

Abstract
Motivation
RNA sequencing (RNA-seq) experiments now span hundreds to thousands of samples. Current spliced alignment software is designed to analyze each sample separately. Consequently, no information is gained from analyzing multiple samples together, and it requires extra work to obtain analysis products that incorporate data from across samples.
Results
We describe Rail-RNA, a cloud-enabled spliced aligner that analyzes many samples at once. Rail-RNA eliminates redundant work across samples, making it more efficient as samples are added. For many samples, Rail-RNA is more accurate than annotation-assisted aligners. We use Rail-RNA to align 667 RNA-seq samples from the GEUVADIS project on Amazon Web Services in under 16 h for US$0.91 per sample. Rail-RNA outputs alignments in SAM/BAM format; but it also outputs (i) base-level coverage bigWigs for each sample; (ii) coverage bigWigs encoding normalized mean and median coverages at each base across samples analyzed; and (iii) exon–exon splice junctions and indels (features) in columnar formats that juxtapose coverages in samples in which a given feature is found. Supplementary outputs are ready for use with downstream packages for reproducible statistical analysis. We use Rail-RNA to identify expressed regions in the GEUVADIS samples and show that both annotated and unannotated (novel) expressed regions exhibit consistent patterns of variation across populations and with respect to known confounding variables.
Availability and Implementation
Rail-RNA is open-source software available at http://rail.bio.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Sequencing has improved rapidly in the last several years (Glenn, 2011; Hayden, 2014). RNA sequencing (RNA-seq) (Ozsolak and Milos, 2010; Wang et al., 2009) has become a standard tool for studying gene expression due to its ability to detect novel transcriptional activity without relying on previously defined gene sequence. The Sequence Read Archive contains data for over 170 000 RNA-seq samples, including over 50 000 from human samples (Leinonen et al., 2010b). Projects like GTEx (Lonsdale et al., 2013) and TCGA (http://cancergenome.nih.gov) are generating RNA-seq data on thousands of samples across normal and malignant tissues derived from hundreds to thousands of individuals.
A common use of RNA-seq is to characterize gene expression across samples and ultimately to identify expression patterns associated with outcomes of interest. At the outset, a read aligner determines where sequencing reads originated with respect to the reference genome or annotated transcriptome. Unlike read alignment for DNA sequencing reads, RNA-seq alignment must be splice-aware to accommodate introns spliced out of mature mRNA transcripts, creating exon–exon junctions. While RNA-seq data is often generated for large groups (hundreds) of biological replicates, past alignment tools were designed to run on each sample separately (Au et al., 2010; Bonfert et al., 2012; Bryant et al., 2010; Cloonan et al., 2009; De Bona et al., 2008; Dobin et al., 2013; Grant et al., 2011; Hu et al., 2012; Huang et al., 2011; Jean et al., 2010; Kim et al., 2013, 2015; Marco-Sola et al., 2012; Philippe et al., 2013; Trapnell et al., 2009; Wang et al., 2010; Wu and Nacu, 2010; Zhang et al., 2012).
We introduce Rail-RNA, an annotation-agnostic spliced read aligner. Uniquely, Rail-RNA is designed to analyze many samples at once to (i) borrow strength for accurate detection of exon–exon junctions, even at low coverage, (ii) avoid effort spent aligning redundant sequences within or across samples, greatly improving scalability and (iii) compute cross-sample results including the normalized mean coverages of each base of the genome across samples. Rail-RNA can be run on a computer cluster at the user’s home institution or on a cluster rented from a commercial cloud computing provider at modest cost per sample. Cloud services offer standardized units of hardware and software, enabling Rail-RNA users to easily reproduce large-scale analyses performed in other labs. Rail-RNA’s outputs are compatible with downstream tools for isoform assembly, quantitation and count- and region-based differential expression.
We demonstrate Rail-RNA is more accurate than other tools. We show Rail-RNA is less susceptible to biases affecting other tools; specifically, (i) Rail-RNA has substantially higher recall of alignments across low-coverage exon–exon junctions and (ii) Rail-RNA is accurate without a gene annotation, avoiding annotation bias resulting from potentially incomplete (Jaffe et al., 2015) or incorrect transcript annotations. We run Rail-RNA on 667 paired-end lymphoblastoid cell line (LCL) RNA-seq samples from the GEUVADIS study (Lappalainen et al., 2013), obtaining results in 15 h and 47 min at a cost of US$0.91 (hereafter, we use the symbol $ to denote USD) per sample. This is a fraction of per-sample sequencing costs, which are $20 or more (Combs and Eisen, 2015). We illustrate the usability of Rail-RNA’s output by performing a region-based differential expression analysis of the GEUVADIS dataset. Our analysis identifies 285 695 expressed regions, including 19 649 in intergenic regions. We show that intergenic and annotated regions exhibit similar patterns of variation across populations and with respect to technical confounders.
Altogether, Rail-RNA is a significant step in the direction of usable software that quickly, reproducibly and accurately analyzes large numbers of RNA-seq samples.
