

Abstract
Objective
Imatinib has been investigated for the treatment of systemic sclerosis (SSc) because of its ability to inhibit the platelet-derived growth factor receptor and transforming growth factor-β signaling pathways, which have been implicated in SSc pathogenesis. In a 12-month open-label clinical trial assessing the safety and efficacy of imatinib in the treatment of diffuse cutaneous SSc (dcSSc), significant improvements in skin thickening were observed. Here, we report our analysis of sera collected during the clinical trial.
Methods
We measured the levels of 46 cytokines, chemokines, and growth factors in the sera of individuals with dcSSc using Luminex and ELISA. Autoantigen microarrays were used to measure immunoglobulin G reactivity to 28 autoantigens. Elastic net regularization was used to identify a signature that was predictive of clinical improvement (reduction in the modified Rodnan skin score ≥ 5) during treatment with imatinib. The signature was also tested using sera from a clinical trial of nilotinib, a tyrosine kinase inhibitor that is structurally related to imatinib, in dcSSc.
Results
The elastic net algorithm identified a signature, based on levels of CD40 ligand, chemokine (C-X-C motif) ligand 4 (CXCL4), and anti-PM/Scl-100, that was significantly higher in individuals who experienced clinical improvement than in those who did not (p = 0.0011). The signature was validated using samples from a clinical trial of nilotinib.
Conclusion
Identification of patients with SSc with the greatest probability of benefit from treatment with imatinib has the potential to guide individualized treatment. Validation of the signature will require testing in randomized, placebo-controlled studies. Clinicaltrials.gov NCT00555581 and NCT01166139. (First Release March 15 2017; J Rheumatol 2017;44:631–8; doi:10.3899/jrheum.160833)
Key Indexing Terms: DIFFUSE CUTANEOUS SYSTEMIC SCLEROSIS, IMATINIB, NILOTINIB, PROTEIN MICROARRAYS, AUTOANTIBODIES
Systemic sclerosis (SSc) is a chronic disease characterized by fibrosis and vascular dysfunction affecting the skin and internal organs. Individuals with diffuse cutaneous SSc (dcSSc) experience substantial morbidity related to widespread skin tightening, development of contractures, and painful tendon friction rubs. In addition, these patients are at high risk for internal organ involvement affecting the lungs, kidneys, gastrointestinal tract, and heart, particularly early in their disease course during the phase of progressive skin tightening1. Currently, there are limited therapeutic options for patients with dcSSc, and there is no approved therapy specifically for SSc. Several fibrotic pathways are implicated in the pathogenesis of SSc, including the platelet-derived growth factor receptor (PDGFR) and transforming growth factor-β (TGF-β) pathways2.
Imatinib is a tyrosine kinase inhibitor that has been shown to antagonize the PDGFR and TGF-β pathways through direct inhibition of PDGFR and c-Abl, a downstream kinase in both pathways3. Seven clinical trials of imatinib in SSc have been conducted with variable results because of different patient populations, dosing regimens, and study designs4,5,6,7,8,9,10. Our group performed a 12-month, single-arm, open-label, phase IIa clinical trial of the safety and efficacy of imatinib mesylate in 30 individuals with dcSSc, and observed significant improvements in skin thickening and forced vital capacity4. Sequential blood samples were collected during the clinical trial for biomarker analyses, in particular to identify markers of clinical improvement that could potentially facilitate individualized treatment11.
Multiple circulating cytokines, chemokines, and growth factors are dysregulated in SSc, and have been associated with disease subsets and features12,13. Measurement of cytokines, chemokines, and growth factors using multiplex platforms, such as Luminex, has the advantage of allowing the generation of multianalyte signatures or scores. These multianalyte metrics have the potential to better identify the clinical heterogeneity of SSc compared with single-analyte tests.
Autoantibodies, including antinuclear antibody, antitopoisomerase I (anti-Scl-70), anti-RNA polymerase III, and anticentromere antibody (ACA) are commonly measured in the clinical setting to support the diagnosis of SSc. Presence of specific autoantibodies has also been related to cutaneous subsets and clinical features of SSc14. Many other autoantibodies have been described in SSc, but corresponding laboratory tests are not widely available for clinical use14. Autoantigen microarrays allow multiplex measurement of autoantibodies15, including less common autoantibodies, making them an ideal platform for identification of novel disease associations.
In our current study, we investigated the cytokines, chemokines, growth factors, and autoantibodies in serum samples from a clinical trial of imatinib for the treatment of dcSSc using Luminex, ELISA, and autoantigen microarrays. We identified a multianalyte signature, based on the baseline levels of CD40 ligand, chemokine (C-X-C motif) ligand 4 (CXCL4), and anti-PM/Scl-100, that was predictive of clinical improvement. The signature was highly accurate in predicting clinical improvement in a validation cohort of serum samples from a clinical trial of nilotinib, a tyrosine kinase inhibitor that is structurally related to imatinib, for the treatment of dcSSc. Use of the signature to identify patients with the greatest likelihood of benefit from treatment with imatinib or nilotinib has the potential to guide individualized treatment of SSc.
