

Abstract
Background
Emerging proteomic technologies using novel affinity-based reagents allow for efficient multiplexing with high sample throughput. To identify early biomarkers of myocardial injury, we recently applied an aptamer-based proteomic profiling platform that measures 1,129 proteins to samples from patients undergoing septal alcohol ablation for hypertrophic cardiomyopathy, a human model of “planned myocardial injury” (PMI). Here we examined the scalability of this approach using a markedly expanded platform to study a far broader range of human proteins in the context of myocardial injury.
Methods
We applied a highly multiplexed, expanded proteomic technique that uses single stranded DNA aptamers to assay 4,783 human proteins (4,137 distinct human gene targets) to derivation and validation cohorts of PMI, to individuals with spontaneous myocardial infarction (SMI), and to at-risk controls.
Results
We found 376 target proteins that significantly changed in the blood after PMI in a derivation cohort (n=20; P < 1.05E-05, one-way repeated measures ANOVA, Bonferroni-threshold). Two hundred forty-seven of these proteins were validated in an independent PMI cohort (n=15; P < 1.33E-04, one-way repeated measures ANOVA); > 90% were directionally consistent and reached nominal significance in the validation cohort. Among the validated proteins that were increased within 1 hour after PMI, 29 were also elevated in patients with spontaneous myocardial infarction (n=63; P < 6.17E-04). Many of the novel markers identified in our study are intracellular proteins not previously identified in the peripheral circulation or have functional roles relevant to myocardial injury. For example, the cardiac LIM protein cysteine and glycine-rich protein 3 (CSRP3) is thought to mediate cardiac mechanotransduction and stress responses while the mitochondrial ATP synthase F0 subunit component (ATP5J) is a vasoactive peptide upon its release from cells. Finally, we performed aptamer-affinity enrichment coupled with mass spectrometry to technically verify aptamer specify for a subset of the new biomarkers.
Conclusion
Our results demonstrate the feasibility of large scale aptamer multiplexing at a level that has not previously been reported and with sample throughput that greatly exceeds other existing proteomic methods. The expanded aptamer-based proteomic platform provides a unique opportunity for biomarker and pathway discovery following myocardial injury.
Keywords: proteins, cardiovascular disease, myocardial infarction
INTRODUCTION
Obstacles for comprehensive blood proteomic profiling include the immense size and structural heterogeneity of circulating proteins, as well as the broad range of abundance levels.1–3 Applications of mass spectrometry based profiling to plasma are limited by complex analytical steps that severely restrict sample throughput.4 By contrast, multiplexing of targeted methods based on capture and detection of specific proteins may afford advantages for large scale efforts to characterize human samples. However, many antibody-based assays are limited by cross-reactivity which precludes large scale multiplexing.5
To address this limitation, investigators have turned to alternative affinity-based reagents, including DNA aptamers. Whether aptamer reagents are well-suited for efficient multiplexing of thousands of proteins at high sample throughput remains to be tested. As a proof of principle, we recently applied a proteomic profiling platform with multiplexed aptamers6 to samples from patients undergoing septal alcohol ablation for hypertrophic cardiomyopathy.6 This human model of “planned” myocardial injury (PMI), in which each individual serves as their own biologic control, reproduces key clinical features of “spontaneous” MI, including chest pain, electrocardiographic changes and wall motion abnormalities, as well as the release of established markers of myocardial injury.7, 8 Upon screening 1,129 proteins on the original platform, we validated 79 proteins that changed in the context of myocardial injury. Here we tested the scalability of this approach with a markedly expanded platform containing ~ 5,000 aptamers targeting a far broader range of analytes. The expanded platform measures many intracellular proteins not previously assayed in the blood, as well as many low-abundance secreted proteins not previously targeted by aptamer-based reagents. Thus, the substantially expanded platform provides an unprecedented opportunity for novel discovery of proteins that are altered following myocardial injury.
METHODS
A complete list of all proteins that change significantly after "planned MI" are included in Supplemental Table 1; a complete list of proteins that differ between spontaneous MI patients and controls are included in Table 1 and Supplemental Table 2. The analytic methods will be made available to other researchers for purposes of reproducing the results or replicating the procedure. However, a subset of the study materials (e.g., DNA aptamers) is proprietary to Novartis and Somalogic and investigators are encouraged to contact the corresponding PIs for questions.
Table 1.
