

Abstract
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disease in the elderly. Zinc (Zn) ion interacts with the pathogenic hallmark, amyloid-β (Aβ), and is enriched in senile plaques in brain of AD patients. To understand Zn-chelated Aβ (ZnAβ) species, here we systematically characterized ZnAβ aggregates by incubating equimolar Aβ with Zn. We found ZnAβ40 and ZnAβ42 both form spherical oligomers with a diameter of ~12–14 nm composed of reduced β-sheet content. Oligomer assembly examined by analytical ultracentrifugation, hydrophobic exposure by BisANS spectra, and immunoreactivity of ZnAβ and Aβ derived diffusible ligands (ADDLs) are distinct. The site-specific 13C labeled solid-state NMR spectra showed that ZnAβ40 adopts β-sheet structure as in Aβ40 fibrils. Interestingly, removal of Zn by EDTA rapidly shifted the equilibrium back to fibrillization pathway with a faster kinetics. Moreover, ZnAβ oligomers have stronger toxicity than ADDLs by cell viability and cytotoxicity assays. The ex vivo study showed that ZnAβ oligomers potently inhibited hippocampal LTP in the wild-type C57BL/6JNarl mice. Finally, we demonstrated that ZnAβ oligomers stimulate hippocampal microglia activation in an acute Aβ-injected model. Overall, our study demonstrates that ZnAβ rapidly form toxic and distinct off-pathway oligomers. The finding provides a potential target for AD therapeutic development.
Introduction
AD is the most common cause of dementia in the elder population after age of 65. All current AD clinical trials have failed due to insignificant beneficial effects or severe adverse effects1,2. The failure of clinical trials suggests that the fundamental molecular mechanism of AD pathogenesis is still not fully understood. Aβ, a pathogenic hallmark in AD, is cleaved from amyloid precursor protein by β- and γ-secretases3,4. Aβ40 and Aβ42 are the two major isoforms that differ in two additional amino acids in the C- terminus of Aβ5,6. Aβ is an intrinsically disordered protein that is prone to aggregate into cross-β-rich fibrils via a nucleation-dependent manner7. The classic amyloid fibrillization consists of a nucleation state followed by fibril elongation and a plateau for mature fibril formation. The major cause of AD is considered to associate with assemble of Aβ into oligomers, which impair synaptic function and lead to activation of a cascade of subsequent detrimental events8,9. Aβ oligomers are previously referred to heterogeneous intermediates in the aggregation, including various types of species, e.g. prefibrillar oligomer, protofibrils, annular protofibrils, paranuclei, globulomers, amylospheroids, ADDLs, and Aβ56*9–12. Despite the intrinsic structural heterogeneity of Aβ oligomeric aggregates, many of their structural features have been unraveled by solid-state NMR13–16. Aβ fibrillization can be monitored by thioflavin T (ThT) that emits fluorescence upon chelating to cross-β-stands in amyloid fibrils, however, the oligomer intermediates showed no or low binding to ThT17.
Although Aβ40 and Aβ42 are the two major Aβ isoforms, they have distinct properties in structure, aggregation, and toxicity. Freshly prepared Aβ40 was reported to be monomer and Aβ42 adopts rapid equilibrium of monomer and trimer/tetramer18. Aβ40 and Aβ42 adopt distinct fibrillization pathways19,20. During the aggregation, Aβ42 forms a pentameric/hexameric paranuclei, whereas, Aβ40 undergoes monomer addition19,20. Ion mobility mass spectrometry showed Aβ40 assembles through tetramer, whereas, Aβ42 forms tetramer and further forms hexamer that stacks into dodecamer prior to protofibril/fibril formation20. The fibril structure of Aβ40 contains two β-strands formed by amino acids 10 to 22 and 30 to 4021–23 and Aβ42 fibrils contain multiple β-sheets adopting the so-called LS-shaped structure24. Aβ42 is demonstrated more detrimental than Aβ40 in vitro and in vivo25,26 despite the amount of Aβ42 is 10 times less than Aβ40 in cerebrospinal fluids27.
Metal ion dyshomeostasis is an existing concept in the AD research28. Abnormally high content of metal ions including Zn2+, Cu2+, Fe3+ have been found in senile plaques of postmortem AD brains29,30. Many evidences further suggested imbalance of Zn2+ or Cu2+ homeostasis was linked with AD pathology31,32 and association of metal ions with Aβ, especially Cu2+ or Zn2+ were reported in a series of studies33–36. Aβ is known to bind with the metal ions31,32. The metal ions, including Zn2+ and Cu2+, were identified to interact with histidine residues at N-terminal of Aβ37,38. The mechanism of interaction of Zn2+ with Aβ was reported to share similar coordination with Cu2+ by three histidines at residue 6, 13, and 14 of Aβ39,40. However, how Zn2+ modulates Aβ conformation is still elusive, that may be due to assemblies of ZnAβ were variable and sensitive due to changes of pH, temperature, concentration, and buffer environment. The morphology of ZnAβ was previously shown as fibrils at 37 °C41, whereas others discovered that ZnAβ formed amorphous aggregates42 or oligomers at room temperature33,35. Postmortem brain analysis and in vivo model of AD both provided evidences to support that Zn2+ might play a role in AD pathologensis43,44. Bush et al. further developed a Cu/Zn ion chelator, PBT2, to provide beneficial effect for AD45,46.
To understand the effect of Zn2+ to Aβ40 and Aβ42, here we systematically characterized the oligomer formed by ZnAβ complex. We found Zn2+ rapidly induced secondary structure changes and retained Aβ in an oligomer state with higher hydrophobic surface. ZnAβ oligomers have distinct immunoreactivity against several anti-Aβ antibodies and possess higher toxicity than ADDLs. Removal of Zn2+ leads to restoration of Aβ fibrillization with a faster kinetics than Aβ alone. In addition, solid-state NMR data suggested that ZnAβ40 oligomers possess β-sheet structure. The electrophysiology result showed that ZnAβ oligomer inhibited hippocampal LTP in the wild-type mice hippocampus slice. In the acute Aβ-injected mice model, increasing of hippocampal microglial activation was found followed by ZnAβ injection.
Results
Zn2+ promotes Aβ40 and Aβ42 oligomer formation
To systematically examine the effect of Zn2+ on Aβ aggregation, we incubated equal molar ratio of Zn2+ ions with Aβ40 and Aβ42 and examined their fibrillization by ThT assay, the secondary structural changes by far-UV CD, and the epitope changes of the assembly by dot blotting. Aβ40 and Aβ42 were prepared in ammonium hydroxide, lyophilized, and then dissolved in 10 mM Tris-HCl buffer, pH 7.4. Fifty µM Aβ and Zn2+ were used in the experiments. In ThT assay, we found that in the absence of Zn2+, Aβ40 fibrillization adapted a classic nucleation-dependent response. It showed a lag phase of ~64 hr, and reached maximal ThT fluorescence at ~94 hr (Fig. 1A). Upon addition of equal molar concentration of Zn2+ during Aβ40 aggregation, we found the ThT signal was retained without much increase. The result showed that there is lower incidence, if any, of amyloid fibril formation in the presence of Zn2+ ions. In the case of Aβ42, Aβ42 alone showed a slow growing phase of ~20 hr, then increased to maximal ThT fluorescence intensity at ~33 hr (Fig. 1F). Whereas, in the presence of Zn2+, ThT fluorescence was significantly inhibited similar to the result obtained from Aβ40. The result demonstrated that Zn2+ suppresses both Aβ40 and Aβ42 fibrillization. We also examined Aβ40 and Aβ42 fibrillization in various concentrations of Zn2+ in ThT assay. Five different concentrations of Zn2+ ranging from 10 to 200 µM were added into the 50 µM Aβ solution. The result showed Zn2+ suppressed Aβ40 and Aβ42 fibrillization in a dose-dependent manner (Fig. S1). To examine the possible precipitation, we examined the turbidity of the samples for 7 days of incubation by absorbance at 340 nm and also quantified the protein concentration at 280 nm. The samples were turbid especially for the ZnAβ samples indicating large soluble aggregate formation, but no precipitation was observed during the experimental time (Fig. S2). To see whether there is any Aβ aggregation in the presence of Zn2+, we imaged the ZnAβ40 and ZnAβ42 aggregates by TEM (Fig. 1B,G, respectively). TEM images showed that in the absence of Zn2+ the end-point products of Aβ40 and Aβ42 both formed straight or curvy amyloid fibrils. In contrast, equal molar ratio of Zn2+ promoted formation of spherical Aβ species, whose diameters in average were ~14 and ~12 nm for ZnAβ40 and ZnAβ42, respectively, as measured from the TEM images (Fig. S3). Furthermore, we added equal amount of Zn2+ into preformed Aβ40 fibrils and studied whether Zn2+ is capable to destabilize preformed fibrils by ThT assay and Fourier transform infrared spectra (FTIR). The results showed that the addition of Zn2+ ions cannot decrease ThT intensity of Aβ fibrils (Fig. S4A) and does not alter β-sheet structure signals at 1,630 cm−1 in FTIR analysis (Fig. S4B). This result suggested that Zn2+ only affects Aβ at the early stage.
Figure 1.