MATERIALS AND METHODS
Patients and controls
As previously described4, our group performed a 1-year, phase IIa, single-arm, open-label clinical trial (ClinicalTrials.gov: NCT00555581) of imatinib mesylate in the treatment of dcSSc. Patients (n = 30) were treated with a target dose of imatinib (400 mg daily by mouth) for 12 months. A reduction in the modified Rodnan skin score (mRSS) ≥ 5 at 12 months was defined as a clinical improvement based on the defined minimally important difference in dcSSc16. Of the 30 patients enrolled, sera from 26 patients were collected at screening (1 month prior) or baseline (0 months). During the course of the trial, 1 patient withdrew by 6 months and 2 patients withdrew by 12 months. They were removed from analysis for those respective timepoints onward. Pretreatment sera from individuals who met the American College of Rheumatology criteria for dcSSc, collected as part of a 12-month trial of nilotinib in dcSSc17 (ClinicalTrials.gov: NCT01166139)18, were used as a validation cohort for our multianalyte signature. Baseline sera from 7 completers (of 10 enrolled) were analyzed. Baseline demographics are shown in Table 1. The institutional review board (IRB) at the Hospital for Special Surgery approved the above protocols (imatinib 27049 and nilotinib 10041).
Table 1.
Baseline patient characteristics of individuals with dcSSc. Values are n (%) unless otherwise specified.
| Characteristics | Imatinib, n = 26 | Nilotinib, n = 8 |
|---|---|---|
| Age, yrs, mean (range) | 48.6 (18–71) | 47.9 (18–69) |
| Women | 21 (80.8) | 6 (75) |
| Race | ||
| White | 22 (84.6) | 5 (62.5) |
| Hispanic | 2 (7.7) | 0 |
| African American | 0 (0) | 3 (37.5) |
| Undefined | 2 (7.7) | 0 (0) |
| Disease duration, yrs, mean (range)* | 3.3 (0.33–8) | 0.75 (0.42–2) |
| mRSS, median (range) | 26.5 (19–46) | 27 (22–44) |
| Scl-70+ | 7 (28) | 2 (25) |
| ANA+ | 16 (83.3) | 7 (100) |
| ACA+ | 0 (0) | 0 (0) |
| RNA Pol III** | 1 (16.7) | 5 (62.5) |
| ILD involvement | 13 (50) | 2 (25) |
| FVC % predicted, mean (range) | 84 (45–128) | 77 (58–98) |
| DLCO % predicted, mean (range) | 79 (45–127) | 71 (55–85) |
Since first non-Raynaud symptom of SSc.
Seven patients in the imatinib trial were tested for RNA Pol III antibodies. mRSS: modified Rodnan skin score; ANA: antinuclear antibody; ACA: anticentromere antibodies; ILD: interstitial lung disease; FVC: forced vital capacity.
The Stanford Chronic Immunologic Disease Registry and Repository provided age- and sex-matched healthy control sera (n = 8). The Stanford IRB approved the study (protocol #14734). Written consent was obtained from all individuals who participated in this study according to the Declaration of Helsinki.
Luminex
The Stanford Human Immune Monitoring Center performed the Luminex 41-plex Human Cytokine/Chemokine Panel analysis with 3 additional analytes: C-reactive protein (CRP), CXCL4, and soluble L-selectin (EMD Millipore), per manufacturer’s instructions. See Supplementary Table 1 for a complete list of analytes (available with the online version of this article). All samples were run in duplicate and the means of raw mean fluorescence intensity (MFI) values were used. The average coefficient of variation (CV) was 7.32%. Samples with a CV ≥ 30% (< 3% of samples) were excluded from the analyses.
Chemokine ELISA
Serum macrophage inflammatory protein-3β (MIP3β) and I-TAC were measured using human DuoSet ELISA kits (R&D Systems). We coated Nunc-Immuno Maxisorp 96-well plates (Thermo Scientific) with goat anti-human capture antibodies or 10 mg/ml bovine serum albumin (BSA) in phosphate buffered saline (PBS) overnight at 4°C. After washing 5× in wash buffer (PBS with 0.05% Tween), plates were blocked with blocking buffer (10 mg/ml BSA in PBS with 0.05% Tween). Plates were washed and probed with patient serum diluted 1:10 in blocking buffer. After washing, plates were probed with biotinylated goat anti-human detection antibodies in blocking buffer. Plates were washed and probed with streptavidin-conjugated Europium diluted in Delfia Assay Buffer (both by Perkin Elmer). Plates were washed, then incubated in Delfia enhancement buffer (Perkin Elmer) for 25 min at 37°C. The time-resolved fluorescence of each well was measured with a Wallac Victor model 1420 Multilabel Counter (Perkin Elmer). All patient samples were run in duplicate. For each sample, the fluorescence counts from the BSA-coated wells were subtracted from the respective analyte.
Autoantigen microarrays
We printed 28 SSc-associated autoantigens and 2 control features (PBS and goat anti-human secondary) on epoxy-coated glass slides (NEXTERION Slide E) using a VersArray ChipWriter Compact microarraying robot (BioRad). See Supplementary Table 2 (available with the online version of this article) for more information on the autoantigens. Microarrays were printed in a 12-pad format, with all features printed in triplicate. The microarrays were blocked with 5% nonfat milk (blotting grade; BioRad) in PBS, and washed 5× with wash buffer (PBS with 0.1% Tween-20). Patient serum diluted 1:200 in probing buffer (5% fetal calf serum in PBS with 0.1% Tween-20) was used to probe the microarrays in duplicate. After probing, the microarrays were washed 5× with wash buffer and stained with AlexaFluor 647-conjugated goat anti-human immunoglobulin G (IgG; Fcγ fragment-specific) secondary (Jackson Immuno-Research). Microarrays were then washed at room temperature as follows: 5 quick washes in wash buffer, 5 min in wash buffer with gentle shaking, and two 5-min washes in PBS with gentle shaking. Finally, slides were dried at 1000× the force of gravity for 3 min.