2 Results
The GEUVADIS consortium performed mRNA sequencing of 465 lymphoblastoid cell line samples derived from CEPH (CEU), Finnish (FIN), British (GBR), Toscani (TSI) and Yoruba (YRI) populations of the 1000 Genomes Project (Lappalainen et al., 2013), giving 667 paired-end samples. Per-sample sequencing depth is summarized in Section S.2. For information on reproducing all our results, including software versions, see Section S.3.
2.1 Design principles of Rail-RNA
Rail-RNA follows the MapReduce programming model, and is structured as an alternating sequence of aggregate steps and compute steps. Aggregate steps group and order data in preparation for future compute steps. Compute steps run concurrently on ordered groups of data. In this framework, it is natural to bring together related data so that decisions can be informed by all samples at once. This affords greater accuracy and efficiency than if samples are analyzed separately. Rail-RNA aggregates across samples at multiple points in the pipeline to increase accuracy (borrowing strength for junction calling) and scalability (through elimination of redundant alignment work).
Rail-RNA can be run in elastic, parallel, or single-computer mode. In single-computer mode, Rail-RNA runs on multiple processors on a single computer. In parallel mode, Rail-RNA runs on any of the variety of cluster setups supported by IPython Parallel (Perez and Granger, 2007). These include Sun Grid Engine (SGE), Message Passing Interface (MPI) and StarCluster.
In elastic mode, Rail-RNA is run using the Amazon Web Services (AWS) Elastic MapReduce (EMR) service, which itself uses Hadoop, an implementation of MapReduce (Dean and Ghemawat, 2008). EMR is specifically for computer clusters rented on demand from Amazon’s Elastic Compute Cloud (EC2). Amazon Simple Storage Service (S3) stores intermediate data and output.
There are advantages and disadvantages to commercial cloud computing services like AWS (Schatz et al., 2010; Stein et al., 2010). One advantage is that it facilitates reproducibility: one researcher can reproduce the same hardware and software setup used by another. Another advantage for Rail-RNA, which stores all intermediate and final results in S3, is that there is no risk of exhausting the cluster’s disk space even for datasets with many samples. Without these facilities, making scalable software that runs easily on large numbers of RNA-seq samples in different laboratories is quite challenging.
Rail-RNA supports both paired-end and unpaired RNA-seq samples. It supports input data consisting of reads of various lengths, and can detect exon–exon junctions involving exons as short as 9 bp by default. Rail-RNA uses Bowtie 2 (Langmead and Salzberg, 2012) to align reads, including reads spanning exon–exon junctions, so it is naturally both indel-aware (with affine gap penalty) and base quality-aware. Rail-RNA is also deterministic; the same input data and parameters will yield the same outputs regardless of number of processors and computers used and regardless of whether it runs in elastic, parallel, or single-computer mode.
2.2 Scalability
We randomly selected 112 paired-end samples from the GEUVADIS study, with 16 coming from each of the 7 laboratories in which sequencing was performed. We also randomly selected subsets of 28 and 56 samples from the 112 to illustrate Rail-RNA’s scalability on EMR (Section S.4).
We use the term ‘instance’ to refer to a computer (or virtualized fraction of one) that can be rented from EC2. An instance can have multiple processing cores. For experiments described in this section: (i) we used c3.2xlarge EC2 spot instances, each with eight processing cores. See Section S.5 for details on this instance type and Section S.6 for details on spot instances; (i) we used S3 to store inputs, intermediate results and final results; (iii) we performed all experiments and stored all S3 data in the EU region (eu-west-1). See Section S.7 for details on cost measurements. For every experiment in this paper, we measure cost by totaling the bid price for the EC2 spot instances (here, $0.11 per instance per hour using spot marketplace) and the Elastic MapReduce surcharges (here, $0.105 per instance per hour). On the one hand, this estimate can be low since it does not account for costs incurred by S3 and related services. On the other, the estimate can be high since the actual price paid depends on the spot market price, which is lower than the bid price. Storage costs can vary dramatically depending on how long intermediate and final results are kept on S3. In the EU region, it currently costs about $0.03 per GB per month to store data on S3. Further, downloading data from S3 to a local cluster costs about $0.09 per GB. Alignment BAMs, coverage bigWigs and junction and indel BEDs together take up about 2 GB per sample with 20 million read pairs. This adds about $0.18 per sample if results are downloaded to a local cluster and $0.06 per sample if results are kept on S3 for a month. Intermediate data deleted immediately after a job flow is complete contributes negligibly to analysis cost per sample.
The 112 selected GEUVADIS samples spanned 2.4 terabytes of compressed FASTQ data. We preprocessed each subset of 28, 56 and 112 samples of the 112 using different Elastic MapReduce clusters, each spanning 21 c3.2xlarge instances. Each cluster downloaded source data from the European Nucleotide Archive FTP server (Leinonen et al., 2010a); see http://www.ebi.ac.uk/ena/data/view/ERP001942 for download URLs. Preprocessing 28 samples took 1 h and 3 min, costing $9.03, preprocessing 56 samples took 1 h and 14 min, costing $9.03 and preprocessing 112 samples took 2 h and 14 min, costing $13.54.