Zn2+ promotes Aβ oligomerization. (A,F) ThT assay of Aβ in the absence or presence of Zn2+. Aβ or Aβ42 (F) at 50 µM was incubated with and without equal molar concentration of Zn2+. The final ThT concentration in the working solution is 5 µM. ThT fluorescence was monitored at 25 °C. (B,G) TEM images of the end-point products of Aβ alone or ZnAβ. The scale bars are 100 nm. (C–E) and (H–J) Far-UV CD spectra of Aβ40 and Aβ42. The spectra were scanned from 250 to 190 nm at the indicated time. Aβ40 in the absence (C) and presence of Zn2+ (D) after different incubation time are shown. The single wavelength changes of Aβ40 at 200 and 220 nm after addition of Zn2+ are shown in panel (E). Aβ42 in the absence (H) and presence of Zn2+ (I) were also monitored. The single wavelength changes of Aβ42 at 200 and 220 nm after addition of Zn2+ were shown in panel (J).
Zn2+ rapidly changes secondary structures of Aβ40 and Aβ42 and retains Aβ in a less β-conformation
To understand how Zn2+ affects the conformation of Aβ, the secondary structure of Aβ40 and Aβ42 with and without Zn2+ was examined by far-UV CD spectroscopy for several days (Fig. 1C–E, H–J). On day 0, freshly-prepared A40 showed a random-coil-like spectrum and the conformation gradually transformed to α-helix-like spectra on day 4 and 5, then finally formed β-sheet-rich structures which minimum wavelength at 217 nm on day 6 (Fig. 1C). For the spectra of Aβ42, the random-coil-like conformation on day 0 quickly moved to β-sheet-rich structure after day 1 (Fig. 1H), this result is consistent with the result of ThT assay showing Aβ42 forms fibrils much faster than Aβ40. When Zn2+ was mixed with Aβ40 and Aβ42, the ellipticity was significantly reduced on and after day 0 (Fig. 1D,I). The result showed that Zn2+ affects the secondary structure of Aβ rapidly and the aggregates contain different secondary structures than Aβ alone. Previously, this reduction was also found in the presence of Al3+ and other metal ions in our previous findings with a different Aβ preparation method33. Interestingly, we found Zn2+ affects Aβ40 much more than Aβ42. The CD spectra of Aβ40 with Zn2+ did not show much secondary structure from day 1 to even after 6 days of incubation, whereas Aβ40 without Zn2+ formed β-sheet fibrils readily. In the presence of Zn2+ with Aβ42, the random-coil-like structure was also reduced at day 0 and transformed to mixed spectra after 1 day (Fig. 1I). On day 2, there is no obvious β-stand formation unlike the Aβ42 alone (Fig. 1H). The single wavelength changes of Aβ40 (Fig. 1E) and Aβ42 (Fig. 1J) at 200 and 220 nm are plotted against time after the addition of Zn2+. The results showed that Zn2+-induced Aβ changes were rapid and complete in minutes.
ZnAβ40 and ZnAβ42 form oligomers that are less heterogeneous and possess higher hydrophobic-exposed surfaces than ADDLs
To estimate the size of ZnAβ oligomers, we analyzed ZnAβ40 and ZnAβ42 oligomers and ADDLs composed of Aβ40 or Aβ42 by sedimentation velocity (SV) in analytical ultracentrifugation (AUC). ADDL is a type of Aβ oligomers commonly reported in the literature that are cytotoxic25,47,48. ADDL was prepared as described in method according to previous literatures25,48,49. The SV result showed that the predominant sedimentation coefficient distribution of ZnAβ40 or ZnAβ42 were similar. They ranged from 10 to 17.5 S corresponding to molecular weight ranging from 152 to 214 kDa (~33 to 49 mer). The peak maxima were situated at 11.3 and 11.9 S, respectively, where the peaks contained ~63% and ~46.2% of total Aβ (Fig. 2A,B). In the condition to form ADDLs by Aβ40, the major distribution were broadly distributed which indicates no distinct oligomer formed in this condition. This result is consistent with the previously report that no Aβ40 oligomer was observed under AFM in ADDL preparation25. In ADDL made by Aβ42, there is a major peak situated at ~10.27 S which corresponds to molecular weight of 159 kDa (~35 mer), containing ~26.09% of total peptide. There are some other assemblies presented under 5 S which corresponds to molecular weight less than ~95 kDa (~21 mer). The friction ratio of ZnAβ40 and ZnAβ42 are 1.4 and 1.2, whereas, ADDL40 and ADDL42 are 1.57 and 1.48, respectively. Since the perfect sphere has a fraction ratio of 1.2, the result indicates that ZnAβ complexes are more spherical. Our AUC data demonstrated that ZnAβ40 and ZnAβ42 form oligomers that are less heterogeneous and more spherical than ADDLs.
Figure 2.

ZnAβ40 or ZnAβ42 assemble into oligomers and contain more exposed hydrophobic surfaces than ADDLs. (A,B) SV in AUC of ZnAβ or ADDLs consist of Aβ40 or Aβ42, respectively. The data were analyzed by a continuous c(s) distribution model and the sedimentation coefficients were calculated and shown. (C,D) Bis-ANS spectra of ZnAβ and ADDL. ZnAβ40, ZnAβ42, ADDL-Aβ40, and ADDL-Aβ42 at 100 µM were prepared. Before the measurement, all Aβ solution were diluted to 50 µM in 10 mM Tris-HCl, pH 7.4, then, mixed with 0.5 µM Bis-ANS dye. Bis-ANS fluorescence spectra were collected. The spectra were subtracted from the respective buffer control.
Besides AUC analysis, we employed Bis-ANS fluorescence to monitor the hydrophobic clusters exposed on Aβ surface18,50,51. The experiments were performed with 50 µM ZnAβ oligomers or ADDLs. The samples were mixed with Bis-ANS readily within 1 min and subjected to fluorescence measurement. The fluorescence spectra of Aβ in the presence of Zn2+ ion was shown to be ~2.8 and ~1.6 fold higher than that of ADDLs Aβ40 or Aβ42, respectively (Fig. 2C,D). This enhancement indicated higher level of hydrophobic surface exposed on the protein surface of ZnAβ oligomers. Besides increasing of fluorescence intensity, the spectra of ZnAβ40 oligomers also showed blue-shift of maximal wavelength from 499 nm to 485 nm, but the shift in the ZnAβ42 is not that obvious, indicating that the presence of Zn2+ altered the Bis-ANS binding environment.
Low immunoreactivity of ZnAβ oligomers reveal distinct oligomeric structure
To further elucidate the effect of Zn2+ on conformation changes of Aβ, we collected time-course Aβ aggregates for dot blotting analysis to monitor epitope changes (Fig. S5). Aβ40 and Aβ42 samples at different time points of incubation were dotted on the nitrocellulose membrane with several replicates and probed by several Aβ antibodies including conformational specific antibodies A11 and OC and sequence specific antibodies like 6E10, 4G8, A3356, and C-terminal antibody. A11 antibody was used to detect common prefibrillar oligomers52, whereas OC antibody recognizes fibrils and fibrillar oligomers, which are structurally and immunologically related to fibrils53,54. The sequence specific antibodies used were 6E10 antibody recognizing N-terminal Aβ residues 1–16, 4G8 recognizing Aβ residues 17–24, A3356 recognizing the middle terminus of Aβ residues 22–35, anti-C-terminal of 42 recognizing the last 6 residues of Aβ42. The peptide loading control is detected by Direct Blue staining. From the different time points collected during Aβ40 and Aβ42 aggregation incubated with or without Zn2+ (Fig. S3A and S3B), we found A11 signals of Aβ40 increased after day 1, and that of ZnAβ40 increased after day 5. Both of the signal saturated after 6 days. In the OC blot, we found the signals of Aβ40 potently increased after day 1, but ZnAβ40 showed almost no OC signal in comparison to Aβ40 alone. Similarly, A11 and OC signals were both much weaker in the ZnAβ42 blot compared with Aβ42 alone. These evidences suggest that ZnAβ oligomers revealed distinct immunological status compared with Aβ aggregates in the absence of Zn2+. The data are consistent with the results from ThT assay and TEM imaging demonstrating no or little fibrillary structure is formed in ZnAβ. Surprisingly, we found that even sequence-dependent antibodies such as 6E10 or 4G8 showed weaker reactivity to recognize ZnAβ40 and ZnAβ42 while we have equal Aβ loading on the blot as shown by Direct Blue staining. Furthermore, we compared the epitopes in ZnAβ oligomers and ADDLs by the different antibodies. We dotted ZnAβ incubated for 1 and 7 days with ADDLs that were prepared in media for 1 day as described in the method. We found that both ZnAβ40 and ZnAβ42 incubated for 1 or 7 days showed weaker immunoreactivity compared with the corresponding ADDLs (Fig. S5C and S5D). This result further demonstrated that ZnAβ oligomers expose distinct epitopes and maintain a quite different conformation than ADDLs and the intermediates in Aβ aggregation without Zn2+.
ZnAβ40 oligomers are structurally more disordered than Aβ40 fibrils
It is well known that the chemical shifts of CO, Cα, and Cβ are sensitive to the backbone conformations of polypeptides55–57. To understand the molecular structure of ZnAβ, we first examined the 13C uniformly labeled ZnAβ40 by solid-state homonuclear 13C correlation spectrum. In contrast to the highly resolved NMR spectra of Aβ40 fibrils58, the resolution of the spectrum acquired for our ZnAβ40 sample was very poor (Fig. S6). It implied that ZnAβ40 still exhibited a significant structural heterogeneity at the molecular level. This finding suggested that the formation of ZnAβ40 aggregates is a kinetically controlled event. To tackle the resolution problems, we prepared a selectively labeled ZnAβ40 sample in which only the residues of V24, A30, I31, G33, and L34 were 13C labeled. The corresponding NMR spectrum is shown in Fig. 3. The assignments of the ZnAβ40 spectrum are given in the Supporting Information (Fig. S7). Accordingly, most of the cross peaks of V24 were significantly attenuated and only the Cβ/Cγ cross peak was barely observed. The Cα/Cβ cross peak of A30 was discernible but that of I31 was not observed. Nonetheless, the cross peaks of the side chain carbons of I31 remained observable. The CO/Cα cross peak of G33 were observed but its line width was relatively large, rendering an accurate determination of its chemical shifts difficult. The cross peaks of L34 were the most intense in the ZnAβ40 spectrum and there were at least two sets of resonances. For comparison, the labeled peptides were also incubated under quiescence conditions to form Aβ40 fibrils. Figure 3 showed the overlay 13C NMR spectrum of ZnAβ40 and Aβ40 fibrils. The chemical shifts and line widths of the backbone carbons of L34 were rather similar between ZnAβ40 and Aβ40 fibrils. On the other hand, the chemical shifts of ZnAβ40 revealed that the residue A30 is largely in the β-sheet conformation, but to a lower extent than Aβ40 fibrils. In addition, the line widths of the A30 signals of ZnAβ40 were larger than those of Aβ40.
