

Key Points
Mutation patterns in blast phase MPN, including paired sample analysis, point to specific mutations with potential pathogenetic relevance.
RUNX1 mutations predict inferior survival in blast phase MPN, independent of specific treatment strategies.
Abstract
Among 248 consecutive patients with blast phase myeloproliferative neoplasm (MPN-BP), DNA collected at the time of blast transformation was available in 75 patients (median age, 66 years; 64% men). MPN-BP followed primary myelofibrosis in 39 patients, essential thrombocythemia in 20 patients, and polycythemia vera in 16 patients. A myeloid neoplasm–relevant 33-gene panel was used for next-generation sequencing. Driver mutation distribution was JAK2 57%, CALR 20%, MPL 9%, and triple-negative 13%. Sixty-four patients (85%) harbored other mutations/variants, including 37% with ≥3 mutations; most frequent were ASXL1 47%, TET2 19%, RUNX1 17%, TP53 16%, EZH2 15%, and SRSF2 13%; relative mutual exclusivity was expressed by TP53, EZH2, LNK, RUNX1, SRSF2, and NRAS/KRAS mutations. Paired chronic-blast phase sample analysis was possible in 19 patients and revealed more frequent blast phase acquisition of ASXL1, EZH2, LNK, TET2, TP53, and PTPN11 mutations/variants. In multivariable analysis, RUNX1 and PTPN11 mutations/variants were associated with shorter survival duration; respective hazard ratios (HRs) (95% confidence interval [CI]) were 2.1 (95% CI, 1.1-3.8) and 3.0 (95% CI, 1.1-6.6). An all-inclusive multivariable analysis confirmed the prognostic relevance of RUNX1 mutations (HR, 1.9; 95% CI, 1.5-5.5) and also showed additional contribution from a treatment strategy that includes transplant or induction of complete or near-complete remission (HR, 0.3; 95% CI, 0.2-0.5). The current study points to specific mutations that might bear pathogenetic relevance for leukemic transformation in MPN and also suggest an adverse survival effect of RUNX1 mutations.
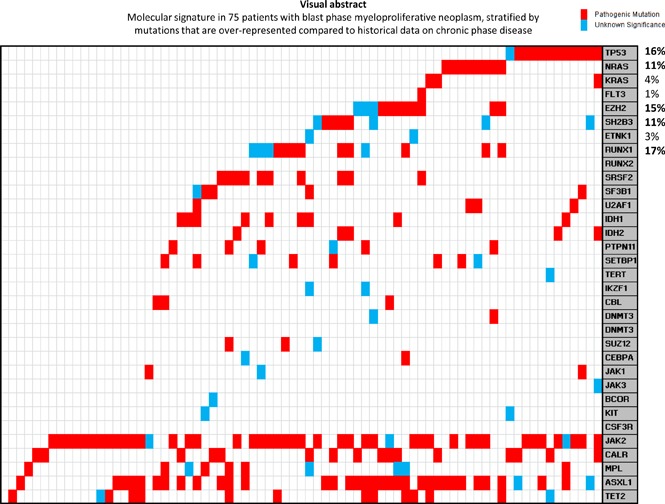
Visual Abstract
Introduction
The morphologic bond between the classic BCR-ABL1–negative myeloproliferative neoplasms (MPNs), namely essential thrombocythemia (ET), polycythemia vera (PV), and primary myelofibrosis (PMF), has recently been strengthened by their common expression of 1 of 3 “driver” mutations, including JAK2, CALR, and MPL.1 This specific molecular profile is also shared by additional morphologic variants of MPNs, including early/prefibrotic PMF (pre-PMF) and “MPN, unclassifiable (MPN-U),”2-4 as well as advanced cases of ET or PV that develop phenotypic resemblance to that of PMF and are respectively referred to as “post-ET” and “post-PV myelofibrosis.”5 All of these MPN variants are at risk for disease progression into acute myeloid leukemia (AML), also known as blast phase MPN (MPN-BP); current diagnostic criteria for MPN-BP are in accordance with those used for AML and include the presence of ≥20% blasts in the peripheral blood or bone marrow (BM).1 The risk of leukemic transformation in MPNs is variable and dependent on the specific MPN variant; 10-year estimates range from <1% in ET to ∼20% in PMF.6,7 Furthermore, the presence of certain clinical or genetic variables has been shown to predict leukemic transformation in PMF (eg, unfavorable karyotype; thrombocytopenia; and ASXL1, SRSF2, IDH1/2, RUNX1, CEBPA, and LNK mutations),8-10 in PV (eg, leukocytosis; older age; abnormal karyotype; treatment with radiophosphorus, chlorambucil, or pipobroman; SRSF2 and IDH2 mutations),11-14 and in ET (eg, pre-PMF morphologic status, anemia, extreme thrombocytosis, older age, leukocytosis, TP53 and EZH2 mutations).14,15
Blast transformation in MPNs is an ominous event without effective therapy.16 In a recent survey of 410 patients with MPN-BP, 2 separate retrospective cohorts from the Mayo Clinic (n = 248) and multiple centers from Italy (n = 162) were considered; median survival time in the Mayo cohort was only 3.6 months with 1-, 3-, and 5-year survival rates of 17%, 6%, and 4%, respectively.17 Median survival time was equally disappointing in the Italian cohort at 3.6 months with 1- and 3-year survival rates of 25% and 11%, respectively.17 In both patient cohorts, outcome had not improved during the last 15 years.17 Also in the particular study, which is the largest of its kind, short-term survival time was significantly better in patients receiving allogeneic hematopoietic stem cell transplant (HCT) or, in the absence of HCT, in patients achieving complete remission (CR) or near-complete remission (CRi); in addition, high-risk karyotype and platelet count <100 × 109/L predicted inferior survival independent of treatment strategy.17 Unfortunately, the benefit of transplantation or achieving CR/CRi was short-lived with respective 5-year survival rates of only 10% and 13%.17 Observations from earlier studies on MPN-BP were also in line with these observations.16 The main objective of the current study was to isolate mutations that are overrepresented in blast-versus-chronic-phase MPNs and those that were more likely to be acquired during blast phase disease; the implication, in this regard, is that such mutations might contribute to the process of leukemic transformation. The second objective for the current study was to examine the effect of mutations on survival after blast transformation and clarify their prognostic interaction with karyotype and other clinical risk factors.
