

Abstract
Motivation
Genome sequencing projects sometimes uncover more organisms than expected, especially for complex and/or non-model organisms. It is therefore useful to develop software to identify mix of organisms from genome sequence assemblies.
Results
Here we present PhylOligo, a new package including tools to explore, identify and extract organism-specific sequences in a genome assembly using the analysis of their DNA compositional characteristics.
Availability and implementation
The tools are written in Python3 and R under the GPLv3 Licence and can be found at https://github.com/itsmeludo/Phyloligo/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The development of sequencing technologies has enabled to target the genome of complex non-model organisms and communities of organisms. Some of these non-model organisms can be challenging to isolate from their environment or cannot be cloned in vitro. They might alternatively be compulsorily associated with cognate commensal or parasitic organisms, or even embedded within a host. Consequently, genome assembly datasets sometimes include DNA from unexpected sources like mixture of untargeted species, but may also contain organelles or even laboratory contaminants. The presence of additional untargeted species was indeed reported in several recent genome assemblies, for instance in the draft assembly of domestic cow (Merchant et al., 2014), in several isolates of the phytopathogenic fungi Magnaporthe oryzae (Chiapello et al., 2015) or recently in the tardigrade genome (Delmont and Eren, 2016). Such mixed assemblies may produce several biases and problems in downstream bioinformatics analyses and raise the need for tools able to deal with mixed-organism DNA assemblies.
Several tools were recently designed or used to detect and filter untargeted organisms from sequence datasets. A first type of approach, used by khmer software (Crusoe et al., 2015), is to compute k-mer frequencies on short reads to pre-process and filter read datasets prior to de novo sequence assembly. Other packages like Blobtools (Kumar et al., 2013) and Anvi’o (Eren et al., 2015) combine sequence properties (GC content, oligonucleotide profile) and additional information such as depth of coverage, similarity to public databases and reference gene sets to identify untargeted species using both raw reads and assembled contigs of a genomic dataset. Finally, a last type of approach is to use software dedicated to metagenomic species read binning, such as CONCOCT (Alneberg et al., 2014) that use sequence composition and coverage across multiple samples to automatically cluster contigs into genomes or Kraken (Wood and Salzberg, 2014) that relies on exact alignment of k-mers to a k-mer reference database to assign taxonomic labels to metagenomic DNA.
Here we present PhylOligo, a toolset designed to explore, segment and subtract untargeted material from assembled sequences using an ab initio alignment-free approach relying only on the intrinsic oligonucleotide signature of an assembled genomic dataset. Compared to existing software, PhylOligo provides several features to explore assemblies, including: (i) a customizable oligonucleotide pattern, including continuous and spaced pattern k-mers (Břinda et al., 2015; Leimeister et al., 2014; Noé and Martin, 2014). (ii) handling bare contig-level assemblies (raw reads and coverage information are not required for detecting untargeted species) (iii) an interactive cladogram-based visualization of the contig signature similarity and cumulative size to explore the signature clusters to profile putative additional materials (iv) an effective sliding window-based partitioning scan of the assembly based on a supervised learning and a double-threshold system asserting that regions are labelled as untargeted organism when meeting two criteria: (i) being distant enough from the host sequence oligonucleotide profile (first threshold) and (ii) being close enough from a cluster of untargeted sequences previously selected by supervised learning (second threshold).
Our strategy present several advantages. (i) Unlike approaches that process short read datasets prior to the de novo sequence assembly and use sequence homology information, PhylOligo allows the identification of potentially uncharacterised and distantly related sequences in already assembled genomic datasets, the handling of any type of genome assembly, shunning the dependency on the availability of raw sequencing reads data, additional data and patchiness of knowledge in databases; (ii) The double-threshold species-specific filtration prevents the removal of HGTs and the subsequent fragmentation of the assembly; (iii) Learning the compositional profile on longer and assembled sequences such as contigs compared to unassembled reads, allows for a refined oligonucleotide profile, unbiased from heterogeneous sequencing depth along the sequence. Moreover, the partitioning process of PhylOligo provides the possibility to detect and split chimeric sequences or mis-scaffolding;
2 Workflow strategy
Our strategy includes 3 main steps: (i) assembly exploration using an interactive tree visualization based on oligonucleotide profiles computed from all genomic contigs, (ii) oligonucleotide profile prototype learning based on contig subsets selected by the user at nodes of the tree and (iii) assembly partitioning to locate organism-specific regions and classify contigs or segments according to the learned prototypes.
