

Abstract
Summary
Rule-based modeling is a powerful approach for studying biomolecular site dynamics. Here, we present SPATKIN, a general-purpose simulator for rule-based modeling in two spatial dimensions. The simulation algorithm is a lattice-based method that tracks Brownian motion of individual molecules and the stochastic firing of rule-defined reaction events. Because rules are used as event generators, the algorithm is network-free, meaning that it does not require to generate the complete reaction network implied by rules prior to simulation. In a simulation, each molecule (or complex of molecules) is taken to occupy a single lattice site that cannot be shared with another molecule (or complex). SPATKIN is capable of simulating a wide array of membrane-associated processes, including adsorption, desorption and crowding. Models are specified using an extension of the BioNetGen language, which allows to account for spatial features of the simulated process.
Availability and implementation
The C ++ source code for SPATKIN is distributed freely under the terms of the GNU GPLv3 license. The source code can be compiled for execution on popular platforms (Windows, Mac and Linux). An installer for 64-bit Windows and a macOS app are available. The source code and precompiled binaries are available at the SPATKIN Web site (http://pmbm.ippt.pan.pl/software/spatkin).
Contact
spatkin.simulator@gmail.com
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
In living cells, information processing is enabled by networks of interacting biomolecules that typically have multiple modification and binding sites. Tracking the time- and condition-dependent states of these sites in a model is challenging because of the high or even infinite number of potentially populated chemical species. This challenge is overcome with rule-based modeling approaches, in which models are formulated in terms of rules for biomolecular interactions. With a rule-based approach, a large reaction network can be concisely represented by a relatively small set of rules, with each rule characterizing an interaction and an associated class of reactions (Chylek et al., 2014). A complete list of reactions implied by rules can be generated before a numerical simulation, as with BioNetGen (Harris et al., 2016), or the rules may be used as event generators in a stochastic simulation algorithm, as with NFsim (Sneddon et al., 2011). The latter approach enables one to analyze models consisting of rules that imply reaction networks too large to be generated.
There are many software packages that enable rule-based modeling but only a handful of them support spatial modeling. Tools that enable spatial rule-based modeling include MCell-R (Tapia Valenzuela, 2016), Simmune (Angermann et al., 2012), Smoldyn (Andrews, 2017), SK (Sorokina et al., 2013), SRsim (Gruenert et al., 2010), SSC (Lis et al., 2009) and VCell (Schaff et al., 2016). Simmune, SSC and VCell depend on network generation, meaning that a list of possible reactions must be derived from rules before a simulation is performed. The remaining three simulators have the advantage of not relying on network generation.
Here, we present SPATKIN, which offers stochastic rule-based modeling capabilities that do not rely on prior network generation. The simulator tracks positions of individual molecules (not accounting for their internal spatial structure) in a two-dimensional space discretized using a triangular lattice. A comparison of SPATKIN with the most similar of the above-mentioned tools is provided in Section 4. Early-development-stage versions of SPATKIN have been used to study how diffusion and the intrinsic noise of biochemical reactions affect the initiation of traveling waves (Kochańczyk et al., 2013; Zuk et al., 2012). The software has been validated by comparing simulation results obtained for very fast diffusion (rendering the simulated reactor well-mixed) with the results obtained with (non-spatial) BioNetGen. For finite diffusivity, it was shown that the effective macroscopic rate constants determined using SPATKIN agree with analytical estimates for these rate constants (Nałecz-Jawecki et al., 2015; Szymańska et al., 2015) and that the traveling wave velocity agrees with that obtained in finite element-based simulations of analogous partial differential equations-defined systems (Kochańczyk et al., 2013).
2 Approach
SPATKIN implements two-dimensional lattice-based Brownian dynamics (BD) and is designed for simulations of reaction–diffusion processes occurring on surfaces, such as the plasma membrane of a cell. Models and simulation protocols are specified using an extension of the BioNetGen language (BNGL), or to precise, a superset of a subset of BNGL. The extended language is referred to as SptBNGL. A complete, formal definition of SptBNGL, along with its comparison with BNGL, are provided in the User Manual. SptBNGL allows for the specification of boundary conditions, the diffusion properties of biomolecules and biomolecular complexes, and other information necessary to initialize SPATKIN simulations (e.g. starting positions of molecules).