Top protein changes in spontaneous MI
| Entrez Symbol |
UniProt ID | Target Protein Name | Median Fold Change |
P |
|---|---|---|---|---|
|
| ||||
| TNNI | P19429 | troponin I, cardiac | 66.7 | 5.93E-16 |
| ADSSL1 | Q8N142 | adenylosuccinate synthase like 1 | 15.3 | 1.73E-12 |
| CKB, CKM | P12277, P06732 | creatine kinase M-type: B-type heterodimer | 10 | 1.80E-13 |
| FABP3 | P05413 | fatty acid binding protein 3, muscle and heart | 8.3 | 3.69E-11 |
| CSRP3 | P50461 | cysteine and glycine-rich protein 3 | 6.36 | 2.55E-11 |
| ATP5J | P18859 | ATP synthase, mitochondrial Fo complex, subunit F6 | 5.27 | 5.30E-11 |
| MDH1 | P40925 | malate dehydrogenase, cytoplasmic | 5.2 | 4.20E-12 |
| CKM | P06732 | creatine kinase M-type | 3.99 | 1.18E-11 |
| CHCHD10 | Q8WYQ3 | coiled-coil-helix-coiled-coil-helix domain containing 10 | 3.78 | 4.01E-09 |
| MDH2 | P40926 | malate dehydrogenase 2, NAD (mitochondrial) | 3.19 | 4.59E-09 |
| TPM3 | P06753 | tropomyosin 3 | 2.93 | 7.06E-07 |
| REXO2 | Q9Y3B8 | REX2, RNA exonuclease 2 homolog | 2.34 | 1.01E-08 |
| DUSP3 | P51452 | dual specificity phosphatase 3 | 2.08 | 4.31E-05 |
| HIST2H2BE | Q16778 | histone cluster 2, H2be | 2.03 | 1.66E-04 |
| LANCL1 | O43813 | LanC lantibiotic synthetase component C-like 1 | 2.03 | 1.83E-06 |
| TPI1 | P60174 | triosephosphate isomerase 1 | 2.01 | 3.16E-06 |
| HDHD2 | Q9H0R4 | haloacid dehalogenase-like hydrolase domain 2 | 1.94 | 1.08E-07 |
| MB | P02144 | myoglobin | 1.94 | 3.87E-07 |
| CRIP2 | P52943 | cysteine-rich protein 2 | 1.89 | 1.07E-07 |
| TKT | P29401 | transketolase | 1.78 | 6.40E-07 |
| PEBP1 | P30086 | phosphatidylethanolamine-binding protein 1 | 1.66 | 6.35E-06 |
| RPS6KA1 | Q15418 | ribosomal protein S6 kinase, 90kDa, polypeptide 1 | 1.66 | 2.34E-05 |
| FCN1 | O00602 | ficolin (collagen/fibrinogen domain containing) 1 | 1.62 | 3.19E-05 |
| PCDHGB1 | Q9Y5G3 | protocadherin gamma subfamily B, 1 | 1.61 | 2.07E-07 |
| GLRX2 | Q9NS18 | glutaredoxin 2 | 1.55 | 1.47E-08 |
| DECR1 | Q16698 | 2,4-dienoyl CoA reductase 1, mitochondrial | 1.51 | 4.64E-04 |
| LST1 | O00453 | leukocyte specific transcript 1 | 1.45 | 3.87E-07 |
| NAP1L2 | Q9ULW6 | nucleosome assembly protein 1-like 2 | 1.27 | 4.10E-04 |
| SUMF2 | Q8NBJ7 | sulfatase modifying factor 2 | 1.24 | 5.05E-05 |
Shown are proteins increased in SMI cases vs. controls with P <6.17E-04, listed in descending fold change from control (Wilcoxon rank-sum on log transformed RFU values). All proteins were also significant and increased within 1 hour after injury in both PMI derivation and validation cohorts (P < 1.05E-05 and P < 1.33E-04, respectively).
Human Studies Participants
A total of thirty-five patients undergoing PMI using alcohol septal ablation for the treatment of symptomatic hypertrophic cardiomyopathy were included in this study (20 in the derivation cohort; 15 in a distinct validation cohort). Inclusion criteria for this cohort and details of the procedure have been previously described.9–11 Blood was drawn at baseline, 10 minutes, 1 hour and 24 hours after injury. To determine the effects of the catheterization procedure alone, including heparin administration, we studied ten individuals who underwent cardiac catheterization without myocardial injury as controls (eight patients undergoing cardiac catheterization for patent foramen ovale closure and two patients undergoing cardiac catheterization for PMI who did not undergo septal ablation for anatomical reasons).
We also enrolled 20 patients undergoing emergent cardiac catheterization for acute ST-segment elevation spontaneous MI within 8 hours of symptom onset. Femoral venous blood samples were obtained in the coronary catheterization suite upon initial presentation. Peripheral blood samples from 43 at-risk patients with negative standard Bruce protocol exercise tests were used as controls for spontaneous MI.12
All blood samples were collected in K2EDTA-treated tubes, centrifuged within 15 minutes at 2000g for 10 min to pellet cellular elements and subsequently stored at −80 °C.
Human study protocols were approved by the Institutional Review Boards of Beth Israel Deaconess Medical Center and Massachusetts General Hospital. Informed consent was obtained from all participants.
Proteomic assay overview
A proteomic profiling platform has been developed that applies 5,034 single-stranded DNA aptamers (validated SOMAmers™) to assay 4,783 human proteins (4,137 distinct human gene targets), 48 non-human vertebrate proteins and 203 bacterial and viral protein targets covering a broad range of protein functional classifications (Supplemental Figure 1). The intra-assay and inter-assay CVs are presented in the Results section. As compared to the prior iteration of the platform, this version covers a broader array of intracellular proteins (approximately 30%).