Figure 3.

Overlaid 13C homonuclear correlation spectra of ZnAβ40 (green) and Aβ40 fibrils (red). The Aβ40 peptides were 13C enriched at V24, A30, I31, G33, and L34. Spectral assignments were given for the spectrum of the Aβ40 fibrils. The dashed elliptical circle highlighted the region of the Cα/Cβ cross peaks of V24 and I31 expected for ZnAβ40. The peaks denoted by asterisks were spinning sidebands.
Removal of Zn2+ restores amyloid fibril formation with a faster kinetics and ZnAβ is unable to serve as fibrillization seeds
Since the presence of Zn2+ promoted Aβ40 or Aβ42 forming stable oligomers, we wonder whether metal ion chelator, such as EDTA, can disrupt the interaction of Zn2+ ions and Aβ. Here, we added three-fold molar concentration of EDTA into the ZnAβ or Aβ solution after plateau phase of ThT intensity at the time point indicated by arrows and monitored the conformation and morphology by far-UV CD and TEM image (Fig. 4). Aβ40 or Aβ42 in the absence or presence of Zn2+ were incubated and monitored by ThT assay (Fig. 4A,E). When the ThT intensity of Aβ alone reached a plateau phase, EDTA were added to both solution. We found that upon adding EDTA to trap Zn2+ ions, ZnAβ40 showed a rapid restoration to form amyloid fibrils with a short lag phase (<~40 hr), whereas ThT intensity of Aβ40 alone was not changed after EDTA addition (Fig. 4A). We also monitored the secondary structural changes by time-course far-UV CD spectra (Fig. 4B). Interestingly, the result showed that removal of Zn2+ ions immediately promoted ZnAβ40 to again show random-coil-like structure (labeled as day 0) similar to the Aβ40 monomer solution without Zn2+. The initial spectra of ZnAβ before addition of EDTA was flat (labeled as dash line). After EDTA treatment, Aβ gradually transformed to β-sheet-rich structure after 4 days of incubation. This transition is similar to conformational transition (6 days) in Aβ alone but faster. We monitored the morphology of the end-point products by TEM (Fig. 4C) and found ZnAβ40 after EDTA treatment has formed mature fibrils similar to the control Aβ without Zn2+. Dot blotting by A11 and OC antibody of the time course ZnAβ40 samples after EDTA treatment also demonstrated that trapping of Zn2+ by EDTA results in restoration of Aβ40 to fibrillization pathway (Fig. 4D). In the case of Aβ42, ThT assay showed that EDTA treatment also promoted ZnAβ42 to immediate formation of amyloid fibrils without a lag phase (Fig. 4E). However, in far-UV CD analysis, we found the spectra removal of Zn2+ ions by EDTA still showed the β-strand content which may be due to unable to monitor the rapid aggregation of Aβ42. Dot blotting and TEM further confirmed the restoration of ZnAβ42 into amyloid fibrils and restoration of the immunoreactivity (Fig. 4G,H).
Figure 4.

ZnAβ40 oligomer can be restored rapidly back to fibrillization pathway by EDTA. (A,E) Aβ and ZnAβ40 (A) or ZnAβ42 (E) aggregation monitored by ThT assay. Three folds of EDTA was added when Aβ fibrillization reached a plateau. The time of addition is indicated by arrows. (B,F) Time-course conformation changes of ZnAβ40 or ZnAβ42 after EDTA treatment measured by far-UV CD. (C,D and G,H) TEM images (C and G) and dot blotting (D,H) of the time course samples in the aggregation. Scale bars in TEM images are 100 nm. ZnAβ was first prepared and incubated for 1 day for ZnAβ oligomer formation prior to EDTA treatment in the CD and dot blotting experiments. Dot blotted membrane was probed with A11 and OC antibody and stained by Direct blue as a dotting control.
Next, we would like to examine the property of Aβ-EDTA and ZnAβ-EDTA being seeded. We performed seeding experiment by addition of fibril seeds generated from preformed Aβ fibrils to soluble Aβ-EDTA and ZnAβ-EDTA solution and monitored the fibrillization kinetics by ThT fluorescence (Fig. 5A,B). The preformed Aβ40 or Aβ42 fibril seeds were sonicated and added in 5% to respective soluble Aβ solution. The result showed that all Aβ-EDTA and ZnAβ-EDTA can be seeded by fibril seeds showing accelerated fibrillization. There is no difference in initial kinetic rate of fibrillization, but the plateau phase of ThT intensity of ZnAβ-EDTA was markedly lower than Aβ-EDTA. This may indicate a distinct ThT binding environment or fibril structure formed from ZnAβ-EDTA fibrillization. Furthermore, we directly compared the fibrillization kinetics of Aβ and Zn-Aβ both treated with EDTA (Aβ-EDTA and ZnAβ-EDTA) by ThT assay (Fig. 5C,D). The result showed that the aggregation kinetics of ZnAβ40-EDTA was notably faster than Aβ40-EDTA with a shorter lag phase (ZnAβ40-EDTA, lag time = 33 hr; Aβ40-EDTA, lag time = 42 hr) (Fig. 5C), whereas, ZnAβ42-EDTA still showed similar kinetic profile as Aβ42-EDTA alone (Fig. 5D). The plateau phase of ThT intensity of both ZnAβ40-EDTA and ZnAβ42-EDTA were markedly lower than the respective Aβ-EDTA. The result is consistent with observing faster aggregation after Zn removal in aforementioned experiments (Fig. 4A,B). It suggests ZnAβ oligomer may facilitate formation of an Aβ conformation that is less heterogeneous and can facilitate the Aβ fibrillization after removal of Zn2+. However, the effect is not easily seen in Aβ42 that may be due to fast aggregation of Aβ42 hindering the observation of the difference.
Figure 5.

EDTA-treated ZnAβ40 showed faster fibrillization rate and ZnAβ oligomers are unable to seed Aβ. (A,B) Fibrillization kinetics of (A) EDTA-treated ZnAβ40 or Aβ40 and (B) EDTA-treated ZnAβ42 or Aβ42 after seeding. Five % sonicated preformed fibril seeds were added into EDTA-treated ZnAβ and Aβ solution. Fibrillization kinetic was measured by ThT fluorescence. ZnAβ alone and buffer containing 5% fibril seeds were also monitored. (C,D) Fibrillization kinetics of Aβ40 (C) and Aβ42 (D) with or without Zn2+ after EDTA treatment. ZnAβ was first prepared and incubate for 1 day for oligomerization, then added EDTA to remove Zn2+ ion. Aβ alone solution was also treated with EDTA for comparison. Fibrillization kinetics was measured by ThT fluorescence. (E,F) Seeding experiment were performed by adding Aβ fibrils or ZnAβ40 (E) or ZnAβ42 (F) as seeds. The respective buffer were added to Aβ solution as negative control.
We further seeded preformed Aβ fibrils and ZnAβ oligomers to Aβ solution to examine the seeding property of ZnAβ in comparison to Aβ fibrils (Fig. 5E,F). Aβ40 and Aβ42 fibril seeds or ZnAβ40 and ZnAβ42 seeds were add 5% to the Aβ solution. The corresponding buffers were also added separately as negative controls. We found that Aβ fibril seeds can greatly accelerate fibrillization as expected. In Aβ40, the ThT signal of Aβ with fibril seeds elongated immediately without a lag phase compared to Aβ without fibril seeds. Whereas, Aβ with ZnAβ40 seeds did not show acceleration but adopted a similar aggregation characteristic of Aβ without seeds (buffer added). Similarly, in Aβ42, Aβ42 with Aβ42 fibril seeds adopted a faster elongation rate in comparison to the Aβ42 without seeds. ZnAβ42 oligomer did not seed Aβ42 solution. The results demonstrate ZnAβ cannot serve as seeds to accelerate fibrillization. Overall, the seeding experiments showed consistent results to indicate off-pathway property of ZnAβ.
