

Abstract
Structure-function relationships of nine rationally designed ritonavir-like compounds were investigated to better understand the ligand binding and inhibitory mechanism in human drug-metabolizing cytochrome P450 3A4 (CYP3A4). The analogs had a similar backbone and pyridine and tert-butyloxycarbonyl (Boc) as the heme-ligating and terminal groups, respectively. N-isopropyl, N-cyclopentyl or N-phenyl were the R1-side group substituents alone (compounds 5a–c) or in combination with phenyl or indole at the R2 position (8a–c and 8d–f subseries, respectively). Our experimental and structural data indicate that (i) for all analogs, a decrease in the dissociation constant (Ks) coincides with a decrease in IC50 but no relation with other derived parameters is observed; (ii) an increase in the R1 volume, hydrophobicity and aromaticity markedly lowers Ks and IC50, whereas addition of aromatic R2 has a more pronounced positive effect on the inhibitory potency than the binding strength; (iii) the ligands’ association mode is strongly influenced by the mutually dependent R1–R2 interplay, but the R1-mediated interactions are dominant and define the overall conformation in the active site; (iv) formation of a strong H-bond with Ser119 is a prerequisite for potent CYP3A4 inhibition; and (v) the strongest inhibitor in the series, the R1-phenyl/R2-indole containing 8f (Ks and IC50 of 0.08 and 0.43 μM, respectively), is still less potent than ritonavir, even under conditions that prevent the mechanism based inactivation of CYP3A4. Crystallographic data were essential for better understanding and interpretation of the experimental results, and suggested how the inhibitor design could be further optimized.
Keywords: CYP3A4, ligand binding, inhibitor design, crystal structure, structure/function relations
Graphical abstract
INTRODUCTION
Cytochrome P450 3A4 (CYP3A4) is the major and most clinically relevant drug-metabolizing enzyme in the human body1 that oxidizes over 50% of the commonly used drugs,2, 3 some of which could also act as inducers, effectors and inhibitors of CYP3A4.4 Inhibition of CYP3A4 could lead to drug toxicity, drug-drug interactions and other adverse effects, and is usually undesired. In some cases, however, CYP3A4 inactivation might be beneficial because it could improve therapeutic efficiency of the quickly metabolized drugs by elevating their plasma levels. Elucidation of structure/function relations and the ligand binding and inhibitory mechanism of CYP3A4 is necessary for the development of safer drugs and more potent pharmacoenhancers. Currently, two CYP3A4 inhibitors, ritonavir (Fig. 1A) and its derivative cobicistat, are used as pharmacoen-hancers in combination therapy for HIV and HCV infections.5–8 Both drugs were developed based on the chemical structure-activity relationship studies rather than the CYP3A4 crystal structure, and their inhibitory mechanism is still under debate.9–14
Figure 1.
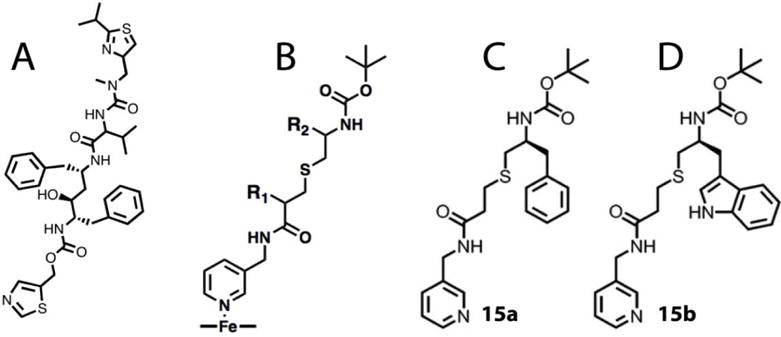
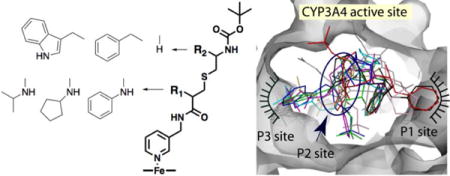
A, Chemical structure of ritonavir. B, Scaffold used as a template for synthesis of CYP3A4 inhibitors: pyridine nitrogen is the heme ligand, R1 and R2 are variable side groups, and Boc is the terminal moiety. C and D, Two most potent inhibitors of CYP3A4 from the first
One complexity in this research area is that ritonavir, the most potent CYP3A4 inhibitor developed so far, also serves as a substrate15, 16 and its reactive metabolites could inactive CYP3A4 via covalent or non-covalent attachment.9–11 Because of ritonavir’s large size and complex chemical structure, identification of its reactive metabolites and the molecular mechanism responsible for the mechanism based inactivation has been challenging. One recent study showed that the ritonavir-dependent inactivation of CYP3A4 proceeds via heme destruction and formation of a heme-protein adduct, with no significant modification of the apoprotein.13 In contrast, the following thorough investigation by Rock et al. ruled out the heme modification/destruction pathway, as well as the non-covalent metabolic inhibitory complex formation.14 Instead, it was demonstrated that the metabolite, supposedly produced upon S-oxidation of the isopropylthiazole, attaches to the peripheral surface residue Lys257. The role of cytochrome b5 in bioactivation of ritonavir was suggested but not tested. Most importantly, there was no evidence that elimination of the Lys257 side chain prevents modification and inactivation of CYP3A4.
Alternatively, our mechanistic and structural studies on highly purified recombinant CYP3A4 suggested that ritonavir-like molecules could inactivate CYP3A4 via strong ligation to the heme iron, which leads to the blockage of the active site and prevents the substrate access to the catalytic center, decreases the heme redox potential, and slows down electron transfer from the redox partner, cytochrome P450 reductase (CPR).12, 17–19 Furthermore, we mapped the active site and built a pharmacophore model for the CYP3A4-specific inhibitor.20 The pharmacophoric determinants, ranked according to their importance, include: I – a strong heme-ligating nitrogen donor (pyridine ring); II – a flexible backbone allowing conformational adjustments; III and IV – hydrophobic/aromatic R1 and R2 side groups, mediating contacts with the cavities above the I-helix and near the heme plane (P1 and P2 sites, respectively); V – an H-bond donor/accepting group capable of interacting with the active site Ser119; and VI – a multi-functional terminal group. Using a general scaffold (Fig. 1B), we designed and tested the first set of inhibitors, lacking R1 and differing in the backbone length and R2 substituents.21 The results of this study supported the pharmacophore model and confirmed the importance of the backbone flexibility, H-bonding to Ser119, and hydrophobic interactions at P1 and P2 sites. Most importantly, two structurally simple compounds from this series, 15a and 15b (phenyl and indole as R2, respectively; Fig. 1C,D), had submicromolar affinity and inhibitory potency for CYP3A4.