Methods
The current study constitutes a retrospective review of consecutive cases of MPN-BP, where diagnoses of leukemic transformation and the antecedent MPNs were confirmed by both clinical and BM examinations, in line with World Health Organization criteria.1 Study patients were recruited from institutional databases of the Mayo Clinic, Rochester, MN, after approval from the Institutional Review Board. This study was conducted in accordance with the Declaration of Helsinki. Clinical and laboratory data, including cytogenetic information, were collected from patients at the time of leukemic transformation. Cytogenetic analysis and reporting were done according to the International System for Human Cytogenetic Nomenclature criteria.18 Cytogenetic risk stratification was according to the recently revised system that included “very high risk”: single/multiple abnormalities of −7, i(17q), inv(3)/3q21, 12p-/12p11.2, 11q-/11q23, or other autosomal trisomies not including +8/+9 (eg, +21, +19); “favorable”: normal karyotype or sole abnormalities of 13q-, +9, 20q-, chromosome 1 translocation/duplication or sex chromosome abnormality including -Y; and “unfavorable”: all other abnormalities.19 Survival was calculated from time of leukemic transformation, commensurate with time of cytogenetic and mutation analysis and collection of clinical and laboratory parameters examined for impact on survival. Standard statistical methods were used to determine significance of differences among groups in the distribution of continuous or nominal variables. Overall survival data were prepared by the Kaplan-Meier method and compared by the log-rank test. The cox proportional hazard regression model was applied for multivariable analysis. P < .05 was considered significant. The JMP Pro 13.0.0 software (SAS Institute, Cary, NC) was used for all calculations.
Target capture-based next-generation sequencing (NGS) was carried out on BM DNA for the complete coding regions of the following 33 genes: TET2, DNMT3A, DNMT3B, JAK1, JAK3, IDH1, IDH2, ASXL1, EZH2, SUZ12, BCOR, TERT, SRSF2, SF3B1, U2AF1, PTPN11, TP53, SH2B3, RUNX1, RUNX2, CBL, NRAS, KRAS, JAK2, CSF3R, FLT3, KIT, CALR, MPL, CEBPA, IKZF1, ETNK1, and SETBP1, by previously described methods.10 Genome_GPS v4.0.1 (formerly named “TREAT”20) analysis pipeline was used to analyze the initial data. For this, FASTQ files were aligned to the hg19 reference genome using bwa-mem (VN:V7.10) with the default options. Realignment and recalibration was performed using GATK (VN:3.4-46)21 best-practices version 3 for each sample separately. Variant calling was performed using the GATK (VN:3.4-46) Haplotype Caller to generate gVCF file. Identified variants were annotated using the BioR22 framework with functional features, impact prediction, and clinical significance using CAVA, ClinVar, HGMD, and Exome Aggregation Consortium population frequencies. Specific variants were included if they were found at <0.1% by the Exome Aggregation Consortium (Broad Institute, Cambridge, MA). These variants were identified as “Unknown” for unknown significance, unless they had been cited by the Catalogue of Somatic Mutations in Cancer database (http://cancer.sanger.ac.uk), in which case they were labeled “Mutated.” Several variants with >0.1% frequency in the normal population and a fivefold to eightfold increased frequency in our cohort were identified and labeled as “Germline Predisposition.”
Results
Clinical and cytogenetic information at time of leukemic transformation
The study cohort included 75 consecutive patients (median age, 66 years; 64% men) with MPN-BP, including 39 (52%) with post-PMF, 20 (27%) with post-ET, and 16 (21%) with post-PV (Table 1); prior to blast transformation, disease progression to myelofibrosis was documented in 12 of the 20 patients with post-ET AML and in 7 of the 16 patients with post-PV AML (Table 1). BM examination morphology reports included 25 patients (33%) with AML, not otherwise specified (AML-NOS); 24 (32%) with AML with myelodysplasia-related changes; 5 patients (7%) each with AML-NOS-M7, AML-NOS-M0, AML-NOS-M2, and AML-NOS-M4; 4 patients (5%) with AML-NOS-M5; and 1 patient each with AML-NOS-M6 and AML with recurrent favorable cytogenetic abnormalities. Among all evaluable patients, at the time of leukemic transformation, 37% displayed the need for red blood cell transfusion; 58% had a platelet count <100 × 109/L; 38% had a leukocyte count >25 × 109/L; 73% exhibited circulating blasts ≥20%; and 87% displayed BM blasts ≥20% (Table 1). Cytogenetic information was available in 62 cases (83%), of which 21 (34%) were classified as very high risk, 22 (35%) as unfavorable, and 19 (31%) as favorable.
Table 1.