ZnAβ oligomers possess higher cytotoxicity than ADDLs
Aβ oligomers have been proposed to be the major cause contributed to neurotoxicity in the Aβ assembly. Here, to understand whether ZnAβ oligomers show difference in the biological effects compared with ADDLs, we examined the cell viability and cytotoxicity contributed from ZnAβ oligomers and ADDLs. Since high Zn2+ ion concentration induces cell death59,60, we exchanged excess Zn2+ in the preparation with 10 mM Tris-HCl buffer, pH 7.4 by MWCO 3 KDa centrifugal filter and re-quantify concentration prior to further test. The remaining Zn concentration after exchanged was 2 µM (data not shown). The cell viability and cytotoxicity of the oligomers were measured in human neuroblastoma BE(2)-C cells by MTT and LDH assays, respectively. After 24 hr of incubation with ZnAβ or ADDL at concentration ranging from 10 to 40 µM, both ZnAβ or ADDL oligomers showed reduction of cell viability. In general, ZnAβ was able to induce dose-dependent cytotoxicity. At 40 µM, ZnAβ40 and ZnAβ42 reduced 54% and 41% of cell survival respectively, whereas, ADDL40 and ADDL42 reduced 15% and 11% of cell survival, respectively (Fig. 6A,B). Besides MTT assay for cell viability, we also used LDH assay to interpret whether these oligomers increased cytotoxicity. The LDH assay showed both ZnAβ significantly increased the percentage of cytotoxicity in a dose-dependent manner. At 40 µM of ZnAβ40 and ZnAβ42 increased cytotoxicity to 71% and 42%, respectively, whereas ADDL40 and ADDL42 increased 15% and 14% of cytotoxicity, respectively (Fig. 6C,D). Besides cell viability and cytotoxicity assays, Ca2+ influx is one of the Aβ oligomer-induced adverse effect that promotes apoptosis61. Here, we employed Ca2+ binding dye, Fluo-3 AM, to detect Ca2+ influx. Fluo-3 AM dye is cell permeable and emits fluorescence by intracellular esterase activity. The cells were subjected to ZnAβ or ADDLs of Aβ40 or Aβ42 at 40 µM and the fluorescence signals were measured after sample addition (Fig. 6E,F). From the result, we found both ZnAβ40 and ZnAβ42 were able to increase Ca2+ influx levels as indicated by the fluorescence intensity and the level are similar to ADDLs. Overall, the result demonstrated that ZnAβ40 and ZnAβ42 are more toxic than ADDLs but they induce similar effect in Ca2+ influx.
Figure 6.

ZnAβ oligomers show stronger reduction to cell viability and are more cytotoxic than ADDLs, but they induce similar Ca2+ influx. (A,B) Cell viability was examined by MTT assay for ZnAβ and ADDLs treatment. ZnAβ and ADDLs in different concentrations were treated to neuroblastoma cells. (C,D) Cytotoxicity assay by LDH for ZnAβ and ADDLs treatment. The percentage of LDH release was calculated by the slope of fluorescence intensity compared with maximum LDH release control. Triton-X 100 treated cell was served as 100% cytotoxicity. (E,F) Intracellular Ca2+ was monitored by Fluo-3 AM dye. ZnAβ samples were filtered prior to experiment. Before treatment, cell was first incubated with dye and measured for basal level of fluorescence intensity for 10 mins. Aβ was added as indicated arrow. The statistical analysis was performed by one-way ANOVA and Turkey Post Hoc Test, where p < 0.05 (*), <0.01 (**), <0.001 (***), <0.0001 (****). The averaged values were shown with standard divisions.
ZnAβ oligomers inhibit hippocampal LTP and increase microglial activation in wild-type mice
Previous studies have demonstrated toxic effect of ADDLs on synaptic plasticity and long-term potentiation (LTP). LTP is a good paradigm to investigate synaptic plasticity and a model of learning and memory62–64. Therefore, to further assess the effect of ZnAβ oligomer on synaptic plasticity, we treated 500 nM ZnAβ42 oligomer for 30 mins on the hippocampal slice from wild-type C57BL/6JNarl mice since Aβ42 is considered the more toxic Aβ species. The ZnAβ42 oligomers were filtered before the experiments to remove excessive Zn ions. In LTP results, we found significant LTP impairment generated by ZnAβ42 oligomers compared to the buffer treated and control one. Hippocampal slices treated with ZnAβ42 showed LTP-impairment for 47 mins after tetanic stimulation (mean amplitude 89.44 ± 13.91%), whereas, control and filtered Zn buffer-treated hippocampal slice induced LTP within the amplitude of 147.5 ± 16.27% and 152 ± 9.29% (Fig. 7). This result indicated the effect of ZnAβ oligomers to interfere synaptic plasticity.
Figure 7.
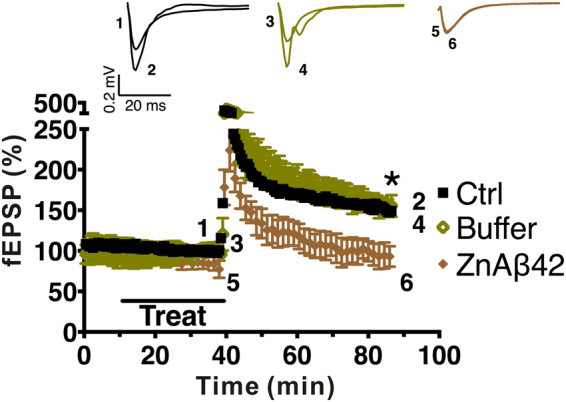
ZnAβ42 oligomers inhibit LTP in wild-type mice hippocampal slice. Filed EPSPs (fEPSP) were first measure for stable base line, then treated with filtered ZnAβ42 or buffer control for 30 min (ctrl, n = 5, buffer, n = 3, ZnAβ42, n = 3). LTP was induced by theta burst stimulation given at baseline intensity after treatment. Insets show example fEPSP traces before and post theta-burst stimulation (stimulus artifact was removed for clarity). The statistical analysis were performed by two-way ANOVA and Turkey Post Hoc Test (*p < 0.05).
To investigate whether ZnAβ oligomers prepared in vitro showed neurotoxicity in the mice brain, we injected 40 µM ZnAβ or ADDL42 oligomers into dorsal hippocampus of the WT mice brain as an acute Aβ-injected model. After 14 days, the mice were sacrificed and the brain slices were subjected to immunohistochemistry stained with Iba1 antibody for microgliosis and GFAP antibody for astrogliosis. The representative Iba1 (Fig. 8A) and GFAP staining (Fig. S8) in the hippocampus were shown and the quantified results were calculated. The results showed that the mice received injection of ZnAβ oligomers have ~20% increase of microglial density and ~30% increase of microglial area in the hippocampus region compared with the respective buffer injected group (Fig. 8B,C). Relatively, ADDL-injected group did not show obvious microglia activation compared with its buffer injected group (Fig. 8D,E). The extent of astrogliosis was also measured by density and area stained by GFAP antibody, however it seems no increase of density and area in the mice received ZnAβ or ADDL42 compared with its respective buffer injected groups (Fig. S8B to S8E).
Figure 8.

Effect of ZnAβ42 oligomer on hippocampal microglia activation in the wild-type mice. (A) The micrograph represents the Iba1+ positive microglia in the hippocampus of the mice after ZnAβ42 or ADDLs injection. The quantitative results of Iba1+ microglia are represented as density (B,D) and area (%) (C,E) (ZnAβ42, n = 19, Zn buffer, n = 23, ADDL42, n = 24, ADDL buffer, n = 17, and n values are from the brain slices of 3 mice of each group). The statistical analysis was performed by unpaired Student’s t test where p < 0.05 (*), <0.01 (**). Scale bar = 800 µm.
Discussion
Zn2+ homeostasis and synaptic Zn2+ levels play crucial role in the AD pathology65,66. In the glutamatergic synapses, storage of Zn2+ is concentrated by the ZnT3 transporter as a pool achieved up to ~300 µM31. In the physiological condition, extracellular Zn2+ concentration is quite low (<1 µM), and may be released up to exceed 100 µM from the synaptic cleft at the peak of neuron activity67. Zinc ion has been reported to play both neuroprotective and neurotoxic roles. In previous literature, the neurotoxicity of Aβ reduces in the presence of Zn2+ at sub-stoichiometric concentration42,68, whereas, the neurotoxicity of Aβ increases in vitro and in vivo at stoichiometric concentration of Zn2+ 68,69 with some exceptions in different experimental conditions70. Besides, knockout of ZnT3 transporter in AD mice have marked reduction of amyloid plaque load65. In the present study, we focused on characterizing biological and structural concepts of Zn2+-induced Aβ oligomerization. We first showed the influence of equal molar concentration of Zn2+ on Aβ40 or Aβ42 that significantly altered the structure and pathway of Aβ fibrillization resulting in oligomerization as evidenced by their low ThT and far-UV CD signals, larger hydrophobic exposed surfaces by Bis-ANS fluorescence, oligomer assembly by AUC, oligomeric morphology in TEM, and altered immunoreactivity. The effect is limited in the initial stage of aggregation but not to the preformed fibrils. Interestingly, removal of Zn by EDTA not only rapidly restored Aβ fibrillization but also adopted a faster kinetics. We also demonstrated the ZnAβ oligomers potently inhibited hippocampal LTP and initiate microglia activation in acute Aβ-injected model. Our result demonstrated that Zn2+ plays a key role in the conformation and oligomerization of Aβ in which the species is highly toxic.
On the basis of the 13C solid-state NMR spectrum of uniformly labeled ZnAβ40, we found that the overall structure of ZnAβ40 was highly disordered. Nonetheless, the spectrum acquired for the selectively labeled ZnAβ40 revealed some site-specific structural insights for ZnAβ40. With reference to the published backbone conformation of Aβ40 fibrils71, the fold of the peptides adopts a motif of β-sheet/turn/β-sheet. The residue V24 is in the turn region, whereas A30 to L34 are in the second β-sheet region. Our chemical shift data of ZnAβ40 suggested that the residue V24 was very structurally disordered and/or the residue might undergo considerable motional dynamics. Interestingly, we found that the residues A30 and L34 of ZnAβ40 largely adopted a β-sheet conformation, although we did not prepare a series of selectively labeled samples to map out the sequence location of the β-sheet structure. Recently, it has been shown by solid-state NMR that Zn2+ ions would modulate the structure of the aggregates of ZnAβ40 and ZnAβ4216,72. In comparison, our chemical shift data at A30 and L34 are consistent with the data reported by Madhu and co-workers for ZnAβ4016, whereas the signal line widths of our data were considerably larger (Fig. 3). Because our ZnAβ40 sample was prepared with a relatively short incubation time, the sample was structurally more heterogeneous than the corresponding fibrillar aggregates, as revealed in the solid-state NMR spectra. At the present stage, it is not clear whether the elevated cytotoxicity of ZnAβ40 is due to the structural flexibility of ZnAβ40, or a small subset of the structural isoform of ZnAβ40 is particularly cytotoxic. In view of the poor spectral resolution for ZnAβ40, we did not attempt to measure the NMR spectrum for ZnAβ42.