Statistical analyses
For the PMI studies, all protein values were log transformed due to nonnormal distributions as determined by the Kolomogorov-Smirnov and Shapiro-Wilk normality tests. For the PMI and cardiac catheterization control studies, one-way repeated measures ANOVA was used to test differences in protein levels across time points (baseline, 10 minutes, 1 hour, and 24 hours). All reported p-values are global p-values, i.e., indicating whether protein levels change significantly between baseline and any of the time points. The multivariate adjustment was used if sphericity was violated. In the derivation cohort, we used a Bonferroni-corrected P threshold < 1.05E-05 (0.05/4783 proteins unaffected in control catheterization, see below) and repeat Bonferroni-corrected P threshold < 1.33E-04 (0.05/376) aptamers in the PMI validation cohort. For the spontaneous myocardial infarction (SMI) case-control analysis, a Wilcoxon rank-sum test was used with a Bonferroni-corrected P threshold < 6.17E-04 (0.05/81 validated proteins shown to increase within 1 hour after myocardial injury in the PMI cohort). All analyses were performed with SAS Software version 9.3 (SAS Institute, Cary, NC).
RESULTS
Protein changes in peripheral plasma of PMI patients
To first assess reproducibility of the platform, we embedded pooled plasma control samples within and across experimental plates to document intra-and inter-experimental coefficients of variance (CVs). The intra-assay and inter-assay CVs (applying performance characteristics for 95% of the probe content in plasma) were 7.45 and 6.49, respectively. Inter-assay CVs were based on plates that were run up to 21 days apart. To assess biological variability, we compared two plasma samples from normal controls collected 10 minutes apart (n=12), which yielded a median intraclass correlation of 0.86 across the entire analytes assayed.
Clinical characteristics of the PMI study patients are detailed in Supplemental Table 3. Using each individual as their own biologic control, we performed proteomic profiling at baseline, 10 minutes, 1 hour and 24 hours after injury. We identified a total of 376 proteins that were significantly changed within 24 hours post injury in a derivation cohort of 20 patients (Bonferroni-adjusted P < 1.05E-05, one-way repeated measures ANOVA). Of note, we identified 2,027 aptamer-protein pairs that were influenced by the catheterization procedure alone with similar directional changes in PMI, likely due to heparin treatment (nominal P < 0.05). These proteins were therefore excluded from the analyses due to possible confounding effects. In a validation cohort of 15 patients, changes in 247 proteins exceeded a repeat Bonferroni threshold (P < 1.33E-04, one-way repeated measures ANOVA). Ninety percent of the proteins changes seen in the derivation cohort were directionally consistent and reach at least nominal significance (P < 0.05) in the smaller validation cohort. Table 2 details a subset of the proteins found to be significant in both derivation and validation cohorts that increased by greater than 30% within 10 minutes after PMI in both cohorts. All validated proteins are listed in Supplemental Table 1. See Supplemental Table 4 for clinical characteristics of the catheterization controls.
Table 2.
Top proteins increased in the peripheral blood after myocardial injury
| Entrez Symbol |
UniProt ID | Target Protein Name | Derivation, n=20, Median % Change | Validation, n=15 Median % Change | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| 10 min vs. Baseline |
1 h vs. Baseline |
24 h vs. Baseline |
P | 10 min vs. Baseline |
1 h vs. Baseline |
24 h vs. Baseline |
P | |||
| ADSSL1 | Q8N142 | adenylosuccinate synthase like 1 | 437.6 | 1407.2 | 108.1 | 8.64E-10 | 473.2 | 1952.1 | 122.2 | 2.81E-09 |
| ARHGAP1 | Q07960 | Rho GTPase activating protein 1 | 49.9 | 48 | 5.1 | 2.24E-07 | 49.2 | 51.1 | 1.7 | 1.02E-10 |