We ran Rail-RNA several times to assess scalability, which we measured with respect to both the number of instances in the cluster and the number of input samples. To measure scalability with respect to cluster size, Rail-RNA was run on a random subset of 28 of the 112 samples using EMR clusters of 10, 20 and 40 c3.2xlarge core instances (Fig. 1a, b). Each EMR cluster has an extra instance called the master instance that coordinates cluster activity; we exclude it from instance counts here because it contributes no workers. Each of the 10- and 20-instance experiments was run exactly once, while the 40-instance experiment was run three times to measure wall-clock time variability. The orange dot representing the experiment on 40 instances is at the mean number of samples analyzed per hour, while the horizontal lines above and below represent the minimum and maximum values. The dashed blue line shows an ideal linear extrapolation from the 10-instance data point. This illustrates how throughput would increase if doubling the number of instances also doubled throughput. Rail-RNA’s scaling falls short of ideal linear scaling, but this is expected due to various sources of overhead including communication and load imbalance. Importantly, Rail-RNA’s throughput continues to grow as the number of instances increases to 40, indicating Rail-RNA can make effective use of hundreds of processors at once.
Fig. 1.

Scaling and cost of Rail-RNA versus cluster size and number of samples. (a) depicts scaling with respect to cluster size. Horizontal axis is the number of instances in the cluster. Vertical axis is throughput measured as number of samples analyzed per hour. The dashed line illustrates ideal linear scaling, extrapolated from the 10-instance result. (b) is a table of per-sample costs for each experiment in a). (c) plots Rail-RNA’s running time on 28, 56 and 112 paired-end GEUVADIS samples. The dashed line represents linear scaling, extrapolating that it takes twice as long to analyze approximately twice as much data. Rail-RNA achieves better-than-linear scaling with respect to number of samples as reflected in the table of costs in (d): cost per sample decreases as number of samples analyzed increases. Per-sample costs in (b) and (d) reported here do not include the cost of preprocessing, which depends on download speed. These costs are reported in the main text. The master instance is also not included in the cluster sizes quoted here
To measure scalability with respect to number of input samples, Rail-RNA was run on 28, 56 and 112 samples using a cluster of 40 instances (Fig. 1c, d). The 40-instance experiment with 28 samples (blue dot and lines) reports the same results as in 1a, but now in terms of running time. The dashed blue line extrapolates linear scaling from the 28-sample data point, assuming doubling the number of samples doubles running time. The 56- and 112-sample points fall well below the line, indicating Rail-RNA achieves better-than-linear scaling of running time with respect to number of samples analyzed. Rail-RNA gets more efficient as more samples are analyzed in part because it identifies and eliminates redundant alignment work within and across samples (Section S.8). Analyzing many samples together is particularly beneficial, with cost per sample dropping from $1.89 for 28 samples to $1.02 for 112 samples (Fig. 1d). In an experiment described in Section 2.4, per-sample cost was reduced to $0.91 per sample.
A breakdown of the time taken by the steps in Rail-RNA’s pipeline is provided in Table S1.
2.3 Accuracy
We simulated 112 RNA-seq samples with 40 million 76-bp paired-end reads each using Flux Simulator (Griebel et al., 2012). Typically, Flux assigns expression levels to transcripts with a model-based approach. Instead, we used FPKM expression levels from the set of 112 randomly selected paired-end samples studied in Section 2.2; they are taken from the GEUVADIS study (Lappalainen et al., 2013) and are available at http://www.ebi.ac.uk/arrayexpress/files/E-GEUV-3/GD660.TrQuantRPKM.txt.gz. See Section S.9 for simulation details.
We compared Rail-RNA’s accuracy to that of TopHat 2 v2.1.0 (Kim et al., 2013), Subjunc from v1.4.6p-v4 of the Subread package (Liao et al., 2013), STAR v2.4.2a (Dobin et al., 2013) and HISAT v0.1.6-beta (Kim et al., 2015). We ran TopHat 2 with (‘Tophat 2 ann’) and without (‘Tophat 2 no ann’) the Gencode v12 annotation (Cunningham et al., 2015) provided. We ran Subjunc using the default values of its command-line parameters. We ran STAR in three ways: in one pass (‘STAR 1 pass’); in one pass with exon–exon junctions from Gencode v12 provided (‘STAR 1 pass ann’); and in two passes (‘STAR 2 pass’). We similarly ran HISAT in three ways: in one pass (‘HISAT 1 pass’); in one pass with exon–exon junctions from Gencode v12 provided (‘HISAT 1 pass ann’); and in two passes (‘HISAT 2 pass’). One-pass methods (‘STAR 1 pass,’ ‘STAR 1 pass ann,’ ‘HISAT 1 pass,’ and ‘HISAT 1 pass ann’) align reads and call exon–exon junctions in one step, whereas two-pass methods (all other protocols) additionally perform a second step that realigns reads in light of exon–exon junction calls from the first. Section S.10 describes the protocols in detail.