Aβ oligomer among Aβ aggregates is suggested as the actual toxic species to impair synapse function in AD pathology8,9,62. The aggregation pathway initiated with misfolded monomer can be oligomerized to on-pathway or off-pathway oligomers. For example, fibrillary oligomers with OC antibody-positive signal are suggested as on-pathway oligomer within the ability to directly serve as fibril nuclei or seeds to elongate by adding free monomers to their ends11. However, monomer might aggregate into prefibrillar oligomer, A11-positive, OC-negative signals, as off-pathway oligomer, which cannot directly elongate into form mature fibrils without first form protofibrils and further conformation changes52,53. EGCG interaction with Aβ promotes the formation of off-pathway non-toxic Aβ oligomers73. Our immunodotting results showed ZnAβ oligomers possess lower affinity with most anti-Aβ antibodies including A11 and OC. The result suggest novel properties of ZnAβ oligomers. As shown in our AUC result for the first time that ZnAβ oligomers are less heterogeneous than ADDLs, the reported culprit in AD that are often used in many biological papers74–76. From EDTA experiments, we found EDTA recovered ZnAβ fibrillization by restoring ZnAβ40 to random coil-like structure and followed by a faster fibrillization kinetics as evidenced by ThT assay, far-UV CD, and dot blotting. The results suggest ZnAβ oligomers are off-pathway species that are trapped by Zn ion. Once Zn is removed, Aβ rapidly restores its properties to undergo fibrillization. Although we only capture ZnAβ40, but not ZnAβ42, returned to random coil structure after Zn removal, the faster kinetics of Aβ42 fibrillization may be the reason hinder the observation. We hypothesized that Zn trapped Aβ oligomer concentrated local Aβ that facilitates the faster fibrillization after Zn removal.
Although both ZnAβs and ADDLs form oligomers, they have substantial amount of differences. Firstly, ZnAβ oligomers expose larger hydrophobic surface area and show lower immunoreactivity against Aβ antibodies than ADDLs. Besides, ZnAβ oligomers are stable oligomers even incubation at room temperature, whereas, only Aβ42 ADDLs but not Aβ40 ADDLs form in a special condition at 4 °C25. Comparing to our data and previously literature74, our AUC result confirmed the narrower distribution of ZnAβ oligomers. The higher toxicity of ZnAβ compared to ADDL also strengthen the pathogenic role of ZnAβ in AD. Zn and Aβ coordination has been proposed for Zn2+ using the three histidines and the N-terminus39,40. Inter-molecular Aβ-Zn coordination has also been reported via histidine-bridges77–80. Although N-terminal Aβ is not considered in the current oligomer and fibril models, manipulation of N-terminal Aβ indeed affected the Aβ aggregation and induced toxicity. For example, genetic variation in alanine 2 in Aβ sequence, or alanine 673 in APP, were reported to have either protective81 or adverse effect82,83 on AD. N-terminal Aβ inhibitor84 or antibody against the N-terminal region85 have been shown to have beneficial effect.
Synaptic activity has been reported as a critical role to control Aβ oligomers formation and targeting at synaptic terminals and Zn2+ was involved in this activity-dependent regulation86. Takeda et al. further supported the critical role of Zn2+ by demonstrating single Aβ injection stimulates Zn2+ influx and causes a short-term cognitive deficit87, while applying Zn2+ chelator can rescue the LTP impairment. They further proposed that interaction of Zn2+ and Aβ42 was essential for the rapid uptake of Zn2+ and Aβ42 into the rat dentate gyrus88. These results support the role of ZnAβ in AD pathogenesis. Clinical trials of passive immunization against Aβ aggregates has been a major therapeutic strategy. Recently, anti-Aβ antibody aducanumab has successfully shown cognitive improvement in mild AD patients in phase Ib clinical trial89. Future Aβ antibody development can also target ZnAβ oligomers. In conclusion, our aggregation and biological studies showed the aggregation mechanism of ZnAβ which facilitate the understanding the possible role of ZnAβ oligomers in the AD and provide a new insight to understand ZnAβ oligomer structure. This study may contribute to identify a potential therapeutic target in AD.
Materials and Methods
Aβ synthesis and preparation
Synthetic Aβ40 or Aβ42 peptides were acquired by solid phase peptide synthesis from the Genomics Research Center, Academia Sinica, Taiwan, as previously described. For selective-13C labeled Aβ40 were synthesized by the department of chemistry, National Taiwan University90. Aβ peptides were solubilized in 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP, Sigma-Aldrich, St. Louis, MO, USA) in 1 mg/mL to dissolve preformed aggregates. HFIP was lyophilized and then treated with 10% ammonium hydroxide (NH4OH) and lyophilized. Finally, lyophilized Aβ40 or Aβ42 were dissolved in 10 mM Tris-HCl buffer, pH 7.4. The Aβ solution was quantified by absorbance at 280 nm (ε = 1280 cm−1 M−1)91 to prepare working concentration. To prepare ADDL oligomers, lyophilized HFIP-treated Aβ40 or Aβ42 were dissolved in DMSO to 5 mM and sonicated for 5 mins. Then the dissolved Aβ was added to DMEM/F12 (GIBCO, Invitrogen). The concentration was quantified by absorbance at 280 nm to prepare 100 µM stock and incubated at 4 °C for 24 hr25,49.
ThT Assay
Fifty µM Aβ solution was prepared in the presence of 5 µM ThT as previously described92. Briefly, the samples were incubated at 25 °C with agitating 60 sec each hr. Fluorescence of ThT was monitored at 485 nm by an ELISA microplate reader SpectraMax M5 (Molecular Devices, Sunnyvale, CA, USA) and excitation wavelength was at 442 nm. The experiments were repeated at least 3 times and averaged and presented as mean ± S.D.
Dot blotting
Two µl of the each Aβ time-course samples were collected at a series of time points and dotted onto nitrocellulose membrane with several duplicates for probing by different antibodies. Membranes were probed with A11 (Invitrogen), OC (Millipore, Temecula, CA, USA) separately. Membrane was also staining with direct blue as a loading control to check equal loading of each dot.
Direct blue staining
The staining solution of Direct blue 71 (Sigma-Aldrich, St. Louis, MO, USA) was prepared with 0.1% (w/v) in the double distilled water containing 40% ethanol, and 10% acetic acid. The nitrocellulose membrane with Aβ samples dotted was directly incubated in the staining solution and incubated for 10 min, then washed by double distilled water to acquire clear signals.
Far-UV CD spectroscopy
Aβ samples were measured at a series of time points. Aβ solution was loaded into a circular quartz cuvette with 1 mm path length (110-QS, Hellma Analytics). Spectra were monitored from 250 to 190 nm by a J-815 CD spectropolarimeter (Jasco Inc., Easton, MD, USA). The data were accumulated 10 times of scans and averaged.
TEM
The samples were dropped onto 400-mesh Formvar carbon-coated copper grids (EMS Electron Microscopy Sciences, Hatfield, PA, USA) and incubated for 5 mins and washed twice by double distilled water, then negatively staining was performed with 2% uranyl acetate for 2 mins and dried at room temperature overnight. Image were observed with a FEG-TEM, FEI Tecnai G2 TF20 Super TWIN transmission electron microscope with an accelerating voltage of 120 kV.
Bis-ANS
Aβ samples were first mixed with 0.5 µM Bis-ANS solution and measured the fluorescence signal. The emission spectra of Bis-ANS were collected from 450 to 550 nm with excitation at 400 nm. Fluorescence emission spectra were obtained using a FluoroMax-3 spectrofluorometer (Horiba Jobin Yvon). The background from buffer control was subtracted.
AUC
SV of AUC experiments were performed on a Beckman Optima XL-I analytical ultracetrifugation (Beckman Coulter, USA). ADDLs at 100 µM in DMEM/F12 or ZnAβ 40 or 42 at 100 µM in 10 mM Tris-HCl (pH 7.4) were centrifuged at 60,000 rpm for 72 hr at 4 °C using an An-60Ti rotor. The moving boundary was monitored continuously by the absorption at 230 nm with scanning recorder every 4 min. Sedimentation velocity data were analyzed using C(S) distribution method by SEDFIT (NIH, Bethesda, MD, USA) and the parameters were calculated using SEDNTERP (NIH, Bethesda, MD, USA).
Solid-state NMR
Uniformly 13C labeled Aβ40 peptides were prepared by bacterial expression following our previous literature93. Briefly, the recombinant Aβ with His tag were prepared in minimal media in the transformed BL21(DE3) E. coli cells. The His-Aβ was purified through two Ni-affinity columns coupled with TEV protease digestion to remove the tag. The selectively 13C labeled peptides were synthesized on an automated Odyssey microwave peptide synthesizer (CEM Corp., Matthews, NC), using a FMOC-Valine-preloaded CLEAR-Acid resin (Peptide International) with 0.44 mequiv/g substitution level, and FMOC chemistry with hydroxybenzotriazole/N, N′-diisopropylcarbodiimide activation. The final products were validated by MALDI-TOF mass spectrometry. Aβ were incubated with equimolar of ZnCl2 for overnight. All NMR experiments were carried out carried out at 13C and 1H frequencies of 201.2 and 800.2 MHz, respectively, on a Bruker Avance III spectrometer equipped with a commercial 3.2 mm E-free probe. 13C chemical shifts were externally referenced to tetramethylsilane (TMS) using adamantane as the secondary reference. Spinning frequency at the magic angle was 10 kHz and the sample temperature was maintained at 268 K.