In this work, we utilized the same scaffold to design the second series of analogs with various R1 functionalities and R1–R2 combinations. The new compounds (Fig. 2) had a cysteine-based backbone, pyridine and tert-butyloxycarbonyl (Boc) as the heme-ligating and terminal moiety, respectively, and N-isopropyl, N-cyclopentyl or N-phenyl as a sole R1 group or with phenyl or indole at R2 position. Our experimental and structural data provided deeper insights into the ligand binding process, helped to more accurately map the CYP3A4 active site, and highlighted strategies for further optimization of the inhibitor design.
Figure 2.
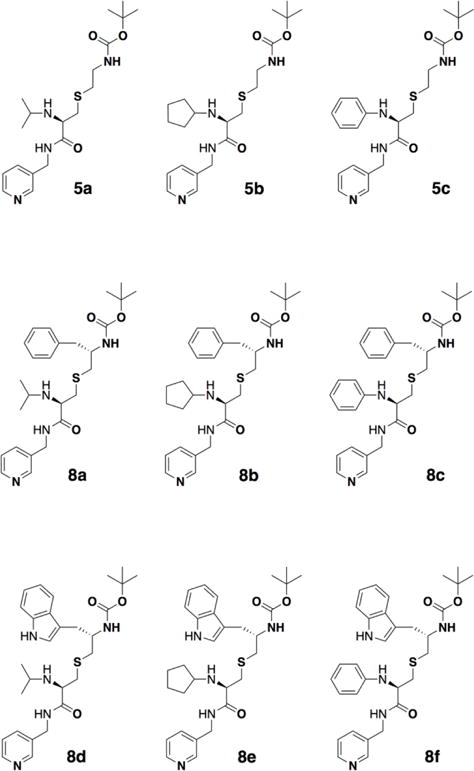
Chemical structures of compounds investigated in this study.
EXPERIMENTAL PROCEDURES
Synthesis, mass spectrometry and NMR data for compounds 5a–8f are included in the Supporting Information. The full length codon-optimized and Δ3-22 non-optimized wild type human CYP3A4 were produced as reported previously.22 Δ3-22 CYP3A4 was used for crystallization, equilibrium titrations, ligand binding kinetics and thermal denaturation assays, whereas the full-length protein was utilized in the functional assays because the N-terminal tail promotes interaction with CPR.23
Spectral Binding Titrations
Ligand binding to CYP3A4 was monitored in a Cary 300 spectrophotometer at ambient temperatures in 0.1M phosphate pH 7.4, containing 20% glycerol and 1 mM dithiothreitol (buffer A). Inhibitors were dissolved in dimethyl sulfoxide and added to a 1.5–2 μM protein solution in small aliquots, with the final solvent concentration <2%. Spectral dissociation constants (Ks) were determined from the hyperbolic or quadratic fits to titration curves.
Kinetics of Ligand Binding
Kinetics of ligand binding to CYP3A4 was monitored at ambient temperatures in 0.1M phosphate, pH 7.4, in a SX.18MV stopped flow apparatus (Applied Photophysics, UK). After mixing 2 μM CYP3A4 with a 20 μM ligand solution, absorbance changes at 427 nm were measured to follow conversion of the heme iron to the low-spin form. Kinetic data were analyzed using manufacturer’s PROKIN software.
Thermal Denaturation and Inhibitory Potency Assays
To compare ligand-dependent changes in CYP3A4 stability, thermal denaturation experiments were conducted in 0.1 M phosphate, pH 7.4, as previously described.18 The inhibitory potency of the investigated compounds on the 7-benzyloxy-4-(trifluoromethyl)coumarin (BFC) O-debenzylation activity of CYP3A4 was evaluated fluorometrically in a soluble, lipid-free reconstituted system. The reaction was carried out at room temperature in 0.1 M phosphate, pH 7.4, containing catalase and superoxide dismutase (2 Units/ml each). A mixture of CYP3A4 and rat CPR (1 μM each) was preincubated at room temperature for 1 hour and then diluted 20-fold with the assay buffer. A 100 μl sample of the 0.05 μM protein mixture was incubated for 2 minutes at room temperature with various concentrations of inhibitors (dissolved in DMSO) before addition of 50 μM BFC. The reaction was initiated with 200 μM NADPH (final concentration) and formation of 7-hydroxy-4-trifluoro methylcoumarin was followed in a Hitachi F100 fluorometer (λex = 430 nm; λem = 500 nm) for two minutes, during which the fluorescence increase was linear. Measurements were performed in triplicates to ensure the accuracy of calculations. The IC50 values were derived from the [% activity] vs. [inhibitor] plots by fitting the data to a four-parameter logistic nonlinear regression equation: y = D + ((A–D)/(1+ 10(X-logC)•B)), where A and D are the maximal and minimal activity values, respectively, B is the slope factor, and C is IC50.
Determination of the X-ray Structures of CYP3A4
CYP3A4 was co-crystallized with the inhibitors by a microbatch method under oil. Protein in buffer A (70–80 mg/ml) was mixed with a 5-to-10-fold excess of the ligand and centrifuged to remove the precipitate. The ligand-bound CYP3A4 (0.4–0.6 μl) was mixed with 0.4–0.6 μl of 6-10% polyethylene glycol 3,350 and 60–90 mM sodium malonate, pH 6.0–7.0, and covered with paraffin oil. After harvesting, crystals were cryoprotected with Paratone-N and frozen in liquid nitrogen. X-ray diffraction data were collected at the Stanford Synchrotron Radiation Lightsource beamline 9-2 and the Advanced Light Source beamlines 5.0.2 and 8.2.2. Crystal structures were solved by molecular replacement with PHASER24 and the 5VCC structure as a search model. The initial models were rebuilt and refined with COOT25 and PHENIX.26 Data collection and refinement statistics are summarized in Tables S1 and S2. The atomic coordinates and structure factors for the 5a-, 5b-, 5c-, 8a-, 8b-, 8c-, 8d-, 8e- and 8f-bound CYP3A4 were deposited in the Protein Data Bank with the ID codes 6BCZ, 6BD5, 6BD6, 6BD7, 6BD8, 6BDH, 6BDI, 6BDK and 6BDM, respectively.
RESULTS AND DISCUSSION
A new series of inhibitors were designed to investigate how changes in the volume, hydrophobicity and aromaticity of the R1 substituent affect ligation to CYP3A4. For this purpose, N-isopropyl, N-cyclopentyl or N-phenyl groups were introduced at R1 position alone (compounds 5a–c, respectively) or in combination with the R2 phenyl or indole (compounds 8a–c and 8d–f, respectively; Fig. 2). R1 was a racemic mixture in 5c, 8c and 8f, and in R-configuration in other compounds. As in the previous series,21 the R2 side group was in S-configuration. To better define structure/function relations, properties of the new compounds were compared to those of the R2-oly-containig 15a and 15b from the first series (Fig. 1C,D)21 and ritonavir,12 a highly potent CYP3A4 inactivator currently used as a pharmacoenhancer for anti-viral drugs.