When an alignment program was run with annotation (‘Tophat 2 ann,’ ‘STAR 1 pass ann,’ and ‘HISAT 1 pass ann’), we provided the same annotation from which Flux Simulator simulated the reads. That is, the provided annotation consisted of a superset of the actual transcripts present. Consequently, protocols where the annotation was provided were given an artificial advantage.
Described below are the three ways in which Rail-RNA was run.
On a single sample (‘Rail single’). Here, Rail-RNA uses reads from the given sample to identify exon–exon junctions. Like in two-pass protocols, reads are then realigned to transcript fragments to handle the case where a read overlaps an exon–exon junction near its 5′ or 3′ end.
On 112 samples with exon–exon junction filter (‘Rail all’). After initial alignment, Rail-RNA compiles a list of exon–exon junctions found in any sample. The list is then filtered; an exon–exon junction passes the filter if (i) it appears in at least 5% of the input samples, or (ii) it is overlapped by at least 5 reads in any sample. The filtered exon–exon junction list is used to build the set of transcript fragments to which each sample is then realigned.
On 112 samples without exon–exon junction filter (‘Rail all NF’). Identical to ‘Rail all’ but with no exon–exon junction filter. This is not a recommended protocol; we include it only to show the filter’s effect.
In none of the three modes does Rail-RNA use a gene annotation: Rail-RNA is consistently annotation-agnostic.
We consider two accuracy measures in the main text: (i) overlap accuracy, measuring precision and recall of overlap events. Each event is an instance where the primary alignment of a read overlaps an exon–exon junction; (ii) exon–exon junction accuracy, measuring precision of exon–exon junctions called by a given aligner and recall of the set of exon–exon junctions within a sample or across samples. We also compute F-score, the harmonic mean of precision and recall. Section S.11 formally defines these measures as well as a measure of overall mapping accuracy.
The ‘Rail all’ and ‘Rail all NF’ protocols were run on all 112 simulations. Other protocols were run on each of the 20 samples randomly selected from the 112. We emphasize that even though protocols labeled ‘Rail all’ analyze all 112 samples at once, we evaluate their output alignments and calls only for the sample of 20. Figure 2 displays overlap and exon–exon junction accuracy measurements. Figure 2a summarizes the accuracy of each tool across the 20 samples.
Fig. 2.

Accuracy comparisons. (a) Means and standard deviations of accuracy measures of various alignment protocols across 20 simulated samples whose FPKMs mimic those of 20 GEUVADIS samples. ‘Rail all’ protocols involve running Rail-RNA on all 112 simulated samples. Overlap accuracy is shown in red, exon–exon junction accuracy in blue. Some tools are run with (top table) and without (bottom table) a provided gene annotation. Note that Rail-RNA never uses an annotation. For Rail-RNA versus unannotated protocols, the best two results in each column are in boldface, and for Rail-RNA versus annotated protocols, the best result in each column is in boldface. (b) Rail-RNA’s accuracy on two sets of exon–exon junctions found across all 112 simulated samples: one before and one after application of the exon–exon junction filter
As illustrated in Figure 2a, Rail-RNA has the highest overlap F-score of the protocols tested, including those using a gene annotation. Rail-RNA’s overlap precision is comparable to the most precise protocol (‘Subjunc’ in this case) and its recall is comparable to the highest of any protocol (‘STAR 1 pass ann’). Further, analyzing many samples at once (‘Rail all’) achieves greater F-score compared to analyzing one (‘Rail single’). This is more pronounced for exon–exon junction accuracy than for overlap accuracy since borrowing strength across replicates is particularly effective at detecting low-coverage junctions (Section S.13). Mean exon–exon junction recall increases from 0.880 to 0.939 (Section S.12), which adds about 10 000 true positives.
Figure 2b demonstrates the efficacy of the exon–exon junction filter in the ‘Rail all’ protocol. Precision/recall are defined similarly as for a single sample, but pooled across all samples. That is, recall is the fraction of exon–exon junctions with at least one simulated spanning read in at least one sample that Rail-RNA detects in at least one sample. The improvement in precision when moving from the unfiltered (0.673) to the filtered (0.964) exon–exon junction list shows that the filter removes a large fraction of incorrect exon–exon junction calls because they are supported in only a few samples. Further, the distribution of filtered exon–exon junctions with certain donor/acceptor motifs matches the expected distribution (GT–AG: 96.5%, GC–AG: 2.6%, AT–AC: 1.0%) much more closely than the same distribution for the unfiltered exon–exon junctions.
Rail-RNA’s protocols also tie ‘STAR 1 pass’ in achieving the highest mean F-scores in an accuracy comparison that considers all alignments, including those falling entirely within exons; see Section S.12.