Aβ seeding assay
Fifty µM Aβ fibril were prepared after shaking at 750 rpm for 5 days at room temperature. The fibril morphology was confirmed by TEM. The fibrils were sonicated in water bath for 10 mins, ZnAβ oligomers (filtered prior to use) and respective buffer were added to Aβ solution in all experiments was 5% (2.5 µM) of the concentration of the Aβ solution. The Aβ solution in the presence of 5 µM ThT were monitored in 384 well plates by ThT assay.
Cytotoxicity assay
MTT assay was aimed to investigate the effect of Aβ on cell viability. The human neuroblastoma BE(2)-C cells (ATCC #CRL-2268) were cultured at 37 °C, 5% CO2 in the RPMI medium containing 10% FBS. Twenty thousand cells were seeding in a 96-well plate each well. ZnAβ or ADDLs diluted in the RPMI medium were added into the well and incubated for 24 hr. Then, ten µl MTT solution (5 mg/ml) was added and incubated for 3 hr for formation of formazan crystals. After incubation, the medium was carefully discarded and 100 µl DMSO was added to lyse the formazan crystals. Absorbance at 570 and 690 nm (background absorbance) were monitored by an ELISA microplate reader SpectraMax M5 (Molecular Devices, Sunnyvale, CA, USA). The difference of absorbance between 570 and 690 nm was calculated. For LDH assay, the assay was followed the instructions from the protocol of LDH assay toxicity kit (Promega, Madison, WI, USA). BE(2)-C cells were seeding the same as MTT ZnAβ or ADDLs were added and incubated for 24 hr. Next day, LDH assay was performed by monitoring the rate of substrate formation, emission at 590 nm while excitation at 560 nm. The results were replicated for 3 times. The cell lysed with 2% Triton X-100 was served as positive control for 100% cytotoxicity.
Calcium influx assay
The fluorescence probe Fluo-3 AM (Invitrogen) was used to detect the intracellular Ca2+ levels. Twenty thousand BE(2)-C cells were seeded in a black 96-well plate with clear bottom. Next day, fluo-3 AM powder dissolved in DMSO was added into culture medium without FBS. The final concentration of 10 µM Fluo-3 AM was incubated for 1 hr within plated cell. The probe-containing medium was replaced by RPMI medium, then incubated at 37 °C for 10 mins, and fluorescence intensity was started to monitor prior to the addition of Aβ samples as stable baseline, and the wavelength of the fluorescence of Fluo-3 AM was excited at 480 nm and recorded emission wavelength at 560 nm. Data were averaged (n = 3).
Animal
All experiments were done in accordance with the National Institutes of Health Guideline for Animal Research (Guide for the Care and Use of Laboratory Animals) and Taiwan Animal Protection Law and were approved by the Academia Sinica Institutional Animal Care and Utilization Committee (IACUC 12–03–340). All of the mice were housed (4–5 per cage) at a stable temperature (23 ± 1 °C), humidity 55 ± 5%, and unrestricted access to food and water during the experimental period.
In vitro electrophysiology
Eight-week-old C57BL/6JNarl male mice were purchased from the National Laboratorial Animal Center and keep at the Animal Facility of the Genomic Research Center (GRC) at Academia Sinica. The mice were housed in GRC animal facility and following the GRC facility housing rule during the period. Animals were housed in a room maintained on a 12-h/12-h light/dark cycle (light on at 8:00 a.m.). Two to four-month-old C57BL/6 J mice were used to examine effect of ZnAβ42 oligomer. Mice were anaesthetized by isoflurane and decapitated. The brain was rapidly removed and immersed in ice-cold ACSF (which contained 119 mM NaCl, 2.5 mM KCl, 1.3 mM MgSO4, 2.5 mM CaCl2, 26.2 mM NaHCO3, 11 mM Glucose and 1.25 mM NaH2PO4). Cronol sections (450 μm) of hippocampal slices were immediately transferred to a chamber containing ACSF and 100 µM picrotoxin. Circulating ACSF was continuously bubbled with a mixture of 95% O2 and 5% CO2 and slices allowed to recover for at least 2 hr at room temperature. For extracellular recordings, the hippocampal regions were placed at the center of an MED–P515A probe (Panasonic International Inc.) with 64 embedded recording electrodes. The slices were randomly assigned to control or ZnAβ42 or Zn buffer treatment and subsequently perfused with ZnAβ42 at 500 nM in oxygenated ACSF for 0.5 hr before LTP induction. fEPSPs were recorded using a MED64 multichannel recording system, and the data were collected from Schaffer collaterals. Test stimuli were given every 30 s (0.033 Hz), and the stimulus intensity was set to give a baseline fEPSP of 10–90% of the maximal response. A stable baseline was recorded for at least 20 min. Filtered ZnAβ42 oligomer (stock solutions were diluted into ACSF to produce concentrations of 500 nM based on the starting weight of Aβ42 monomer), the sample was perfused for 30 mins prior to induction of LTP. LTP was induced by theta burst stimulation (10 bursts of 4 stimuli at 100 Hz, with an interburst interval of 200 ms) given at baseline intensity according to previously described74. The ACSF was recycled using peristaltic pumps ensuring that the ZnAβ42 was present for the duration of the experiment. LTP is expressed as the mean ± SEM % of baseline fEPSP slope. Statistical comparisons used two-way ANOVA with post hoc Bonferroni test. In addition, control/ZnAβ42/Zn2+ buffer was performed on the same day to avoid any temporal bias.
Acute Aβ-injected mice models
The experiment procedure of acute Aβ-injected-models was followed and modified from previous protocols94–98. Eight-week-old C57BL/6 JNarl male mice were first intraperitoneally anesthetized by zoletilTM (100 mg/kg) plus xylazine hydrochloride (10 mg/ kg, Sigma-Aldrich, St. Louis, MO, USA). Forty µM filtered ZnAβ42/ADDLs oligomer or their respective buffer was bilaterally injected into the hippocampus at following stereotaxic coordinated: 2 mm posterior to the bregma, 2.1 mm left/right to the midline, and 1.8 mm ventral to the skull surface. The volume of injection was 2 µl and lasted for 5 mins, and remain in injected region for 5 mins followed by injection to avoid Aβ reflux along the needle tract, 14 days were allowed for the development of Aβ-induced damage.
Immunohistochemistry
Prior to processing immunohistochemistry, mice were anesthetized and perfused with 0.9% ice cold NaCl. After removing the brains, left hemispheres were stored at −80 °C, and the right hemispheres were subjected to fixed in 10% neutral buffered formalin solution (Sigma-Aldrich, St. Louis, MO, USA) at 4 °C for 24 hrs, dehydrated and cyroprotected in 30% sucrose solution then embedded in OCT (Leica, Mannheim, Germany). The brains were further cut-into 25 µm-thick coronal sections on a cryostat (Leica CM3050S, Mannheim, Germany). The sections were washed in PBS and mounted on poly-L lysine-coated slides (Thermo Fisher Scientific, Waltham, MA, USA), dried overnight. Then, the coronal sections were washed, antigen retrieval heated at 80 °C in sodium citrate buffer for 30 mins, washed, treated with 1% H2O2 to inactivate endogenous peroxidase activity, blocking in 3% BSA for 30 mins. The sections were further stained with following primary antibodies: anti-Iba1 (1:1000, Millipore, Temecula, CA, USA) for microglia. To analyze protein levels, images were captured by Aperio AT2 Digital Pathology Scanner (Leica Biosystem, Mannheim, Germany). For microglia activation, we selected coronal sections 360 μm apart between bregma −1.34 mm to −3.80 mm, which encompassed hippocampal region and further analyzed by Image J (NIH, Bethesda, MD, USA). To quantify density and surface area of microglia and astrocyte, we first optimized brightness and contrast of control group, then manually set the best threshold value. The area of Iba1+ was measured with the signal which was higher than background. For the density, cells with clear and identifiable cell bodies were counted. The densities were represented by dividing cell numbers by the area of the hippocampal region.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Electronic supplementary material
Acknowledgements
We thank peptide synthesis core, Genomics Research Center, Academia Sinica for peptide synthesis, Mr. Yu-Jen Chang for Aβ peptide purification, Mr. Tai-Lang Lin (Institute of Cellular and Organismic) for assisting TEM, Dr. Sin-Jhong Cheng (Neuroelectrophysiology Core at Neuroscience Program of Academia Sinica, Taiwan) for technique support of electrophysiology.