2.1 Assembly exploration
PhylOligo allows for a visual exploration of the compositional similarity distribution and structure of the contigs in an assembly based on either continuous (k-mers) or spaced-pattern oligonucleotide frequencies. The oligonucleotide profile of each contig is computed and a pairwise distance matrix based on metrics including Euclidean or Jensen-Shannon is produced (Fig. 1A) to generate an interactive Neighbour-Joining tree. Branch width is drawn proportional to the cumulated length of the contigs in a clade, allowing the user to track where the main part of the assembly clusters (assumed to correspond to the targeted organisms) and what significant clades branch out as hint for separate organisms (see Fig. 1B). Thanks to the Ape package (Paradis et al., 2004), sequences from a clade are interactively selected on the tree and exported to fasta files to learn a prototype of their oligonucleotide profile. An alternative unsupervised clustering method relying on HDBSCAN (Campello et al., 2013) and t-SNE (van der Maaten and Hinton, 2008) for visualization is also implemented (Fig. 1C).
Fig. 1.
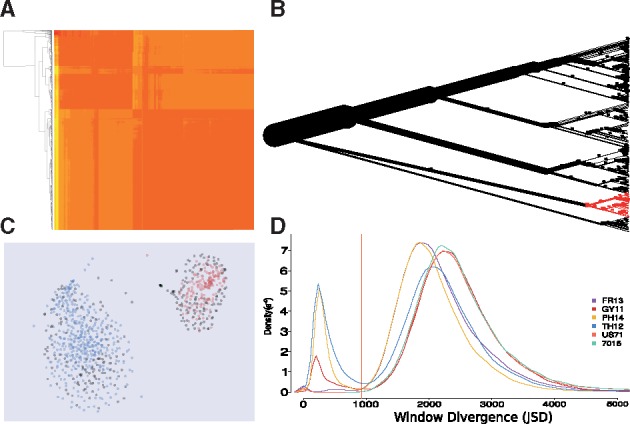
Visualization and interactive exploration of assemblies. (A) Pairwise compositional divergence of contigs produced by PhylOligo. Contigs are reordered by hierarchical clustering. (B) Contig tree produced by PhylOligo on the tardigrade genome. The clade in red is the current selection pointed by the user. (C) Contigs clustered by HDBSCAN on oligonucleotide frequencies, Data from Magnaporthe oryzae. Red and blue are predicted clusters, grey are unclassified. The hyperspace is reduced to 2 dimensions with t-SNE. (D) Determination of the untargeted threshold in ContaLocate based on the distribution of distances between the untargeted clade and the scanning windows over the whole assembly (Color version of this figure is available at Bioinformatics online.)
2.2 Prototype learning
ContaLocate then allows the learning of oligonucleotide profiles from the main and presumed additional organisms identified by user-selected subsets from the previous step. These subsets must cumulate at least 50 Kb in order to generate an accurate prototype sufficiently representative of an organism. Learning subsets can be generated or complemented from public sequences, specialised databases (Ménigaud et al., 2012) or other tools (Alneberg et al., 2014; Eren et al., 2015; Kumar et al., 2013).
2.3 Assembly partitioning
The assembly is then scanned with sliding windows to locate organism-specific regions using oligonucleotide divergences computed against the targeted and the additional profiles. The distribution of the divergence against both is used to establish two thresholds best separating the different modes in the density functions (see Fig. 1D). The thresholds are visually validated by the user and can also be adjusted manually. Genomic regions with a divergence simultaneously crossing respective thresholds to the targeted and to the additional profiles are labelled as part of the additional organism and exported as a GFF file.