SPATKIN does not rely on network generation. Rather, rules are directly used to generate reaction events. The event-generation procedure is a generalization of that used to generate reaction events in a conventional BD simulation, with use of a list of reactions being replaced by use of a list of rules. Immediately after simulation of a reaction or diffusion event, lists of possible events consistent with model rules are updated locally, which is efficient due to the hybrid use of hierarchical space-partitioning trees and hash tables. The Monte Carlo method is rejection-free unless there are lattice subdomains of diminished diffusivity, which can be introduced to model traps or membrane heterogeneity (Kochańczyk et al., 2013). As with other rule-based modeling software tools, the biomolecules and biomolecular complexes considered in a SPATKIN simulation are associated with graphs, with nodes representing functional components of biomolecules and edges representing bonds between components. The nodes may have textual attributes that represent internal states of biomolecular sites. Thus, SPATKIN differs from conventional BD simulators in that the molecular substructures of chemical species are explicitly tracked. However, the internal graph data structures of SPATKIN are not used to represent spatial configurations of complexes. Molecules and complexes are each taken to occupy a single lattice site, to the exclusion of other unconnected molecules.
There are two kinds of chemical entities: molecules, which represent membrane-tethered proteins and occupy hexagonal cells of a lattice, and binders, which can occupy the sites of a dual lattice (see Fig. 1). The main difference between these two types of entities is that regular molecules can assume different internal states and are capable of binding other molecules, whereas binders have no internal state and can only bind regular molecules. The concept of binders, which are always taken to be immobile, was introduced into SPATKIN to model immunogenic ligands that induce receptor clustering and immobilization. A single lattice node may contain no molecules, or a single (whole) molecule, or an (entire) complex of several (connected) molecules. Because of the constraints of a triangular lattice, any molecule can have up to six other molecules and six binders as neighbors, and any binder can be adjacent to a maximum of three molecules (see Fig. 1). The temporal evolution of a simulated system can be viewed as analogous to that of a cellular automaton in which a single event occurs in each time step.
Fig. 1.
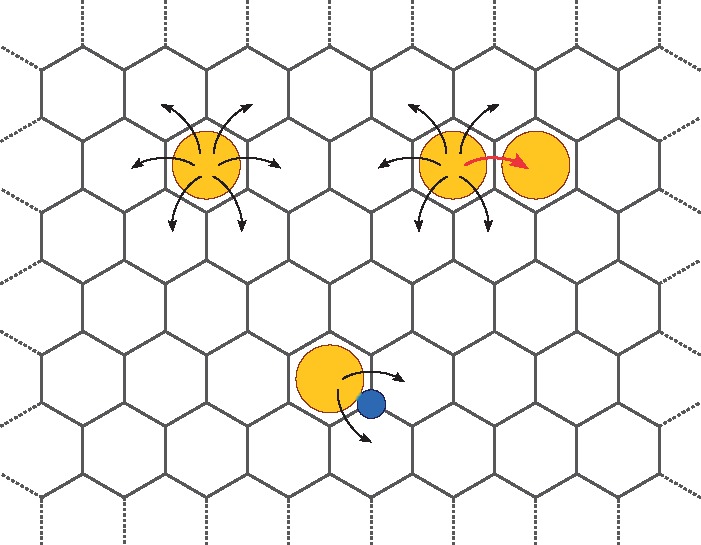
Lattice confinement of (regular) molecules and binders. Top left: A molecule (orange) in a site of a triangular lattice can hop to one of unoccupied adjacent lattice sites. Top right: A molecule can jump to an occupied adjacent lattice site (red arrow) only when a complex formation reaction is allowed (and such an event has been selected). Bottom: Movements of a molecule bound to an (immobile) binder (blue), which is placed in a node of a dual lattice, are constrained so that the bond is not broken
We recommend using SPATKIN in conjunction with BioNetGen. As the latter tool implements an efficient version of Gillespie’s direct method for well-mixed systems, it can be used to determine behavior in the infinite-diffusion limit and find starting parameter estimates for more expensive SPATKIN simulation studies. A model formulated in BNGL can be easily recast as a spatial model defined using SptBNGL. From this point, SPATKIN can be applied to investigate spatial effects, such as the possible consequences of diffusion-limited reaction rates or spatially varying diffusivity.
3 Implementation
SPATKIN comprises a simulator equipped with a parser for interpreting plain-text input files, which define models and simulation settings using the conventions of SptBNGL, and a tool for visualizing snapshots of system configurations. Both tools can be invoked from the command line or used within a GUI, which facilitates creation, editing and debugging of input files, and visualization of simulation results in the form of X–Y plots and lattice snapshots (see Fig. 2). The source code is written in C ++; the build process is managed by CMake. The Spirit Parser Framework was used to develop the SptBNGL parser. The Cairo programming library was used to develop SPATKIN’s visualization capabilities. The Qt framework was used to build the GUI. The GUI provides a multiple document interface (MDI), which enables running multiple stochastic simulations in parallel.
Fig. 2.

SPATKIN GUI with (A) code editor as an MDI window and separate windows for viewing simulations results as (B) X–Y plots and (C) snapshots of system configurations
Supplementary Material online contains commented example input files. A detailed User Manual is available at the SPATKIN Web site.