Microarrays were scanned at 635 nm using a G2505C Microarray Scanner (Agilent). Images were gridded using GenePix Pro 7.0 software (Molecular Devices) and the MFI minus background fluorescence measurements were used for analysis. The mean MFI of triplicate features was averaged across duplicate samples. Three samples (of 33) were removed from the analysis because their positive control feature (goat anti-human secondary) was negative.
Statistics
Calculations were performed with R version 3.0.219. The R package samr was used to perform significance analysis of microarrays (SAM) on the Luminex and microarray data20. The 2-class unpaired Wilcoxon test statistic was used with 1000 permutations. The pheatmap package was used to create hierarchically clustered heatmaps21. The Mann-Whitney U tests were used for comparison of chemokine and auto-antibody levels between individuals with dcSSc and healthy controls, and between clinical improvement groups. No adjustments were made for multiple testing in Supplementary Figure 1 or Supplementary Figure 2 (available with the online version of this article).
The elastic net algorithm was used to create a predictive signature of clinical improvement during imatinib treatment. Elastic net fits linear models in a stepwise progression along a sequence of values that penalize model complexity (magnitude of the coefficient vector)22. Baseline Luminex, autoantigen microarray, and ELISA measurements as well as clinical history (Supplementary Table 3, available with the online version of this article) from the time of sampling from individuals with dcSSc in the imatinib clinical trial (n = 23 completers) were used as input variables. Prior to performing elastic net, missing values were imputed using k-nearest neighbors (k = 4) from the impute package23. Clinical measurement of ACA was removed prior to imputation because it was measured for only 8 individuals in the imatinib trial. Less than 6% of the combined Luminex, autoantigen microarray, and clinical values required imputation. RNAP (RNA polymerase III antibody) and Scl-70 were measured both clinically and using autoantigen microarrays. Measurements from both platforms were included as input for elastic net because the algorithm is capable of handling data where the number of predictors (p) is greater than the number of observations (n), as well as highly correlated predictors22. Continuous variables were log2 transformed prior to elastic net analysis.
The glmnet package was used to perform elastic net (α = 0.5) with a binomial response type24. Cross-validation (k-fold) was used to estimate the ideal elastic net penalty value (selecting the largest λ value within 1 standard error of the minimum mean squared error), and the coefficients at this value were used to calculate the signature for each individual patient with dcSSc. Variables with coefficients of 0 were removed from the model, which resulted in selection of relevant variables.
Receiver-operating characteristic (ROC) curve analysis was performed using the ROCR package25. For all patients who completed the trial but did not provide sera at a given timepoint, we used last observation carried forward.
RESULTS
Luminex and ELISA analysis of sera from individuals with dcSSc
We used Luminex to measure the levels of 44 cytokines, chemokines, and growth factors in the baseline serum samples of individuals with dcSSc (n = 26) from the clinical trial of imatinib (and healthy controls, n = 8). See Supplementary Table 1 (available with the online version of this article) for a list of the Luminex analytes. To check the reliability of our measurements, we identified inflammatory mediators at significantly different levels between individuals with dcSSc at baseline and healthy controls using SAM (q < 0.001, fold-change > 2), and compared our results with published reports26. There was a high level of agreement between our measurements and the previous studies because we identified 18 analytes that were at significantly higher levels in dcSSc (Figure 1), including CRP, eotaxin, granulocyte macrophage colony-stimulating factor, interleukin (IL)-6, IL-8 (CXCL8), 10 kDa interferon γ-induced protein (IP10, CXCL10), monocyte chemotactic proteins 1 and 3 (MCP1, CCL2 and MCP3, CCL7), and vascular endothelial growth factor, all of which had been previously observed27,28,29,30. Four individuals with dcSSc formed a relatively distinct cluster on the left side of the heatmap. Analysis of their clinical variables showed that they had significantly greater disease duration and significantly lower hemoglobin than the other patients with dcSSc.
Figure 1.
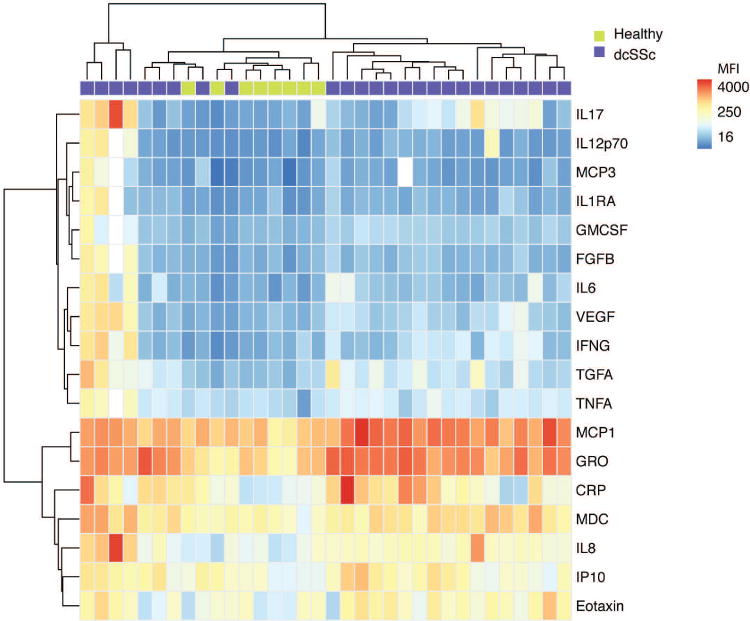
Luminex identifies increased levels of multiple inflammatory proteins in the sera of individuals with dcSSc. The levels of 44 inflammatory proteins were measured in the sera of individuals with dcSSc at baseline (n = 26) and healthy controls (n = 8) by Luminex bead-based immunoassays. SAM was used to identify serum proteins at significantly different levels between groups (q < 0.001, fold-change > 2). A hierarchically clustered (unsupervised, Euclidean distance) heatmap of the SAM-positive proteins is shown. The raw MFI of all proteins were log2 transformed prior to clustering. dcSSc: diffuse cutaneous systemic sclerosis; SAM: significance analysis of microarrays; MFI: mean fluorescence intensity; IL17: interleukin 17; MCP3: monocyte chemotactic protein 3; IL1RA: interleukin 1 receptor antagonist; GMCSF: granulocyte macrophage colony-stimulating factor; FGFB: fibroblast growth factor-β; VEGF: vascular endothelial growth factor; IFNG: interferon γ; TGFA: transforming growth factor-α; TNFA: tumor necrosis factor-α; GRO: growth-related oncogene (CXCL1); CRP: C-reactive protein; MDC: macrophage-derived chemokine (CCL22); IP10: interferon γ-induced protein 10.
