

By feeding prostate-specific Pten−/− mice an AIN93M, a 0.2% α-T or a 0.2% δ-T supplemented diet, we found δ-T reduced prostate adenocarcinoma multiplicity. δ-T-specific cancer preventive activity is mediated through its inhibition of the AKT activation, beyond its antioxidant activity.
Abstract
The PTEN/PI3K/AKT axis plays a critical role in regulating cell growth, differentiation and survival. Activation of this signaling pathway is frequently found in human cancers. Our previous studies demonstrated that δ-tocopherol (δ-T) attenuates the activation of AKT by growth factor in prostate cancer cell lines, leading to inhibition of proliferation and induction of apoptosis. Herein, we investigated whether δ-T inhibits the development of prostate adenocarcinoma in prostate-specific Pten−/− (Ptenp−/−) mice in which the activation of AKT is the major driving force for tumorigenesis. By feeding Ptenp−/− mice with AIN93M or 0.2% δ-T supplemented diet starting at the age of 6 or 12 weeks, we found that δ-T treatment reduced prostate adenocarcinoma multiplicity at the age of 40 weeks by 53.3 and 42.7%, respectively. Immunohistochemical (IHC) analysis demonstrated that the phosphorylation of AKT (T308) was reduced in the prostate of the mice administered the δ-T diet. Consistently, proliferation was reduced and apoptosis was increased in prostate lesions of mice on the δ-T diet. Oxidative stress, as determined by IHC staining of 8-OH-dG, was not altered during prostate tumorigenesis, nor was it affected by administration of δ-T. In contrast, α-tocopherol (α-T) at 0.2% in the diet did not affect prostate adenocarcinoma multiplicity in the Ptenp−/− mice. This finding is consistent with data from our previous study that δ-T, but not α-T, inhibits the activation of AKT and the growth of prostate cancer cells. Together, these results demonstrate that δ-T inhibits the development of prostate adenocarcinoma in Ptenp−/− mice, mainly through inhibition of AKT activation.
Introduction
Tocopherols, the major forms of vitamin E, are widely found in foods such as vegetable oils, eggs, nuts, corn, soybean and whole grains (1). They contain a chromanol ring structure and a phytyl chain of 16 carbons. The phytyl chain can serve as a hydrophobic tail inserting itself into the lipid bilayer of the cell membrane. The hydroxyl group on the six position of the chromanol ring can quench free radicals such as reactive oxygen species by one electron reduction to produce a tocopherol phenoxy radical (2,3). The phenolic group can be regenerated through reduction by ascorbic acid or glutathione, constituting the most important physiological antioxidant mechanism to protect the integrity of the cell membrane. The number and position of the methyl groups on the chromanol ring define the different forms of tocopherols: α-tocopherol (α-T) is trimethylated at the five, seven and eight positions of the chromanol ring, β-tocopherol is dimethylated at the five and eight positions, γ-tocopherol (γ-T) is dimethylated at the seven and eight positions and δ-tocopherol (δ-T) is methylated at the eight position (2,3). While all tocopherols can function as antioxidants to trap reactive oxygen species, γ-T and δ-T can also effectively trap reactive nitrogen species, resulting in production of 5-nitro-γ-T, 5-nitro-δ-T, 7-nitro-δ-T and 5,7-dinitro-δ-T (4–7). American dietary sources such as vegetable oil and nuts are rich in α-T and γ-T with γ-T being more abundant than α-T. δ-T is also abundant in certain oils and nuts. All forms of tocopherols are absorbed into the liver, but hepatic α-T transfer protein preferentially transfers α-T to the blood over γ-T and δ-T, resulting in that blood and tissue levels of α-T are higher than the levels of γ-T and δ-T (2,8). Due to its higher blood level and superior activity in the classical ‘fertility-restoration assay’ over other tocopherols, α-T is recognized as the major vitamin E form. Therefore, most of the studies on vitamin E have used α-T or its synthetic derivative α-tocopheryl acetate.
Epidemiological studies have shown that higher dietary intake or blood levels of vitamin E are associated with lower risk for prostate cancers (9–11). To evaluate the potential application of vitamin E in cancer prevention, large scale interventional trials were launched. For example, the Physicians’ Health Study II Randomized Control Trial (12) and the Selenium and Vitamin E Cancer Prevention Trial (SELECT) (13) were conducted with the goal to generate data on the efficacy of vitamin E in cancer prevention. However, the supplementation with high doses of α-T did not prevent cancer in prostate and other organs in these trails. Results from animal studies on different forms of tocopherols have not been consistent especially when α-T was used in different experimental models in cancer prevention studies (3,14,15). Our recent studies with a γ-T-rich mixture of tocopherols (γ-TmT; containing 130 mg α-T, 15 mg β-tocopherol, 243 mg δ-T and 563.8 mg γ-T/g) have demonstrated the effective inhibitory action of γ-TmT in several transplanted and chemically or genetically induced rodent tumorigenesis models (16–22). The superior cancer preventive activities of γ-T over α-T have also been demonstrated by others (23–25). Using purified α-T, γ-T and δ-T, we have found that δ-T is more effective in inhibiting AOM-induced colon carcinogenesis in rats, lung tumor growth in a mouse xenograft model and PhIP/DSS-induced colon carcinogenesis in mice (26–28). Using prostate cancer cells, we have further demonstrated that δ-T is more effective than γ-T and α-T in inhibiting cell proliferation and inducing apoptosis (29). Since δ-T is readily available through our diet and dietary supplements, there is great potential for the application of δ-T in human cancer prevention. Before launching clinical trials, both better understanding of the molecular mechanisms of δ-T action and a hypothesis-driven investigation in relevant animal models are needed.
In the present study, we investigated the effects of δ-T on the development of prostate adenocarcinoma in prostate-specific Pten−/− (Ptenp−/−) mice. The reason for selecting this mouse prostate cancer model is that the neoplastic growth of prostate gland epithelial cells and the development of prostate lesions, such as high-grade prostatic intraepithelial neoplasia (HG-PIN) and adenocarcinoma, are driven by the elevated phospho-AKT (pAKT) resulted from the loss of PTEN in the prostate (30,31). Since the activation of PTEN/PI3K/AKT signaling is one of the most important human cancer promoting pathway, and loss of heterozygosity of PTEN occurs frequently in many advanced sporadic cancers, including ~60% of advanced prostate cancers (32), studying the prostate cancer inhibitory effect of δ-T in Ptenp−/− mice would further justify the potentials of δ-T in human prostate cancer prevention. In addition, murine models of prostate cancer induced by the loss of Pten and the combination of the loss of Pten with other genetic alterations best recapitulate the entire spectrum of prostate cancer from initiation, through progression, and, finally, to metastasis (33). Therefore, Ptenp−/− mice are suitable for examining whether the inhibition of AKT activation by δ-T found in our previous study using prostate cancer cell lines is an effective mechanism to prevent prostate cancer in vivo. We found that feeding Ptenp−/− mice with 0.2% δ-T diet reduced prostate adenocarcinoma multiplicity by ~40%. Consistently, we also found reduced pAKT and cell proliferation, as well as increased apoptosis, in the prostate, suggesting that δ-T effectively prevents prostate tumorigenesis through inhibiting the activation of AKT in vivo.