Binding Affinity
Upon binding to CYP3A4, all compounds induced a low-spin (type II) shift in the Soret band, indicative of the pyridine nitrogen ligation to the heme iron, with subtle differences in the position and amplitude of the absorption peak (red spectra in Fig. 3). Reduction of the ligand-bound forms with sodium dithionite led to formation of the 442–443 nm band, characteristic for the ritonavir-like inhibitors (green spectra in Fig. 3). Based on the previous studies,12, 17–19, 21 the amplitude of the 442 nm band was suggested to correlate with the inhibitory potency of the bound compound: the higher the peak the stronger CYP3A4 inactivation. To quantify and compare spectral perturbations, we calculated ratios between the absorption maxima of the ferric/ferrous ligand-bound and ferric ligand-free forms (A421/417 and A443/417, respectively; Table 1). The maximal 421 and 443 nm absorption was observed for the 5a-/8d- and 8f-bound CY3A4, respectively, but the largest A442/417 value was derived for the ritonavir-bound CYP3A4 (Table 1 and Fig. S1).
Figure 3.
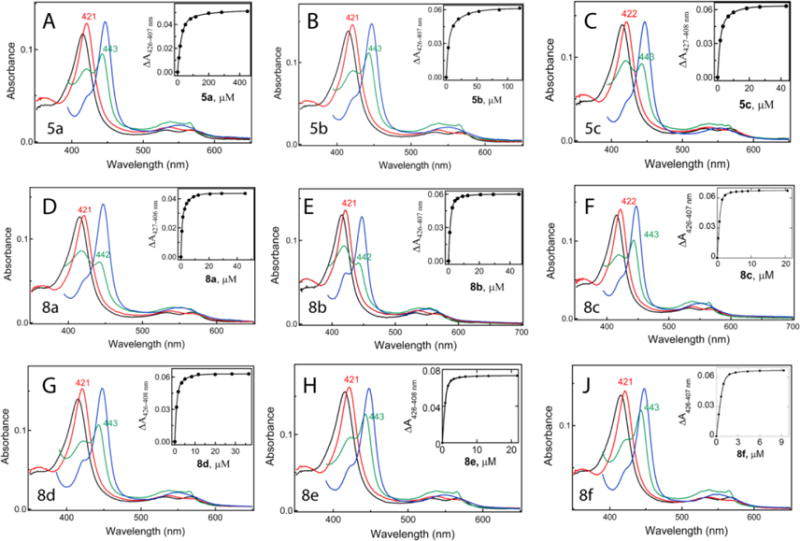
Spectral changes induced in CYP3A4 by the investigated compounds. Spectra of the ferric ligand-free CYP3A4 are in black, and those of the ferric ligand-bound, ferrous and ferrous CO bound forms measured at the end of the titration are in red, green and blue, respectively. Insets are equilibrium titration plots and hyperbolic or quadratic fittings, from which spectral dissociation constants (Ks) were calculated (listed in Table 1).
Table 1.
Properties of the CYP3A4 inhibitors
| compound | λmax (nm) | A421/417a | A443/417b | Ksc μM | IC50d μM | IC50/Ks | ΔTme °C | kfastf s−1 | fast phaseg % | ΔA427nmh % | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ferric | ferrous | ||||||||||
| 5a | 421 | 443 | 1.12 | 0.82 | 12 ± 2 | 27.8 ± 2.6 | 2.3 | 0.2 | 1.4±0.2 | 41 | 38 |
| 5b | 421 | 443 | 1.07 | 0.81 | 5 ± 1 | 10.8 ± 0.5 | 2.2 | 0.7 | 1.4±0.3 | 50 | 55 |
| 5c | 422 | 443 | 1.03 | 0.66 | 1.8 ± 0.2 | 2.3 ± 0.1 | 1.3 | 0.5 | 1.6±0.1 | 55 | 74 |
| 8a | 421 | 442 | 1.02 | 0.58 | 0.92 ± 0.02 | 3.9 ± 0.2 | 4.2 | 0.4 | 1.0±0.1 | 55 | 64 |
| 8b | 421 | 442 | 1.05 | 0.56 | 0.42 ± 0.03 | 1.96 ± 0.08 | 4.7 | 0.9 | 1.4±0.1 | 48 | 78 |
| 8c | 422 | 443 | 1.07 | 0.76 | 0.11 ± 0.01 | 0.72 ± 0.03 | 6.5 | 0.8 | 1.9±0.2 | 59 | 96 |
| 8d | 421 | 443 | 1.12 | 0.77 | 0.78 ± 0.03 | 0.97 ± 0.06 | 1.2 | 1.0 | 1.6±0.2 | 53 | 93 |
| 8e | 421 | 443 | 1.05 | 0.81 | 0.11 ± 0.01 | 0.66 ± 0.03 | 6.0 | 1.4 | 1.6±0.1 | 55 | 94 |
| 8f | 421 | 443 | 1.05 | 0.87 | 0.08 ± 0.01 | 0.43 ± 0.02 | 5.4 | 1.1 | 2.4±0.3 | 54 | 100 |
| ritonavir | 421 | 442 | 1.08 | 1.10 | 0.017± 0.003 | 0.32 ± 0.04 | 18.8 | 0.9 | 1.2±0.1 | 60 | 100 |
| 15ai | 421 | 442 | 1.02 | 0.81 | 0.9 | 0.52 | 0.6 | −1.0 | 1.7 | 55 | n.d.j |
| 15bi | 421 | 442 | 1.00 | 0.87 | 0.5 | 0.21 | 0.4 | 0.2 | 8.0 | 57 | n.d. |
Ratios between the absorbance maxima for the ferric and ferrous ligand-bound forms and the Soret band of ferric ligand-free CYP3A4, respectively.
The dissociation constant for the CYP3A4-inhibitor complex determined from the titration plots shown in Insets in Figure 3.
Determined for the BFC debenzylase activity of recombinant CYP3A4 in a reconstituted system with CPR.
Ligand-dependent changes in the melting temperature of CYP3A4.
The rate constant for the fast and slow kinetic phase of the CYP3A4-ligand binding reaction determined at saturating ligand concentration.
Percentage of the absorbance change taking place during the fast kinetic phase.
Percentage of the maximal absorbance change observed at the end of the reaction.
Characterized previously.21
Not determined.