| ATP5J | P18859 | ATP synthase, mitochondrial Fo complex, subunit F6 | 1492.2 | 1044 | 215.9 | 1.41E-09 | 1913.2 | 1693.6 | 220.4 | 6.58E-20 |
| C1orf185 | Q5T7R7 | chromosome 1 open reading frame 185 | 40.9 | 55.1 | 8.9 | 2.36E-07 | 44 | 45.5 | 13.7 | 1.11E-10 |
| CHCHD10 | Q8WYQ3 | coiled-coil-helix-coiled-coil-helix domain containing10 | 258.7 | 271.4 | 41.4 | 3.41E-06 | 310.1 | 423.9 | 47.8 | 6.05E-06 |
| CKB,CKM | P12277 P06732 | creatinine kinase, brain/muscle-type heterodimer | 117.4 | 326.9 | 389.6 | 1.19E-08 | 122.6 | 475 | 275.7 | 9.12E-08 |
| CKM | P06732 | creatine kinase, muscle | 31.7 | 124.9 | 186.8 | 6.24E-09 | 35.7 | 163.8 | 224.6 | 4.95E-08 |
| COL23A1 | Q86Y22 | collagen, type XXIII, alpha 1 | 51.9 | 48.7 | −1.3 | 2.33E-10 | 50.1 | 40.2 | −2.8 | 4.34E-16 |
| CRABP2 | P29373 | cellular retinoic acid binding protein 2 | 62.3 | 64.8 | 16.8 | 1.08E-06 | 64.1 | 67.3 | 7.5 | 5.62E-18 |
| CRIP2 | P52943 | cysteine-rich protein 2 | 45.3 | 74.1 | −7.4 | 3.12E-06 | 38.3 | 74.2 | −10.3 | 2.34E-05 |
| CSRP3 | P50461 | cysteine and glycine-rich protein 3 | 496.5 | 841.5 | 65.5 | 1.82E-10 | 492.9 | 1179.8 | 71 | 5.46E-22 |
| DAG1 | Q14118 | dystrophin-associated glycoprotein 1 | 43.4 | 25.9 | −9.4 | 2.71E-06 | 55.6 | 35.5 | −0.2 | 1.95E-08 |
| DDR1 | Q08345 | discoidin domain receptor tyrosine kinase 1 | 55 | 84.1 | 15.4 | 5.03E-06 | 53.4 | 67 | 25.5 | 6.73E-07 |
| DECR1 | Q16698 | 2,4-dienoyl CoA reductase 1, mitochondrial | 568.6 | 584.4 | −0.6 | 6.63E-07 | 779.2 | 781.6 | −26.5 | 3.95E-18 |
| FABP3 | P05413 | fatty acid binding protein 3, muscle and heart | 127.4 | 439.3 | 35.3 | 3.18E-21 | 147.7 | 536.6 | 37.6 | 7.84E-18 |
| FCN1 | O00602 | ficolin (collagen/fibrinogen domain containing) 1 | 31.8 | 40.9 | 13.6 | 6.60E-08 | 30.5 | 37 | 12.7 | 5.02E-06 |
| FKBP2 | P26885 | FK506 binding protein 2, 13kDa | 39.3 | 40.5 | 8.7 | 2.60E-07 | 48.6 | 43.9 | 17.3 | 1.23E-10 |
| GLRX2 | Q9NS18 | glutaredoxin 2 | 34.4 | 51.3 | 12.4 | 9.34E-06 | 38.6 | 91.1 | 17.1 | 4.02E-06 |
| H1FX | Q92522 | H1 histone family, member X | 772.6 | 457.8 | 54.5 | 7.11E-08 | 1004.2 | 926.7 | 25 | 1.63E-19 |
| MAZ | P56270 | MYC-associated zinc finger protein | 35.2 | 50.7 | 18.3 | 1.57E-06 | 47 | 45.5 | 15.3 | 1.30E-07 |
| MB | P02144 | myoglobin | 52.9 | 113.7 | 37.8 | 9.24E-11 | 51.8 | 135.9 | 43.6 | 4.45E-08 |
| MDH1 | P40925 | malate dehydrogenase 1, NAD (soluble) | 43.7 | 170.7 | 132.7 | 2.30E-11 | 43.8 | 186.4 | 99.6 | 9.87E-09 |
| MDH2 | P40926 | malate dehydrogenase 2, NAD (mitochondrial) | 551.8 | 1037.7 | 211.7 | 9.61E-11 | 893.5 | 1652.7 | 173.7 | 4.60E-08 |
| NDUFV2 | P19404 | NADH dehydrogenase (ubiquinone) flavoprotein 2 | 315 | 422.5 | 52 | 2.02E-08 | 406.2 | 700.3 | 57.8 | 8.01E-06 |
| OLFML3 | Q9NRN5 | olfactomedin-like 3 | 36.4 | −3.4 | −24.9 | 5.87E-10 | 34.7 | −7.8 | −19.2 | 4.69E-06 |
| PCDHGB1 | Q9Y5G3 | protocadherin gamma subfamily B, 1 | 71.6 | 79.2 | 25.8 | 1.90E-09 | 81 | 70.2 | 15.9 | 1.62E-10 |
| PEBP1 | P30086 | phosphatidylethanolamine binding protein 1 | 33.6 | 72.9 | −9.4 | 5.24E-06 | 36.6 | 78.2 | −12 | 8.84E-11 |
| PKLR | P30613 | pyruvate kinase, liver and RBC | 59.7 | 49.1 | 33.6 | 9.83E-07 | 53.5 | 73.2 | 17.8 | 1.63E-10 |
| REXO2 | Q9Y3B8 | REX2, RNA exonuclease 2 homolog | 31.7 | 53.5 | 54.4 | 5.08E-07 | 32.5 | 79.9 | 32.6 | 3.96E-09 |
| TKT | P29401 | transketolase | 55.4 | 79.5 | 11.4 | 6.04E-07 | 34.7 | 65.2 | −0.5 | 3.14E-10 |
| TNNI3 | P19429 | troponin I type 3 (cardiac) | 347.7 | 909.8 | 4614.8 | 2.02E-13 | 361.6 | 1279.9 | 5926.6 | 3.36E-12 |
| TPI1 | P60174 | triosephosphate isomerase 1 | 54.1 | 87.7 | 14.6 | 4.96E-07 | 30.6 | 97.5 | 0.7 | 4.22E-12 |
Proteins found to be increased by greater than 30% within 10 minutes after PMI in both derivation and validation cohorts (global P < 1.05E-05 and P < 1.33E-04, respectively, for change within 24-hour period by one-way repeated measures ANOVA on log transformed RFU values). Change values denote median percent change. Proteins listed in alphabetical order.