Author Contributions
M.C.L. conducted the biochemical and cellular experiments. W.C.Y. and Z.H.G. prepared the 13C enriched samples, S.J.H. conducted NMR experiments. C.Y.C., performed the AUC experiments. C.Y.C., M.C.L. analyzed the AUC data. M.C.L. and Y.H.S. conducted electrophysiology and animal experiments. J.C.C.C. directed the NMR work and edited the manuscript and figures. M.C.L., and Y.R.C. planed the experiments, managed the works, prepared figures, and wrote the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-23122-x.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Selkoe, D. J. & Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med (2016). [DOI] [PMC free article] [PubMed]
- 2.Harrison JR, Owen MJ. Alzheimer’s disease: the amyloid hypothesis on trial. Br J Psychiatry. 2016;208:1–3. doi: 10.1192/bjp.bp.115.167569. [DOI] [PubMed] [Google Scholar]
- 3.Kang J, et al. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 4.Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bibl M, et al. Blood-based neurochemical diagnosis of vascular dementia: a pilot study. J Neurochem. 2007;103:467–474. doi: 10.1111/j.1471-4159.2007.04763.x. [DOI] [PubMed] [Google Scholar]
- 6.Schoonenboom NS, et al. Amyloid beta 38, 40, and 42 species in cerebrospinal fluid: more of the same? Ann Neurol. 2005;58:139–142. doi: 10.1002/ana.20508. [DOI] [PubMed] [Google Scholar]
- 7.Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid beta-protein assembly and Alzheimer disease. J Biol Chem. 2009;284:4749–4753. doi: 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 9.Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behavioural brain research. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamley IW. The amyloid beta peptide: a chemist’s perspective. Role in Alzheimer’s and fibrillization. Chemical reviews. 2012;112:5147–5192. doi: 10.1021/cr3000994. [DOI] [PubMed] [Google Scholar]
- 11.Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 13.Chimon S, et al. Evidence of fibril-like beta-sheet structures in a neurotoxic amyloid intermediate of Alzheimer’s beta-amyloid. Nat Struct Mol Biol. 2007;14:1157–1164. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]
- 14.Liang C, et al. Kinetic intermediates in amyloid assembly. J Am Chem Soc. 2014;136:15146–15149. doi: 10.1021/ja508621b. [DOI] [PubMed] [Google Scholar]
- 15.Parthasarathy S, et al. Structural Insight into an Alzheimer’s Brain-Derived Spherical Assembly of Amyloid beta by Solid-State NMR. J Am Chem Soc. 2015;137:6480–6483. doi: 10.1021/jacs.5b03373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandra B, et al. Curcumin Dictates Divergent Fates for the Central Salt Bridges in Amyloid-beta40 and Amyloid-beta42. Biophys J. 2017;112:1597–1608. doi: 10.1016/j.bpj.2017.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biancalana M, Koide S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochimica et biophysica acta. 2010;1804:1405–1412. doi: 10.1016/j.bbapap.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen YR, Glabe CG. Distinct early folding and aggregation properties of Alzheimer amyloid-beta peptides Abeta40 and Abeta42: stable trimer or tetramer formation by Abeta42. J Biol Chem. 2006;281:24414–24422. doi: 10.1074/jbc.M602363200. [DOI] [PubMed] [Google Scholar]
- 19.Bitan G, et al. Amyloid beta -protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize through distinct pathways. Proc Natl Acad Sci USA. 2003;100:330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernstein SL, et al. Amyloid-beta protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nature chemistry. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tycko R. Molecular structure of amyloid fibrils: insights from solid-state NMR. Q Rev Biophys. 2006;39:1–55. doi: 10.1017/S0033583506004173. [DOI] [PubMed] [Google Scholar]
- 22.Torok M, et al. Structural and dynamic features of Alzheimer’s Abeta peptide in amyloid fibrils studied by site-directed spin labeling. J Biol Chem. 2002;277:40810–40815. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- 23.Williams AD, et al. Mapping abeta amyloid fibril secondary structure using scanning proline mutagenesis. J Mol Biol. 2004;335:833–842. doi: 10.1016/j.jmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 24.Gremer L, et al. Fibril structure of amyloid-beta(1-42) by cryo-electron microscopy. Science. 2017;358:116–119. doi: 10.1126/science.aao2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dahlgren KN, et al. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. The Journal of biological chemistry. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 26.McGowan E, et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seubert P, et al. Isolation and quantification of soluble Alzheimer’s beta-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 28.Ayton S, Lei P, Bush AI. Metallostasis in Alzheimer’s disease. Free Radic Biol Med. 2013;62:76–89. doi: 10.1016/j.freeradbiomed.2012.10.558. [DOI] [PubMed] [Google Scholar]
- 29.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci. 1998;158:47–52. doi: 10.1016/S0022-510X(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 30.Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci. 2005;6:449–462. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- 31.Bush AI. The metallobiology of Alzheimer’s disease. Trends Neurosci. 2003;26:207–214. doi: 10.1016/S0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- 32.Zatta P, Drago D, Bolognin S, Sensi SL. Alzheimer’s disease, metal ions and metal homeostatic therapy. Trends Pharmacol Sci. 2009;30:346–355. doi: 10.1016/j.tips.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 33.Chen WT, Liao YH, Yu HM, Cheng IH, Chen YR. Distinct effects of Zn2+, Cu2+, Fe3+, and Al3+ on amyloid-beta stability, oligomerization, and aggregation: amyloid-beta destabilization promotes annular protofibril formation. J Biol Chem. 2011;286:9646–9656. doi: 10.1074/jbc.M110.177246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tougu V, Tiiman A, Palumaa P. Interactions of Zn(II) and Cu(II) ions with Alzheimer’s amyloid-beta peptide. Metal ion binding, contribution to fibrillization and toxicity. Metallomics. 2011;3:250–261. doi: 10.1039/c0mt00073f. [DOI] [PubMed] [Google Scholar]
- 35.Solomonov I, et al. Zn2+ -Abeta40 complexes form metastable quasi-spherical oligomers that are cytotoxic to cultured hippocampal neurons. J Biol Chem. 2012;287:20555–20564. doi: 10.1074/jbc.M112.344036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen WT, et al. Amyloid-beta (Abeta) D7H mutation increases oligomeric Abeta42 and alters properties of Abeta-zinc/copper assemblies. PloS one. 2012;7:e35807. doi: 10.1371/journal.pone.0035807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minicozzi V, et al. Identifying the minimal copper- and zinc-binding site sequence in amyloid-beta peptides. J Biol Chem. 2008;283:10784–10792. doi: 10.1074/jbc.M707109200. [DOI] [PubMed] [Google Scholar]
- 38.Nair NG, Perry G, Smith MA, Reddy VP. NMR studies of zinc, copper, and iron binding to histidine, the principal metal ion complexing site of amyloid-beta peptide. J Alzheimers Dis. 2010;20:57–66. doi: 10.3233/JAD-2010-1346. [DOI] [PubMed] [Google Scholar]
- 39.Danielsson J, Pierattelli R, Banci L, Graslund A. High-resolution NMR studies of the zinc-binding site of the Alzheimer’s amyloid beta-peptide. FEBS J. 2007;274:46–59. doi: 10.1111/j.1742-4658.2006.05563.x. [DOI] [PubMed] [Google Scholar]
- 40.Syme CD, Viles JH. Solution 1H NMR investigation of Zn2+ and Cd2+ binding to amyloid-beta peptide (Abeta) of Alzheimer’s disease. Biochim Biophys Acta. 2006;1764:246–256. doi: 10.1016/j.bbapap.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 41.Klug GM, et al. Beta-amyloid protein oligomers induced by metal ions and acid pH are distinct from those generated by slow spontaneous ageing at neutral pH. Eur J Biochem. 2003;270:4282–4293. doi: 10.1046/j.1432-1033.2003.03815.x. [DOI] [PubMed] [Google Scholar]
- 42.Garai K, Sahoo B, Kaushalya SK, Desai R, Maiti S. Zinc lowers amyloid-beta toxicity by selectively precipitating aggregation intermediates. Biochemistry. 2007;46:10655–10663. doi: 10.1021/bi700798b. [DOI] [PubMed] [Google Scholar]
- 43.Ayton S, Lei P, Bush AI. Biometals and their therapeutic implications in Alzheimer’s disease. Neurotherapeutics. 2015;12:109–120. doi: 10.1007/s13311-014-0312-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bush AI. The metal theory of Alzheimer’s disease. J Alzheimers Dis. 2013;33(Suppl 1):S277–281. doi: 10.3233/JAD-2012-129011. [DOI] [PubMed] [Google Scholar]
- 45.Faux NG, et al. PBT2 rapidly improves cognition in Alzheimer’s Disease: additional phase II analyses. Journal of Alzheimer’s disease: JAD. 2010;20:509–516. doi: 10.3233/JAD-2010-1390. [DOI] [PubMed] [Google Scholar]
- 46.Lannfelt L, et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008;7:779–786. doi: 10.1016/S1474-4422(08)70167-4. [DOI] [PubMed] [Google Scholar]
- 47.Chromy BA, et al. Self-assembly of Abeta(1-42) into globular neurotoxins. Biochemistry. 2003;42:12749–12760. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- 48.Reinke AA, Seh HY, Gestwicki JE. A chemical screening approach reveals that indole fluorescence is quenched by pre-fibrillar but not fibrillar amyloid-beta. Bioorg Med Chem Lett. 2009;19:4952–4957. doi: 10.1016/j.bmcl.2009.07.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen RJ, Chang WW, Lin YC, Cheng PL, Chen YR. Alzheimer’s amyloid-beta oligomers rescue cellular prion protein induced tau reduction via the Fyn pathway. ACS chemical neuroscience. 2013;4:1287–1296. doi: 10.1021/cn400085q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brand L, Gohlke JR. Fluorescence probes for structure. Annu Rev Biochem. 1972;41:843–868. doi: 10.1146/annurev.bi.41.070172.004211. [DOI] [PubMed] [Google Scholar]