Clinical and laboratory characteristics of 75 patients with blast phase MPNs with targeted gene sequencing information at the time of leukemic transformation, stratified by driver mutations
| Variables | All patients (n = 75) | Patients with JAK2 mutation (n = 43, 57.3%) | Patients with CALR mutation (n = 15, 20%) | Patients with MPL mutation (n = 7, 9.3%) | Patients with triple-negative (n = 10, 13.3%) | P |
|---|---|---|---|---|---|---|
| Age, median (range), y | 66 (44-86) | 66 (44-86) | 63 (46-81) | 72 (52-80) | 69 (61-76) | .5 |
| Age >65 y, n (%) | 40 (53) | 22 (51) | 6 (40) | 5 (71) | 7 (70) | .4 |
| Male sex, n (%) | 48 (64) | 29 (67) | 7 (47) | 3 (43) | 9 (90) | .09 |
| Transfusion dependent, n (%); “N” evaluable = 71 (95%) | 26 (37) | 13 (31) | 3 (25) | 4 (57) | 6 (60) | .2 |
| Hemoglobin, median (range), g/dL; “N” evaluable = 69 (92%) | 9 (5.8-18.1) | 8.9 (5.8-18.1) | 9.1 (7.5-10.7) | 8.6 (7-10.2) | 10.4 (6.6-11.1) | .5 |
| Platelets, median (range), × 109/L; “N” evaluable = 69 (92%) | 73 (4-1246) | 73 (4-1246) | 80 (24-464) | 143 (10-430) | 69 (14-417) | .9 |
| Platelets < 100 × 109/L, n (%); “N” evaluable = 69 (92%) | 40 (58) | 24 (59) | 7 (54) | 3 (50) | 6 (67) | .9 |
| Leukocytes, median (range), × 109/L; “N” evaluable = 69 (92%) | 18.4 (0.8-130.1) | 12.5 (1.3-90.9) | 23.3 (3.7-130.1) | 17.4 (1.4-66) | 18.7 (0.8-57.4) | .7 |
| Leukocytes > 25 × 109/L, n (%); “N” evaluable = 69 (92%) | 26 (38) | 15 (37) | 6 (46) | 2 (33) | 3 (33) | .9 |
| Circulating blasts %, median (range); “N” evaluable = 70 (93%) | 30 (0-89) | 27 (1-89) | 30 (1-75) | 25 (11-46) | 32 (0-74) | .9 |
| Circulating blasts ≥20%, n (%); “N” evaluable = 70 (93%) | 51 (73) | 30 (73) | 10 (71) | 4 (66) | 7 (78) | .9 |
| BM blasts %, median (range); “N” evaluable = 61 (81%) | 33 (2-91) | 33 (2-91) | 25 (3-72) | 36 (10-68) | 45 (22-90) | .4 |
| BM blasts ≥20%, n (%); “N” evaluable = 61 (81%) | 53 (87) | 31 (84) | 10 (91) | 5 (83) | 7 (100) | .7 |
| ANC, median (range), × 109/L; “N” evaluable = 69 (92%) | 4.4 (0.1-38.7) | 4.4 (0.1-38.7) | 8.6 (0.9-38.3) | 4.6 (0.2-13.4) | 2.9 (0.1-23.5) | .5 |
| AMC, median (range), × 109/L; “N” evaluable = 68 (91%) | 0.5 (0-24.3) | 0.4 (0-18.6) | 0.5 (0-24.3) | 1.2 (0.05-18.5) | 1.1 (0.2-7) | .4 |
| MPN variant, n (%) | .0006 | |||||
| Post-PMF AML | 39 (52) | 22 (51) | 5 (33) | 4 (57) | 8 (80) | |
| Post-ET AML | 20 (27) | 6 (14) | 10 (67) | 2 (29) | 2 (20) | |
| Post-PV AML | 16 (21) | 15 (35) | 0 (0) | 1 (14) | 0 (0) | |
| Karyotype, n (%); “N” evaluable = 62 (83%) | .2 | |||||
| Very high-risk karyotype | 21 (34) | 15 (41) | 3 (30) | 1 (17) | 2 (22) | |
| Unfavorable karyotype | 22 (35) | 13 (35) | 5 (50) | 3 (50) | 1 (11) | |
| Favorable karyotype | 19 (31) | 9 (24) | 2 (20) | 2 (33) | 6 (67) | |
| NGS, n (%) | ||||||
| ASXL1 mutated | 35 (47) | 14 (33) | 9 (60) | 5 (71) | 7 (70) | .04 |
| TET2 mutated | 14 (19) | 9 (21) | 1 (7) | 2 (29) | 2 (20) | .6 |
| TP53 mutated | 12 (16) | 7 (16) | 3 (20) | 1 (14) | 1 (10) | .9 |
| SRSF2 mutated | 10 (13) | 7 (16) | 1 (7) | 2 (29) | 0 (0) | .3 |
| RUNX1 mutated | 13 (17) | 11 (26) | 0 (0) | 1 (14) | 1 (10) | .1 |
| EZH2 mutated | 11 (15) | 4 (9) | 4 (27) | 1 (14) | 2 (20) | .4 |
| IDH1 mutated | 9 (12) | 4 (9) | 3 (20) | 2 (29) | 0 (0) | .2 |
| NRAS mutated | 8 (11) | 4 (9) | 0 (0) | 0 (0) | 4 (40) | .008 |
| SETBP1 mutated | 8 (11) | 4 (9) | 2 (13) | 1 (14) | 1 (10) | .9 |
| SH2B3 mutated | 8 (11) | 5 (12) | 1 (7) | 0 (0) | 2 (20) | .6 |
| PTPN11 mutated | 6 (8) | 3 (7) | 1 (7) | 1 (14) | 1 (10) | .9 |
| IDH2 mutated | 5 (7) | 5 (12) | 0 (0) | 0 (0) | 0 (0) | .3 |
| SF3B1 mutated | 5 (7) | 2 (5) | 2 (13) | 1 (14) | 0 (0) | .4 |
| U2AF1 mutated | 4 (5) | 4 (9) | 0 (0) | 0 (0) | 0 (0) | .4 |
| SUZ12 mutated | 3 (4) | 2 (5) | 0 (0) | 1 (14) | 0 (0) | .4 |
| CBL mutated | 3 (4) | 0 (0) | 1 (7) | 1 (14) | 1 (10) | .2 |
| KRAS mutated | 3 (4) | 2 (5) | 1 (7) | 0 (0) | 0 (0) | .8 |
| JAK1 mutated | 3 (4) | 3 (7) | 0 (0) | 0 (0) | 0 (0) | .5 |
| DNMT3A mutated | 2 (3) | 1 (2) | 0 (0) | 0 (0) | 1 (10) | .4 |
| CEBPA mutated | 2 (3) | 0 (0) | 0 (0) | 2 (29) | 0 (0) | .0002 |
| ETNK1 mutated | 2 (3) | 1 (2) | 1 (7) | 0 (0) | 0 (0) | .7 |
| IKZF1 mutated | 2 (3) | 1 (2) | 1 (7) | 0 (0) | 0 (0) | .7 |
| KIT mutated | 2 (3) | 0 (0) | 1 (7) | 1 (14) | 0 (0) | .1 |