Materials and methods
Tocopherols and animal diet
δ-T and α-T were purified from commercial tocopherol sources to ≥97% purity by use of an automated flash chromatography system (Teledyne ISCO CombiFlash Companion XL; Teledyne Isco, Inc., Lincoln, NE, USA) with RediSep Rf Gold high performance flash silica gel column (20–40 μmol/L in particle size) as described in previous publication (26). The commercial sources were d-forms of α-T (with 69.7% α-T, 2.6% γ-T and 0.2% δ-T) and δ-T (with 94% δ-T, 5.5% γ-T, and 0.5% α-T) purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). The final purified δ-T and α-T products did not have detectable levels of other forms of tocopherols. Semi-purified rodent diet (AIN93M) and diet containing 0.2% α-T or 0.2% δ-T were prepared by Research Diets, Inc. (New Brunswick, NJ, USA). All diets were stored in double-sealed plastic bags flashed with nitrogen at 4°C.
Animal studies
Male Ptenp−/− mice (Pten-loxP+/+:pB-Cre+; in Fvb background) were produced by breeding male Pten-loxP+/+:pB-Cre+ mice with female Pten-loxP+/+ mice (kindly provided by Dr. Ronald DePinho at MD Anderson Cancer Center) (31). The pB-Cre+ mice (34) were originally from the Jackson Laboratory (Bar Harbor, ME, USA). The mice were housed at room temperature (20 ± 2°C) with a relative humidity of 50 ± 10% and an alternating 12 h light/dark cycle. Water and diet were provided ‘ad libitum’. All animal studies were conducted in accordance to approved animal study protocol (No. 91-024) by the Institutional Animal Care and Use of Rutgers University. Twenty breeding pairs of same age mice were maintained to produce male Ptenp−/− mice. Male pups were genotyped for Cre at the age of 3 weeks. Male Ptenp−/− mice of the same age (born within 3 days) were grouped randomly starting at the age of 5 or 11 weeks and fed AIN93M diet. After 1 week, mice were continuously fed AIN93M diet (as control) or switched to a purified diet supplemented with 0.2% δ-T or α-T. At the age of ~16 weeks, due to incompatability, male Ptenp−/− mice were single housed until the experiment’s end point. The diet was replenished on a weekly basis. Body weight and food consumption were recorded weekly. At the end point of the study, mice were euthanized by CO2 asphyxiation. Blood was collected by cardiac puncture for serum preparation. The prostate from each mouse was excised as a whole and fixed in 10% PBS-buffered formalin (Thermo Fisher Scientific, Waltham, MA, USA) for preparation of paraffin-embedded tissue blocks.
Histopathological characterization
To prepare the prostate for histological characterization, fixed prostate was dissected and different lobes were collected for preparing paraffin blocks. This process was done by following the detailed procedure described in a recent publication (35). In brief, seminal vesicles, male reproductive tract and bladder were removed from the prostate under a dissecting microscope. Then, the anterior lobes were carefully separated from the rest of the prostate. The urethra originally attached with the rest of the prostate was cut to ~1 mm allowing the residue part to be used for orienting the position of the tissue in the paraffin blocks for precise cross sectioning. For the purpose of monitoring the progression, each prostate was prepared in two separate blocks: one for anterior lobes, and the other for ventral, dorsal and lateral lobes. At least five sections (4 μm), approximately 20 sections apart, were stained with hematoxylin and eosin (H&E) for histological characterization. For scoring the tocopherol-treated samples collected from Ptenp−/− mice at 12 and 25 weeks, ventral lobes were further separated from dorsal-lateral lobes; therefore, each prostate was prepared in three separate blocks, anterior lobe-, ventral lobe- and dorsal-lateral lobe-blocks. Each block was then sectioned and scored separately (dorsal and lateral lobes were attached together but could be clearly recognized under the microscope). Histopathological analysis of each prostate was conducted on two H&E stained sections, approximately 20 sections apart, from the samples taken from the middle of the tissues. For scoring 40 week samples, only anterior lobes were separated from the rest of the prostate because ventral, dorsal and lateral lobes were tightly attached and could not be easily recognized and separated under the dissecting microscope; therefore, each prostate was prepared in two blocks, one for anterior lobes and the other for the rest of the prostate. When preparing paraffin block, the ventral-dorsal-lateral prostate was cross cut to top and bottom halves in the middle. Two halves were then positioned together with the smooth cut faces oriented towards the bottom in the embedding mold, thereby, creating two samples in each section. Histopathological analysis of each prostate was conducted on two H&E stained sections, approximately 20 sections apart, taken from the middle of the prostate. PIN and adenocarcinoma were identified according to consensus criteria established by the panel of human and veterinary pathologists for scoring prostate lesions in different transgenic mouse models (36,37), as well as by the previous histopathological characterization of Ptenp−/− mouse prostate (38). Low-grade PIN (LG-PIN) is characterized by the neoplastic growth of luminal epithelial cells forming multiple layers in part of the luminal epithelium along with loss of polarity in some lesioned cells and the intact basal membrane. In the LG-PIN gland, the non-lesioned area in luminal epithelium retains the single layer of polarized luminal epithelial cells. HG-PIN is characterized by the neoplastic growth of luminal epithelial cells in the majority of the luminal epithelium and the appearance of lesioned cells with loss of polarity, large nuclei of relatively uniform size and prominent nucleoli. The basal membrane in the HG-PIN gland is normal, although some areas of the basal membrane can become thicker with the appearance of multiple layers of basal cells. Adenocarcinoma is characterized by the disruption or loss of the basal membrane and evidence of tumor cell invasion into stroma. Histopathological score was performed for both the left and right lobes.
Immunohistochemistry staining and quantification
Immunohistochemical (IHC) analyses for the activation of AKT, the proliferation and apoptosis of tumor cells, and the oxidative stress in prostate tissues were carried out using a standard avidin–biotin peroxidase complex method described in a previous publication (39) using the following antibodies: anti-pan-cytokeratin (mouse monoclonal; Abcam, Cambridge, MA, USA), anti-pAKT (T308) (rabbit polyclonal; Abcam), anti-Ki67 (rabbit polyclonal, Abcam), cleaved-Caspase 3 (C-Caspase 3; Abcam), anti-8-OH-dG (mouse monoclonal; JaICA; Fukuroi, Shizuoka, Japan) and anti-nitrotyrosine (rabbit polyclonal; EMD Millipore, Billerica, MA, USA). The IHC staining was carried out using biotinylated secondary antibody (1:200), followed by streptavidin-biotin peroxidase conjugate (Vector Laboratories, Burlingame, CA, USA), and development using 3,3′-diaminobenzidine substrate (Vector Laboratories). The slides were then counterstained with hematoxylin. For negative controls, the incubation with primary antibody was omitted in the procedure. The IHC staining was quantified using the Aperio ScanScope GL system (Vista, CA, USA). For pAKT, 8-OH-dG and nitrotyrosine, the immunostaining intensity was normalized by the total number of cells (counted by nuclei; >3000 cells in 10–20 glands per slide) and was presented as intensity per cell. For Ki67 and C-Caspase 3, the number of positively stained cells was counted and normalized by the total number of cells (counted by nuclei; >3000 cells in 10–20 glands per slide), then presented as the percentage of positive stained cells. For these quantification analyses, stromal cells were not included and, for each sample, the quantification result was the average of the two sections.
Statistical analysis
Results were analysed using the GraphPad Prism software (GraphPad, CA, USA). Student’s t-test was performed to determine the differences between two groups. One-way ANOVA followed by Tukey’s post hoc test was used to compare multiple groups. Differences were considered to be statistically significant when the calculated P-value was less than 0.05.
Results
The histological progression of prostate lesions in Ptenp−/− mice.
