of acute promyelocytic leukemia (APL) driven by the PML-RARα fusion oncogene encoded by the t(15;17) translocation, and previous studies have proposed that the interaction between Hhex and Pml may enforce a differentiation block in this disease,29 we sought to determine whether Hhex may block myeloid differentiation via a direct interaction with Pml.
To assess this, LSK cells from Pml-knockout mice16 were transduced with MIG-Hhex to assess the effects of Hhex on myeloid differentiation in the absence of Pml. This showed that Pml-knockout LSK cells were capable of terminal differentiation into granulocytes, macrophages, and mast cells with similar kinetics to control LSK cells (Figure 4C). Moreover, Hhex overexpression was capable of inducing a block in myeloid differentiation of Pml-knockout cells to a similar extent to wild-type cells, and readily generated promyelocytic cell lines (Figure 4C and data not shown). Thus, the ability of Hhex to block promyelocyte differentiation and induce myeloid transformation is independent of its interaction with Pml.
Our previous studies have indicated that Hhex promotes myeloid leukemia in MLL-ENL– or Hoxa9/Meis1-driven models by repressing the p16Ink4a and p19Arf tumor suppressors encoded by the Cdkn2a locus, with repression of p19Arf being particularly important in maintaining leukemic growth.15 Moreover, a recent study has reported that loss of Cdkn2a can promote self-renewal of myeloid progenitors.34 Therefore, we tested whether deletion of Cdkn2a enhances the self-renewal of myeloid progenitors both in the presence and absence of Hhex overexpression. To test this, we purified lineage-depleted BM cells from mice lacking either p19Arf alone, or both p16Ink4a and p19Arf.17,18 These were transduced with control (MIG) or MIG-Hhex retroviruses and cultured in vitro in the presence of IL-3. We found that Lin− progenitors lacking both p16Ink4a and p19Arf exhibited growth arrest at day 14 of culture, similarly to wild-type progenitors (Figure 5A). Unexpectedly, progenitors lacking p19Arf showed markedly less proliferation, however, these cultures also arrested by day 14 of culture. In contrast, progenitors of all 3 genotypes were readily transformed by Hhex overexpression, leading to the rapid selection of GFP+Mac1+ promyelocytic cell lines (Figure 5). Together, these data indicate that the ability of Hhex to promote promyelocyte self-renewal is independent of its ability to repress the Cdkn2a-encoded tumor suppressors, p16Ink4a and p19Arf.
Figure 5.

Hhex-induced promyelocyte transformation is independent of p16Ink4aand p19Arf. (A) Growth curves showing expansion of wild-type (WT), Cdkn2a-knockout (p16/19KO), or p19-knockout (p19 KO) Lin− myeloid progenitors, infected with either control (MIG) or Hhex-overexpressing (Hhex) retroviruses and cultured in vitro in the presence of IL-3. (B) FACS analysis of GFP and Mac-1 expression in myeloid cultures as in panel A at day 28 postinfection.
Hhex overexpression represses myeloid developmental genes in hematopoietic progenitors
To further understand the molecular basis of the Hhex-induced myeloid differentiation block, we performed RNA-sequencing (RNA Seq) analysis on LSK cells 2 days after retroviral transduction with MIG-Hhex or MIG vector control (n = 3 per group). Differential gene expression analysis revealed that 639 genes were significantly differentially expressed more than twofold, using a false discovery rate cutoff of 0.1 in MIG-Hhex–transduced LSKs, with 431 being upregulated and 208 being repressed (supplemental Table 2). As expected, Hhex expression was significantly elevated more than 10-fold in MIG-Hhex–transduced cells (Figure 6A-B; supplemental Table 2) whereas endogenous 3′ Hhex noncoding sequences were repressed, indicating that overexpression of MIG-Hhex represses transcription from the endogenous Hhex gene (Figure 6A).
Figure 6.