4 Comparison to similar tools
Among the three spatial and network-free rule-based modeling tools mentioned in Introduction, SK has the coarsest spatial resolution and SRSim has the finest spatial resolution. SK implements a next-subvolume method, in which reaction compartments are divided into subvolumes that are taken to be locally well mixed; translocations between subvolumes are treated as reactions. MCell-R integrates NFsim and MCell, providing a tool for off-lattice stochastic simulations of BD and diffusion-controlled reactions defined by rules specified in BNGL. Biomolecules and biomolecular complexes are treated as point particles in both SK and MCell-R. SRSim accounts for the space occupied by biomolecules (i.e. excluded-volume effects) and thus requires specification of the coarse structures of biomolecules, as well as geometric constraints on interactions. SRSim performs molecular dynamics simulations, requiring the user to specify a force field that governs biomolecular interactions.
In comparison to SK, SPATKIN offers greater spatial resolution, with the disadvantage of higher simulation cost. In contrast to MCell-R, SPATKIN is specialized to 2-D reaction systems. In comparison to SRSim, which performs force field-based simulations, SPATKIN, in which simulations are based on excluded-volume BD and chemical kinetics, allows for easier comparison of results against those produced by commonly used non-spatial simulators, making it possible to unambiguously identify diffusion effects.
There are two recently developed simulators, not mentioned thus far, that also consider excluded-volume effects: ReaDDy (Schöneberg and Noé, 2013) and SpringSaLaD (Michalski and Loew, 2016). These tools treat molecules as collections of beads. In ReaDDy, a force field governs diffusive bead movements. In SpringSaLaD, links between beads, modeled as springs, impose constraints on the relative positions of beads. These tools are distinct from SPATKIN, SK, MCell-R and SRSim in that they do not enable model formulation in terms of rules for interactions using a formal language.
Funding
This work was supported by Polish National Science Centre (NCN) grant 2014/13/B/NZ2/03840 and NIH/NIGMS grant GM111510. MK was supported by 2013/09/N/NZ2/02631 (decision NCN-KR-0011/253/2/13).
Conflict of Interest: none declared.
Supplementary Material
References
- Andrews S.S. (2017) Smoldyn: particle-based simulation with rule-based modeling, improved molecular interaction, and a library interface. Bioinformatics, 33, 710–717. [DOI] [PubMed] [Google Scholar]
- Angermann B.R. et al. (2012) Computational modeling of cellular signaling processes embedded into dynamic spatial contexts. Nat. Methods, 9, 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chylek L.A. et al. (2014) Rule-based modeling: a computational approach for studying biomolecular site dynamics in cell signaling systems. WIREs Syst. Biol. Med., 6, 13–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenert G. et al. (2010) Rule-based spatial modeling with diffusing, geometrically constrained molecules. BMC Bioinformatics, 11, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris L.A. et al. (2016) BioNetGen 2.2: advances in rule-based modeling. Bioinformatics, 32, 3366–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochańczyk M. et al. (2013) Stochastic transitions in a bistable reaction system on the membrane. J. R. Soc. Interface, 10, 20130151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis M. et al. (2009) Efficient stochastic simulation of reaction–diffusion processes via direct compilation. Bioinformatics, 25, 2289–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski P.J., Loew L.M. (2016) SpringSaLaD: a spatial, particle-based biochemical simulation platform with excluded volume. Biophys. J., 110, 525–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nałecz-Jawecki P. et al. (2015) Effective reaction rates for diffusion-limited reaction cycles. J. Chem. Phys., 143, 215102. [DOI] [PubMed] [Google Scholar]
- Schaff J.C. et al. (2016) Rule-based modeling with Virtual Cell. Bioinformatics, 32, 2880–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöneberg J., Noé F. (2013) ReaDDy – a software for particle-based reaction-diffusion dynamics in crowded cellular environments. PLoS One, 8, e74261.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneddon M.W. et al. (2011) Efficient modeling, simulation and coarse-graining of biological complexity with NFsim. Nat. Methods, 8, 177–183. [DOI] [PubMed] [Google Scholar]
- Sorokina O. et al. (2013) A simulator for spatially extended kappa models. Bioinformatics, 29, 3105–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymańska P. et al. (2015) Effective reaction rates in diffusion-limited phosphorylation-dephosphorylation cycles. Phys. Rev. E, 91, 022702. [DOI] [PubMed] [Google Scholar]
- Tapia Valenzuela J.J. (2016) A study on systems modeling frameworks and their interoperability. Ph.D. Thesis, University of Pittsburgh.
- Zuk P.J. et al. (2012) Dynamics of a stochastic spatially extended system predicted by comparing deterministic and stochastic attractors of the corresponding birth–death process. Phys. Biol., 5, 055002.. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.