An earlier report suggested that Ptenp−/− mice develop adenocarcinoma at the age of ~12 weeks (40); however, careful characterizations of this model by several other groups support that HG-PIN develops in Ptenp−/− mice at the age of ~6–12 weeks and progresses very slowly to adenocarcinoma (30,31,38). Although the majority of glands in these mice developed HG-PIN quickly, the incidence of adenocarcinoma and the timeline of its development depend on the genetic background of the Ptenp−/− mice (41). Therefore, it is necessary to obtain a detailed timeline for the histological progression of prostate lesions in the Fvb background Ptenp−/− mice in order to design an experimental plan of a cancer prevention study. For this purpose, we collected the prostate from Ptenp−/− mice aged 6 to 50 weeks at 2 week intervals, using at least two mice at each time point. We then characterized the histology of prostate lesions found in all lobes. The male littermates without Cre recombinase (i.e. Pten-loxP+/+:Cre- mice) were used as the wild-type (WT) controls.
In the WT controls, the normal prostate glands in all lobes consist of a single layer of polarized gland epithelial cells, featured with a mononucleus and uniformly oriented to the underlying topography, and a basal membrane, formed by a single layer of basal cells, separating gland epithelium from the stroma. The stroma between glands mainly consists of connective tissue and contains no infiltration of inflammatory cells. The histology of normal prostate glands remains unchanged through the various ages of the mice, although anterior lobe glands in the older mice are larger. Representative images of normal prostate glands in the 6- and 40-week-old mice are shown in Supplementary Figure 1, available at Carcinogenesis Online.
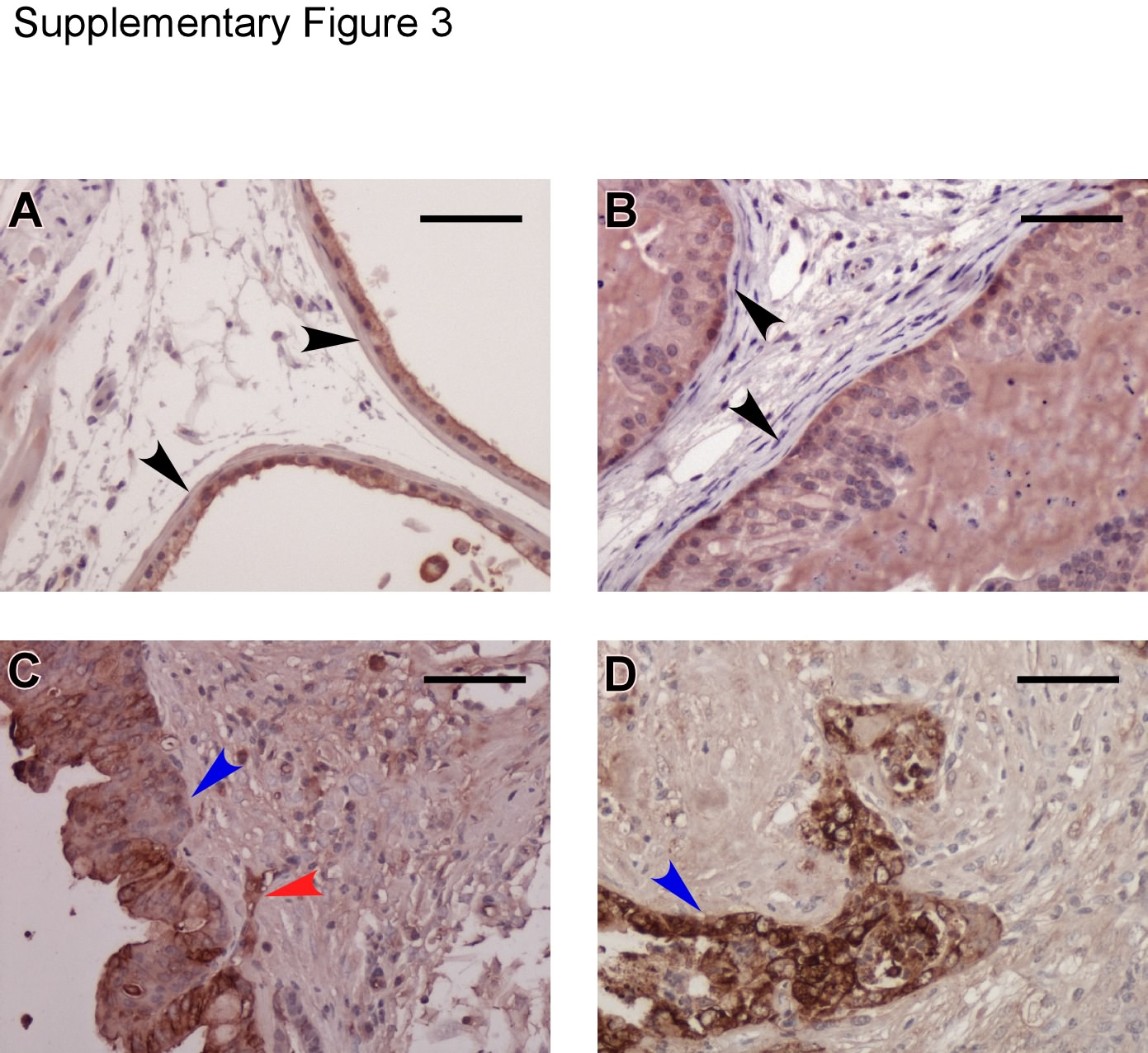
In Ptenp−/− mice, we found that LG-PIN lesions begin to develop in all lobes of the prostate at the age of 6–8 weeks, >80% glands develop HG-PIN at 12 weeks, some HG-PINs progress to lesions with micro-invasive features (i.e. basal membrane rupture and local invasion of lesioned cells) at 36 weeks, and lesions begin to progress to adenocarcinoma after 36 weeks. LG-PIN glands feature the neoplastic growth of luminal epithelial cells in a small area of gland epithelium, whereas the rest of epithelium remains formed by a single layer of polarized luminal epithelial cells. In the neoplastic area, lesioned cells lose epithelial cell polarity. LG-PINs are typically found in early ages. HG-PINs are the glands with the aggressive growth of neoplastic cells in the majority of the epithelium. In HG-PIN, almost all cells in gland epithelium lose polarity and become pleomorphic. These lesioned cells frequently occupy the majority space of the lumen and display nuclear atypia (i.e. enlarged nuclei, prominent nucleoli and nuclear hyperchromasia), but the basal layer remains intact. Along with further progression through the ages, the Ptenp−/− mouse stromal tissues become infiltrated with a significant amount of inflammatory cells. In the older Ptenp−/− mice, it is common to find a thicker basal layer consisting of multiple layers of basal cells. When mice are over 36 weeks, their basal membranes become disrupted in the dorsal, lateral and ventral lobes; in these disrupted areas, neoplastic cells invade into stroma, suggesting the progression to adenocarcinoma. At the age of 40 weeks, adenocarcinoma can be commonly found in the prostate dorsal, lateral and ventral lobes. Typical pictures of prostate lesions in the dorsal, lateral and ventral lobes are presented in Supplementary Figure 2, available at Carcinogenesis Online. While the invading cells can be recognized through H&E stained slides, they can also be identified by IHC staining using anti-cytokeratin antibody as they are epithelial cells origin and are positive for cytokeratin (Supplementary Figure 3, available at Carcinogenesis Online). However, the progression of the anterior lobe lesions is different. Anterior lobes become enlarged and the glands in each anterior lobe fuse forming a cyst full of fluid after 25 weeks. In our study, no invasiveness was found in the anterior lobes. Typical micrographs of the anterior lobe are presented in Supplementary Figure 4, available at Carcinogenesis Online.