RNA sequencing analysis of Hhex overexpression in LSK cells. (A) RNA Seq trace showing expression of Hhex in LSK cells 2 days postinfection with control (MIG) or Hhex-expressing (MIG-Hhex) retroviruses. Units are reads per million mapped reads (RPM). (B) Waterfall plots showing RNA Seq analysis of the top 20 downregulated genes (left panel) and upregulated genes (right panel) in LSK cells 2 days postinfection with control or Hhex-expressing retroviruses (n = 3). (C-D) Inverse correlation between Hhex-overexpressing and -knockout LSK cells. Genes that are significantly upregulated (C) and downregulated (D) following Hhex deletion in LSK cells in vivo were compared in control (MIG) and Hhex-overexpressing (MIG-Hhex) LSK cells using gene set enrichment analysis. In panel C, the enrichment plot (left) demonstrates that genes upregulated in Hhex-knockout LSK cells are repressed in Hhex-overexpressing (MIG-Hhex) LSK cells, whereas the heat map (right) shows the relative expression of the top 30 genes that are both upregulated following Hhex deletion and downregulated following Hhex overexpression. In panel D, the enrichment plot (right) demonstrates that genes downregulated in Hhex-knockout LSK cells are upregulated in Hhex-overexpressing LSK cells, whereas the heat map (right) shows the relative expression of the top 30 genes that are both downregulated following Hhex deletion and upregulated following Hhex overexpression. FDR, false detection rate; NES, normalized enrichment score.
To determine whether Hhex overexpression regulates a similar set of genes to endogenous Hhex, we compared genes that were differentially expressed in MIG-Hhex–transduced LSKs to those differentially expressed following deletion of Hhex in LSK cells in vivo.11 Using gene set enrichment analysis, there was a significant inverse correlation between Hhex loss and Hhex overexpression in LSK cells, with genes upregulated in Hhex-knockout LSK cells being repressed MIG-Hhex–transduced LSK cells; genes repressed in Hhex-knockout LSK cells were overexpressed in MIG-Hhex–transduced LSK cells (Figure 6C-D). These results indicate that Hhex regulates a specific set of target genes that are dispensable for normal myeloid differentiation,9,10 but block myeloid differentiation upon Hhex overexpression.
As Hhex is thought to operate primarily as a transcriptional repressor, we focused on genes that are repressed upon Hhex overexpression. Analysis using Metascape26 revealed significant downregulation of the neutrophil degranulation pathway within the Reactome database (supplemental Figure 2A-B). Accordingly, among the genes most downregulated following Hhex overexpression were those encoding Myeloperoxidase (Mpo), a key component of neutrophilic granules, along with the neutrophil granule protein Ym1 (Chil3) and myeloid inhibitory C-type lectin (Clec12a) (Figure 6B). Together, these data imply that Hhex overexpression impairs myeloid development by repression of key developmental genes.
Hhex cooperates with factor independence to elicit rapid promyelocytic leukemia in mice
As Hhex-induced promyelocyte cell lines require growth factors to survive in vitro, we tested whether overcoming their growth factor requirements would be sufficient to allow these lines to elicit leukemia in vivo. To achieve this, we retrovirally transduced Hhex-overexpressing cell lines with a constitutively active IL-3 receptor β subunit (βcV449E), which we have previously shown to elicit growth factor independence of myeloid cell lines.35 Expression of βcV449E in Hhex-transformed promyelocytic lines readily allowed them to grow continuously in the absence of growth factors (data not shown). We then tested the leukemogenic potential of 2 independent MIG-Hhex βcV449E+ cell lines, along with their factor-dependent MIG-Hhex parental line controls, by injection into sublethally irradiated congenic (Ly5.1) recipient mice. Strikingly, mice injected with MIG-Hhex βcV449E+ cell lines all succumbed to disease within 50 days, unlike mice injected with the parental factor-dependent MIG-Hhex cell lines, which remained healthy (Figure 7A). Autopsies of MIG-Hhex βcV449E+ injected mice at the time of illness revealed development of promyelocytic leukemia, as evident from elevated white blood cell counts, accumulation of GFP+ cells in the peripheral blood, with infiltration of spleen and BM, accompanied by anemia and thrombocytopenia (Figure 7B-C). Histological sections of spleen, liver, and lungs from these mice showed infiltration of these organs by promyelocytes (Figure 7D); flow cytometric analysis of peripheral blood, spleen, and BM showed that the majority of cells were donor-derived (CD45.2+) myeloid cells (Mac-1+) expressing Hhex retrovirus (GFP+) (supplemental Figure 3A-C). Together, these data indicate that Hhex overexpression combined with growth factor independence is sufficient to elicit rapid promyelocytic leukemia in vivo.