Our Luminex analysis showed there were increased levels of multiple interferon-regulated chemokines in the sera of individuals with dcSSc, including MCP1, MCP3, and IP10. We used ELISA to evaluate the levels of 2 additional interferon-regulated chemokines, MIP3β (CCL19) and interferon-inducible T-cell α chemoattractant (ITAC, CXCL11), and found that they were also significantly elevated compared with healthy controls (Supplementary Figure 1, available with the online version of this article)29.
Autoantigen microarray analysis of sera from individuals with dcSSc
We profiled the autoantibodies present in the baseline sera of individuals with dcSSc from the clinical trial of imatinib (n = 24) and healthy controls (n = 7) using autoantigen microarrays featuring 28 known SSc autoantigens and controls (Supplementary Table 2, available with the online version of this article). Our measurements were of high quality because the SAM algorithm identified significantly higher IgG reactivity to multiple known SSc autoantigens, including Scl-70 (topoisomerase I), PM/Scl-75, RNA polymerase III, and nucleophosmin 1, in individuals with dcSSc compared with healthy controls (q < 0.001, fold-change > 2; Figure 2). Further, a high level of agreement was observed between the microarray results and clinical antibody tests (Supplementary Figure 2, available with the online version of this article). One healthy control clustered with individuals who have dcSSc. Other groups observed this previously31, and we concluded that it reflected variability in self-reactivity in the healthy population.
Figure 2.
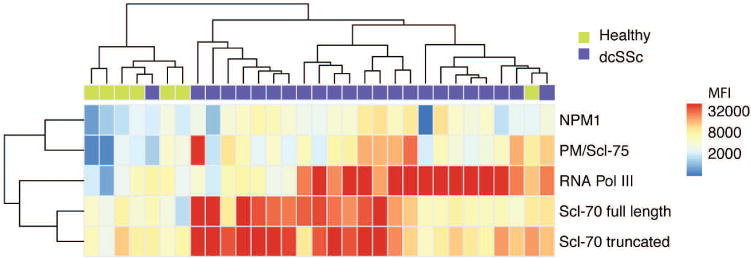
Autoantigen microarray analysis of baseline sera from individuals with dcSSc. Sera from patients with dcSSc (n = 24) and healthy controls (n = 7) were used to probe microarrays with 28 known autoantigens. SAM was used to identify autoantigens with significantly different reactivity levels between groups (q < 0.001, fold-change > 2). A hierarchically clustered (unsupervised, Euclidean distance) heatmap of the SAM-positive autoantigens is shown. All features were log2 transformed prior to clustering. dcSSc: diffuse cutaneous systemic sclerosis; SAM: significance analysis of microarrays; NPM1: nucleophosmin 1; MFI: mean fluorescence intensity.
Identification of a multianalyte signature associated with clinical improvement during treatment with imatinib
We used the elastic net multiple regression technique to identify a multianalyte signature that is predictive of clinical improvement in dcSSc during treatment with imatinib22. Input variables used were baseline Luminex, autoantigen microarray, and ELISA measurements as well as clinical history (Supplementary Table 3, available with the online version of this article) from the time of sampling of individuals with dcSSc in the imatinib clinical trial (n = 23 completers). Clinical improvement in skin thickness (reduction in mRSS ≥ 5 at 12 mos) was used as the outcome of interest. A signature, corresponding to the following linear equation based on serum measurements of CD40L, CXCL4, and anti-PM/Scl-100, was identified by elastic net (coefficients have been rounded to 3 decimal places; Supplementary Figure 3, available with the online version of this article):
The equation was used to calculate scores for each individual patient with dcSSc [CD40L and CXCL4 represent Luminex values, and anti-PM/Scl-100 (PM) was measured by microarray]. Scores were significantly higher in individuals with dcSSc who had a clinical improvement after 12 months of treatment with imatinib than in those who did not (Figure 3A). ROC curve analysis revealed that the signature was highly accurate [area under curve (AUC) = 0.91, 95% CI 0.77–1.0; Figure 3B]. Using the median score as a threshold, we found that individuals with high scores (> 0.69) were more than twice as likely to experience a clinical improvement than individuals with low scores [relative risk (RR) 2.3, 95% CI 1.1–5.0; p = 0.0069].
Figure 3.