3 Results
3.1 Synthetic datasets
We evaluated the performances of PhylOligo by generating artificial contaminations on 32 contig datasets generated by GRINDER (Angly et al., 2012) from real Refseq genome data (see section 6.1 of Supplementary Material for detailed protocol). The species were chosen to cover the main domain of life (archea, bacteria, fungi, protozoa and vertebrate) and different degrees of genome complexity, content, length and composition. We benchmarked the automatic version of PhylOligo that uses the unsupervised HDBSCAN clustering and evaluated performances by using three indicators: (i) the cluster specificity i.e. the maximum fraction of contaminant in a cluster, (ii) the cluster sensitivity, i.e. the fraction of the whole contaminant aggregated in the cluster and (iii) an hybrid score, which indicated the best computed value of the product of cluster specificity and sensitivity. We used PhylOligo default parameters on all the combinations of the 32 simulated genomes assemblies. We also evaluated the impact of the k-mer parameter by panelling continuous and spaced-pattern k-mers on a focus subset of ten pairs (see results in Table 1). Complete results are detailed in section 6.2 of Supplementary Material. Overall, the benchmark demonstrates a great ability to discriminate contaminant clusters with very high specificity and good sensitivity, suited with the requirements for supervised learning and partitioning. Concerning k parameter impact, we obtained best results according to our hybrid score for two spaced patterns: 11001 (mean:0.8133, median: 0.9499) and 110101 (mean:0.8344, median:0.9459). Continuous k-mers of length 4 and 5 also performed well but with slightly lower scores (median scores of 0.7909 and 0.9305 for k = 4 and 5 respectively).
Table 1.
Impact of k-mer pattern on the hybrid score (best computed value of the product of cluster specificity and sensitivity) for 10 pairs of simulated data
| K-mer pattern | ||||||
|---|---|---|---|---|---|---|
| Species mix | 111 | 1111 | 11111 | 11001 | 110101 | 111001 |
| S.enterica in A.fumigatus | 0.39 | 0.79 | 0.94 | 0.45 | 0.93 | 0.97 |
| B.cereus in C.canadensis | 0.99 | 0.99 | 0.98 | 0.99 | 0.98 | 0.99 |
| B.mallei in H.sapiens | 1.00 | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 |
| A.fulgidus in P.tetraurelia | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 |
| A.fumigatus in P.tigris | 0.96 | 0.96 | 0.93 | 0.95 | 0.95 | 0.95 |
| G.intestinalis in X.tropicalis | 0.95 | 0.99 | 0.95 | 0.99 | 0.96 | 0.98 |
| S.enterica in T.vaginalis | 0.41 | 0.50 | 0.70 | 0.72 | 0.71 | 0.72 |
| B.cereus in A.australis | 0.73 | 0.73 | 0.71 | 0.72 | 0.71 | 0.72 |
| S.cerevisiae in T.vaginalis | 0.60 | 0.56 | 0.49 | 0.57 | 0.51 | 0.58 |
| S.pombe in T.vaginalis | 0.65 | 0.61 | 0.43 | 0.58 | 0.47 | 0.58 |
| Mean | 0.69 | 0.74 | 0.75 | 0.81 | 0.83 | 0.78 |
| Median | 0.73 | 0.79 | 0.93 | 0.95 | 0.95 | 0.95 |
| Min | 0.01 | 0.01 | 0.15 | 0.45 | 0.47 | 0.05 |
| Max | 1.00 | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 |
3.2 Real datasets
PhylOligo has been successfully applied to identify untargeted large bacterial regions in four out of nine fungal genomic datasets of Magnaporthe (Chiapello et al., 2015). GOHTAM (Ménigaud et al., 2012) taxonomical assignment of these additional regions confirmed their homogeneity and origin from Burkholderiales. Targeted Blast comparisons indicated that some of these supplementary regions were almost identical to Burkholderia fungorum sequences (100% identity for 16S, recA and gyrB genes) suggesting an origin or relatedness to one or several bacterial isolate(s) of this species. PhylOligo was applied to filter genome assemblies, validated with BUSCO (Simão et al., 2015) and DOGMA (Dohmen et al., 2016) (see Supplementary Material) and allowed to continue further bioinformatics analyses without rebuilding the costly initial genome assembly and annotation processes.