Figure 7.

Growth factor independence combines with Hhex overexpression to elicit lethal promyelocytic leukemia in vivo. (A) Survival of irradiated congenic (Ly5.1) recipient mice transplanted with factor-dependent or factor-independent (V449E-expressing) MH8 and MH9 cell lines. (B) Peripheral blood (PB) profiles of leukemic mice injected with factor-independent lines (+V449E) vs control mice injected with factor-dependent lines (control) at time of sacrifice. (C) Spleen weight, splenocyte counts, and percentage of GFP+ splenocytes and BM cells in leukemic and control mice as in panel B. (D) Histological sections stained with hematoxylin and eosin showing myeloid infiltration in spleen, liver, and lung of mice transplanted with factor-independent cell lines (MH8-V449E, MH9-V449E) but not factor-dependent control (MH8). Images are at ×4 (spleen) and ×10 magnification (liver, lung). Insets are images of the same tissues at ×100 magnification.
Discussion
Hematopoietic development requires the coordinated regulation of transcription factors to enable lineage specification, whereas the downregulation of stem cell–associated transcription factors is critical for differentiation and to prevent leukemia. Here, we have examined the effect of sustained overexpression of the stem-cell–associated transcription factor Hhex on myeloid differentiation. We found that sustained Hhex expression enables serial replating of myeloid progenitors and rapid development of myeloid cell lines. Upon becoming growth factor independent, these lines could cause rapid promyelocytic leukemia in mice. Together with our recent studies demonstrating that Hhex is overexpressed in human AML and is required to repress the Cdkn2a tumor suppressor locus to enable AML initiation and maintenance,15 this indicates that Hhex overexpression can contribute to myeloid leukemia by multiple mechanisms, including epigenetic repression of tumor suppressors and differentiation blockade.
Of note was the mature stage of differentiation of Hhex-induced cell lines. Along with their promyelocytic morphology, these cells showed a Mac1+Gr1− surface profile and high motility in semisolid cultures suggesting developmental arrest at a more mature stage than cells transformed by other homeobox transcription factors such as Hoxa9.36,37 Our previous work has shown that the requirement for Hhex in MLL-ENL–driven AML is dependent on Cdkn2a, indicating that Hhex is not essential for the differentiation block conferred by this fusion oncoprotein.15 However, Hoxa genes along with their cofactor Pbx3 remain overexpressed in the absence of Hhex,15 suggesting that these homeobox transcription factors may compensate for Hhex in blocking myeloid differentiation. Thus, although a requirement for Hhex to repress Cdkn2a may be a general feature of AML, its importance in blocking differentiation is likely to be most relevant in cases lacking overexpression of HOXA genes.
Our biochemical studies showed that continuous nuclear localization is required for Hhex to transform promyelocytes, suggesting that it does so via transcriptional regulation. Notably, in humans, HHEX is a target of a rare chromosomal translocation [t(10;11)(q23;p15)] that results in the generation of a NUP98-HHEX fusion oncogene in AML.14 However, in mouse models, this fusion protein leads to expansion of immature Kit+ cells and transcriptional activation of a number of target genes that are in common with other NUP98 homeobox fusion proteins, including Hoxa5, Hoxa9, Flt3, and Pbx3. In contrast, our studies show that Hhex leads to immortalization at a later stage of myeloid differentiation and appears to deregulate a distinct gene expression program, suggesting a different leukemic mechanism from that of NUP98-HHEX. Nevertheless, these studies suggest that Hhex overexpression may play a more widespread role in myeloid leukemia than previously appreciated.
In accordance with its established role as a transcriptional repressor, an Hhex mutant lacking the homeodomain, or a point mutant with abolished DNA-binding activity, were unable to block myeloid differentiation. In addition, a separate N-terminal domain was critical for this function. Although it encompassed a region shown previously to bind the Pml,29 Hhex was still able to transform promyelocytes from Pml-knockout mice, indicating that this interaction is dispensable for Hhex’s ability to block differentiation and suggesting other important interacting partners at the N-terminal region. Nevertheless, as HHEX expression is elevated in APL,15 HHEX and PML have been shown to associate in APL cells,29 and HHEX blocks differentiation at a promyelocytic stage resembling the phenotype of APL blasts; it remains possible that this association enforces HHEX function to promote the promyelocytic differentiation block seen in this disease.