Spectral dissociation constants for the CYP3A4-inhibitor complexes (Ks; a measure of the binding affinity) were determined from the equilibrium titration plots (Insets in Fig. 3; Table 1). Compared to the side-group-lacking compound 4 from the first series,21 introduction of a short branched side chain at R1 position led to a 9-fold decrease in Ks: from 105 to 12 μM for 5a. The binding affinity was increased by several-fold when cyclopentyl or phenyl was the R1 substituent (Ks of 5 and 1.8 μM for 5b and 5c, respectively). Addition of phenyl or indole at R2 position led to a further and more drastic decrease in Ks (16-to-45-fold). In all subseries, compounds with the aromatic R1 were the strongest binders. The lowest Ks (0.08 μM) and the highest A443/417 ratio were derived for the R1-phenyl/R2-indole containing 8f but, overall, there was no correlation between these parameters (Table 1).
Kinetics of Ligand Binding
Presteady-state kinetics of the ligand binding to CYP3A4 was investigated by stopped flow spectrophotometry by monitoring conversion of the heme iron to a low-spin form (Fig. 4). Under studied conditions, all reactions were biphasic, with ~40–60% of the absorbance change taking place during the fast phase. The lowest and the highest kfast values were derived for 8a and 8f (1.0 and 2.4 s−1, respectively; Table 1); kslow remained within the 0.13–0.23 s−1 range for all compounds. The relative percentage of the fast phase and the maximal absorbance change observed at the end of the reaction (ΔA427nm) are given in Table 1. It is evident from the kinetic data that (i) regardless of the R2 presence, the aromatic R1 group promotes and drives the ligation reaction to completion, (ii) the enhancing effect is maximal when indole is at R2 position, (iii) the R1-phenyl/R2-indole combination is most optimal for the initial complex formation with CYP3A4, and (iv) there is interference between the bulky side groups that complicates and slows down the ligand association (kfast for 8d–f is 4-fold lower than for 15b).
Figure 4.
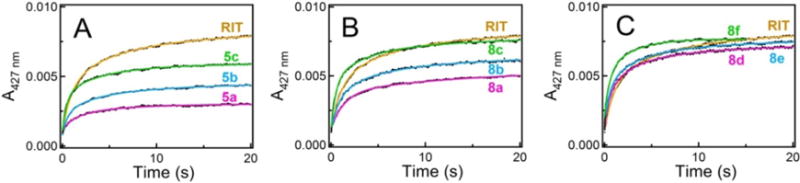
Kinetics of ligand binding to CYP3A4. Kinetic traces were recorded at ambient tem peratures after mixing 2 μM CYP3A4 with 20 μM solutions of the investigated compounds or ritonavir (RIT); fitting curves are shown as solid colored lines. All reactions were biphasic within the studied time interval. The rate constants for the fast phase (kfast) are listed in Table 1.
Melting Temperature and Inhibitory Potency
Tightly bound ligands are known to increase protein stability, which could manifest through an increase in the melting temperature (Tm). The first series of rationally designed inhibitors had a negative or slightly positive impact on Tm of CYP3A4.21 In contrast, all new compounds increased protein stability, although to a different extent (Table 1). Binding of 5a caused the smallest ΔTm change (0.2°C), whereas association of 8d–f increased Tm by 1.0–1.4°C. Interestingly, in each subseries, cyclopentyl rather than phenyl at R1 position led to a maximal stabilization of CYP3A4. Thermal denaturation experiments indicate also that interactions mediated by the side groups rather than the poly-functional tail portion have a greater impact on the protein stability (compare ΔTm for 8d–f and ritonavir).
The inhibitory potency of new compounds on the catalytic activity of CYP3A4 was determined in a lipid-free reconstituted system with CPR and BFC as a substrate. Soluble CYP3A4 is ~100-fold less active than the membrane-bound enzyme.22, 23, 27 However, exclusion of lipids from the assay allows to standardize experimental conditions by preventing unequal partition of the investigated compounds into the lipid bilayer. Introduction of isopropyl, cyclopentyl or phenyl at R1 position led to a notable decrease in IC50 (inhibitor concentration required for a 50% decrease in the enzyme activity): from 75 μM for the side-group-lacking compound 421 to 28, 11 and 2 μM for 5a–c, respectively (Table 1). The IC50 values were further decreased and reached submicromolar levels when aromatic R2 was added.
Comparison of the respective Ks and IC50 values shows that, for all investigated compounds, tighter binding leads to more potent inhibition of CYP3A4 (Table 1). However, this positive correlation is not strong, as the IC50:Ks ratio varies by several fold (from 1.3 to 6.5) and reaches the extreme values for ritonavir and the R2-only-containing compounds 15a and 15b (19 and 0.4–0.6, respectively). This could be due to a different impact of the side groups on Ks and IC50 and/or because of variations in the assay conditions. For instance, glycerol, present in the titration buffer, can bind and partially block the active site,22 whereas CPR, a redox partner used for the inhibitory assays, alters the CYP3A4 active site topology through protein-protein interactions.28 Both ingredients could influence Ks and IC50 in a ligand-dependent manner. On the other hand, the contribution of the mechanism based inactivation of CYP3A4 in our reconstituted system is minimal, if any, because (i) NADPH was the last to be added to the inhibitory assay mixture, and (ii) our compounds lack thiazole and isopropylthiazole moieties, the likely sites of bioactivation in ritonavir.9, 14, 29
Crystal Structures of the Inhibitor-Bound CYP3A4
To get direct insights on how changes in the R1 substituent and R1–R2 combination affect the ligand binding mode, CYP3A4 was co-crystallized with each investigated compound. The X-ray structures were determined to 2.23–2.67 Å resolution (Tables S1 and S2) and structural features of the CYP3A4-inhibitor complexes are compared in Table 2. During structural refinement with PHENIX,26 the R1 chirality in 5c, 8c and 8f was not specified but, for all three compounds, the S-stereoisomer of R1 was automatically chosen. Manual examination confirmed that this enantiomer fits better into electron density maps, and the respective structures could be refined to the lower R/Rfree factors (a measure of the agreement between the model and the experimental X-ray diffraction data). This implies that CYP3A4 selects and preferentially binds the S-configuration of R1-phenyl.
Table 2.