- 51.LeVine H., 3rd 4,4(′)-Dianilino-1,1(′)-binaphthyl-5,5(′)-disulfonate: report on non-beta-sheet conformers of Alzheimer’s peptide beta(1-40) Arch Biochem Biophys. 2002;404:106–115. doi: 10.1016/S0003-9861(02)00246-1. [DOI] [PubMed] [Google Scholar]
- 52.Kayed R, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 53.Kayed R, et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. 2007;2:18. doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu JW, et al. Fibrillar oligomers nucleate the oligomerization of monomeric amyloid beta but do not seed fibril formation. The Journal of biological chemistry. 2010;285:6071–6079. doi: 10.1074/jbc.M109.069542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saito H, Ando I, Ramamoorthy A. Chemical shift tensor - the heart of NMR: Insights into biological aspects of proteins. Progress in nuclear magnetic resonance spectroscopy. 2010;57:181–228. doi: 10.1016/j.pnmrs.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wishart DS. Advances in metabolite identification. Bioanalysis. 2011;3:1769–1782. doi: 10.4155/bio.11.155. [DOI] [PubMed] [Google Scholar]
- 57.Wishart DS. Interpreting protein chemical shift data. Progress in nuclear magnetic resonance spectroscopy. 2011;58:62–87. doi: 10.1016/j.pnmrs.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 58.Bertini I, Gonnelli L, Luchinat C, Mao J, Nesi A. A new structural model of Abeta40 fibrils. J Am Chem Soc. 2011;133:16013–16022. doi: 10.1021/ja2035859. [DOI] [PubMed] [Google Scholar]
- 59.Adamo AM, Oteiza PI. Zinc deficiency and neurodevelopment: the case of neurons. Biofactors. 2010;36:117–124. doi: 10.1002/biof.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim YH, Kim EY, Gwag BJ, Sohn S, Koh JY. Zinc-induced cortical neuronal death with features of apoptosis and necrosis: mediation by free radicals. Neuroscience. 1999;89:175–182. doi: 10.1016/S0306-4522(98)00313-3. [DOI] [PubMed] [Google Scholar]
- 61.Lal R, Lin H, Quist AP. Amyloid beta ion channel: 3D structure and relevance to amyloid channel paradigm. Biochim Biophys Acta. 2007;1768:1966–1975. doi: 10.1016/j.bbamem.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lambert MP, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang HW, et al. Soluble oligomers of beta amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain research. 2002;924:133–140. doi: 10.1016/S0006-8993(01)03058-X. [DOI] [PubMed] [Google Scholar]
- 64.Cullen WK, Suh YH, Anwyl R, Rowan MJ. Block of LTP in rat hippocampus in vivo by beta-amyloid precursor protein fragments. Neuroreport. 1997;8:3213–3217. doi: 10.1097/00001756-199710200-00006. [DOI] [PubMed] [Google Scholar]
- 65.Lee JY, Cole TB, Palmiter RD, Suh SW, Koh JY. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc Natl Acad Sci USA. 2002;99:7705–7710. doi: 10.1073/pnas.092034699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu WH, Lukiw WJ, Bergeron C, Niznik HB, Fraser PE. Metallothionein III is reduced in Alzheimer’s disease. Brain Res. 2001;894:37–45. doi: 10.1016/S0006-8993(00)03196-6. [DOI] [PubMed] [Google Scholar]
- 67.Xie XM, Smart TG. A physiological role for endogenous zinc in rat hippocampal synaptic neurotransmission. Nature. 1991;349:521–524. doi: 10.1038/349521a0. [DOI] [PubMed] [Google Scholar]
- 68.Lovell MA, Xie C, Markesbery WR. Protection against amyloid beta peptide toxicity by zinc. Brain research. 1999;823:88–95. doi: 10.1016/S0006-8993(99)01114-2. [DOI] [PubMed] [Google Scholar]
- 69.Bishop GM, Robinson SR. The amyloid paradox: amyloid-beta-metal complexes can be neurotoxic and neuroprotective. Brain Pathol. 2004;14:448–452. doi: 10.1111/j.1750-3639.2004.tb00089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cuajungco MP, et al. Evidence that the beta-amyloid plaques of Alzheimer’s disease represent the redox-silencing and entombment of abeta by zinc. J Biol Chem. 2000;275:19439–19442. doi: 10.1074/jbc.C000165200. [DOI] [PubMed] [Google Scholar]
- 71.Petkova AT, et al. A structural model for Alzheimer’s beta -amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mithu VS, et al. Zn(++) binding disrupts the Asp(23)-Lys(28) salt bridge without altering the hairpin-shaped cross-beta Structure of Abeta(42) amyloid aggregates. Biophys J. 2011;101:2825–2832. doi: 10.1016/j.bpj.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ehrnhoefer DE, et al. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol. 2008;15:558–566. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- 74.Freir DB, et al. Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nature communications. 2011;2:336. doi: 10.1038/ncomms1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.De Felice FG, et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging. 2008;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Minicozzi V, et al. Identifying the Minimal Copper- and Zinc-binding Site Sequence in Amyloid-{beta} Peptides. J. Biol. Chem. 2008;283:10784–10792. doi: 10.1074/jbc.M707109200. [DOI] [PubMed] [Google Scholar]
- 78.Syme CD, Viles JH. Solution 1H NMR investigation of Zn2+ and Cd2+ binding to amyloid-beta peptide (A[beta]) of Alzheimer’s disease. Biochimica et Biophysica Acta (BBA) - Proteins & Proteomics. 2006;1764:246–256. doi: 10.1016/j.bbapap.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 79.Danielsson J, Pierattelli R, Banci L, Graslund A. High-resolution NMR studies of the zinc-binding site of the Alzheimer’s amyloid beta peptide. FEBS Journal. 2007;274:46–59. doi: 10.1111/j.1742-4658.2006.05563.x. [DOI] [PubMed] [Google Scholar]
- 80.Stellato F, et al. Metal binding in amyloid β-peptides shows intra- and inter-peptide coordination modes. European Biophysics Journal. 2006;35:340–351. doi: 10.1007/s00249-005-0041-7. [DOI] [PubMed] [Google Scholar]
- 81.Jonsson T, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 82.Di Fede G, et al. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science. 2009;323:1473–1477. doi: 10.1126/science.1168979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Giaccone G, et al. Neuropathology of the recessive A673V APP mutation: Alzheimer disease with distinctive features. Acta Neuropathol. 2010;120:803–812. doi: 10.1007/s00401-010-0747-1. [DOI] [PubMed] [Google Scholar]
- 84.Lin TW, et al. Alzheimer’s amyloid-beta A2T variant and its N-terminal peptides inhibit amyloid-beta fibrillization and rescue the induced cytotoxicity. PLoS One. 2017;12:e0174561. doi: 10.1371/journal.pone.0174561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Basi GS, et al. Structural correlates of antibodies associated with acute reversal of amyloid beta-related behavioral deficits in a mouse model of Alzheimer disease. The Journal of biological chemistry. 2010;285:3417–3427. doi: 10.1074/jbc.M109.045187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Deshpande A, Kawai H, Metherate R, Glabe CG, Busciglio J. A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J Neurosci. 2009;29:4004–4015. doi: 10.1523/JNEUROSCI.5980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Takeda A, et al. Amyloid beta-mediated Zn2+ influx into dentate granule cells transiently induces a short-term cognitive deficit. PloS one. 2014;9:e115923. doi: 10.1371/journal.pone.0115923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takeda A, et al. Extracellular Zn2+ Is Essential for Amyloid beta1-42-Induced Cognitive Decline in the Normal Brain and Its Rescue. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2017;37:7253–7262. doi: 10.1523/JNEUROSCI.0954-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sevigny J, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature. 2016;537:50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 90.Burdick D, et al. Assembly and aggregation properties of synthetic Alzheimer’s A4/beta amyloid peptide analogs. J Biol Chem. 1992;267:546–554. [PubMed] [Google Scholar]
- 91.Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 92.Chang YJ, Chen YR. The coexistence of an equal amount of Alzheimer’s amyloid-beta 40 and 42 forms structurally stable and toxic oligomers through a distinct pathway. The FEBS journal. 2014;281:2674–2687. doi: 10.1111/febs.12813. [DOI] [PubMed] [Google Scholar]
- 93.Liao YH, Chen YR. A novel method for expression and purification of authentic amyloid-beta with and without (15)N labels. Protein expression and purification. 2015;113:63–71. doi: 10.1016/j.pep.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 94.Tsai KJ, Tsai YC, Shen CK. G-CSF rescues the memory impairment of animal models of Alzheimer’s disease. J Exp Med. 2007;204:1273–1280. doi: 10.1084/jem.20062481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Morroni F, Sita G, Tarozzi A, Rimondini R, Hrelia P. Early effects of Abeta1-42 oligomers injection in mice: Involvement of PI3K/Akt/GSK3 and MAPK/ERK1/2 pathways. Behavioural brain research. 2016;314:106–115. doi: 10.1016/j.bbr.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 96.Chen JH, Ke KF, Lu JH, Qiu YH, Peng YP. Protection of TGF-beta1 against neuroinflammation and neurodegeneration in Abeta1-42-induced Alzheimer’s disease model rats. PloS one. 2015;10:e0116549. doi: 10.1371/journal.pone.0116549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tsukuda K, et al. Cognitive deficit in amyloid-beta-injected mice was improved by pretreatment with a low dose of telmisartan partly because of peroxisome proliferator-activated receptor-gamma activation. Hypertension. 2009;54:782–787. doi: 10.1161/HYPERTENSIONAHA.109.136879. [DOI] [PubMed] [Google Scholar]
- 98.Cascella R, et al. Extracellular chaperones prevent Abeta42-induced toxicity in rat brains. Biochimica et biophysica acta. 2013;1832:1217–1226. doi: 10.1016/j.bbadis.2013.04.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.