2.4 Analysis of GEUVADIS RNA-seq samples
We demonstrate the utility of Rail-RNA’s outputs by performing a novel analysis of 667 paired-end GEUVADIS RNA-seq samples from lymphoblastoid cell lines (LCLs) (Lappalainen et al., 2013). Starting from FASTQ inputs, Rail-RNA produced bigWig files (Kent et al., 2010) encoding genomic coverage; per-sample BED files recording exon–exon junctions, insertions and deletions; and sorted, indexed BAM (Li et al., 2009) files recording the alignments. We downloaded and preprocessed all the GEUVADIS data using a cluster of 21 c3.2xlarge Amazon EC2 instances in 7 h and 29 min, costing a total of $36.12. Rail-RNA then ran on 61 c3.8xlarge Amazon EC2 instances (see S.5) spanning 1952 processors. The run completed in 15 h and 47 min and cost $605.12 ($0.91 per sample). Including preprocessing, the cost per sample was $0.96.
We analyzed bigWig outputs using the derfinder Bioconductor package (Collado-Torres et al., 2015) based on algorithms described in (Frazee et al., 2014; Jaffe et al., 2015). derfinder identified contiguous genomic stretches where average coverage across all samples was above a genome-wide threshold (see Section S.14). Adjacent bases above the threshold were grouped into ‘expressed regions’ (ERs). We identified 285 695 ERs in this way; median ER length was 70 bp (interquartile range, IQR: 7–145). While gene annotation/structure is not used to identify ERs, the regions can be overlapped with a gene annotation to assess novel transcriptional activity, as shown in Supplementary Figure S1a. Relative to Ensembl v75 (Cunningham et al., 2015), we found that 151 581 ERs (53.1%) were within exons (median length: 93 bp, IQR: 21–155) and another 38 784 (13.9%) overlapped both exonic and intronic sequence (median length: 132 bp, IQR: 76–269) perhaps representing alternative ends to known exons. We also found 72 367 regions (25.3%) contained within intronic sequence (median length: 9 bp, IQR: 2–37) and another 19 649 regions (6.9%) within intergenic sequence (median length: 15 bp, IQR: 3–71). These intronic and intergenic ERs could represent novel (polyadenylated, since the data was generated using polyA+ prep) non-coding RNA.
We also reproduced the variance component modeling illustrated in Figure 3 of Ac’t Hoen et al. (2013). At each of the 285 695 ERs, we modeled -adjusted expression levels as a function of many largely technical variables related to the RNA (RIN value, RNA concentration and RNA quantity), the sequencing libraries (library preparation date, library primer index for each sample, method then target and actual quantities of library concentrations, and library size) and the sequencing itself (cluster kit, sequencing kit, cluster density and sequencing lane). We further included the ethnicity of each sample as a variable because it appeared to explain moderate variability in expression levels at many ERs. Lastly, we calculated the amount of residual variability not explained by any of the variables we considered. Interestingly, we found little difference in the amounts of variability explained by each of the variables studied when we stratified the ERs by the annotation categories described above (Supplementary Fig. S1b–e), suggesting that the technical factors affect expression of previously unannotated (i.e. intronic and intergenic) features in a similar manner as they do annotated gene structures.
Fig. 3.
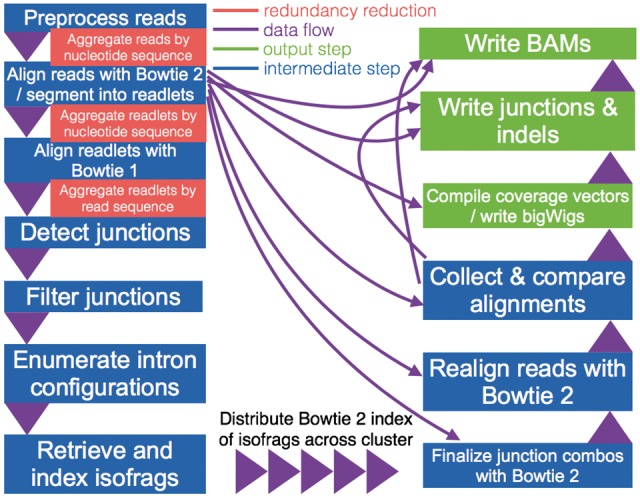
Rail-RNA pipeline. Blue boxes are steps, and green boxes are output steps, which are optional depending on which deliverables are requested by the user. Purple indicators illustrate the flow of intermediate data, with triangles specifying the order in which steps are executed. Aggregations that eliminate redundant alignment work are highlighted in red (Color version of this figure is available at Bioinformatics online.)
3 Methods
For each read, Rail-RNA seeks the (possibly spliced) alignment maximizing the alignment score, which measures similarity between the read and reference. Bowtie 2’s local-mode scoring function (see http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml) is used: matched bases add a bonus while mismatched bases and gaps incur a penalty. Gap penalties are affine. Ties for best alignment score are broken as follows. Each alignment i among the highest-scoring alignments overlaps some number n(i) of exon–exon junctions, where n(i) = 0 for an alignment wholly contained in a single exon. If the smallest n(i) among the highest-scoring alignments is attained by exactly one alignment, that alignment is chosen. If the smallest n(i) is attained by more than one alignment, the tie is broken in a weighted random fashion where alignments overlapping high-coverage junctions are preferred to alignments overlapping low-coverage junctions (Section S.15).