The kinetic changes of several representative proteins are illustrated in Figure 1. Using the expanded platform, we confirmed increases in well-established clinical markers of myocardial injury including troponin I and CK-MB, as well as other biomarkers previously identified by our group and others such as fatty acid binding protein (FABP)13, 14 and malate dehydrogenase 1 (MDH1).6 Many of these protein changes were novel in the context of early myocardial injury (see Table 2 and Supplemental Table 1), including a mitochondrial ATP synthase F0 subunit component (ATP5J),15 mitochondrial 2–4-dienoyl-CoA reductase 1 that is important in fatty acid beta oxidation (DECR1),16 and a muscle-specific adenylosuccinate synthase that plays a role in purine nucleotide metabolism (ADSSL1).17
Figure 1. Protein markers that are increased early after the onset of planned myocardial injury (PMI) and in spontaneous myocardial injury (SMI).
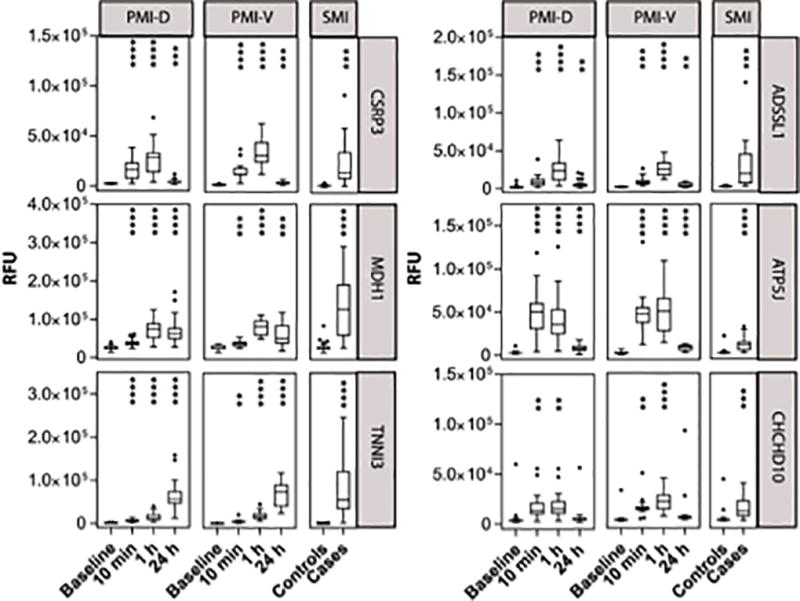
Data presented are from PMI derivation (PMI-D) and validation (PMI-V) cohorts of selected proteins that increased in both a) derivation (n=20) and validation (n=15) cohorts (P< 1.05E-05) and b) SMI cohorts (n=63) P < 6.17–04. P values calculated by one-way repeated measures ANOVA performed on log transformed RFU values. Edges of boxes denote 25th and 75th percentiles, lines denote median, and whiskers are plotted using Tukey method. **** P < 0.0001, *** P <0.001, ** P < 0.01; where P represents significance of change from baseline.
Validation of candidate protein markers in spontaneous myocardial infarction
To better establish the clinical relevance of the observed changes, we profiled a cohort of spontaneous myocardial infarction (SMI) patients (n=20), as well as at-risk individuals without ischemia as determined by exercise stress testing (n=43, clinical characteristics in Supplemental Table 4). Because the timing of sample collection relative to SMI onset was variable (6.0 ± 1.9 h), we focused on the PMI-derived candidate protein markers that were elevated within one hour after injury, the time point in the PMI series closest to the time of SMI presentation. Of the 81 proteins that were significant and elevated within 1 hour after PMI in both the derivation and validation cohorts, 29 were also elevated in SMI (repeat Bonferroni adjusted P < 6.17E-04; WRS, Table 1). Among the many novel findings that were concordantly increased in both PMI and SMI were ADSSL1, ATP5J, DECR1 as discussed above, as well as Coiled-Coil-Helix-Coiled-Coil-Helix Domain Containing 10 (CHCHD10), a mitochondrial protein that may play a role in oxidative phosphorylation,18 and cysteine and glycine rich protein 3 (CSRP3), a muscle LIM domain protein. Representative data for known biomarkers and several novel proteins are demonstrated in Figure 1.
In additional exploratory analysis, we also compared SMI versus control samples without filtering for proteins that were changed in PMI. This analysis yielded an additional 58 proteins elevated in SMI vs. the control cohort, (Supplemental Table 2), which included the “rediscovery” of IL1RL1(ST2), as well as many novel proteins including the mitochondrial iron-sulfur cluster assembly factor BolA family member 3 (BOLA3) and the chloride intracellular channel 4 (CLIC4).
Technical validation by mass spectrometry
To rigorously verify the specificity of observed changes in several novel putative biomarkers including ADSSL1, olfactomedin like 3 (OLFML3) and glucose-6-phosphate isomerase (GPI), we performed technical validation studies using orthogonal mass spectrometry based techniques as detailed in Supplemental Methods. The representative data in Supplemental Figure 2 using aptamer enriched-multiple reaction monitoring (MRM) demonstrates a striking concordance with findings from the expanded aptamer platform and provides accurate quantitation of our protein findings. Supplemental Table 5 details the peptide sequences, precursor masses and transition ions selected for MRM analysis.