Prostate-specific knockout of Pten is processed by Probasin promoter-driven Cre recombinase to excise Pten-loxP in male Pten-loxP+/+:pB-Cre+ mice. This is expected to occur at the age of ~5 weeks when the mice become mature and the Probasin promoter-driven Cre is activated. By monitoring the progression, we demonstrate that the Fvb background Ptenp−/− mice quickly develop HG-PIN, but there is a long latency, ~30 weeks, for HG-PIN to progress to adenocarcinoma in prostate dorsal, lateral and ventral lobes (as illustrated in Supplementary Figure 5, available at Carcinogenesis Online). This timeline is consistent with the long latency period of the progression to adenocarcinoma, reported previously (31).
Feeding 0.2% δ-T diet starting at the age of 12 weeks reduces prostate adenocarcinoma multiplicity in Ptenp−/− mice
Since Ptenp−/− mice at the age of 12 weeks develop HG-PIN in most prostate glands and as these lesions need ~28 weeks to progress to adenocarcinoma, starting treatment in these mice at the age of 12 weeks offers an opportunity to determine whether δ-T is able to inhibit the activation of AKT and the development of HG-PIN to adenocarcinoma. Therefore, we conducted an experiment to determine the inhibitory activity of δ-T, in which we fed Ptenp−/− mice a 0.2% δ-T diet starting at the age of 12 weeks. This experiment consisted of 26 male Ptenp−/− mice born within 3 days who were fed AIN93M diet at the age of 11 weeks. At the age of 12 weeks, the mice were randomly separated into three groups and were maintained on an AIN93M, a 0.2% δ-T diet or a 0.2% α-T diet for 28 weeks (illustrated in Figure 1A). The average body weight of three groups of mice showed no difference during the experiment period, suggesting that the tocopherol diets did not affect animal growth. At the age of 40 weeks, all mice were sacrificed and their prostates were fixed for histopathological characterization. In all prostate samples, the majority of glands in the dorsal, ventral and lateral lobes developed HG-PIN. There were also significant instances of adenocarcinomas featured with the rupture or loss of basal membrane and the presence of neoplastic cells invasion into stroma (Supplementary Figure 2, available at Carcinogenesis Online). The prostate anterior lobes were enlarged and glands in each anterior lobe were fused, forming a cyst full of fluid (Supplemental Figure 4). No invasiveness was observed in anterior lobes. The percentages of glands that developed adenocarcinomas in the Ptenp−/− mice on an AIN93M, a 0.2% δ-T diet and a 0.2% α-T diet were 24.1 ± 8% (n = 8), 13.8 ± 4.1% (n = 10) and 21.9 ± 4.7% (n = 8), respectively (Figure 1B) (only the glands in dorsal, ventral and lateral lobes were counted). Compared with the AIN93M diet group, the group fed a δ-T diet showed reduced prostate adenocarcinoma multiplicity (glands developed adenocarcinoma per 100 glands counted) by an average of 42.7%, whereas the group fed an α-T diet showed no difference, thus concluding that the α-T diet was ineffective (P-value = 0.001; one-way ANOVA). This result demonstrates that δ-T, but not α-T, inhibited the development of adenocarcinoma in Ptenp−/− mice.
Figure 1.

The percentage of prostate glands developing adenocarcinoma in 40-week-old Ptenp−/−mice fed AIN93M, diet supplemented with 0.2% δ-T or 0.2% α-T starting at the age of 12 weeks. (A) Illustration of the experimental procedure for mice fed different diets starting from the age of 12 weeks. (B) The adenocarcinoma multiplicity (glands developed adenocarcinoma/100 glands counted) in dorsal, ventral and lateral lobes in mice at the age of 40 weeks fed AIN93M (n = 8), 0.2% δ-T diet (n = 10) or 0.2% α-T diet (n = 8). Data are presented as mean ± SD. One-way ANOVA followed by Tukey’s post hoc test is used to compare the percentages of prostate glands developing adenocarcinoma among the three groups (*P = 0.001).
Feeding 0.2% δ-T diet starting at the age of 12 weeks reduces pAKT level and cell proliferation while increasing apoptosis in prostate of Ptenp−/− mice
Treating cultured prostate cancer cell lines with δ-T reduces cell proliferation and increases apoptosis by inhibiting AKT activation (29). To assess whether this mechanism was involved in reducing adenocarcinoma multiplicity in Ptenp−/− mice fed a 0.2% δ-T diet, we performed IHC staining for pAKT (T308) on the prostate samples. We found that Ptenp−/− mouse prostate tissue displayed strong positive staining for pAKT (T308), while the WT mouse prostate showed a minimal level of pAKT (T308) staining (Figure 2A and B). This result is consistent with the fact that the loss of Pten leads to elevated pAKT in this mouse model (30,31,40). The lesioned cells in HG-PINs and adenocarcinomas from the same prostate samples displayed the same level of pAKT staining. Prostate from Ptenp−/− mice fed a 0.2% δ-T diet displayed weaker pAKT staining (Figure 2B–D). Quantification of the pAKT staining from all prostate samples using ScanScope revealed that a 0.2% δ-T diet reduced pAKT levels by an average of 28.8%, whereas a 0.2% α-T diet had no effect on these levels (P-value = 0.037; one-way ANOVA) (Figure 2M).
Figure 2.

Dietary δ-T reduced pAKT, suppressed proliferation and increased apoptosis in prostate tissues. Ptenp−/− mice fed either an AIN93M (n = 8), a 0.2% δ-T diet (n = 10) or a 0.2% α-T diet (n = 8) and the WT mice fed an AIN93M diet (n = 7) were analyzed for the active AKT, proliferation and apoptosis using IHC staining for pAKT (T308), Ki67 and cleaved-Caspase 3 (C-Caspase 3), respectively. Images of representative IHC staining for these mouse prostate samples are shown in (A–D, pAKT; E–H, Ki67; I–L, C-Caspase 3). The scale bar in the figures represents 50 µm. Quantified results of pAKT (T308), Ki67 and C-Caspase 3 IHC staining for the four groups of mice were determined using the Aperio ScanScope and summarized in (M), (N) and (O), respectively. Data are presented as mean ± SD. One-way ANOVA followed by Tukey’s post hoc test is used to compare the IHC staining results in the groups (*P-value = 0.037, 0.018 and 0.031 in M, N and O, respectively).
To assess the effect of the 0.2% δ-T diet on cell proliferation, we performed IHC staining for Ki67. We found that the majority of prostate gland cells were negative for Ki67 in the WT mice, but stained positive for Ki67 in Ptenp−/− mice (Figure 2E and F). Within the Ptenp−/− mice, there was no obvious difference in the percentage of Ki67-positive cells between HG-PIN and adenocarcinoma from the same prostate. Prostates of the Ptenp−/− mice on the 0.2% δ-T diet had less Ki67-positive cells (Figure 2F–H). Using ScanScope, quantification of Ki67-positive cells within all prostate samples revealed that a 0.2% δ-T diet reduced the number of Ki67-positive cells in the prostate by an average of 30.5%, whereas a 0.2% α-T diet had no significant effect (P-value = 0.018; one-way ANOVA) (Figure 2N).
Apoptosis in prostate samples was analyzed by IHC staining for C-Caspase 3. Overall, we found that the number of apoptotic cells was less than 1% in the normal prostate gland epithelium, and this number was increased in Ptenp−/− prostate, consistent with a previous report (30) (Figure 2I and J). There was no obvious difference in the percentage of C-Caspase 3 positively stained cells between HG-PIN and adenocarcinoma from samples of the same prostate. Prostate of Ptenp−/− mice fed a 0.2% δ-T diet showed an increased number of C-Caspase 3-positive cells (Figure 2J–L). Quantification of the positive C-Caspase 3 staining of all samples using ScanScope revealed that the 0.2% δ-T diet increased the number of apoptotic cells in the prostate, whereas the 0.2% α-T diet had no significant effect (P-value = 0.031; one-way ANOVA) (Figure 2O).