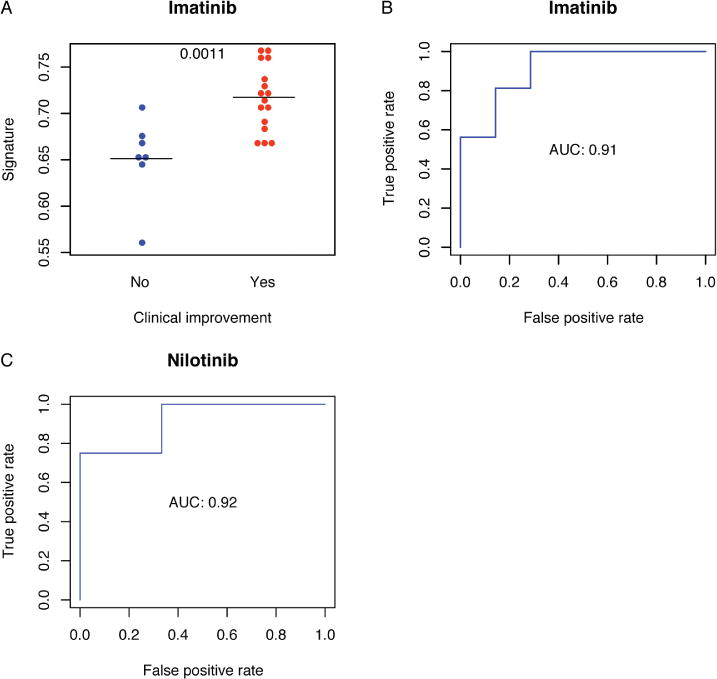
Multianalyte signature predicts improvement in mRSS during treatment with imatinib and nilotinib. The elastic net algorithm was used to identify a signature, based on serum levels of CD40L, CXCL4, and PM/Scl-100 at baseline, that was predictive of clinical improvement during treatment with imatinib (defined as a decrease in mRSS ≥ 5 at 12 mos). (A) The signature was calculated for each individual patient in the imatinib trial at baseline, and was compared between clinical improvement groups. Bars represent group medians and the p values of Mann-Whitney U tests are shown. (B) ROC curve analysis was used to evaluate the accuracy of the baseline signature in predicting clinical response at 12 months in the imatinib trial. (C) Serum samples from a clinical trial of nilotinib for the treatment of dcSSc were used as an independent cohort to validate the signature. Baseline levels of CD40L, CXCL4, and PM/Scl-100 were used to calculate the signature for each individual patient, and ROC curve analysis was performed to assess its accuracy at predicting clinical improvement (reduction in mRSS ≥ 5) at 6 months. mRSS: modified Rodnan skin score; CXCL4: chemokine (C-X-C motif) ligand 4; ROC: receiver-operating characteristic; AUC: area under the curve.
To validate the signature, independent serum samples from a clinical trial of nilotinib for the treatment of dcSSc were assessed (n = 7). Measurements of CD40L, CXCL4, and anti-PM/Scl-100 were used to calculate the signature for each baseline sample in a blinded fashion. Change in the mRSS at 6 months was a primary endpoint in the nilotinib clinical trial, and so we defined clinical improvement as a reduction in mRSS ≥ 5 at 6 months (compared with 12 mos in the imatinib trial)18. Four of 7 individuals in the trial experienced clinical improvement at 6 months. ROC analysis showed that the model was also highly accurate (AUC = 0.92, 95% CI 0.69–1.0) in the validation cohort (Figure 3C). Similar to the imatinib trial, individuals with high scores were more than twice as likely to experience a clinical improvement than individuals with low scores (RR 2.4).
DISCUSSION
The use of imatinib in SSc has been investigated in 7 clinical trials with highly variable results4,5,6,7,8,9,10. Identification of patients with SSc with the greatest probability of benefit from imatinib treatment has the potential to guide individualized treatment for patients with diffuse cutaneous disease11. We tested whether baseline measurement of cytokines, chemokines, growth factors, or autoantibodies was predictive of clinical improvement (decrease in mRSS ≥ 5) during treatment with imatinib. While the levels of individual analytes were not significantly associated with clinical improvement (data not shown), the elastic net algorithm identified a multianalyte signature, based on baseline levels of CD40L, CXCL4, and anti-PM/Scl-100, that was predictive and accurate (AUC = 0.91).
Our group performed a 12-month, single-arm, open-label clinical trial of the safety and efficacy of nilotinib in 10 individuals with dcSSc, and observed significant improvements in skin thickening at 6 months18. Nilotinib is a tyrosine kinase inhibitor that is structurally related to imatinib. It is a more potent inhibitor of the Abl kinase domain than imatinib, and inhibits PDGFR and other kinases32. We used sera collected during the clinical trial to validate the signature in an independent cohort and found that it was robust (AUC = 0.92).
Limitations of our study include the relatively small sample size and the lack of placebo control groups in the original trials. It is possible that the signature identifies patients who have a good prognosis in terms of skin thickness rather than response to tyrosine kinase inhibitors. Testing the signature in a placebo-controlled trial would clarify whether the signature’s association with clinical improvement is specific to drug treatment. It will also be interesting to investigate whether the signature is predictive of clinical improvement in clinical trials of other drugs targeting different pathways in dcSSc in the future.