Structural features of the CYP3A4-inhibitor complexes
| compound | Fe-N bond
|
pyridine ring rotation (°)b | I-helix displacement (Å)c | F-F′ loop conformation | H-bonds
|
Boc-interacting residuesd | ||
|---|---|---|---|---|---|---|---|---|
| distance (Å) | angle (°)a | atoms | length (Å) | |||||
| 5a | 2.28 | 10 | 22 | 0.37–0.45 | ordered | O12-Arg212:NH2 O13-Arg212:NH2 O12-Ala370:O O19-Ser119:OG |
2.64 3.17 3.19 2.90 |
304, 37, 372 |
| 5b | 2.26 | 0 | 30 | 0.58–0.70 | traceable | none | 215, 373, 374 | |
| 5c | 2.23 | 1 | 30 | 1.64–1.43 | traceable | O22-Ser119:OG | 2.75 | heme, 105, 119 |
| 8a | 2.21 | 13 | 2 | 0.54–0.63 | ordered | O19-Ala370:O O26-Ser119:OG |
3.13 2.45 |
217, 370, 372, 374 |
| 8b | 2.22 | 2 | 30 | 1.56–1.65 | traceable | none | 212, 213, 304 | |
| 8c | 2.37 | 5 | 25 | 1.95–1.94 | disordered | O22-Ser119:OG | 2.73 | 304, 369, 482 |
| 8d Ae | 2.27 | 13 | 7 | 0.50–0.63 | ordered | O29 – Ser119:OG N13-Arg372:O |
2.37 2.70 |
212, 309, 369, 370 |
| Be | 2.30 | 10 | 7 | 0.50–0.63 | ordered | O29 – Ser119:OG N13-Arg372:O |
2.28 2.70 |
212, 309, 369, 370 |
| 8e | 2.37 | 25 | 5 | 0.54–0.68 | traceable | O21 – Ser119:OG N33-Arg372:O |
2.47 3.20 |
212, 309, 369, 370 |
| 8f | 2.21 | 4 | 30 | 1.81–2.40 | disordered | O22 – Ser119:OG | 2.53 | 106. 108, 115, 119, 120 |
| ritonavirf | 2.16 | 8 | 45g | 1.99–2.18 | traceable | O24: Ser119:OG O41: Ser119:OG |
2.92 2.84 |
|
Deviation from perpendicularity.
Angle between the planes passing through the pyridine ring and the NB-ND heme atoms
Distance between the Cα-atoms of Phe304 and Ala305 in the inhibitor-bound and the ligand-free CYP3A4 structure (PDB ID 5VCC).
Residues within the Van der Waals distance from the terminal Boc group.
Two alternative conformation with the occupancy of 60 and 40%, respectively.
Parameters derived from the 5VC0 structure.
Respective angle for the thiazole ring.
Structures of the 5a-, 5b- and 5c-bound CYP3A4
Conformations of 5a–c varied significantly because of differences in the R1 orientation (Fig. 5). Only 5c phenyl could be inserted into the pocket above the central I-helix (P1 site) to form π-π stacking and hydrophobic interactions with Phe304 and the neighboring Leu210, Phe213, Phe241 and Ile301 residues (Fig. 5C). The backbone of 5c bends sharply in order to place Boc near the pyridine and heme rings (P2 site) and promote hydrophobic interactions. This binding manner leads to the I-helix displacement and enables formation of a strong H-bond between O22 and Ser119. In contrast, the isopropyl and cyclopentyl groups cannot insert into the pocket and are positioned near the adjacent phenylalanine cluster, a unique feature of CYP3A4. As a result, 5a–b adapt a more extended conformation and do not distort the I-helix (Fig. 5A,B,D). The mid-portions of their backbone fill in P2 site, and the tails are less ordered and point toward the polar surface formed by the β-domain residues 370–373 (will be referred to as P3 site). 5a forms three H-bonds: with Ser119 via O19 and with Arg212 and Ala370 via Boc. None of these stabilizing contacts are established by 5b, because its cyclopentane ring is pulled closer to the Phe-cluster and the backbone curvature is altered (Fig. 5D,F). Compared to 5a–b, the pyridine ring of 5c is at a different angle and closer to the heme (Fig. 5E; Table 2). These structural dissimilarities and a more optimal stereochemistry of R1-phenyl could, in part, explain why 5c binds to CYP3A4 stronger than the counterparts.
Figure 5.
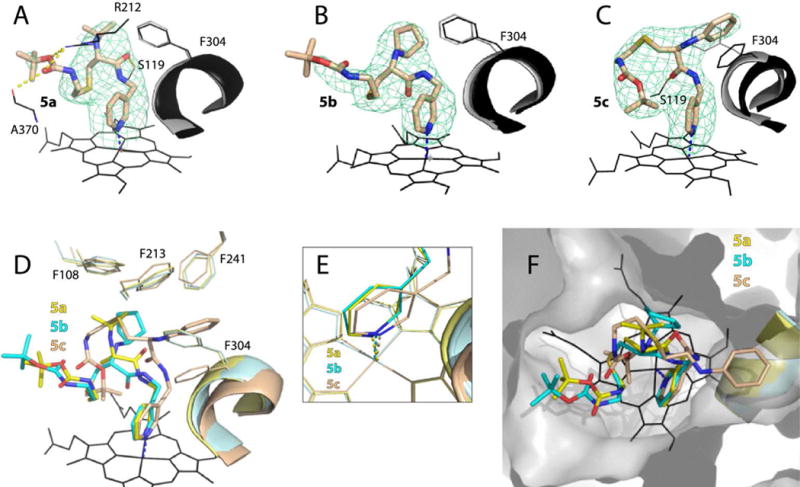
Crystal structures of CYP3A4 bound to the 5a–c analogs. A–C, The binding mode of 5a, 5b and 5c, respectively. The Phe304-containing portions of the I-helix in the inhibitor-bound and ligand-free (5VCC) structures of CYP3A4 are displayed in black and gray, respectively. Polder omit maps contoured at 4σ level are shown as green mesh; yellow dashed lines are hydrogen bonds. D–F, Structural overlays showing the extent of the I-helix displacement, position of the heme-ligating pyridine, and the ligands’ orientation in the active site cavity (top view), respectively.
Structures of the 8a-, 8b- and 8c-bound CYP3A4
The binding manner of 8a–c is shown in Figure 6. In 8a, R2-phenyl is positioned at P2 site and has virtually no effect on R1. The ligand conformation is stabilized by aromatic stacking interactions between the R2-phenyl and pyridine rings, and H-bonding to Ser119 and Ala370 (Fig. 6A). In 8b and 8c, there is a larger overlap between the R2-phenyl and pyridine rings, which not only strengthens π-π stacking interactions but also alters the R1 orientation (Fig. 6B,C). The cyclopentyl moiety of 8b moves closer and partially inserts into P1 pocket parallel to the Phe304 ring. These hydrophobic interactions compensate, to some extent, for the inability of 8b to form stabi-lizing Hbonds. A similar trend is observed for 8c, whose R1-phenyl is pushed deeper into P1 site and stacks with the parallel Phe304 and Phe241 rings (Fig 6D), triggering reorganization in the adjacent F–G fragment (Fig. S2A). Thus, there is mutual influence between the R2-phenyl and R1-cyclopentyl/phenyl groups that helps to optimize and strengthen protein-ligand interactions.
Figure 6.