| BCOR mutated | 1 (1) | 0 (0) | 1 (7) | 0 (0) | 0 (0) | .3 |
| TERT mutated | 1 (1) | 0 (0) | 1 (7) | 0 (0) | 0 (0) | .3 |
| JAK3 mutated | 1 (1) | 1 (2) | 0 (0) | 0 (0) | 0 (0) | .9 |
| FLT3 mutated | 1 (1) | 1 (2) | 0 (0) | 0 (0) | 0 (0) | .9 |
| Allogeneic SCT, n (%) | 6 (8) | 4 (10) | 1 (7) | 0 (0) | 1 (10) | .8 |
| Follow-up, median (range), mo | 4.1 (0.5-70.7) | 4.7 (0.6-65.7) | 8.1 (0.5-32.5) | 3.7 (0.6-70.7) | 3.4 (0.5-35.3) | .3 |
| Deaths, n (%) | 74 (99) | 42 (98) | 15 (100) | 7 (100) | 10 (100) | .9 |
Post-PV AML patients (n = 16): post-PV AML without myelofibrosis phase (n = 9); post-PV AML with myelofibrosis phase (n = 7). Post ET-AML patients (n = 20): post-ET AML without myelofibrosis phase (n = 8); post-ET AML with myelofibrosis phase (n = 12). No mutations identified in CSF3R, RUNX2, and DNMT3A. Bold indicates significant values.
AMC, absolute monocyte count; ANC, absolute neutrophil count.
Mutation information at time of leukemic transformation and during paired chronic-blast phase sample analysis
Driver mutational status was JAK2 in 43 patients (57%), CALR in 15 (20%), MPL in 7 (9%), and triple-negative in 10 (13%) patients; in addition, 6 patients were dual-mutated “JAK2+ CALR” (n = 2), “JAK2+ MPL” (n = 2), and “CALR+ MPL” (n = 2). Furthermore, among CALR-mutated patients, 67% were type 1/like and 33% type 2/like. Among all DNA changes detected, other than driver mutations, 141 were classified as being “pathogenic” and 33 as “variants of unknown significance (VUS)” (Figure 1). No pathogenic mutations were detected affecting KIT, CSF3R, RUNX2, DNMT3B, TERT, BCOR, JAK3, ETNK1, or IKZF1 genes, whereas only single cases were noted for FLT3, CEBPA, and DNMT3A mutations (Figure 1). The frequencies of pathogenic mutations/VUS are listed in Table 1 and Figure 1 and included the following as the most frequent: ASXL1 47%, TET2 19%, RUNX1 17%, TP53 16%, EZH2 15%, SRSF2 13%, and IDH1 12%; furthermore, as illustrated in Figure 1B, relative mutual exclusivity was demonstrated for TP53, NRAS/KRAS, EZH2, LNK, RUNX1, and SRSF2 mutations/VUS. In general, mutations other than JAK2, CALR, or MPL were detected in 64 (85%) of the 75 study patients; 20 patients (13%) harbored 1 mutation, 25 (33%) harbored 2 mutations, thirteen (17%) harbored 3 mutations, 9 (12%) harbored 4 mutations, 6 (8%) harbored 5 mutations, and 1 patient (1%) harbored 6 mutations. We found no difference in the number of mutations/VUS across different driver mutational states (P = .6), whereas significant associations were noted between CEBPA and MPL (P < .001) and NRAS and triple-negative state (P = .008) (Table 1).
Figure 1.

Molecular signature in 75 patients with blast phase MPN. (A) stratification headed by driver mutations followed by most frequent mutations. (B) Stratification headed by mutations that are overrepresented compared with historical data on chronic phase disease.
Paired samples at both chronic and blast phase disease were available in 19 patients (Figure 2). Mutations with 2 or more instances of acquisition in blast phase disease included TP53, EZH2, LNK, ASXL1, PTPN11, and TET2 (Figure 2). Among the 19 patients with paired samples, both cases of TP53 mutations were detected only during blast phase disease and the percentage of cases with blast phase mutation acquisition was 67% for EZH2, 67% for PTPN11, 60% for LNK, 50% for TET2, and 33% for ASXL1 (Figure 2). In contrast, no blast phase acquisitions were evident for the majority of other mutations, including the JAK2/CALR/MPL driver mutations, SRSF2, and U2AF1 mutations and chronic phase-only mutation detection was generally infrequent; mutations other than JAK2/CALR/MPL were not seen in 6 (32%) of the 19 patients with paired samples, whereas only 1 such mutation was seen in 7 patients (37%). Also as shown in Figure 2, among mutations acquired at the time of blast phase disease, relative mutual exclusivity was once again demonstrated for EZH2 and LNK whereas co-segregation was suggested for ASXL1 and EZH2. Of the driver-mutated cases (JAK2 [n = 12], MPL [n = 1], and CALR [n = 4]), JAK2 allele burden declined by at least 50% (n = 1) or disappeared (n = 1) in 2 cases whereas one of the 4 CALR-mutated cases became homozygous at the time of transformation and another one lost the CALR mutation.