Methods used in the statistical analysis of the GEUVADIS dataset are described in Section S.14. Steps of the Rail-RNA pipeline are in boldface and described in the following paragraphs. The term ‘workers’ refers to computer processes under Rail-RNA’s control. Usually many workers operate simultaneously, distributed across several computers. Each step writes either intermediate or final output data. Depending on the outputs requested by the user, some steps may be omitted. Details of how Rail-RNA is implemented in various distributed environments are given in Section S.16. Figure 3 illustrates the full Rail-RNA pipeline, highlighting where redundant alignment work is eliminated.
As input to the preprocess reads step, the user provides a manifest file containing a URL pointer to each input FASTQ file. Two URLs are specified for each paired-end sample, one for a single-end sample. URLs point to the local filesystem, the web, or on Amazon’s Simple Storage Service (S3). Input reads are downloaded as necessary and preprocessed into a format that facilitates parallelism (Section S.17).
Duplicate reads and readlets both within and across samples lead to redundant alignment work. To eliminate redundant work, in the align reads step Rail-RNA groups duplicate reads so that a worker operates on all reads having the same nucleotide sequence at once. Afterwards, two passes of alignment are performed using Bowtie 2. In the first, each unique read sequence is aligned to the genome. If there is exactly one highest-scoring alignment and it has no gaps, mismatches or soft-clipped bases, all reads with the same nucleotide sequence are assigned that alignment. If the alignment is not perfect or if there is more than one highest-scoring alignment, all reads with the same nucleotide sequence are run through a second pass of Bowtie 2 to ensure that quality sequences are taken into consideration when scoring alignments or ties are broken. Some read sequences with imperfect alignments are divided into short overlapping substrings called readlets. These sequences are searched for whether they overlap exon–exon junctions in a later step. See Section S.18 for further details.
In the align readlets step, Rail-RNA groups duplicate readlets so that a worker operates on all readlets across samples with the same nucleotide sequence at once. Each distinct readlet sequence is aligned to the genome with Bowtie (Langmead et al., 2009) exactly once, eliminating redundant alignment work. Rail-RNA searches for several possible alignments, up to 30 by default using command-line parameters -a -m 30. Readlets with the same nucleotide sequence are then each associated with the same set of alignments.
In the detect exon–exon junctions using readlet alignments step, Rail-RNA uses correlation clustering and maximum clique finding to detect exon–exon junctions spanned by each distinct read sequence, as detailed in Section S.19. The algorithm is reminiscent of the seed-and-vote strategy of Subread/subjunc (Liao et al., 2013), and we note similarities and differences in Section S.20. The step outputs exon–exon junctions and the number of reads covering each junction in each sample.
In simulations we observed that junctions detected in only a small fraction of samples tend to be false positives (Fig. 2b). Consequently, the global list of exon–exon junctions is quickly dominated by false positives as the number of samples increases. To keep precision high, in the filter exon–exon junctions step, Rail-RNA borrows strength across samples to remove junctions not meeting one of these criteria:
The exon–exon junction appears in at least K% of samples.
The exon–exon junction is covered by at least J reads in at least one sample.
by default, but both are configurable. See Section S.21 for a discussion of how this filter was chosen. In the enumerate intron configurations step, Rail-RNA enumerates the ways that multiple exon–exon junctions detected on the same strand in the same sample can be overlapped by a read segment s(readlet_config_size) spanning readlet_config_size bases; we call a way a read or readlet can overlap multiple exon–exon junctions an ‘intron configuration.’ Intron configurations for readlets are obtained and output as described in Section S.22.
In the retrieve and index isofrags step, each worker operates on an intron configuration at a time, concatenating the exonic bases surrounding its introns to form a transcript fragment of size readlet_config_size. This is termed an ‘isofrag.’ Care is taken to avoid including intronic bases in isofrags (Section S.23). Subsequently, a single worker uses bowtie2-build to build a single Bowtie 2 index for all enumerated isofrags. Later, Bowtie 2 uses the index to realign reads in the next step.
In the finalize combinations of exon–exon junctions overlapped by read sequences step, read sequences that failed to align perfectly in the first step are aligned to isofrags using Bowtie 2 in local mode with a minimum score threshold of 48 by default. Local alignment is used since indexed sequences are of length readlet_config_size, shorter than the read length. Rail-RNA runs Bowtie 2 with the parameter -k 30 by default so that many alignments are reported per read sequence. From these alignments Rail-RNA derives a list of exon–exon junctions the read could possibly overlap. A graph algorithm enumerates the combinations of exon–exon junctions the read sequence might simultaneously overlap; see Section S.24 for details.
In the realign reads step, read sequences that failed to align perfectly in the first step are realigned to a set S of transcript fragments. Each transcript fragment in S overlaps a different combination of exon–exon junctions found in the previous step. All the exon–exon junction combinations found for the read’s nucleotide sequence are spanned by a subset of S. Moreover, several distinct read sequences may overlap transcript fragments in S. A given worker performs realignment as follows.