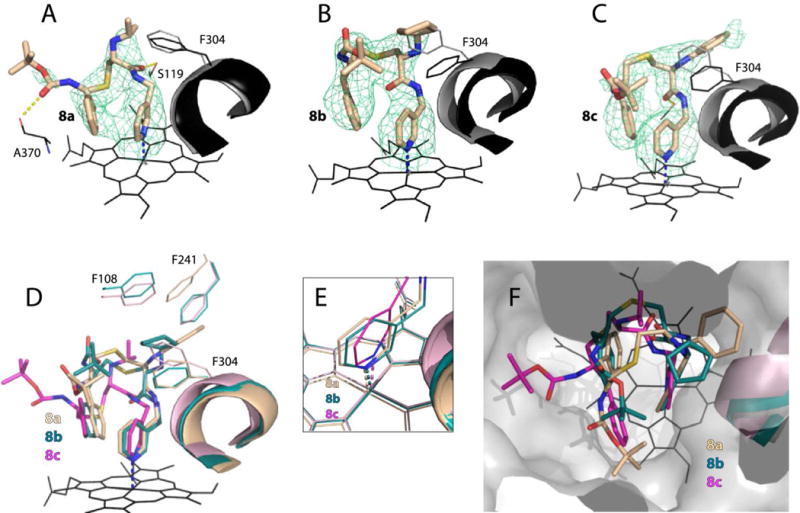
Crystal structures of CYP3A4 bound to 8a–c. A–C, The binding mode of 8a, 8b and 8c, respectively. The Phe304-containing parts of the I-helix in the inhibitor-bound and ligand-free (5VCC) structures of CYP3A4 are displayed in black and gray, respectively. Polder omit maps contoured at 4σ level are shown as green mesh; yellow dashed lines are hydrogen bonds. D–F, Structural overlays showing the extent of the I-helix displacement, positioning of the heme-ligating pyridine, and the ligands’ orientation in the active site cavity (top view), respectively.
Structures of the 8d-, 8e- and 8f-bound CYP3A4
8f binds in a traditional conformation, with R1-phenyl stacked between Phe304 and Phe241 at P1 site and R2-indole parallel to pyridine at P2 site (Fig. 7C,D). The backbone bending maximizes the indole/pyridine overlap and allows Boc to form Van der Waals interactions with Arg105 and Phe108. In contrast, 8d and 8e have a distinct conformation, with the R2-indole occupying P3 rather than P2 site (Fig. 7A,B,D). This orientation is stabilized by an H-bond between the indole nitrogen and the carbonyl oxygen of Arg372. Another notable feature is the alternative conformations of the 8d isopropyl, which can turn toward or away from the Phe-cluster (~40 and 60% occupancy, respectively; Fig. 7A). Importantly, a strong H-bond with Ser119 is formed by all three compounds but only 8f distorts the I-helix and F-F′ loop (Table 2).
Figure 7.
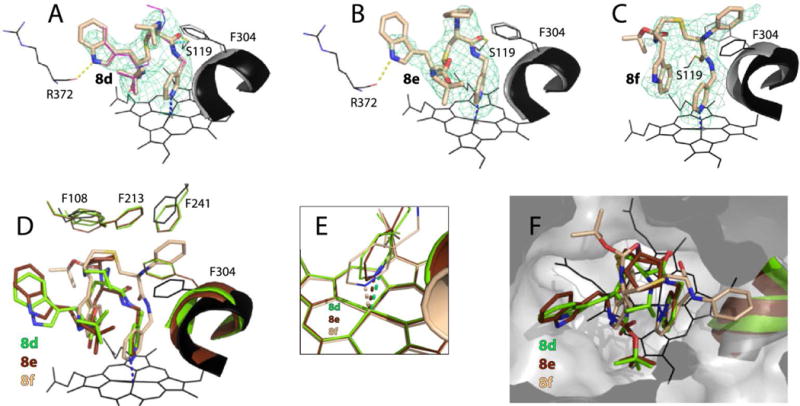
Crystal structures of CYP3A4 bound to 8d–f. A–C, The binding mode of 8d, 8e and 8f, respectively. The Phe304-containing parts of the I-helix in the inhibitor-bound and ligand-free (5VCC) structures of CYP3A4 are displayed in black and gray, respectively. Polder omit maps contoured at 4σ level are shown as green mesh; yellow dashed lines are hydrogen bonds. D–F, Structural overlays showing the extent of the I-helix displacement, position of the heme-ligating pyridine, and the ligands’ orientation in the active site cavity (top view), respectively.
Comparison of the counterparts from different subseries shows that the ligand binding mode is largely controlled by the R1-mediated interactions (Fig. 8). Regardless of the presence and chemical nature of the R2 substituent, R1-isopropyl never inserts into P1 pocket (Fig. 8A) but R1-phenyl always does (Fig. 8C). Even so, addition of aromatic R2 does influence the R1 orientation, especially in the cyclopentyl-containing 8b and 8e (Fig. 8B). Thus, there is interdependence and mutual influence of the side group functionalities that optimizes the ligand binding mode.
Figure 8.
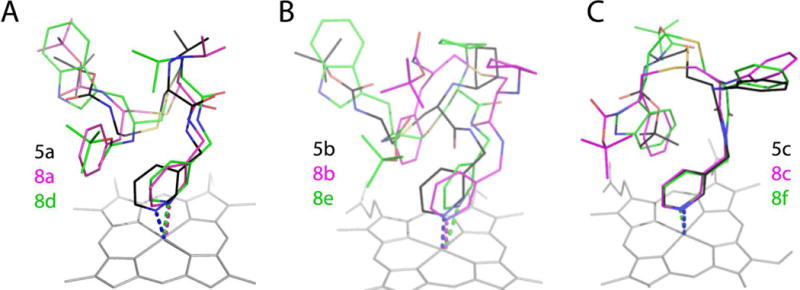
Structural overlays of three subseries of inhibitors.
Comparison of the 8c–15a and 8f–15b pairs
To better understand the side group interplay, we compared 8c and 8f with the R2-only containing 15a and 15b (7D58 and 7D4D structures, respectively). As reported previously,21 the latter two compounds do not adapt a typical ritonavir-like fold upon binding to CYP3A4 but, instead, rotate around the pyridine nitrogen by ~180° to place Boc at P1 site and R2-indole in the P2 area. Comparison of 8c and 15a shows that, despite differences in the backbone folding, R1-phenyl/Boc and R2-phenyls occupy the same space (Fig. 9A). Although R2-phenyls are perpendicular to each other, they orient similarly relative to the heme and pyridine rings and, hence, could contribute equally to the binding energy. If this assumption is true, then the distinct interactions at P1 site, considerably weaker in the 15a-bound structure, would largely be responsible for the 9-fold difference in Ks (Table 1).
Figure 9.