Figure 2.

Molecular signature in paired samples from chronic phase (CP) and blast phase (BP) disease in 19 patients with MPNs.
Overall management strategies and predictors of treatment response
Treatment records were reviewed in detail for all 75 patients and included AML-like induction chemotherapy in 20 patients (32%), hypomethylating agents in 7 (9%), other chemotherapy or clinical trial participation in 15 (20%), and supportive care ± hydroxyurea therapy in 21 (33%). CR or CRi was documented in 14 patients (9 CR and 5 CRi) and occurred only in the setting of AML-like induction (n = 13) or a single case of CRi in a clinical trial. Among the 20 patients receiving AML-like induction chemotherapy, neither the clinical and laboratory variables tested nor cytogenetic risk categories showed significant correlation with the achievement of CR/CRi. In a similar manner, with the caveat that sample size was severely compromised by the small number of informative cases, we were not able to demonstrate significant associations between CR/CRi and driver mutational status (P = .24), individual mutations other than JAK2/CALR/MPL (P > .1 in all instances with the exception of SETBP1 with P = .03), number of mutations (P = .9), or presence or absence of adverse mutations (P = .22; see “Survival analysis and risk factors” for information regarding adverse mutations).
Survival analysis and risk factors
For analysis of the survival effect of mutations/VUS, only those with a minimum of 3 events were considered (Table 1; listed down from ASXL1 to JAK1); accordingly, multivariable analysis that included only mutation information identified RUNX1 (hazard ratio [HR], 2.1; 95% confidence interval [CI], 1.1-3.8) and PTPN11 (HR, 3.0; 95% CI, 1.1-6.6) as independent predictors of inferior survival (Table 2). In univariate analysis, the only other predictor of survival was whether or not patients achieved CR/CRi or received HCT (Table 3). Survival was similar in patients receiving HCT (n = 6) and in those achieving CR/CRi but did not receive HCT afterward (n = 9), and in both instances, it was superior to that of patients who received supportive care only or failed chemotherapy (P < .001). Accordingly, for the purposes of additional all-inclusive multivariable analysis, patients were categorized into 2 treatment groups: (1) patients receiving HCT or achieving CR/CRi but never received transplant (n = 15) and (2) patients who neither received transplant nor achieved CR/CRi (n = 60); the latter group included patients treated by supportive care only and whose outcome was similar to those who did not respond to chemotherapy; HRs were 2.9 (95% CI, 1.5-6.8) for supportive care/chemotherapy failed vs CR/CRi without transplantation, 2.8 (95% CI, 1.3-7.5) for supportive care/chemotherapy failed vs transplant, and 1.0 (95% CI, 0.3-3.0) for transplantation vs CR/CRi without transplantation.
Table 2.
Univariate and multivariable analysis of mutation effect on survival in 75 patients with blast phase MPNs
| Variables | Univariate analysis | Multivariable analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | |
| JAK2 mutated | 1.1 | 0.7-1.8 | .6 | |||
| ASXL1 mutated | 1.2 | 0.7-1.8 | .5 | |||
| CALR mutated | 0.8 | 0.5-1.5 | .5 | |||
| TET2 mutated | 1.7 | 0.9-3.1 | .08 | |||
| TP53 mutated | 1.3 | 0.7-2.4 | .5 | |||
| SRSF2 mutated | 0.8 | 0.4-1.5 | .5 | |||
| RUNX1 mutated | 2.1 | 1.1-3.8 | .02 | 2.1 | 1.1-3.8 | .03 |
| EZH2 mutated | 1.3 | 0.7-2.6 | .4 | |||
| IDH1 mutated | 0.7 | 0.3-1.4 | .3 | |||
| NRAS mutated | 1.6 | 0.7-3.3 | .2 | |||
| SETBP1 mutated | 1.1 | 0.5-2.4 | .8 | |||
| SH2B3 mutated | 1.3 | 0.6-2.7 | .5 | |||
| MPL mutated | 0.7 | 0.3-1.4 | .3 | |||
| PTPN11 mutated | 3.0 | 1.2-7.0 | .01 | 3.0 | 1.1-6.6 | .03 |
| IDH2 mutated | 0.9 | 0.3-2.6 | .9 | |||
| SF3B1 mutated | 0.4 | 0.2-1.0 | .05 | |||
| U2AF1 mutated | 0.6 | 0.2-1.8 | .4 | |||
| SUZ12 mutated | 0.8 | 0.2-2.6 | .7 | |||
| CBL mutated | 2.0 | 0.6-6.5 | .2 | |||
| KRAS mutated | 2.2 | 0.7-6.9 | .2 | |||
| JAK1 mutated | 1.5 | 0.5-4.9 | .5 | |||
Only mutations with at least 3 incident cases are included in the survival analysis. Bold indicates significant values.
Table 3.