Hhex has previously been shown to be a T-cell oncogene in mice, and our previous studies have shown that Hhex can block T-cell differentiation and cause thymocyte self-renewal.12,13 Accordingly, Hhex expression is strongly associated with early T-cell precursor-like acute lymphoblastic leukemia in humans.38 Our present results suggest that Hhex overexpression may block differentiation and promote self-renewal in myeloid and T-cell leukemias by similar mechanisms.
Upon being rendered growth factor independent via constitutively active IL-3 receptor βc, Hhex-induced cell lines caused rapid leukemia in mice. Although the βc mutant used has previously been shown to be capable of leukemogenesis when expressed in a mouse BM transplantation model,39 this occurred rarely (in 5 of 17 mice) and with long latency (∼20 weeks), suggesting a requirement for cooperating mutations in leukemogenesis. This is in contrast to this study, in which all mice succumbed within 7 weeks, indicating synergy between activated βc and Hhex in eliciting leukemia.
Although our previous studies have identified that HHEX is overexpressed in a variety of AML subtypes including those with MLL, RUNX1, CBFB-MYH11, and PML-RARA chromosomal translocations,15 the molecular mechanisms mediating its upregulation are presently unclear. Notably, Hhex has been shown to be regulated by an intronic enhancer by a number of oncogenic transcription factors including Erg, Fli1, Gata2, Tal1, and Lmo2,40-42 which are frequently deregulated in leukemia. Moreover, a recent study has identified that HHEX is particularly highly expressed in AML with FLT3–internal tandem duplication (ITD).43 This study also found that HHEX is a transcriptional target of RUNX1 in FLT3-ITD–associated AML and cooperates with FLT3-ITD to induce myeloid leukemia in vivo. These data validate our finding that Hhex overexpression cooperates with growth factor signaling to elicit myeloid leukemia, and suggest that this process also occurs in certain subsets of human AML. However, as HHEX overexpression is not confined to FLT3-ITD+ AML, it is likely that similar mechanisms converge on Hhex overexpression as a mediator of differentiation blockade. Future studies should aim to identify critical targets and protein interactions required for Hhex to both suppress Cdkn2a expression and block myeloid differentiation in AML. As Hhex is dispensable for HSC function in the resting state, inhibiting these processes via interfering with Hhex function should lead to effective and nontoxic treatments for this debilitating disease.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Matt Ritchie for bioinformatics analysis, Ross Dickins for the p19Arf-knockout mice, Jason Corbin and Jasmine McManus for blood analysis, the Alfred Medical Research and Education Precinct (AMREP) flow cytometry facility, and AMREP animal services. The authors thank Tom Gonda for the RUFneo-βcV449E retroviral construct and Markus Müschen for the MSCV-ERT2-GFP construct. The authors acknowledge P. P. Pandolfi for generating the PML-knockout mice.
This work was supported by project grants (628386, 1003391 [M.P.M.], 1126772 [B.J.S. and M.P.M.]) and the Independent Research Institute’s Infrastructure Support Scheme from the Australian Government’s National Health and Medical Research Council, a Future Fellowship from the Australian Research Council (M.P.M.), and a Victorian State Government Operational Infrastructure Support grant.
Footnotes
The data reported in this article have been deposited in Gene Expression Omnibus database (accession number GSE109793).
Authorship
Contribution: J.T.J. and B.J.S. performed research; J.T.J., S.H., Y.H., and M.P.M. designed research; J.T.J., M.P.M., and A.P.N. analyzed data; B.J.S., S.H., and Y.H. contributed vital new reagents; and J.T.J. and M.P.M. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Matthew P. McCormack, Australian Centre for Blood Diseases, Monash University, Level 1, AMREP Building, 75 Commercial Rd, Melbourne, VIC 3004, Australia; e-mail: matthew.mccormack@monash.edu.