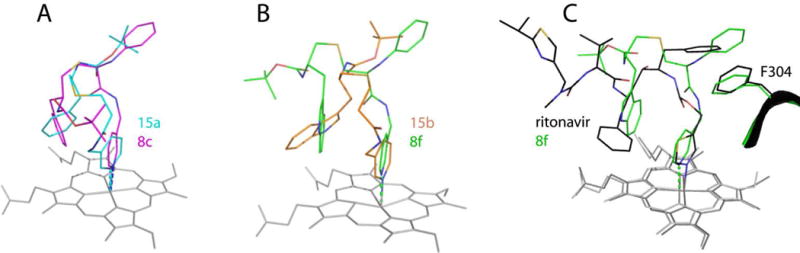
Comparison of 8c and 8f with the R2-only containing 15a and 15b (4D78 and 4D7D structures, respectively) and ritonavir (5VC0).
In 8f and 15b, the R2 groups are also perpendicular (Fig. 9B). However, the 8f indole is positioned ideally for π-π stacking interactions with the parallel pyridine ring, whereas that of 15b overlaps equally with the heme and pyridine. Further, 8f establishes strong aromatic interactions at P1 site and extensive Van der Waals interactions via Boc but, nevertheless, it inhibits CYP3A4 less potently than 15b (Table 1). This implies that the R2-mediated contacts, especially with the heme, preferably impact the inhibitory potency, whereas the binding affinity is largely modulated by R1.
Comparison of 8f and ritonavir
Compound 8f, the strongest CYP3A4 inhibitor we designed so far, is smaller and less complex than ritonavir. Because of variations in the chemical structures, differences in the inhibitors’ binding strength cannot be solely explained by the side group orientation. For instance, we reported previously that pyridine is a stronger heme ligand than thiazole, and that the desoxyritonavir backbone is more flexible and enables a better fit into the CYP3A4 active site.18, 19 There is another important dissimilarity in the backbone spacing between the functional groups (Fig. 9C). In 8f and its analogs, the pyridine-R1 distance is one carbon shorter and R1–R2 one carbon longer than in ritonavir. Further, unlike 8f, ritonavir has R1-phenyl in the R-configuration which, as this study suggested, might be unfavorable for the binding to CYP3A4. These structural variations could impact the ligand association process and, consequently, the binding strength. Thus, one strategy for further improvement of the inhibitor design would be determination of the optimal spacing and stereochemistry of the side group functionalities.
SUMMARY AND CONCLUSIONS
This work was a logical continuation of our previous studies on the ligand binding and inhibitory mechanism of human drug-metabolizing CYP3A4.12, 17–19, 21 Structure-function relationships of nine rationally designed inhibitors were investigated to better understand how the side groups affect the binding affinity and ability to inhibit CYP3A4.
Together, our spectral, kinetic, functional and structural data demonstrate that, regardless of the R2 presence, an increase in the volume, hydrophobicity and aromaticity of R1 strengthens interactions at P1 site and markedly enhances the binding affinity and inhibitory potency for CYP3A4. For all investigated compounds, a decrease in Ks led to a decrease in IC50 but no relation with other parameters, such as the amplitude of the 443 nm band, ΔTm, kfast, ΔA427 nm, the Fe-N bond length/angle, relative orientation of the pyridine and heme rings, the extent of the I-helix displacement and F-F′ loop disorder, or the number of the protein-ligand H-bonds, was observed (Tables 1 and 2). Formation of a strong H-bond between the ligand and Ser119, on the other hand, was a prerequisite for potent inhibition.
Determination of the inhibitor-bound crystal structures of CYP3A4 was essential for better understanding and explanation of the experimental results. Moreover, without structural insights, the unique conformation of 8d and 8e would be impossible to predict. Comparison of the new and previously investigated analogs showed that the CYP3A4-ligand association is strongly influenced by the R1-mediated interactions and R1–R2 interplay. Regardless of the presence and chemical nature of R2, the R1 group always docks near or at P1 site. In contrast, the R2 position depends on the R1 substituent and switches from P2 to the newly identified P3 site if a stabilizing H-bond could be established. Although the side groups cumulatively enhance the binding strength, the R1-mediated aromatic interactions at P1 site are dominant and largely contribute to the binding affinity, whereas the inhibitory strength is preferably affected by the R2-mediated interactions, especially with the heme.
Despite significant improvements in the binding and inhibitory strength, neither compound from this series inhibited CYP3A4 more potently than ritonavir, even under conditions that minimize/prevent the mechanism based inactivation. However, structural analysis suggests that further improvement in the inhibitor design could be achieved upon optimization of the backbone spacing and stereochemistry of the functional groups.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Grant ES025767 and involves research carried out at the Stanford Synchrotron Radiation Lightsource and the Advanced Light Source. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Abbreviations
- CPR
cytochrome P450 reductase
- CYP3A4
cytochrome P450 3A4
- BFC
7-benzyloxy-4-(trifluoromethyl)coumarin
- Boc
tert-butyloxycarbonyl
Footnotes
Supporting Information
Chemical synthesis, mass spectrometry and NMR data, spectral changes induced in CYP3A4 by ritonavir, and structural overlays of the ligand-bound structures of CYP3A4. This material is available free of charge via the Internet at http://pubs.acs.org.
ORCID: 0000-0002-4498-6057
Notes
The authors declare no competing financial interest.