Univariate and multivariable overall survival analysis of 75 patients with blast phase myeloproliferative neoplasms
| Variables | Univariate analysis | Multivariable analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | |
| Age, y | .05 | |||||
| Age >65 y | 1.5 | 0.9-2.4 | .09 | |||
| Male sex | 1.3 | 0.8-2.1 | .3 | |||
| Transfusion-dependent; “N” evaluable = 71 (95%) | .14 | 0.8-2.4 | .1 | |||
| Hemoglobin, g/dL; “N” evaluable = 69 (92%) | .5 | |||||
| Platelets, × 109/L; “N” evaluable = 69 (92%) | .4 | |||||
| Platelets < 100 × 109/L; “N” evaluable = 69 (92%) | 1.5 | 0.9-2.5 | .09 | |||
| Leukocytes, × 109/L; “N” evaluable = 69 (92%) | .2 | |||||
| Leukocytes > 25 × 109/L; “N” evaluable = 69 (92%) | 1.4 | 0.8-2.3 | .2 | |||
| Circulating blasts %; “N” evaluable = 70 (93%) | .8 | |||||
| Circulating blasts ≥20%; “N” evaluable = 70 (93%) | 0.9 | 0.6-1.7 | .9 | |||
| BM blasts; “N” evaluable = 61 (81%) | .8 | |||||
| BM blasts ≥20%; “N” evaluable = 61 (81%) | 1.0 | 0.5-2.1 | .9 | |||
| MPN variant | ||||||
| Post-PMF AML | 0.9 | 0.5-1.7 | .8 | |||
| Post-ET AML | 0.9 | 0.4-1.7 | .7 | |||
| Post-PV AML (reference) | — | — | — | |||
| Karyotype; “N” evaluable = 62 (83%) | ||||||
| Very high-risk karyotype | 1.8 | 0.9-3.6 | .07 | |||
| Unfavorable karyotype | 1.2 | 0.6-2.3 | .6 | |||
| Favorable karyotype (reference) | — | — | — | |||
| RUNX1 mutated | 2.1 | 1.1-3.8 | .02 | 2.9 | 1.5-5.5 | <.001 |
| PTPN11 mutated | 3.0 | 1.2-7.0 | .01 | |||
| Received transplant or achieved CR/CRi | 0.3 | 0.2-0.6 | <.001 | 0.3 | 0.2-0.5 | <.001 |
Bold indicates significant values.
Table 3 outlines univariate and multivariable analysis of these 2 prognostically relevant mutations, in the context of other confounding factors, including age, cytogenetic risk, and treatment strategy; the all-inclusive multivariable analysis confirmed the independent adverse effect of RUNX1 mutation (HR, 2.9; 95% CI, 1.5-5.5) and highlighted the previously recognized favorable influence from HCT or achievement of CR/CRi, even if not transplanted (HR, 0.3; 95% CI, 0.2-0.5). Figure 3 shows survival data stratified by both RUNX1 mutations and treatment group (achieved CR/CRi or received transplant vs neither); median survival time (1-year survival rate) was 9 months (38%) for patients without mutation but with documented CR/CRi or HCT, 1.7 months (0%) for patients with mutation but without CR/CRi or HCT (HR, 11.1; 95% CI, 4.4-28.3), 3.8 months (6%) for patients without mutation and without CR/CRi or HCT (HR, 2.7; 95% CI, 1.4-5.4), and 12 months (0%) for patients with mutation and with documented CR/CRi or HCT (HR, 1.0; 95% CI, 0.2-3.9). Survival differences were also significant between patients with mutation but without CR/CRi or HCT and patients without mutation and without CR/CRi or HCT (HR 4.1, 95% CI, 1.9-8.4) and between the latter group and patients without mutation but with documented CR/CRi or HCT (HR, 2.7; 95% CI, 1.4-5.4).
Figure 3.

Survival data in 75 patients with blast phase MPN stratified by RUNX1 mutation and treatment strategy.
Discussion
The dismal prognosis of MPN-BP has recently been reiterated in multiple publications.16,17 Also, it is generally accepted that drug therapy alone is ineffective in securing long-term survival, although patients achieving CR/CRi, with or without HCT consolidation, might have fared better in the short-term, compared with those receiving supportive care only or fail to respond to chemotherapy. In a most recent study, the respective 1-, 3-, and 5-year survival rates in MPN-BP were 66%, 32%, and 10% for patients receiving HCT; 37%, 19%, and 13% for nontransplant patients who otherwise achieved chemotherapy-induced CR/Cri; and 8%, 1%, and 1% in patients receiving supportive care only or failing to respond to chemotherapy.17 The differences in outcome, in the particular study, did not necessarily favor transplantation vs CR/CRi without transplantation, although both resulted in significantly longer survival times, compared with that of patients who failed to respond to chemotherapy or received supportive care only.17 Furthermore, the possibility of CR/CRi appeared to be limited to patients receiving AML-like induction chemotherapy.17 The findings in the current study were consistent with these previously recognized observations. However, it should be noted that patients receiving transplant or AML-like induction chemotherapy were more likely to be younger and display better performance status, which might have contributed to their superior short-term survival. Other studies have suggested more promising value from HCT in MPN-BP16; for example, the 2-year survival rate in a nationwide survey was 29% with median follow-up of 5.5 years for surviving patients,23 whereas a European registry study reported a 3-year survival rate of 33% with additional value from CR at the time of transplantation.24 Regardless, the fact remains that a dire need exists for additional insight into the molecular events that lead to leukemic transformation in MPNs; identification of patients who are likely to live longer or benefit from HCT; and, most importantly, molecular guidance for the development of more effective targeted therapy, especially considering the limited value of JAK2 inhibitors in MPN-BP.25
The current study suggests overrepresentation of RUNX1, TP53, EZH2, LNK, and PTPN11 during blast phase disease; the respective mutational frequencies were 17%, 16%, 15%, 11%, and 8%. The corresponding figures in our previous NGS studies of chronic phase MPN were 4%, 1%, 1%, 6%, and 2% in PMF (n = 182)10; 2%, 1%, 0%, 9%, and 0% in PV (n = 133)14; and 2%, 2%, 3%, 3%, and 2% in ET (n = 183).14 Noteworthy is that paired-sample analysis identified the very same mutations as being frequently acquired during blast phase disease, and the pattern of their distribution during both chronic and blast phase disease suggested mutual exclusivity. Furthermore, we have previously shown significant associations between leukemic transformation and RUNX1, TP53, EZH2, and LNK mutations in PMF and ET. Taken together, these observations suggest that a set of mutations, and not necessarily a single mutation, might be responsible for some instances of leukemic transformation in MPNs, and candidates in this regard might include RUNX1, TP53, EZH2, and LNK. Consistent with these findings, a previous study had reported increased prevalence of TP53 (27%) and PTPN11 (7%) mutations in blast phase MPN, but also of ASXL1 (47%), IDH2 (31%), and SRSF2 (22%) mutations.26 The particular study also showed complete co-segregation of TP53 and JAK2V617F mutations.26 In contrast, the distribution of TP53 mutations across different driver mutational states was similar in the current study: 8% in JAK2, 4% in CALR, 1% in triple-negative cases, and 3% in dual-mutated cases (supplemental Data). The 2 studies also differed in their emphasis of RUNX1 mutations, although such mutations were prevalent in both.26 The study by Rampal et al26 also showed clonal dominance of JAK2/TP53 mutated leukemic cells and induction of AML in Tp53-null mice transduced with JAK2V617F, suggesting pathogenically relevant cooperation between the 2 mutations.