Together, these results demonstrate that Ptenp−/− mice fed a 0.2% δ-T diet leads to reduced activation of AKT and cell proliferation and increased apoptosis, whereas an α-T diet is ineffective in these actions.
Feeding 0.2% δ-T diet starting at the age of 6 weeks reduces prostate adenocarcinoma multiplicity in Ptenp−/− mice
As shown in the above experiment, a 0.2% δ-T diet was found to be effective in inhibiting pAKT and the prostate adenocarcinoma multiplicity in Ptenp−/− mice when the feeding began at the age of 12 weeks. Intrigued by this finding, we wondered whether starting the feeding earlier would be more effective. We conducted another set of experiments by feeding mice either an AIN93M or a δ-T diet at the age of 6 weeks, before the proposed development of HG-PIN. We then collected samples at multiple time points: the age of 12, 25 and 40 weeks for the determination of the diet’s effect on the development of HG-PIN, as well as adenocarcinoma. At 12 weeks of age, PIN lesions developed in more than half of the prostate glands, and there was no notable difference between mice fed the AIN93M or the 0.2% δ-T diet (data not shown). At 25 weeks of age, the majority of lesions progressed to HG-PIN; however, we found no significant difference in the percentage of HG-PIN glands within each lobe (Figure 3A). The percentage of HG-PIN glands in the ventral lobes of mice fed the 0.2% δ-T diet appeared to be lower, but this difference was not statistically significant. At 40 weeks of age, the Ptenp−/− mice developed multiple adenocarcinomas in the dorsal-lateral-ventral lobes as described in the above experiment. We counted the numbers of adenocarcinomas and found the adenocarcinoma multiplicity in the mice fed the AIN93M and the 0.2% δ-T diet were 46.9 ± 15% (n = 9) and 21.9 ± 8.6% (n = 10), respectively (Figure 3B). The δ-T diet effectively reduced the percentage of prostate glands that developed adenocarcinoma by an average of 53.3% (P-value = 0.0045; Student’s t-test). This finding is consistent with the results from the previous experiment using mice fed a 0.2% δ-T diet starting at the age of 12 weeks.
Figure 3.

The percentage of prostate glands developed HG-PIN and adenocarcinoma in Ptenp−/−mice fed experimental diet at the age of 6 weeks. (A) At the age of 25 weeks, Ptenp−/− mice fed an AIN93M (n = 8) and a 0.2% δ-T diet (n = 8) from the age of 6 weeks were sacrificed to collect prostate tissues for histopathological characterization. The percentages of prostate glands developing HG-PIN in each lobe of the two groups of mice were scored. Data are presented as mean ± SD. There is no statistical significant difference in the percentage of HG-PIN glands in each lobe between the mice fed an AIN93M and a 0.2% δ-T diet (two-tailed Student’s t-test). (B) At the age of 40 weeks, Ptenp−/− mice fed either an AIN93M (n = 9) or a 0.2% δ-T diet (n = 10) were sacrificed to collect prostate tissues for histopathological characterization. The adenocarcinoma multiplicity (glands developed adenocarcinoma/100 glands counted) in dorsal, ventral and lateral lobes of the two groups of mice are presented in the figure. Data are presented as mean ± SD (*P = 0.0045).
Feeding 0.2% δ-T diet effectively reduces the pAKT level and cell proliferation while increasing apoptosis only at the later stage of prostate tumorigenesis
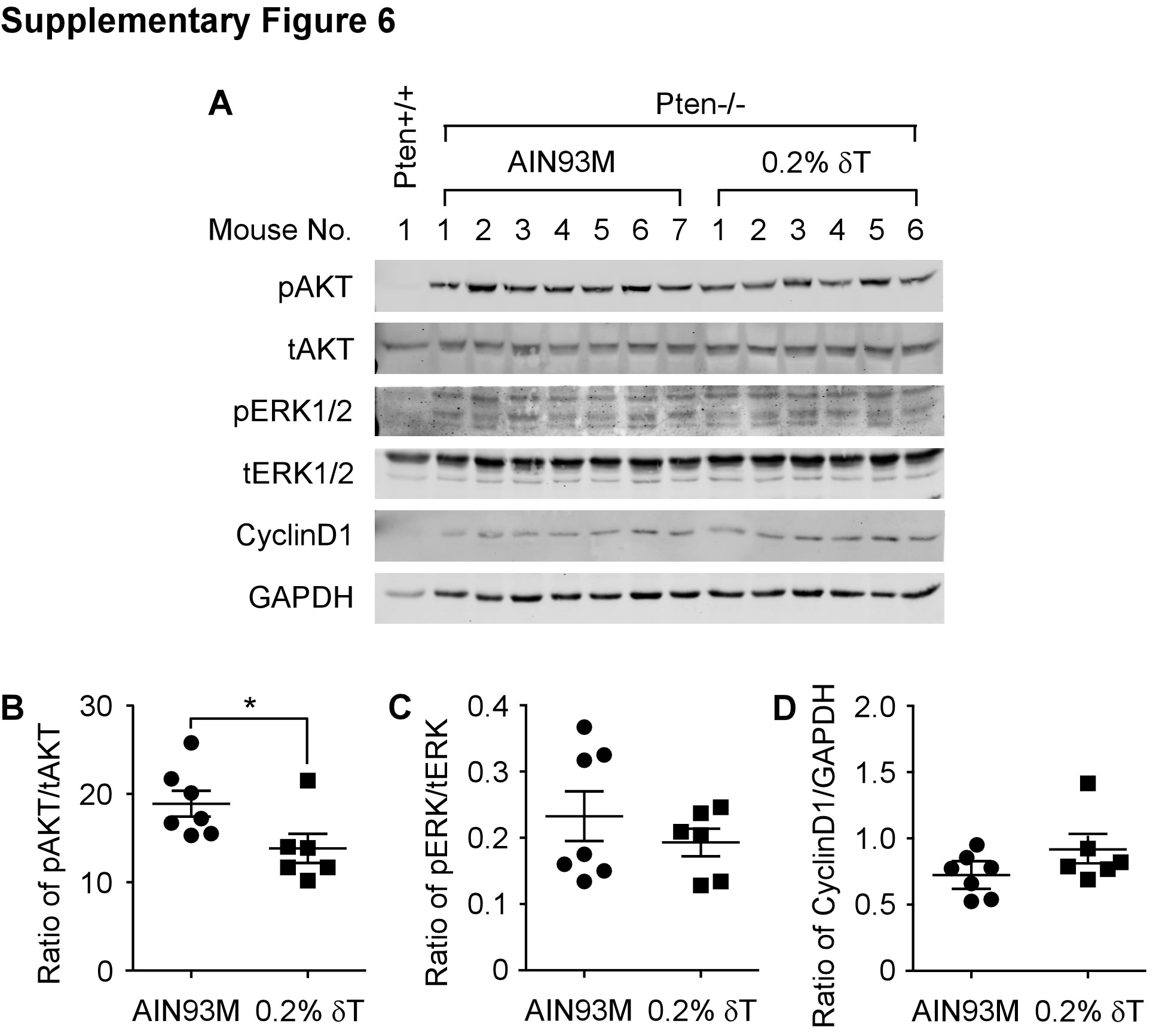
By IHC staining, we assessed pAKT (T308) levels in the samples collected at the three time points along with the same aged WT mice fed an AIN93M diet. As expected, we found that at all three ages, pAKT was induced in the Ptenp−/− mouse prostate but not in the WT mouse prostate (Figure 4). For mice on a 0.2% δ-T diet, the pAKT level was reduced by an average of 24.5% (P-value = 0.033), 24.7% (P-value = 0.017) and 28.3% (P-value = 0.023) at the ages of 12, 25 and 40 weeks, respectively (Figure 4J). To validate the inhibitory activity of δ-T, we conducted an additional experiment in which the 6-week-old mice were fed an AIN93M or a 0.2% δ-T diet for 2 weeks, and prostate samples were then collected for Western blot analysis for pAKT and other related signaling events. We found that pAKT was reduced in the Ptenp−/− mouse prostate by an average of 26.8% (P-value = 0.409; Supplementary Figure 6 and 7, available at Carcinogenesis Online). In addition, the levels of pERK1/2, pFOXO, Cyclin D1, p27, E2F and androgen receptor were also analysed, but there was no statistical significance difference found (Supplementary Figure 6, available at Carcinogenesis Online and data not shown). Together, these data suggest that 0.2% δ-T diet effectively attenuates the activation of AKT and a continuous action can lead to the inhibition on the development of adenocarcinoma.