Multiple lines of evidence suggest that the analytes in our signature are biologically plausible as biomarkers of clinical improvement during treatment with imatinib or nilotinib. Circulating levels of CXCL4 were identified as a biomarker of SSc (especially diffuse cutaneous disease) associated with skin fibrosis and pulmonary disease and predictive of progression in skin and lung fibrosis, as well as pulmonary arterial hypertension33. High serum levels of CXCL4 would contribute to an increased multianalyte signature, suggesting that these patients may benefit from treatment with imatinib or nilotinib. A caveat to our findings is that we measured CXCL4 in serum rather than plasma. CXCL4 levels in serum may not reflect physiological conditions because of platelet activation during serum preparation. Activated T cells appear to be involved in SSc pathogenesis34. Levels of CD40L on the surface of activated CD4+ T lymphocytes35 and circulating levels of soluble CD40L are elevated in SSc compared with controls36,37. T cells from mice lacking Abl kinases, a molecular target of imatinib and nilotinib, exhibit defective activation in response to T cell receptor stimulation38. In a 6-month clinical trial of imatinib for the treatment of dcSSc, Pope, et al found that the change in plasma levels of soluble CD40L was significantly negatively correlated with change in the physician’s global assessment39. This suggests that individuals with increased serum CD40L (and multianalyte signatures) may benefit from imatinib and nilotinib treatment. PM/Scl-100 antibodies are found in 4.9%–7.1% of individuals with SSc40,41. In agreement with our findings, patients with PM/Scl antibodies were previously found to respond favorably to moderate immunosuppression42.
This study builds on our previous work in which we identified a transcriptional signature of imatinib-responsive genes in the lesional skin of 2 individuals with dcSSc2,10. The current signature has the advantages of being predictive of clinical improvement, and measured using serum, which is less invasive than skin biopsy.
We identified a multianalyte signature in individuals with dcSSc, based on serum levels of CD40L, CXCL4, and anti-PM/Scl-100 that is predictive of clinical improvement during treatment with imatinib. We tested the signature’s performance in an independent trial of nilotinib for the treatment of dcSSc and found that it was highly accurate. Further testing in randomized, placebo-controlled studies will be required to fully validate the signature.
Supplementary Material
Acknowledgments
We thank the individuals with diffuse cutaneous systemic sclerosis and the healthy volunteers who participated in this research. We also thank Yael Rosenberg-Hasson, Rohit Gupta, and the Stanford Human Immune Monitoring Center staff. Finally, we thank the members of the Utz laboratory, especially Alex Kuo and Peggie Cheung, for discussions related to the manuscript.
Supported by Novartis (investigator-initiated grant), the Rudolph Rupert Scleroderma Program at the Hospital for Special Surgery, the Donald E. and Delia B. Baxter Foundation (Career Development Award to PJU), the Floren Family Trust (gift to PJU), the Ben May Charitable Trust (gift to PJU), the National Heart Lung and Blood Institute (Proteomics Contract 268201000034C to PJU), the National Institutes of Health (grant numbers T32GM007365, U19-AI082719, U19-AI110491, UH2-AR067676, UM2-AR067678, UM1-AI110498, and U19-AI090019 all to PJU), the Alliance for Lupus Research (grant number 21858 to PJU), the Canadian Institutes for Health Research (Fellowship to DJH), the Stanford Translational Research and Applied Medicine Program (Pilot Grant to DJH), the Scleroderma Research Foundation (to LSC), the Scleroderma Foundation (to LSC and New Investigator Grant to JKG), the Scleroderma Clinical Trials Consortium (to LSC), the Hospital for Special Surgery/Kellen Family Foundation (Clinician Scientist Development Award to JKG), and the US National Institute of Arthritis and Musculoskeletal and Skin Diseases (5U19AI056363-10 to RFS). The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 261382.
Footnotes
ONLINE SUPPLEMENT
Supplementary material accompanies the online version of this article.
Contributor Information
D. James Haddon, Research Associate, Immunology and Rheumatology, Division of Immunology and Rheumatology, Stanford University School of Medicine, Stanford.
Hannah E. Wand, Genetic Counseling Candidate, Division of Immunology and Rheumatology, Stanford University School of Medicine, Stanford.
Justin A. Jarrell, PhD Candidate, Immunology, Division of Immunology and Rheumatology, Stanford University School of Medicine, Stanford, New York, New York, USA
Robert F. Spiera, Professor of Clinical Medicine, Rheumatology and Director, Vasculitis and Scleroderma Program, Department of Rheumatology, Hospital for Special Surgery, New York, New York, USA.
Paul J. Utz, Professor of Medicine, Immunology and Rheumatology, Institute for Immunity, Transplantation and Infection, Stanford University School of Medicine, Stanford.
Jessica K. Gordon, Assistant Professor of Medicine, Rheumatology, Department of Rheumatology, Hospital for Special Surgery, New York.
Lorinda S. Chung, Department of Rheumatology, Palo Alto VA Health Care System, Palo Alto, California.