In an even more recent study, Venton et al27 performed NGS in 56 patients with MPN-BP and found TP53 (36% vs 16% in the current study), ASXL1 (25% vs 47%), TET2 (20% vs 19%), SRSF2 (16% vs 13%), DNMT3A (14% vs 3%), NRAS/KRAS (14% vs 15%), IDH1/2 (13% vs 19%), EZH2 (13% vs 15%), and RUNX1 (13% vs 17%) to be the most frequent. In the latter study,27 ASXL1 and SRSF2 mutations were more likely to occur in post-PMF AML and TP53 in post-PV/ET AML; we did not see a similar repartition pattern in the current study (P = .2 for TP53, P = .4 for ASXL1, and P = .4 for SRSF2). As was the case in the current study, and unlike the observation from Rampal et al,26 Venton et al27 also witnessed coexpression of CALR and TP53 mutations.
The prognostic component of our study identified RUNX1 mutations as the main predictor of inferior survival duration, independent of specific treatment strategies, including HCT; the only other independent predictor of survival was whether or not patients received HCT or achieved CR/CRi, although not consolidated with transplantation. Obviously, because of the relatively small number of informative cases, we may have missed similar prognostic relevance from other mutations, including PTPN11 and TET2, which showed borderline significance on univariate analysis; SF3B1 mutations were associated with favorable prognosis, although the borderline significance in this regard was lost during multivariable analysis. In the study by Venton et al,27 multivariable analysis identified TP53, TET2, and SRSF2 mutations to be associated with shorter survival times. The observations from the aforementioned 3 studies, including the current study, were clearly not always concordant and, in some instances, were overtly conflicting. This makes it hard to be certain of the biological or practical implications of the findings from the current study and those of the aforementioned studies,26,27 and we instead favor their consideration as preliminary observations that require further investigation.
Finally, it is evident from this and other studies that the type of mutations frequently seen in primary AML is different than that seen in MPN-BP; mutational frequencies observed in a representative AML study28 vs the current study were FLT3 37% vs 1%, NPM1 29% vs 0%, DNMT3A 23% vs 3%, NRAS 10% vs 11%, CEBPA 9% vs 3%, TET2 8% vs 19%, IDH2 8% vs 7%, IDH1 7% vs 12%, ASXL1 3% vs 47%, RUNX1 5% vs 17%, TP53 2% vs 16%, and EZH2 0% vs 15%, respectively. In primary AML, an adverse prognostic effect has been consistently demonstrated for FLT3 and a favorable effect for NPM1 and CEBPA mutations. Also, in a large study involving 2439 patients with AML, RUNX1 mutations were found in 10% of patients and were associated with older age, a complex mutation cluster, secondary AML arising from myelodysplastic syndrome, and inferior outcome.29 These latter observations are in line with those of the current study and further support the prognostic relevance of RUNX1 mutations in MPN-BP and possibly other secondary AMLs.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported in part by the Mayo Clinic Harvey-Yulman Charitable Foundation for Myelofibrosis Tissue Bank; the Clinical Database of Molecular and Biological Abnormalities; and the Henry J. Predolin Foundation for Research in Leukemia, Mayo Clinic, Rochester, MN.
Authorship
Contribution: A.T. designed and sponsored the study, contributed patients, collected clinical data and patient samples, reviewed the molecular data, performed statistical analysis, and wrote the manuscript; T.L.L. participated in study design, performed and analyzed the molecular analysis, prepared key tables and figures, and participated in writing the manuscript; C.M.F. performed the molecular analysis; M.M. and N.S. participated in data collection, statistical analysis, and preparation of tables; C.A.H. performed pathology review; R.P.K. performed cytogenetics review; K.H.B. and M.M.P. contributed patients; N.G. contributed patients and collected clinical data; and A.P. contributed patients and reviewed the molecular data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ayalew Tefferi, Department of Internal Medicine, Division of Hematology, Mayo Clinic College of Medicine, 200 First St SW, Rochester, MN 55905; e-mail: tefferi.ayalew@mayo.edu.