DISCUSSION
We previously reported the application of a 1,129-plex aptamer proteomic assay to individuals undergoing planned myocardial injury.1 Here we markedly expanded the scope of prior investigations. While testing the feasibility of large scale multiplexing with a platform that now measures ~5,000 analytes, we provide novel biological insights into the response to injury in humans. Concordant with the ~4-fold increased breadth of the platform, we identified ~ 3-fold number of new protein changes. It is notable that greater than 75% of our prior aptamer findings1 were recapitulated in this study with concordant directionality (P < 0.05), though here we studied more catheterization controls and excluded many additional proteins that appear to be influenced by heparin treatment. Further, twenty-two of the proteins described in two recent mass spectrometry based proteomic profiling papers were confirmed using this new technique;9, 10 an additional ~150 novel changes were identified in the present investigation.
As compared to the prior iteration of the platform, this version covers a broader array of intracellular proteins (approximately 30%), many of which are reproducibly identified in human plasma and changed in the setting of myocardial injury. We were interested to find an enrichment of these proteins following myocardial injury, as 19 of the 33 (58%) most significantly changed proteins are intracellular. Amongst the novel markers identified in both planned and spontaneous injury, we found many proteins, including CHCHD10, DECR1, and MDH1and MDH2, suggesting the release of mitochondrial proteins early after myocardial injury. CSRP3, which was increased after SMI and PMI, is hypothesized to mediate cardiac mechanosensation19 and has been associated with familial and idiopathic dilated and hypertrophic cardiomyopathy.20
In addition to identifying low abundance intracellular proteins not previously assayed in the blood, the expanded platform also provides insight into biologically active proteins which are released following myocardial injury. For example, adenylosuccinate synthetase 1 (ADSSL1) is found in cardiac and skeletal muscle and demonstrates greater than 14-fold increase in circulating levels within 1 hour after myocardial injury. It is a muscle-specific enzyme that plays a role in purine nucleotide cycle by catalyzing the first step in the conversion of inosine monophosphate to adenosine monophosphate, and mutations in the gene have been reportedly associated with myopathy.21 Of particular interest are proteins that may have hormone-like functions on other tissues. The mitochondrial ATP synthase-coupling factor 6 (ATP5J), the most substantially increased protein (by 10 minutes) following myocardial injury, is reported to be a vasoactive peptide. It binds to plasma membrane ATP synthase, leading to vasoconstriction, intracellular acidosis, inhibition of prostacyclin and nitric oxide generation resulting in blood pressure elevation.22
Our study has several limitations. While this expanded aptamer-based proteomics platform provides very broad coverage with high throughput, it is agnostic to changes of analytes not targeted. Exclusion of nearly 2000 proteins that changed in the catheterization control samples translates into a very conservative effort to limit potential confounding factors, possibly excluding proteins that may be changed in myocardial injury as well as catheterization. Additionally, because groups of proteins cluster within biological pathways, the assumption of independent statistical tests used in our Bonferroni-corrected P values is overly stringent, increasing the possibility of false negative results. Similarly, while we have emphasized characteristics of individual markers in our derivation and validation analyses, we anticipate that a multi-marker approach might ultimately be used clinically. Of note, we evaluated temporal co-variance as a method to condense the number of proteins for diagnostic value. In a pilot analysis that focused on proteins that were significantly elevated at one or more time points from baseline following PMI, we identified six temporal protein groupings (Supplemental Figure 3). Carrying forward a subset of proteins with the largest effect size from these groupings (excluding Troponin I and CK-MB), we found 39 principal components that accounted for 85% of the variance in the SMI group versus controls. This approach modestly separated the groups with P = 0.00077 (Supplemental Figure 4). One might hypothesize that specific markers would be most useful when patients are stratified by time to presentation, though this would necessitate future studies in larger numbers of patients. Finally, given that the total number of patients in our cohorts is relatively small, additional samples will also be necessary to identify markers specific to hypertrophic cardiomyopathy patients or proteins that are associated with specific medications or clinical traits.
In summary, we demonstrate the feasibility of large scale aptamer multiplexing at a scale that has not previously been reported. Further, sample throughput of this platform allows one to assess more than 4800 proteins in hundreds of samples within a week, greatly exceeding the sample throughput approachable by other strategies. To provide additional support of these proof-of-concept findings, efforts are ongoing aimed at both additional analytical validation using orthogonal techniques as well as clinical validation in large, heterogeneous cohorts.
Supplementary Material
Clinical Perspective.
What is new?
Like antibodies, DNA aptamers can be generated as affinity reagents for proteins.
Emerging data suggest that they can be used to measure blood protein levels in clinical cohorts; however the technology remains in its infancy.
Here we tested the scalability of this approach with a markedly expanded platform containing ~ 5,000 aptamers targeting a far broader range of analytes than previously examined using this technology.
We applied the platform to a cohort of individuals undergoing “planned myocardial injury” (PMI) for hypertrophic cardiomyopathy.
In addition to confirming findings from prior studies, we identified nearly 150 additional putative markers of myocardial injury.
What are the clinical applications?
Our study suggests that the platform can be applied to identify very early markers of myocardial injury.