References
- 1.Medsger TA., Jr Natural history of systemic sclerosis and the assessment of disease activity, severity, functional status, and psychologic well-being. Rheum Dis Clin North Am. 2003;29:255–73, vi. doi: 10.1016/s0889-857x(03)00023-1. [DOI] [PubMed] [Google Scholar]
- 2.Chung L, Fiorentino DF, Benbarak MJ, Adler AS, Mariano MM, Paniagua RT, et al. Molecular framework for response to imatinib mesylate in systemic sclerosis. Arthritis Rheum. 2009;60:584–91. doi: 10.1002/art.24221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gordon JK, Spiera RF. Targeting tyrosine kinases: a novel therapeutic strategy for systemic sclerosis. Curr Opin Rheumatol. 2010;22:690–5. doi: 10.1097/BOR.0b013e32833f1105. [DOI] [PubMed] [Google Scholar]
- 4.Spiera RF, Gordon JK, Mersten JN, Magro CM, Mehta M, Wildman HF, et al. Imatinib mesylate (Gleevec) in the treatment of diffuse cutaneous systemic sclerosis: results of a 1-year, phase IIa, single-arm, open-label clinical trial. Ann Rheum Dis. 2011;70:1003–9. doi: 10.1136/ard.2010.143974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pope J, McBain D, Petrlich L, Watson S, Vanderhoek L, de Leon F, et al. Imatinib in active diffuse cutaneous systemic sclerosis: results of a six-month, randomized, double-blind, placebo-controlled, proof-of-concept pilot study at a single center. Arthritis Rheum. 2011;63:3547–51. doi: 10.1002/art.30549. [DOI] [PubMed] [Google Scholar]
- 6.Khanna D, Saggar R, Mayes MD, Abtin F, Clements PJ, Maranian P, et al. A one-year, phase I/IIa, open-label pilot trial of imatinib mesylate in the treatment of systemic sclerosis-associated active interstitial lung disease. Arthritis Rheum. 2011;63:3540–6. doi: 10.1002/art.30548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prey S, Ezzedine K, Doussau A, Grandoulier AS, Barcat D, Chatelus E, et al. Imatinib mesylate in scleroderma-associated diffuse skin fibrosis: a phase II multicentre randomized double-blinded controlled trial. Br J Dermatol. 2012;167:1138–44. doi: 10.1111/j.1365-2133.2012.11186.x. [DOI] [PubMed] [Google Scholar]
- 8.Distler O, Distler JH, Varga J, Denton CP, Lafyatis RA, Wigley FM, et al. A multi-center, open-label, proof of concept study of imatinib mesylate demonstrates no benefit for the treatment of fibrosis in patients with early, diffuse systemic sclerosis [abstract] Arthritis Rheum. 2010;62(Suppl 10):560. [Google Scholar]
- 9.Fraticelli P, Gabrielli B, Pomponio G, Valentini G, Bosello S, Riboldi P, et al. Imatinib in Scleroderma Italian Study Group Low-dose oral imatinib in the treatment of systemic sclerosis interstitial lung disease unresponsive to cyclophosphamide: a phase II pilot study. Arthritis Res Ther. 2014;16:R144. doi: 10.1186/ar4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung L, Ruiz P, Wood T, Shoor S, Robinson W, Whitfield M, et al. Evaluation of an imatinib response gene signature in patients with systemic sclerosis [abstract] Arthritis Rheum. 2010;62(Suppl 10):S239. [Google Scholar]
- 11.Bournia VK, Evangelou K, Sfikakis PP. Therapeutic inhibition of tyrosine kinases in systemic sclerosis: a review of published experience on the first 108 patients treated with imatinib. Semin Arthritis Rheum. 2013;42:377–90. doi: 10.1016/j.semarthrit.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Beirne P, Pantelidis P, Charles P, Wells AU, Abraham DJ, Denton CP, et al. Multiplex immune serum biomarker profiling in sarcoidosis and systemic sclerosis. Eur Respir J. 2009;34:1376–82. doi: 10.1183/09031936.00028209. [DOI] [PubMed] [Google Scholar]
- 13.Scala E, Pallotta S, Frezzolini A, Abeni D, Barbieri C, Sampogna F, et al. Cytokine and chemokine levels in systemic sclerosis: relationship with cutaneous and internal organ involvement. Clin Exp Immunol. 2004;138:540–6. doi: 10.1111/j.1365-2249.2004.02642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung L, Utz PJ. Antibodies in scleroderma: direct pathogenicity and phenotypic associations. Curr Rheumatol Rep. 2004;6:156–63. doi: 10.1007/s11926-004-0061-9. [DOI] [PubMed] [Google Scholar]
- 15.Robinson WH, DiGennaro C, Hueber W, Haab BB, Kamachi M, Dean EJ, et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat Med. 2002;8:295–301. doi: 10.1038/nm0302-295. [DOI] [PubMed] [Google Scholar]
- 16.Khanna D, Furst DE, Hays RD, Park GS, Wong WK, Seibold JR, et al. Minimally important difference in diffuse systemic sclerosis: results from the D-penicillamine study. Ann Rheum Dis. 2006;65:1325–9. doi: 10.1136/ard.2005.050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gordon JK, Martyanov V, Magro C, Wildman HF, Wood TA, Huang WT, et al. Nilotinib (Tasigna™) in the treatment of early diffuse systemic sclerosis: an open-label, pilot clinical trial. Arthritis Rheum. 2015;17:S213. doi: 10.1186/s13075-015-0721-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.US National Institutes of Health. Nilotinib in the treatment of systemic sclerosis. NCT01166139. [Internet. Accessed February 28, 2017] Available from: https://clinicaltrials.gov/ct2/results?term=NCT01166139&Search=Search.
- 19.R Core Team. The R project for statistical computing. [Internet. Accessed February 1, 2017] Available from: www.R-project.org.
- 20.Tibshirani R, Chu G, Narasimhan B, Li J. samr: SAM: significance analysis of microarrays. (2.0) [Internet. Accessed February 1, 2017.] Available from: cran.r-project.org/web/packages/samr/index.html.
- 21.Kolde R. Pretty heatmaps. (0.7.7) [Internet. Accessed February 1, 2017.] Available from: cran.r-project.org/web/packages/pheatmap/index.html.