References
- 1.Soufi A, Jayaraman PS. PRH/Hex: an oligomeric transcription factor and multifunctional regulator of cell fate. Biochem J. 2008;412(3):399-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaston K, Tsitsilianos MA, Wadey K, Jayaraman PS. Misregulation of the proline rich homeodomain (PRH/HHEX) protein in cancer cells and its consequences for tumour growth and invasion. Cell Biosci. 2016;6(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crompton MR, Bartlett TJ, MacGregor AD, et al. . Identification of a novel vertebrate homeobox gene expressed in haematopoietic cells. Nucleic Acids Res. 1992;20(21):5661-5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bedford FK, Ashworth A, Enver T, Wiedemann LM. HEX: a novel homeobox gene expressed during haematopoiesis and conserved between mouse and human. Nucleic Acids Res. 1993;21(5):1245-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hromas R, Radich J, Collins S. PCR cloning of an orphan homeobox gene (PRH) preferentially expressed in myeloid and liver cells. Biochem Biophys Res Commun. 1993;195(2):976-983. [DOI] [PubMed] [Google Scholar]
- 6.Martinez Barbera JP, Clements M, Thomas P, et al. . The homeobox gene Hex is required in definitive endodermal tissues for normal forebrain, liver and thyroid formation. Development. 2000;127(11):2433-2445. [DOI] [PubMed] [Google Scholar]
- 7.Keng VW, Yagi H, Ikawa M, et al. . Homeobox gene Hex is essential for onset of mouse embryonic liver development and differentiation of the monocyte lineage. Biochem Biophys Res Commun. 2000;276(3):1155-1161. [DOI] [PubMed] [Google Scholar]
- 8.Hallaq H, Pinter E, Enciso J, et al. . A null mutation of Hhex results in abnormal cardiac development, defective vasculogenesis and elevated Vegfa levels. Development. 2004;131(20):5197-5209. [DOI] [PubMed] [Google Scholar]
- 9.Jackson JT, Nasa C, Shi W, et al. . A crucial role for the homeodomain transcription factor Hhex in lymphopoiesis. Blood. 2015;125(5):803-814. [DOI] [PubMed] [Google Scholar]
- 10.Goodings C, Smith E, Mathias E, et al. . Hhex is required at multiple stages of adult hematopoietic stem and progenitor cell differentiation. Stem Cells. 2015;33(8):2628-2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackson JT, Shields BJ, Shi W, et al. . Hhex regulates hematopoietic stem cell self-renewal and stress hematopoiesis via repression of Cdkn2a. Stem Cells. 2017;35(8):1948-1957. [DOI] [PubMed] [Google Scholar]
- 12.George A, Morse HC III, Justice MJ. The homeobox gene Hex induces T-cell-derived lymphomas when overexpressed in hematopoietic precursor cells. Oncogene. 2003;22(43):6764-6773. [DOI] [PubMed] [Google Scholar]
- 13.McCormack MP, Young LF, Vasudevan S, et al. . The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self-renewal. Science. 2010;327(5967):879-883. [DOI] [PubMed] [Google Scholar]
- 14.Jankovic D, Gorello P, Liu T, et al. . Leukemogenic mechanisms and targets of a NUP98/HHEX fusion in acute myeloid leukemia. Blood. 2008;111(12):5672-5682. [DOI] [PubMed] [Google Scholar]
- 15.Shields BJ, Jackson JT, Metcalf D, et al. . Acute myeloid leukemia requires Hhex to enable PRC2-mediated epigenetic repression of Cdkn2a. Genes Dev. 2016;30(1):78-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang ZG, Delva L, Gaboli M, et al. . Role of PML in cell growth and the retinoic acid pathway. Science. 1998;279(5356):1547-1551. [DOI] [PubMed] [Google Scholar]
- 17.Kamijo T, Zindy F, Roussel MF, et al. . Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91(5):649-659. [DOI] [PubMed] [Google Scholar]
- 18.Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85(1):27-37. [DOI] [PubMed] [Google Scholar]
- 19.Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41(10):e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923-930. [DOI] [PubMed] [Google Scholar]
- 21.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ritchie ME, Phipson B, Wu D, et al. . limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. [DOI] [PubMed] [Google Scholar]