Such markers might hasten the diagnosis of myocardial injury, as well as the administration of appropriate therapy.
Our data also provide new insights into the human response to injury as well as new potential targets for therapeutic intervention.
Finally, the improvements in breadth and throughput of this new platform suggest that it can be applied to large patient cohorts to address a variety of important clinical questions.
Acknowledgments
JJ, DN, LF, MDB, TM, LLJ and REG contributed to study design. JJ, DN, SG, MK, TM, LLJ, and REG analyzed and interpreted the data. NF and RP conducted mass spectrometry experiments, acquired and analyzed data. JJ, DN, RP, LLJ and REG wrote the manuscript.
SOURCES OF FUNDING: These studies were supported by NIH grants HHSN268201000033C, R01HL132320 and R01HL133870 to REG, T32HL007208 to MDB and K01GM103817 to DN.
Footnotes
DISCLOSURES: JJ, SG, NF, RP, TM and LLJ are employees of Novartis and some are also stockholders of Novartis.
References
- 1.Anderson NL, Polanski M, Pieper R, Gatlin T, Tirumalai RS, Conrads TP, Veenstra TD, Adkins JN, Pounds JG, Fagan R, Lobley A. The human plasma proteome: A nonredundant list developed by combination of four separate sources. Mol Cell Proteomics. 2004;3:311–326. doi: 10.1074/mcp.M300127-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Fagerberg L, Oksvold P, Skogs M, Algenas C, Lundberg E, Ponten F, Sivertsson A, Odeberg J, Klevebring D, Kampf C, Asplund A, Sjostedt E, Al-Khalili Szigyarto C, Edqvist PH, Olsson I, Rydberg U, Hudson P, Ottosson Takanen J, Berling H, Bjorling L, Tegel H, Rockberg J, Nilsson P, Navani S, Jirstrom K, Mulder J, Schwenk JM, Zwahlen M, Hober S, Forsberg M, von Feilitzen K, Uhlen M. Contribution of antibody-based protein profiling to the human chromosome-centric proteome project (c-hpp) J Proteome Res. 2013;12:2439–2448. doi: 10.1021/pr300924j. [DOI] [PubMed] [Google Scholar]
- 3.Ping P, Vondriska TM, Creighton CJ, Gandhi TK, Yang Z, Menon R, Kwon MS, Cho SY, Drwal G, Kellmann M, Peri S, Suresh S, Gronborg M, Molina H, Chaerkady R, Rekha B, Shet AS, Gerszten RE, Wu H, Raftery M, Wasinger V, Schulz-Knappe P, Hanash SM, Paik YK, Hancock WS, States DJ, Omenn GS, Pandey A. A functional annotation of subproteomes in human plasma. Proteomics. 2005;5:3506–3519. doi: 10.1002/pmic.200500140. [DOI] [PubMed] [Google Scholar]
- 4.Gerszten RE, Accurso F, Bernard GR, Caprioli RM, Klee EW, Klee GG, Kullo I, Laguna TA, Roth FP, Sabatine M, Srinivas P, Wang TJ, Ware LB. Challenges in translating plasma proteomics from bench to bedside: Update from the nhlbi clinical proteomics programs. Am J Physiol Lung Cell Mol Physiol. 2008;295:L16–22. doi: 10.1152/ajplung.00044.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Juncker D, Bergeron S, Laforte V, Li H. Cross-reactivity in antibody microarrays and multiplexed sandwich assays: Shedding light on the dark side of multiplexing. Curr Opin Chem Biol. 2014;18:29–37. doi: 10.1016/j.cbpa.2013.11.012. [DOI] [PubMed] [Google Scholar]
- 6.Ngo D, Sinha S, Shen D, Kuhn EW, Keyes MJ, Shi X, Benson MD, O'Sullivan JF, Keshishian H, Farrell LA, Fifer MA, Vasan RS, Sabatine MS, Larson MG, Carr SA, Wang TJ, Gerszten RE. Aptamer-based proteomic profiling reveals novel candidate biomarkers and pathways in cardiovascular disease. Circulation. 2016;134:270–285. doi: 10.1161/CIRCULATIONAHA.116.021803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lakkis NM, Nagueh SF, Dunn JK, Killip D, Spencer WH., 3rd Nonsurgical septal reduction therapy for hypertrophic obstructive cardiomyopathy: One-year follow-up. J Am Coll Cardiol. 2000;36:852–855. doi: 10.1016/s0735-1097(00)00767-1. [DOI] [PubMed] [Google Scholar]
- 8.Lakkis NM, Nagueh SF, Kleiman NS, Killip D, He ZX, Verani MS, Roberts R, Spencer WH., 3rd Echocardiography-guided ethanol septal reduction for hypertrophic obstructive cardiomyopathy. Circulation. 1998;98:1750–1755. doi: 10.1161/01.cir.98.17.1750. [DOI] [PubMed] [Google Scholar]