Figure 4.

Dietary δ-T reduced the levels of pAKT in prostate of Ptenp−/− mice. Prostates of the WT mice fed an AIN93M and Ptenp−/− mice fed either an AIN93M or a 0.2% δ-T diet were analyzed for pAKT (T308) levels using IHC staining. Images of representative IHC staining for these mouse prostate samples at the age of 12, 25 and 40 weeks are shown (A–I). The scale bar represents 50 µm. The quantified results of pAKT (T308) levels for the three groups of mice were determined using the Aperio ScanScope and are summarized in (J). Data are presented as mean ± SD (the number of WT mice is six for each time point; the number of Ptenp−/− mice on the AIN93M diet is 7, 7 or 9 for 12, 25 or 40 weeks, respectively and the number of Ptenp−/− mice on the 0.2% δ-T diet is 7, 7 or 10 for 12, 25 or 40 weeks, respectively). *P = 0.033, **P = 0.017 and ***P = 0.023.
Cell proliferation at these time points was characterized by IHC staining for Ki67. Consistent with the pAKT levels, prostate glands of the WT mice displayed a minimal number of Ki67-positive cells. The glands of the Ptenp−/− mice, however, had a large number of cells positively stained for Ki67. The number of the positively stained cells was reduced by the δ-T diet (Figure 5). Compared with the AIN93M diet, the 0.2% δ-T diet decreased the number of Ki67-positive cells in the prostate of Ptenp−/− mice at ages 12, 25 and 40 weeks by an average of 29.0% (P-value = 0.029), 27.2% (P-value = 0.013) and 25.9% (P-value = 0.01), respectively (Figure 5J).
Figure 5.

Dietary δ-T reduced cell proliferation in prostate of Ptenp−/− mice. Prostates of the WT mice fed an AIN93M and Ptenp−/− mice fed either an AIN93M or a 0.2% δ-T diet were analyzed for cellular proliferation using IHC stainingfor Ki67. Images of representative IHC staining for the mouse prostate samples at the ages of 12, 25 and 40 weeks are shown (A–I). The scale bar represents 50 µm. The quantified results of positive Ki67 stained cells in the three groups of mice were determined using the Aperio ScanScope and are summarized in (J). Data are presented as mean ± SD (Experimental conditions are the same as Figure 4). *P = 0.029, **P = 0.013 and ***P = 0.01.
Apoptosis in the samples was determined by IHC staining for C-Caspase 3. The WT mouse prostate at three different ages, 12, 25 and 40 weeks, showed appearance of less than 1% apoptotic cells, whereas this number in Ptenp−/− mice was increased to ~3% (Figure 6), as found in a previous report (30). A significant increase in the number of prostate apoptotic cells in the Ptenp−/− mice fed 0.2% δ-T diet was only seen in the mice at 40 weeks of age [from 3.28% to 5.16% (P-value = 0.011); Figure 6J].
Figure 6.

Dietary δ-T increased apoptosis in prostate of Ptenp−/− mice. Prostates of the WT mice fed an AIN93M and Ptenp−/− mice fed either an AIN93M or a 0.2% δ-T diet were analyzed for cellular apoptosis using C-Caspase 3 IHC staining. Images of representative IHC staining for the mouse prostate samples at the ages of 12, 25 and 40 weeks are shown (A–I). The scale bar represents 50 µm. The quantified results of the numbers of cells positively stained for C-Caspase 3 in the three groups of mice were determined using the Aperio ScanScope and are summarized in (J). Data are presented as mean ± SD (Experimental conditions are the same as Figure 4). *P = 0.011.
Oxidative stress in prostate of Ptenp−/− mice
In the dietary carcinogen-treated CYP1A-humanized mice, we demonstrated previously that oxidative stress plays a critical role in PIN development, and that the prevention activity of γ-TmT in inhibiting PIN development is associated with its antioxidant activity (16). To determine whether the inhibition of δ-T on the development of adenocarcinoma in this study involves its antioxidant activity, we assessed the oxidative stress in all collected prostate samples from the above experiments using IHC staining for 8-OH-dG, the biomarker commonly used for oxidative stress-induced DNA damage (42). In WT mouse prostate samples collected at the age of 12, 25 and 40 weeks, we detected almost no 8-OH-dG positively stained cells. Similar results were seen in the prostate of Ptenp−/− mice fed either an AIN93M or a 0.2% δ-T diet (Supplementary Figure 4, available at Carcinogenesis Online). We also performed IHC staining for nitrotyrosine in these samples using anti-nitrotyrosine antibody to assess whether there was an effect on reactive nitrogen species. We found no difference in all samples (data not shown). These findings suggest that oxidative stress is not significantly altered during prostate tumorigenesis driven by the loss of Pten, nor is it affected by δ-T.
Discussion
Multiple mechanisms are proposed for the cancer preventive activity of tocopherols. These include antioxidant activity of all tocopherols (2,3,16,28,43), inhibition of COX-2 by γ-T and its metabolites (24), upregulation of PPARγ by γ-T (44,45), modulation of the activity of estrogen receptor by γ-T (46,47) and inhibition of the mTOR-mediated activation of AKT by γ-T through recruiting phosphatase PHLPP (48). Protection of cells from oxidative stress-induced DNA damage by γ-T and δ-T, presumably mediated through trapping reactive oxygen species and reactive nitrogen species, in carcinogen-induced colon and mammary gland carcinogenesis has been demonstrated previously (16,28,43,49). In these rodent carcinogenesis models, the activation of AKT, induced by carcinogen and/or oxidative stress, has also been found to be effectively reduced by γ-TmT, δ-T and γ-T (16,43,49). These mechanisms partially explain the higher cancer preventive activities of δ-T and γ-T compared with α-T. Using the kinase array, we recently demonstrated that the molecular basis for the even higher inhibitory activity of δ-T over γ-T involves the δ-T-specific inhibition of the receptor tyrosine kinase (e.g. EGFR)-induced activation of AKT (29). This results in the inhibition of cell cycle and induction of apoptosis in prostate cancer cell lines. δ-T, therefore, carries cancer preventive activity that is beyond its antioxidant roles and can be effective in preventing cancer driven by elevated pAKT levels. In the current study, we demonstrate that δ-T is effective in preventing the development of prostate adenocarcinoma in Ptenp−/− mice. Further examinations revealed that δ-T reduces the activation of AKT, suppresses cell proliferation and increases apoptosis. The results from this investigation using Ptenp−/− mice, in which the elevated AKT is the driver of prostate tumorigenesis, validate that the inhibition on the activation of AKT is an important mechanism of δ-T in prostate cancer prevention.