PhylOligo was also used to explore the scaffolds of the tardigrade assembly (Boothby et al., 2015), for which a multiple contamination was previously proposed (Delmont and Eren, 2016; Koutsovoulos et al., 2016). We compared the the topology of the compositional cladograms established with PhylOligo on both the initial and the filtered assembly obtained with Anvi’o (Eren et al., 2015). Our results showed that the cladogram produced with PhylOligo exhibited a topology where the curated assembly was monophyletic, with a sequence subset and topology highly concordant with the results of Anvi’o (see Supplementary Material Section 5.2).
3.3 PhylOligo performances
PhylOligo handles assembly contigs up to a count of several dozen thousand on a modern workstation within minutes and up to few hundred thousand on a high-memory server. Input sequences can be quality- filtered or sub-sampled with a preprocessing tool to allow for improved signal and quick tests. Several parallel computation optimizations and data compression methods including HDF5 are available to improve performance on larger datasets.
Supplementary Material
Acknowledgements
We thank the INRA bioinformatics platforms MIGALE and Genotoul-bioinfo for resources and Thomas Schiex and Nathalie Villa-Vialaneix for their input.
Conflict of Interest: none declared.
References
- Alneberg J. et al. (2014) Binning metagenomic contigs by coverage and composition. Nat. Methods, 11, 1144–1146. Brief Communication. [DOI] [PubMed] [Google Scholar]
- Angly F.E. et al. (2012) Grinder: a versatile amplicon and shotgun sequence simulator. Nucleic Acids Res., 40, e94.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Břinda K. et al. (2015) Spaced seeds improve k-mer-based metagenomic classification. Bioinformatics, 31, 3584.. [DOI] [PubMed] [Google Scholar]
- Boothby T.C. et al. (2015) Evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proc. Natl. Acad. Sci. USA, 112, 15976–15981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campello R.J.G.B. et al. (2013). Density-Based Clustering Based on Hierarchical Density Estimates. Springer, Berlin, Heidelberg, pp. 160–172. [Google Scholar]
- Chiapello H. et al. (2015) Deciphering genome content and evolutionary relationships of isolates from the fungus Magnaporthe oryzae attacking different host plants. Genome Biol. Evol., 7, 2896–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crusoe M.R. et al. (2015) The khmer software package: enabling efficient nucleotide sequence analysis. F1000Research, 4, doi:10.12688/f1000research.6924.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmont T.O., Eren A.M. (2016) Identifying contamination with advanced visualization and analysis practices: metagenomic approaches for eukaryotic genome assemblies. PeerJ, 4, e1839.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohmen E. et al. (2016) Dogma: domain-based transcriptome and proteome quality assessment. Bioinformatics, 32, 2577.. [DOI] [PubMed] [Google Scholar]
- Eren A.M. et al. (2015) Anvi’o: an advanced analysis and visualization platform for ’omics data. PeerJ, 3, e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsovoulos G. et al. (2016) No evidence for extensive horizontal gene transfer in the genome of the tardigrade hypsibius dujardini. Pro. Natl. Acad. Sci. USA, 113, 5053–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S. et al. (2013) Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated gc-coverage plots. Front. Genet., 4, 237.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leimeister C.-A. et al. (2014) Fast alignment-free sequence comparison using spaced-word frequencies. Bioinformatics, 30, 1991.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant S. et al. (2014) Unexpected cross-species contamination in genome sequencing projects. PeerJ, 2, e675.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ménigaud S. et al. (2012) Gohtam: a website for ‘genomic origin of horizontal transfers, alignment and metagenomics’. Bioinformatics, 28, 1270–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noé L., Martin D.E.K. (2014) A coverage criterion for spaced seeds and its applications to support vector machine string Kernels and k-mer distances. J. Comput. Biol., 21, 28.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E. et al. (2004) Ape: analyses of phylogenetics and evolution in r language. Bioinformatics, 20, 289.. [DOI] [PubMed] [Google Scholar]
- Simão F.A. et al. (2015) Busco: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31, 3210.. [DOI] [PubMed] [Google Scholar]
- van der Maaten L., Hinton G.E. (2008) Visualizing high-dimensional data using t-sne. J. Mach. Learn. Res., 9, 2579–2605. [Google Scholar]
- Wood D.E., Salzberg S.L. (2014) Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol., 15, R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.