- 9.Keshishian H, Burgess MW, Gillette MA, Mertins P, Clauser KR, Mani DR, Kuhn EW, Farrell LA, Gerszten RE, Carr SA. Multiplexed, quantitative workflow for sensitive biomarker discovery in plasma yields novel candidates for early myocardial injury. Molecular & cellular proteomics : MCP. 2015;14:2375–2393. doi: 10.1074/mcp.M114.046813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Addona TA, Shi X, Keshishian H, Mani DR, Burgess M, Gillette MA, Clauser KR, Shen D, Lewis GD, Farrell LA, Fifer MA, Sabatine MS, Gerszten RE, Carr SA. A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nature biotechnology. 2011;29:635–643. doi: 10.1038/nbt.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keshishian H, Addona T, Burgess M, Mani DR, Shi X, Kuhn E, Sabatine MS, Gerszten RE, Carr SA. Quantification of cardiovascular biomarkers in patient plasma by targeted mass spectrometry and stable isotope dilution. Mol Cell Proteomics. 2009;8:2339–2349. doi: 10.1074/mcp.M900140-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis GD, Farrell L, Wood MJ, Martinovic M, Arany Z, Rowe GC, Souza A, Cheng S, McCabe EL, Yang E, Shi X, Deo R, Roth FP, Asnani A, Rhee EP, Systrom DM, Semigran MJ, Vasan RS, Carr SA, Wang TJ, Sabatine MS, Clish CB, Gerszten RE. Metabolic signatures of exercise in human plasma. Sci Transl Med. 2010;2:33ra37. doi: 10.1126/scitranslmed.3001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okamoto F, Sohmiya K, Ohkaru Y, Kawamura K, Asayama K, Kimura H, Nishimura S, Ishii H, Sunahara N, Tanaka T. Human heart-type cytoplasmic fatty acid-binding protein (h-fabp) for the diagnosis of acute myocardial infarction. Clinical evaluation of h-fabp in comparison with myoglobin and creatine kinase isoenzyme mb. Clinical chemistry and laboratory medicine : CCLM / FESCC. 2000;38:231–238. doi: 10.1515/CCLM.2000.034. [DOI] [PubMed] [Google Scholar]
- 14.Glatz JF, van der Vusse GJ, Simoons ML, Kragten JA, van Dieijen-Visser MP, Hermens WT. Fatty acid-binding protein and the early detection of acute myocardial infarction. Clinica chimica acta; international journal of clinical chemistry. 1998;272:87–92. doi: 10.1016/s0009-8981(97)00255-6. [DOI] [PubMed] [Google Scholar]
- 15.Collinson IR, Runswick MJ, Buchanan SK, Fearnley IM, Skehel JM, van Raaij MJ, Griffiths DE, Walker JE. Fo membrane domain of atp synthase from bovine heart mitochondria: Purification, subunit composition, and reconstitution with f1-atpase. Biochemistry. 1994;33:7971–7978. doi: 10.1021/bi00191a026. [DOI] [PubMed] [Google Scholar]
- 16.Fillgrove KL, Anderson VE. The mechanism of dienoyl-coa reduction by 2,4-dienoyl-coa reductase is stepwise: Observation of a dienolate intermediate. Biochemistry. 2001;40:12412–12421. doi: 10.1021/bi0111606. [DOI] [PubMed] [Google Scholar]
- 17.Sun H, Li N, Wang X, Chen T, Shi L, Zhang L, Wang J, Wan T, Cao X. Molecular cloning and characterization of a novel muscle adenylosuccinate synthetase, adssl1, from human bone marrow stromal cells. Mol Cell Biochem. 2005;269:85–94. doi: 10.1007/s11010-005-2539-9. [DOI] [PubMed] [Google Scholar]
- 18.Martherus RS, Sluiter W, Timmer ED, VanHerle SJ, Smeets HJ, Ayoubi TA. Functional annotation of heart enriched mitochondrial genes gbas and chchd10 through guilt by association. Biochem Biophys Res Commun. 2010;402:203–208. doi: 10.1016/j.bbrc.2010.09.109. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Li Q, Adhikari N, Hall JL. A role for muscle lim protein (mlp) in vascular remodeling. Journal of molecular and cellular cardiology. 2006;40:503–509. doi: 10.1016/j.yjmcc.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Geier C, Gehmlich K, Ehler E, Hassfeld S, Perrot A, Hayess K, Cardim N, Wenzel K, Erdmann B, Krackhardt F, Posch MG, Osterziel KJ, Bublak A, Nagele H, Scheffold T, Dietz R, Chien KR, Spuler S, Furst DO, Nurnberg P, Ozcelik C. Beyond the sarcomere: Csrp3 mutations cause hypertrophic cardiomyopathy. Hum Mol Genet. 2008;17:2753–2765. doi: 10.1093/hmg/ddn160. [DOI] [PubMed] [Google Scholar]
- 21.Wen HY, Xia Y, Young ME, Taegtmeyer H, Kellems RE. The adenylosuccinate synthetase-1 gene is activated in the hypertrophied heart. J Cell Mol Med. 2002;6:235–243. doi: 10.1111/j.1582-4934.2002.tb00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Osanai T, Mikami K, Kitajima M, Urushizaka M, Kawasaki K, Tomisawa T, Itaki C, Noto Y, Magota K, Tomita H. Nutritional regulation of coupling factor 6, a novel vasoactive and proatherogenic peptide. Nutrition. 2017;37:74–78. doi: 10.1016/j.nut.2016.07.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.