We demonstrated previously that the reduction of oxidative stress by γ-TmT, γ-T and δ-T plays an important role in inhibiting carcinogen-induced carcinogenesis in the prostate (16) and colon (28). In these models, oxidative stress plays a critical role in carcinogenesis. Oxidative stress-induced DNA damage is effectively reduced in mice treated with γ-TmT, γ-T and δ-T. Consequentially, tumor multiplicity is also significantly reduced. This finding suggests that antioxidants can be effective in cancer prevention in case where oxidative stress is an important risk factor. Our data showed that, in Ptenp−/− mice, oxidative stress is not a major event in prostate tumorigenesis (Supplemental Figure 8), and the antioxidant activity is not expected to play a role in prostate cancer prevention by δ-T. Therefore, we have proposed that the cancer preventive activity of δ-T is mediated by altering lipid membrane property, leading to an interference on the ligand-induced internalization of growth factor receptor (i.e. EGFR). This alteration attenuates the growth factor-induced activation of AKT (29). As shown in this study, the development of prostate cancer adenocarcinoma, induced by the elevation of pAKT levels in Ptenp−/− mice, is inhibited by δ-T through its inhibition on the activation of AKT. In this case, δ-T functions as an inhibitor of receptor tyrosine kinase in activating AKT. α-T and γ-T do not carry such an activity (29). Therefore, cancer prevention by δ-T can be effectively mediated through a mechanism beyond its antioxidant activity. Results from intervention studies (e.g. SELECT) using high levels of α-T did not show a preventive effect (13) but did raise concerns about the application of vitamin E in cancer prevention (50). From the previous and current experimental studies, it is clear that different tocopherol forms possess both distinct and common activities, and their effectiveness in cancer prevention is context dependent. Decisions regarding the use of vitamin E chemicals in preventing cancer and other diseases should be carefully evaluated based on knowledge of the molecular mechanism of the disease and the activities of these chemicals.
The challenge in exploring the molecular mechanism of dietary preventive agents, including tocopherols, is that these chemicals are often involved in more than one pathway, due to the lack of specific targets. All of these mechanisms presumably contribute to the overall cancer preventive activity of tocopherols at different levels. However, the relative importance of each mechanism depends on the specific situation of the model as discussed above. Because prostate tumorigenesis in Ptenp−/− mice is driven by an elevation in pAKT levels resulting from the loss of Pten, the inhibition of the AKT activation by δ-T is expected to play a key role in preventing the development of prostate adenocarcinoma. However, the contribution of other mechanisms requires further investigation.
A thorough understanding of the action and biological consequence of δ-T in cancer prevention is of great importance in our study in order to dissect the detailed pathway and/or targets that may play a critical role in the progression of HG-PIN to adenocarcinoma and to determine which are modulated by δ-T. These details, regardless of whether they are dependent or independent of the inhibition on AKT, could serve as a lead to develop novel prevention and therapeutic strategies. However, the molecular mechanism for HG-PIN progression to adenocarcinoma remains less clear. The development of prostate adenocarcinoma can be promoted in Ptenp−/− mice by introducing genetic events such as the loss of Smad4 (31), the reactivation of telomerase (51), the loss of tumor suppressors (e.g. p53) or the activation of oncogenes (e.g. K-ras) [reviewed in refs (33,52)]. These data demonstrate the importance of additional genetic events in the development of adenocarcinoma. It is expected that random genetic and epigenetic errors can be accumulated in the HG-PIN cells through uncontrolled DNA replication and cell division in a long latency period, which eventually reprograms the HG-PIN cells, leading to the development of adenocarcinoma. In addition, activation of the stromal microenvironment is suggested to be a critical step in prostate adenocarcinoma growth and progression (53). As noted, prostate stroma in the older Ptenp−/− mice is infiltrated with a significant amount of inflammatory cells, indicating the generation of a pro-tumorigenic microenvironment. Therefore, the progression of HG-PIN to adenocarcinoma involves both intrinsic and extrinsic factors. But, the causes of the spontaneous progression to adenocarcinoma in such mice remain unknown. Future studies using genomic and epigenomic approaches will be informative when addressing this issue.
In the current study, we conducted two sets of long term animal experiments by starting to feed Ptenp−/− mice with either an AIN93M or a purified diet supplemented with either 0.2% α-T or δ-T at the age of 6 (Experiment 1) and 12 weeks (Experiment 2). The results from both experiments are consistent and show that 0.2% δ-T diet effectively inhibits the development of prostate adenocarcinoma. However, our data shows that the overall prostate adenocarcinoma occurrence in Experiment 2 was more than that in Experiment 1. For instance, the percentage of prostate glands that developed adenocarcinoma in the control groups was increased from 24.1 ± 8% in Experiment 1 to 46.9 ± 15% in Experiment 2. This difference in development occurrence could be due to the protective effect of the lab chow diet (54). In Experiment 1, the mice were kept on the chow diet until 11 weeks of age, while in Experiment 2, the mice were kept on the chow diet until 5 weeks of age.
In summary, we demonstrate that 0.2% δ-T in the diet effectively inhibits the development of prostate adenocarcinomas in Ptenp−/− mice. Since an elevated level of active AKT is the driver in this tumorigenesis model, these data are consistent with our previous finding that δ-T attenuates the AKT activation in prostate cancer cell lines more effectively than α-T and γ-T (29). Along with the reduced pAKT levels, tumor cell proliferation is significantly decreased, and apoptosis is significantly increased by δ-T in the Ptenp−/− mice, as well as in prostate cancer cell lines. These results suggest that δ-T supresses the development of prostate adenocarcinoma through inhibition of AKT activation in vivo. Since genetic alteration of PTEN is one of the most common key drivers of human prostate cancer (55,56), our finding elucidates the molecular mechanism of action and potential use of δ-T in human prostate cancer prevention.
Supplementary material
Supplementary data can be found at Carcinogenesis online.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Funding
This work was supported by the National Institutes of Health (RO1 CA122474 and RO1 CA133021) as well as the shared facilities funded by CA72720 and ES05022.
Acknowledgements
We thank Dr. Ronald DePinho for kindly providing Fvb background prostate-specific Pten knockout mice. We thank Ms. Helen Yuhai Sun for technical assistance.
Conflict of Interest Statement: None declared.