References
- 1.Guengerich FP. Cytochrome P-450 3A4: Regulation and Role in Drug Metabolism. Annu Rev Pharmacol Toxicol. 1999;39:1–17. doi: 10.1146/annurev.pharmtox.39.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Guengerich FP, Shimada T. Oxidation of Toxic and Carcinogenic Chemicals by Human Cytochrome P-450 Enzymes. Chem Res Toxi-col. 1991;4(4):391–407. doi: 10.1021/tx00022a001. [DOI] [PubMed] [Google Scholar]
- 3.Li AP, Kaminski DL, Rasmussen A. Substrates of Human Hepatic Cytochrome P450 3A4. Toxicology. 1995;104(1-3):1–8. doi: 10.1016/0300-483x(95)03155-9. [DOI] [PubMed] [Google Scholar]
- 4.Zhou SF. Drugs Behave as Substrates, Inhibitors and Inducers of Human Cytochrome P450 3A4. Curr Drug Metab. 2008;9(4):310–322. doi: 10.2174/138920008784220664. [DOI] [PubMed] [Google Scholar]
- 5.Kempf DJ, Marsh KC, Kumar G, Rodrigues AD, Denissen JF, McDonald E, Kukulka MJ, Hsu A, Granneman GR, Baroldi PA, Sun E, Pizzuti D, Plattner JJ, Norbeck DW, Leonard JM. Pharmacokinetic Enhancement of Inhibitors of the Human Immunodeficiency Virus Protease by Coadministration with Ritonavir. Antimicrob Agents Chemother. 1997;41(3):654–660. doi: 10.1128/aac.41.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu L, Desai MC. Pharmacokinetic Enhancers for HIV Drugs. Curr Opin Investig Drugs. 2009;10(8):775–786. [PubMed] [Google Scholar]
- 7.Xu L, Liu H, Murray B, Callebaut C, Lee MS, Hong A, Strickley RG, Tsai LK, Stray KM, Wang Y, Rhodes GR, Desai MC. Cobicistat (GS-9350): A Potent and Selective Inhibitor of Human CYP3A as a Novel Pharmacoenhancer. ACS Med Chem Lett. 2010;1:209–213. doi: 10.1021/ml1000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brayer SW, Reddy KR. Ritonavir-Boosted Protease Inhibitor Based Therapy: A New Strategy in Chronic Hepatitis C Therapy. Expert Rev Gastroenterol Hepatol. 2015;9:547–558. doi: 10.1586/17474124.2015.1032938. [DOI] [PubMed] [Google Scholar]
- 9.Koudriakova T, Iatsimirskaia E, Utkin I, Gangl E, Vouros P, Storozhuk E, Orza D, Marinina J, Gerber N. Metabolism of the Human Immunodeficiency Virus Protease Inhibitors Indinavir and Ritonavir by Human Intestinal Microsomes and Expressed Cytochrome P4503A4/3A5: Mechanism-Based Inactivation of Cytochrome P4503A by Ritonavir. Drug Metab Dispos. 1998;26(6):552–561. [PubMed] [Google Scholar]
- 10.von Moltke LL, Durol AL, Duan SX, Greenblatt DJ. Potent Mechanism-Based Inhibition of Human CYP3A in Vitro by Amprenavir and Ritonavir: Comparison with Ketoconazole. Eur J Clin Pharmacol. 2000;56(3):259–261. doi: 10.1007/s002280000125. [DOI] [PubMed] [Google Scholar]
- 11.Ernest CS2nd, Hall SD, Jones DR. Mechanism-Based Inactivation of CYP3A by HIV Protease Inhibitors. J Pharmacol Exp Ther. 2005;312(2):583–591. doi: 10.1124/jpet.104.075416. [DOI] [PubMed] [Google Scholar]
- 12.Sevrioukova IF, Poulos TL. Structure and Mechanism of the Complex Between Cytochrome P4503A4 and Ritonavir. Proc Natl Acad Sci U S A. 2010;107(43):18422–18427. doi: 10.1073/pnas.1010693107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin HL, D’Agostino J, Kenaan C, Calinski D, Hollenberg PF. The Effect of Ritonavir on Human CYP2B6 Catalytic Activity: Heme Modification Contributes to the Mechanism-Based Inactivation of CYP2B6 and CYP3A4 by Ritonavir. Drug Metab Dispos. 2013;41(10):1813–1824. doi: 10.1124/dmd.113.053108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rock BM, Hengel SM, Rock DA, Wienkers LC, Kunze KL. Characterization of Ritonavir-Mediated Inactivation of Cytochrome P450 3A4. Mol Pharmacol. 2014;86(6):665–674. doi: 10.1124/mol.114.094862. [DOI] [PubMed] [Google Scholar]
- 15.Kumar GN, Rodrigues AD, Buko AM, Denissen JF. Cytochrome P450-Mediated Metabolism of the HIV-1 Protease Inhibitor Ritonavir (ABT-538) in Human Liver Microsomes. J Pharmacol Exp Ther. 1996;277(1):423–431. [PubMed] [Google Scholar]
- 16.Hsu A, Granneman GR, Bertz RJ. Ritonavir. Clinical Pharmacokinetics and Interactions with Other Anti-HIV Agents. Clin Pharmacokinet. 1998;35(4):275–291. doi: 10.2165/00003088-199835040-00002. [DOI] [PubMed] [Google Scholar]
- 17.Sevrioukova IF, Poulos TL. Interaction of Human Cytochrome P4503A4 with Ritonavir Analogs. Arch Biochem Biophys. 2012;520(2):108–116. doi: 10.1016/j.abb.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sevrioukova IF, Poulos TL. Pyridine-Substituted Desoxyritonavir is a More Potent Cytochrome P450 3A4 Inhibitor than Ritonavir. J Med Chem. 2013;56(9):3733–3741. doi: 10.1021/jm400288z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sevrioukova IF, Poulos TL. Dissecting Cytochrome P450 3A4-Ligand Interactions Using Ritonavir Analogues. Biochemistry. 2013;52(26):4474–4481. doi: 10.1021/bi4005396. [DOI] [PubMed] [Google Scholar]
- 20.Sevrioukova IF, Poulos TL. Ritonavir Analogues as a Probe for Deciphering the Cytochrome P450 3A4 Inhibitory Mechanism. Curr Top Med Chem. 2014;14(11):1348–1355. doi: 10.2174/1568026614666140506120647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaur P, Chamberlin R, Poulos TL, Sevrioukova IF. Structure-Based Inhibitor Design for Evaluation of a CYP3A4 Pharmacophore Model. J Med Chem. 2016;59(9):4210–4220. doi: 10.1021/acs.jmedchem.5b01146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sevrioukova IF. High-Level Production and Properties of the Cysteine-Depleted Cytochrome P450 3A4. Biochemistry. 2017;56(24):3058–3067. doi: 10.1021/acs.biochem.7b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sevrioukova IF, Poulos TL. Anion-Dependent Stimulation of CYP3A4 Monooxy-genase. Biochemistry. 2015;54(26):4083–4096. doi: 10.1021/acs.biochem.5b00510. [DOI] [PubMed] [Google Scholar]
- 24.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser Crystallographic Software. J Appl Crystallogr. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and Development of Coot. Acta Crystallogr Section D. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davydov DR, Davydova NY, Sineva EV, Kufareva I, Halpert JR. Pivotal Role of P450-P450 Interactions in CYP3A4 Allostery: The Case of alpha-Naphthoflavone. Biochem J. 2013;453(2):219–230. doi: 10.1042/BJ20130398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamaguchi Y, Khan KK, He YA, He YQ, Halpert JR. Topological Changes in the CYP3A4 Active Site Probed with Phenyldiazene: Effect of Interaction with NADPH-Cytochrome P450 Reductase and Cytochrome b5 and of Site-Directed Mutagenesis. Drug Metab Dispos. 2004;32(1):155–161. doi: 10.1124/dmd.32.1.155. [DOI] [PubMed] [Google Scholar]
- 29.Li F, Lu J, Ma X. Metabolomic Screening and Identification of the Bioactivation Pathways of Ritonavir. Chem Res Toxicol. 2011;24:2109–2114. doi: 10.1021/tx2004147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.