Transcript fragments in S are recorded and indexed with bowtie2-build.
Reads are realigned to the new index using Bowtie 2 in –local mode. These are reads that are in the same index bin referenced in Section S.18.
In the collect and compare read alignments step, Bowtie 2 alignments of reads accumulated in previous steps, except for those that aligned perfectly in the ‘align reads to genome’ step, are collected here and partitioned by read name. A worker operates on all alignments of a given read at once. For each read, if there is exactly one highest-scoring alignment for that read, it is chosen as the primary alignment. Otherwise, Rail-RNA attempts to break the tie by selecting the alignment spanning the fewest exon–exon junctions. If there is still a tie, it is broken by a random draw weighted by the number of uniquely aligned reads supporting each exon–exon junction, as described by (3) in Section S.15.
By default, in the write BAMs step, all primary alignments, including perfect alignments from the ‘align reads’ step, are output. The user may disable this output or instead specify the -k X parameter to ask Rail-RNA to output up to X highest-scoring alignments per read. Alignments are written as sorted, indexed BAM files. By default, one BAM file is output per sample per chromosome. In elastic mode, all BAM files and their indexes are uploaded to S3. Tools such as the UCSC genome browser (Kent et al., 2002) allow users to visualize portions of BAM files without having to download them first.
By default, in the compile coverage vectors and write bigWigs step, Rail-RNA records vectors encoding depth of coverage at each position in the reference genome. The user may disable this output. Two bigWig files are produced per sample: one records coverage of the genome by reads for which each has exactly one highest-scoring alignment, and the other records coverage of the genome by primary alignments. bigWig files encoding mean and median coverage of the genome across samples are also written. The contributions of each sample to the mean and median are normalized by the number of mapped reads in the sample. In elastic mode, bigWig files are uploaded to S3. For example, the analysis in Section 2.4 was performed on bigWig files on S3. Further, tools such as the UCSC genome browser (Kent et al., 2002) allow users to visualize portions of bigWig files without having to download them first. These methods are detailed in Section S.25.
In the write exon–exon-junctions and indels step, Rail-RNA writes a set of TSV files. Each file contains a table with rows corresponding to samples and columns to distinct features. The (i, j)th element is the number of reads in the ith sample containing the jth feature. Three TSVs are written per sample, one where features are insertions, one for deletions and one for exon–exon junctions. Also, three BED files are written per sample: one with exon–exon junctions, one with insertions and one with deletions. These are formatted identically to TopHat 2’s analogous output. In elastic mode, these files are uploaded to S3, where they can be analyzed as soon as Rail-RNA completes. The user may disable any outputs of this step.
4 Discussion
While it is challenging to design software that runs naturally on many samples at once, Rail-RNA demonstrates that this comes with unique and substantial advantages. Rail-RNA achieves better-than-linear growth in throughput (and consequently reduction in cost) per sample as the number of samples grows. By using information from all samples when analyzing data from a given sample, Rail-RNA achieves superior accuracy, with its accuracy advantage growing as samples are added. Rail-RNA also substantially resolves two biases affecting other tools: bias against low-coverage junctions and annotation bias. Rail-RNA results obtained by one investigator can be reliably reproduced by another since Rail-RNA computer clusters rented from commercial cloud services have standardized hardware and software.
We demonstrated Rail-RNA by re-analyzing a 667-sample dataset in 15 h and 40 min at a per-sample cost of $0.91, far lower than sequencing cost (Combs and Eisen, 2015). We analyzed region-level differential expression by simply passing Rail-RNA’s bigWig outputs to standard downstream tools (e.g. the derfinder Bioconductor packages) for further analysis. Users can reproduce this analysis using resources rented from a commercial cloud provider, without having to download any large datasets. Altogether, this is an important step in the direction of usable software that quickly, reproducibly and accurately analyzes large numbers of RNA-seq samples at once.
We used the simple ‘5 reads or 5% of samples’ filter for calling exon–exon junctions to avoid overfitting to our simulations. Further investigation is needed to understand how the filter should be adjusted to optimize accuracy and how to account for other factors like sequencing depth or variability in junction profiles between samples. The filter also contributes to an ‘N + 1’ problem: if Rail-RNA is used to analyze N replicates, but an replicate arrives later, it is difficult to analyze just the and produce the same output as if the N + 1 arrived together. These are areas for future work, discussed further in Section S.21 and Section S.26.
Funding
AN, LCT, JTL and BL were supported by NIH/NIGMS grant 1R01GM105705 to JTL. AN was supported by a seed grant from the Institute for Data Intensive Engineering and Science (IDIES) at Johns Hopkins University to BL and JTL. LCT was supported by Consejo Nacional de Ciencia y Tecnología México 351535. BL and JP were supported by a Sloan Research Fellowship to BL. JAH was supported by Fundación UNAM. BL was supported by National Science Foundation grant IIS-1349906. Experiments using Amazon Web Services were supported by two AWS in Education grants to BL.
Conflict of Interest: none declared.