- 26.Tripathi S, Pohl MO, Zhou Y, et al. . Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe. 2015;18(6):723-735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feil R, Brocard J, Mascrez B, LeMeur M, Metzger D, Chambon P. Ligand-activated site-specific recombination in mice. Proc Natl Acad Sci USA. 1996;93(20):10887-10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Topisirovic I, Guzman ML, McConnell MJ, et al. . Aberrant eukaryotic translation initiation factor 4E-dependent mRNA transport impedes hematopoietic differentiation and contributes to leukemogenesis. Mol Cell Biol. 2003;23(24):8992-9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Topcu Z, Mack DL, Hromas RA, Borden KL. The promyelocytic leukemia protein PML interacts with the proline-rich homeodomain protein PRH: a RING may link hematopoiesis and growth control. Oncogene. 1999;18(50):7091-7100. [DOI] [PubMed] [Google Scholar]
- 30.Swingler TE, Bess KL, Yao J, Stifani S, Jayaraman PS. The proline-rich homeodomain protein recruits members of the Groucho/Transducin-like enhancer of split protein family to co-repress transcription in hematopoietic cells. J Biol Chem. 2004;279(33):34938-34947. [DOI] [PubMed] [Google Scholar]
- 31.Desjobert C, Noy P, Swingler T, Williams H, Gaston K, Jayaraman PS. The PRH/Hex repressor protein causes nuclear retention of Groucho/TLE co-repressors. Biochem J. 2009;417(1):121-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guiral M, Bess K, Goodwin G, Jayaraman PS. PRH represses transcription in hematopoietic cells by at least two independent mechanisms. J Biol Chem. 2001;276(4):2961-2970. [DOI] [PubMed] [Google Scholar]
- 33.Kasamatsu S, Sato A, Yamamoto T, et al. . Identification of the transactivating region of the homeodomain protein, hex. J Biochem. 2004;135(2):217-223. [DOI] [PubMed] [Google Scholar]
- 34.Rossi A, Ferrari KJ, Piunti A, et al. . Maintenance of leukemic cell identity by the activity of the Polycomb complex PRC1 in mice. Sci Adv. 2016;2(10):e1600972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCormack MP, Gonda TJ. Novel murine myeloid cell lines that exhibit a differentiation switch in response to IL-3 or GM-CSF, or to different constitutively active mutants of the GM-CSF receptor beta subunit. Blood. 2000;95(1):120-127. [PubMed] [Google Scholar]
- 36.Schnabel CA, Jacobs Y, Cleary ML. HoxA9-mediated immortalization of myeloid progenitors requires functional interactions with TALE cofactors Pbx and Meis. Oncogene. 2000;19(5):608-616. [DOI] [PubMed] [Google Scholar]
- 37.Bach C, Buhl S, Mueller D, García-Cuéllar MP, Maethner E, Slany RK. Leukemogenic transformation by HOXA cluster genes. Blood. 2010;115(14):2910-2918. [DOI] [PubMed] [Google Scholar]
- 38.McCormack MP, Shields BJ, Jackson JT, et al. . Requirement for Lyl1 in a model of Lmo2-driven early T-cell precursor ALL. Blood. 2013;122(12):2093-2103. [DOI] [PubMed] [Google Scholar]
- 39.McCormack MP, Gonda TJ. Myeloproliferative disorder and leukaemia in mice induced by different classes of constitutive mutants of the human IL-3/IL-5/GM-CSF receptor common beta subunit. Oncogene. 1999;18(51):7190-7199. [DOI] [PubMed] [Google Scholar]
- 40.Donaldson IJ, Chapman M, Kinston S, et al. . Genome-wide identification of cis-regulatory sequences controlling blood and endothelial development. Hum Mol Genet. 2005;14(5):595-601. [DOI] [PubMed] [Google Scholar]
- 41.Oram SH, Thoms JA, Pridans C, et al. . A previously unrecognized promoter of LMO2 forms part of a transcriptional regulatory circuit mediating LMO2 expression in a subset of T-acute lymphoblastic leukaemia patients. Oncogene. 2010;29(43):5796-5808. [DOI] [PubMed] [Google Scholar]
- 42.Migueles RP, Shaw L, Rodrigues NP, et al. . Transcriptional regulation of Hhex in hematopoiesis and hematopoietic stem cell ontogeny. Dev Biol. 2017;424(2):236-245. [DOI] [PubMed] [Google Scholar]
- 43.Behrens K, Maul K, Tekin N, et al. . RUNX1 cooperates with FLT3-ITD to induce leukemia. J Exp Med. 2017;214(3):737-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.