References
- 1.Arber DA, Orazi A, Hasserjian R, et al. . The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. [DOI] [PubMed] [Google Scholar]
- 2.Kim SY, Im K, Park SN, Kwon J, Kim JA, Lee DS. CALR, JAK2, and MPL mutation profiles in patients with four different subtypes of myeloproliferative neoplasms: primary myelofibrosis, essential thrombocythemia, polycythemia vera, and myeloproliferative neoplasm, unclassifiable. Am J Clin Pathol. 2015;143(5):635-644. [DOI] [PubMed] [Google Scholar]
- 3.Guglielmelli P, Pacilli A, Rotunno G, et al. ; AGIMM Group. Presentation and outcome of patients with 2016 WHO diagnosis of prefibrotic and overt primary myelofibrosis. Blood. 2017;129(24):3227-3236. [DOI] [PubMed] [Google Scholar]
- 4.Mudireddy M, Shah S, Lasho T, et al. . Prefibrotic versus overtly fibrotic primary myelofibrosis: clinical, cytogenetic, molecular and prognostic comparisons [published online ahead of print 5 July 2017]. Br J Haematol. doi:10.1111/bjh.14838. [DOI] [PubMed] [Google Scholar]
- 5.Barosi G, Mesa RA, Thiele J, et al. ; International Working Group for Myelofibrosis Research and Treatment (IWG-MRT). Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia. 2008;22(2):437-438. [DOI] [PubMed] [Google Scholar]
- 6.Cerquozzi S, Tefferi A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J. 2015;5(11):e366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tefferi A, Guglielmelli P, Larson DR, et al. . Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014;124(16):2507-2513, quiz 2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gangat N, Caramazza D, Vaidya R, et al. . DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392-397. [DOI] [PubMed] [Google Scholar]
- 9.Vannucchi AM, Lasho TL, Guglielmelli P, et al. . Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861-1869. [DOI] [PubMed] [Google Scholar]
- 10.Tefferi A, Lasho TL, Finke CM, et al. . Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016;1(2):105-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tefferi A, Rumi E, Finazzi G, et al. . Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27(9):1874-1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kiladjian JJ, Chevret S, Dosquet C, Chomienne C, Rain JD. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol. 2011;29(29):3907-3913. [DOI] [PubMed] [Google Scholar]
- 13.Berk PD, Goldberg JD, Silverstein MN, et al. . Increased incidence of acute leukemia in polycythemia vera associated with chlorambucil therapy. N Engl J Med. 1981;304(8):441-447. [DOI] [PubMed] [Google Scholar]
- 14.Tefferi A, Lasho TL, Guglielmelli P, et al. . Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016;1(1):21-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barbui T, Thiele J, Passamonti F, et al. . Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study. J Clin Oncol. 2011;29(23):3179-3184. [DOI] [PubMed] [Google Scholar]
- 16.Yogarajah M, Tefferi A. Leukemic transformation in myeloproliferative neoplasms: a literature review on risk, characteristics, and outcome. Mayo Clin Proc. 2017;92(7):1118-1128. [DOI] [PubMed] [Google Scholar]
- 17.Tefferi A, Mudireddy M, Mannelli F, et al. . Blast phase myeloproliferative neoplasm: Mayo-AGIMM study of 410 patients from two separate cohorts [published online ahead of print 2 February 2018]. Leukemia. doi:10.1038/s41375-018-0019-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGowan-Jordan J, Simons A, Schmid M ISCN 2016: An International System for Human Cytogenomic Nomenclature (2016). Basel, Switzerland: Karger; 2016. [Google Scholar]
- 19.Tefferi A, Nicolosi M, Mudireddy M, et al. . Revised cytogenetic risk stratification in primary myelofibrosis: analysis based on 1002 informative patients. Leukemia. doi:10.1038/s41375-018-0018-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Asmann YW, Middha S, Hossain A, et al. . TREAT: a bioinformatics tool for variant annotations and visualizations in targeted and exome sequencing data. Bioinformatics. 2012;28(2):277-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKenna A, Hanna M, Banks E, et al. . The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kocher JP, Quest DJ, Duffy P, et al. . The Biological Reference Repository (BioR): a rapid and flexible system for genomics annotation. Bioinformatics. 2014;30(13):1920-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takagi S, Masuoka K, Uchida N, et al. . Allogeneic hematopoietic cell transplantation for leukemic transformation preceded by Philadelphia chromosome-negative myeloproliferative neoplasms: a nationwide survey by the Adult Acute Myeloid Leukemia Working Group of the Japan Society for Hematopoietic Cell Transplantation. Biol Blood Marrow Transplant. 2016;22(12):2208-2213. [DOI] [PubMed] [Google Scholar]
- 24.Alchalby H, Zabelina T, Stübig T, et al. ; Chronic Malignancies Working Party of the European Group for Blood and Marrow Transplantation. Allogeneic stem cell transplantation for myelofibrosis with leukemic transformation: a study from the Myeloproliferative Neoplasm Subcommittee of the CMWP of the European Group for Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2014;20(2):279-281. [DOI] [PubMed] [Google Scholar]
- 25.Eghtedar A, Verstovsek S, Estrov Z, et al. . Phase 2 study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including postmyeloproliferative neoplasm acute myeloid leukemia. Blood. 2012;119(20):4614-4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rampal R, Ahn J, Abdel-Wahab O, et al. . Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc Natl Acad Sci USA. 2014;111(50):E5401-E5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venton G, Courtier F, Charbonnier A, et al. . Impact of gene mutations on treatment response and prognosis of acute myeloid leukemia secondary to myeloproliferative neoplasms. Am J Hematol. 2018;93(3):330-338. [DOI] [PubMed] [Google Scholar]
- 28.Patel JP, Gönen M, Figueroa ME, et al. . Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaidzik VI, Teleanu V, Papaemmanuil E, et al. . RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia. 2016;30(11):2160-2168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.