- 22.Zou H, Hastie T. Regularization and variable selection via the elastic net. J R Statist Soc B. 2005;67:301–20. [Google Scholar]
- 23.Hastie T, Tibshirani R, Narasimhan B, Chu G. impute: Imputation for microarray data. (3.2) [Internet. Accessed February 1, 2017.] Available from: bioconductor.org/packages/release/bioc/html/impute.html.
- 24.Tibshirani R. Regression shrinkage and selection via the lasso. J R Statist Soc B. 1996;58:267–88. [Google Scholar]
- 25.Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: visualizing the performance of scoring classifiers. (1.0-7) doi: 10.1093/bioinformatics/bti623. [Internet Accessed February 1, 2017.] Available from: cran.r-project.org/web/packages/ROCR/index.html. [DOI] [PubMed]
- 26.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Mayes MD, Pedroza C, Draeger HT, Gonzalez EB, Harper BE, et al. Does C-reactive protein predict the long-term progression of interstitial lung disease and survival in patients with early systemic sclerosis? Arthritis Care Res. 2013;65:1375–80. doi: 10.1002/acr.21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muangchan C, Harding S, Khimdas S, Bonner A, Canadian Scleroderma Research group. Baron M, et al. Association of C-reactive protein with high disease activity in systemic sclerosis: results from the Canadian Scleroderma Research Group. Arthritis Care Res. 2012;64:1405–14. doi: 10.1002/acr.21716. [DOI] [PubMed] [Google Scholar]
- 29.Liu X, Mayes MD, Tan FK, Wu M, Reveille JD, Harper BE, et al. Correlation of interferon-inducible chemokine plasma levels with disease severity in systemic sclerosis. Arthritis Rheum. 2013;65:226–35. doi: 10.1002/art.37742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bandinelli F, Del Rosso A, Gabrielli A, Giacomelli R, Bartoli F, Guiducci S, et al. CCL2, CCL3 and CCL5 chemokines in systemic sclerosis: the correlation with SSc clinical features and the effect of prostaglandin E1 treatment. Clin Exp Rheumatol. 2012;30(Suppl 71):S44–9. [PubMed] [Google Scholar]
- 31.Li QZ, Karp DR, Quan J, Branch VK, Zhou J, Lian Y, et al. Risk factors for ANA positivity in healthy persons. Arthritis Res Ther. 2011;13:R38. doi: 10.1186/ar3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manley PW, Drueckes P, Fendrich G, Furet P, Liebetanz J, Martiny-Baron G, et al. Extended kinase profile and properties of the protein kinase inhibitor nilotinib. Biochim Biophys Acta. 2010;1804:445–53. doi: 10.1016/j.bbapap.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 33.van Bon L, Affandi AJ, Broen J, Christmann RB, Marijnissen RJ, Stawski L, et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med. 2014;370:433–43. doi: 10.1056/NEJMoa1114576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Reilly S, Hügle T, van Laar JM. T cells in systemic sclerosis: a reappraisal. Rheumatology. 2012;51:1540–9. doi: 10.1093/rheumatology/kes090. [DOI] [PubMed] [Google Scholar]
- 35.Valentini G, Romano MF, Naclerio C, Bisogni R, Lamberti A, Turco MC, et al. Increased expression of CD40 ligand in activated CD4+ T lymphocytes of systemic sclerosis patients. J Autoimmun. 2000;15:61–6. doi: 10.1006/jaut.2000.0387. [DOI] [PubMed] [Google Scholar]
- 36.Allanore Y, Borderie D, Meune C, Lemaréchal H, Weber S, Ekindjian OG, et al. Increased plasma soluble CD40 ligand concentrations in systemic sclerosis and association with pulmonary arterial hypertension and digital ulcers. Ann Rheum Dis. 2005;64:481–3. doi: 10.1136/ard.2003.020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Komura K, Sato S, Hasegawa M, Fujimoto M, Takehara K. Elevated circulating CD40L concentrations in patients with systemic sclerosis. J Rheumatol. 2004;31:514–9. [PubMed] [Google Scholar]
- 38.Gu JJ, Ryu JR, Pendergast AM. Abl tyrosine kinases in T-cell signaling. Immunol Rev. 2009;228:170–83. doi: 10.1111/j.1600-065X.2008.00751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pope J, Walker KM, de Leon F, Vanderhoek L, Seney S, Summers KL. Correlations between changes in cytokines and clinical outcomes for early phase (proof of concept) trials in active diffuse systemic sclerosis using data from an imatinib study. Rheumatology. 2014;53:1830–4. doi: 10.1093/rheumatology/keu216. [DOI] [PubMed] [Google Scholar]
- 40.Mehra S, Walker J, Patterson K, Fritzler MJ. Autoantibodies in systemic sclerosis. Autoimmun Rev. 2013;12:340–54. doi: 10.1016/j.autrev.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 41.Hanke K, Brückner CS, Dähnrich C, Huscher D, Komorowski L, Meyer W, et al. Antibodies against PM/Scl-75 and PM/Scl-100 are independent markers for different subsets of systemic sclerosis patients. Arthritis Res Ther. 2009;11:R22. doi: 10.1186/ar2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marguerie C, Bunn CC, Copier J, Bernstein RM, Gilroy JM, Black CM, et al. The clinical and immunogenetic features of patients with autoantibodies to the nucleolar antigen PM-Scl. Medicine. 1992;71:327–36. doi: 10.1097/00005792-199211000-00001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.