Supplementary Material
References
- Ac’t Hoen P. et al. (2013) Reproducibility of high-throughput mRNA and small RNA sequencing across laboratories. Nat. Biotechnol, 31, 1015–1022. [DOI] [PubMed] [Google Scholar]
- Au K.F. et al. (2010) Detection of splice junctions from paired-end RNA-seq data by splicemap. Nucleic Acids Res., 38, 4570–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfert T. et al. (2012) A context-based approach to identify the most likely mapping for RNA-seq experiments. BMC Bioinf., 13, S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant D.W. et al. (2010) Supersplat spliced RNA-seq alignment. Bioinformatics, 26, 1500–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloonan N. et al. (2009) RNA-mate: a recursive mapping strategy for high-throughput RNA-sequencing data. Bioinformatics, 25, 2615–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado-Torres L. et al. (2015) derfinder: software for annotation-agnostic RNA-seq differential expression analysis. bioRxiv, 015370. [Google Scholar]
- Combs P.A., Eisen M.B. (2015) Low-cost, low-input RNA-seq protocols perform nearly as well as high-input protocols. PeerJ PrePrints, 3, e869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F. et al. (2015) Ensembl 2015. Nucleic Acids Res., 43, D662–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bona F. et al. (2008) Optimal spliced alignments of short sequence reads. BMC Bioinf., 9, O7. [DOI] [PubMed] [Google Scholar]
- Dean J., Ghemawat S. (2008) Mapreduce: simplified data processing. Commun. ACM Large Clusters, 51, 107–113. [Google Scholar]
- Dobin A. et al. (2013) Star: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazee A.C. et al. (2014) Differential expression analysis of RNA-seq data at single-base resolution. Biostatistics, kxt053.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn T.C. (2011) Field guide to next-generation DNA sequencers. Mol. Ecol. Resources, 11, 759–769. [DOI] [PubMed] [Google Scholar]
- Grant G.R. et al. (2011) Comparative analysis of RNA-seq alignment algorithms and the RNA-seq unified mapper (rum). Bioinformatics, 27, 2518–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griebel T. et al. (2012) Modelling and simulating generic RNA-seq experiments with the flux simulator. Nucleic Acids Res., 40, 10073–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden E.C. (2014) Is the $1,000 genome for real? Nat. News, 10, 1038. [Google Scholar]
- Hu J. et al. (2012) Osa: a fast and accurate alignment tool for RNA-seq. Bioinformatics, 28, 1933–1934. [DOI] [PubMed] [Google Scholar]
- Huang S. et al. (2011) Soapsplice: genome-wide ab initio detection of splice junctions from RNA-seq data. Front. Genet., 2, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe A.E. et al. (2015) Developmental regulation of human cortex transcription and its clinical relevance at single base resolution. Nat. Neurosci, 18, 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean G. et al. (2010) RNA-seq read alignments with palmapper. Curr. Protoc. Bioinf., 11–16. [DOI] [PubMed] [Google Scholar]
- Kent W.J. et al. (2002) The human genome browser at UCSC. Genome Res., 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J. et al. (2010) Bigwig and bigbed: enabling browsing of large distributed datasets. Bioinformatics, 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. et al. (2013) Tophat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol., 14, R36.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. et al. (2015) Hisat: a fast spliced aligner with low memory requirements. Nat. Methods, 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. (2012) Fast gapped-read alignment with bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. et al. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol., 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T. et al. (2013) Transcriptome and genome sequencing uncovers functional variation in humans. Nature, 501, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R. et al. (2010a) The European nucleotide archive. Nucleic Acids Res., gkq967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R. et al. (2010b) The sequence read archive. Nucleic Acids Res., gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2009) The sequence alignment/map format and samtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y. et al. (2013) The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res., 41, e108e108.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale J. et al. (2013) The genotype-tissue expression (gtex) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco-Sola S. et al. (2012) The gem mapper: fast, accurate and versatile alignment by filtration. Nat. Methods, 9, 1185–1188. [DOI] [PubMed] [Google Scholar]
- Ozsolak F., Milos P.M. (2010) RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet., 12, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez F., Granger B.E. (2007) Ipython: a system for interactive scientific computing. Comput. Sci. Eng., 9, 21–29. [Google Scholar]
- Philippe N. et al. (2013) Crac: an integrated approach to the analysis of RNA-seq reads. Genome Biol., 14, R30.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz M.C. et al. (2010) Cloud computing and the DNA data race. Nat. Biotechnol., 28, 691–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein L.D. et al. (2010) The case for cloud computing in genome informatics. Genome Biol., 11, 207.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. et al. (2009) Tophat: discovering splice junctions with RNA-seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. et al. (2009) Rna-seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet., 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. et al. (2010) Mapsplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res., gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.D., Nacu S. (2010) Fast and snp-tolerant detection of complex variants and splicing in short reads. Bioinformatics, 26, 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. (2012) Passion: a pattern growth algorithm-based pipeline for splice junction detection in paired-end RNA-seq data. Bioinformatics, 28, 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.