Abbreviations
- α-T
α-tocopherol
- δ-T
δ-tocopherol
- γ-T
γ-tocopherol
- γ-TmT
γ-T-rich mixture of tocopherols
- H&E
hematoxylin and eosin
- HG-PIN
high-grade prostatic intraepithelial neoplasia
- IHC
Immunohistochemical
- pAKT
phospho-AKT
- WT
wild-type
References
- 1. Eitenmiller R.R., et al. (2005)Vitamin E: Food Chemistry, Composition, and Analysis. CRC Press, Marcel Dekker, Inc., New York, NY. [Google Scholar]
- 2. Jiang Q. (2014)Natural forms of vitamin E: metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med., 72, 76–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ju J., et al. (2010)Cancer-preventive activities of tocopherols and tocotrienols. Carcinogenesis, 31, 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Patel A., et al. (2007)Vitamin E chemistry. Nitration of non-alpha-tocopherols: products and mechanistic considerations. J. Org. Chem., 72, 6504–6512. [DOI] [PubMed] [Google Scholar]
- 5. Christen S., et al. (1997)gamma-tocopherol traps mutagenic electrophiles such as NO(X) and complements alpha-tocopherol: physiological implications. Proc. Natl Acad. Sci. USA., 94, 3217–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cooney R.V., et al. (1993)Gamma-tocopherol detoxification of nitrogen dioxide: superiority to alpha-tocopherol. Proc. Natl Acad. Sci. USA., 90, 1771–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jiang Q., et al. (2002)Gamma-tocopherol supplementation inhibits protein nitration and ascorbate oxidation in rats with inflammation. Free Radic. Biol. Med., 33, 1534–1542. [DOI] [PubMed] [Google Scholar]
- 8. Qian J., et al. (2005)Intracellular trafficking of vitamin E in hepatocytes: the role of tocopherol transfer protein. J. Lipid Res., 46, 2072–2082. [DOI] [PubMed] [Google Scholar]
- 9. Weinstein S.J., et al. (2012)Serum α-tocopherol and γ-tocopherol concentrations and prostate cancer risk in the PLCO Screening Trial: a nested case-control study. PLoS One, 7, e40204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang H.Y., et al. (2003)Prospective study of antioxidant micronutrients in the blood and the risk of developing prostate cancer. Am. J. Epidemiol., 157, 335–344. [DOI] [PubMed] [Google Scholar]
- 11. Key T.J., et al. (2015)Carotenoids, retinol, tocopherols, and prostate cancer risk: pooled analysis of 15 studies. Am. J. Clin. Nutr., 102, 1142–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gaziano J.M., et al. (2009)Vitamins E and C in the prevention of prostate and total cancer in men: the Physicians’ Health Study II randomized controlled trial. JAMA, 301, 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lippman S.M., et al. (2009)Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA, 301, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang C.S., et al. (2013)Cancer prevention by different forms of tocopherols. Top. Curr. Chem., 329, 21–33. [DOI] [PubMed] [Google Scholar]
- 15. Yang C.S., et al. (2016)Lessons learned from cancer prevention studies with nutrients and non-nutritive dietary constituents. Mol. Nutr. Food Res., 60, 1239–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen J.X., et al. (2016)Dietary tocopherols inhibit PhIP-induced prostate carcinogenesis in CYP1A-humanized mice. Cancer Lett., 371, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Das Gupta S., et al. (2014)Tocopherols inhibit oxidative and nitrosative stress in estrogen-induced early mammary hyperplasia in ACI rats. Mol. Carcinog., 2014, 22164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ju J., et al. (2009)A gamma-tocopherol-rich mixture of tocopherols inhibits colon inflammation and carcinogenesis in azoxymethane and dextran sulfate sodium-treated mice. Cancer Prev. Res. (Phila), 2, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lambert J.D., et al. (2009)Inhibition of lung cancer growth in mice by dietary mixed tocopherols. Mol. Nutr. Food Res., 53, 1030–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu G., et al. (2010)A gamma-tocopherol-rich mixture of tocopherols inhibits chemically induced lung tumorigenesis in A/J mice and xenograft tumor growth. Carcinogenesis, 31, 687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zheng X., et al. (2011)Inhibitory effect of a γ-tocopherol-rich mixture of tocopherols on the formation and growth of LNCaP prostate tumors in immunodeficient mice. Cancers (Basel), 3, 3762–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee H.J., et al. (2009)Mixed tocopherols prevent mammary tumorigenesis by inhibiting estrogen action and activating PPAR-gamma. Clin. Cancer Res., 15, 4242–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barve A., et al. (2009)Gamma-tocopherol-enriched mixed tocopherol diet inhibits prostate carcinogenesis in TRAMP mice. Int. J. Cancer, 124, 1693–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang Q., et al. (2000)gamma-tocopherol and its major metabolite, in contrast to alpha-tocopherol, inhibit cyclooxygenase activity in macrophages and epithelial cells. Proc. Natl Acad. Sci. USA, 97, 11494–11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takahashi S., et al. (2009)Suppression of prostate cancer in a transgenic rat model via gamma-tocopherol activation of caspase signaling. Prostate, 69, 644–651. [DOI] [PubMed] [Google Scholar]
- 26. Guan F., et al. (2012)δ- and γ-tocopherols, but not α-tocopherol, inhibit colon carcinogenesis in azoxymethane-treated F344 rats. Cancer Prev. Res. (Phila), 5, 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li G.X., et al. (2011)δ-tocopherol is more active than α- or γ-tocopherol in inhibiting lung tumorigenesis in vivo. Cancer Prev. Res. (Phila), 4, 404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen J.X., et al. (2016) δ- and γ-Tocopherols inhibit PhIP/DSS-induced colon carcinogenesis by protection against early cellular and DNA damages. Mol. Carcinog., 13, 22481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang H., et al. (2015) δ-Tocopherol inhibits receptor tyrosine kinase-induced AKT activation in prostate cancer cells. Mol. Carcinog., 14, 22422. [DOI] [PubMed] [Google Scholar]
- 30. Chen Z., et al. (2005)Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature, 436, 725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ding Z., et al. (2011)SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature, 470, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li J., et al. (1997)PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science, 275, 1943–1947. [DOI] [PubMed] [Google Scholar]
- 33. Shen M.M., et al. (2010)Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev., 24, 1967–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu X., et al. (2001)Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech. Dev., 101, 61–69. [DOI] [PubMed] [Google Scholar]
- 35. Drost J., et al. (2016)Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc., 11, 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ittmann M., et al. (2013)Animal models of human prostate cancer: the Consensus Report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res., 73, 2718–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shappell S.B., et al. (2004)Prostate pathology of genetically engineered mice: definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res., 64, 2270–2305. [DOI] [PubMed] [Google Scholar]
- 38. Svensson R.U., et al. (2011)Slow disease progression in a C57BL/6 pten-deficient mouse model of prostate cancer. Am. J. Pathol., 179, 502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li G., et al. (2012)Dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine-induced prostate carcinogenesis in CYP1A-humanized mice. Cancer Prev. Res. (Phila), 5, 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang S., et al. (2003)Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell, 4, 209–221. [DOI] [PubMed] [Google Scholar]
- 41. Freeman D., et al. (2006)Genetic background controls tumor development in PTEN-deficient mice. Cancer Res., 66, 6492–6496. [DOI] [PubMed] [Google Scholar]
- 42. Valavanidis A., et al. (2009)8-hydroxy-2’ -deoxyguanosine (8-OHdG): a critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health. C. Environ. Carcinog. Ecotoxicol. Rev., 27, 120–139. [DOI] [PubMed] [Google Scholar]
- 43. Das Gupta S., et al. (2015)Tocopherols inhibit oxidative and nitrosative stress in estrogen-induced early mammary hyperplasia in ACI rats. Mol. Carcinog., 54, 916–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Campbell S.E., et al. (2009)Gamma tocopherol upregulates the expression of 15-S-HETE and induces growth arrest through a PPAR gamma-dependent mechanism in PC-3 human prostate cancer cells. Nutr. Cancer, 61, 649–662. [DOI] [PubMed] [Google Scholar]
- 45. Campbell S.E., et al. (2003)Gamma (gamma) tocopherol upregulates peroxisome proliferator activated receptor (PPAR) gamma (gamma) expression in SW 480 human colon cancer cell lines. BMC Cancer, 3, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Das Gupta S., et al. (2015)Dietary γ-tocopherol-rich mixture inhibits estrogen-induced mammary tumorigenesis by modulating estrogen metabolism, antioxidant response, and PPARγ. Cancer Prev. Res. (Phila), 8, 807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smolarek A.K., et al. (2013)Dietary tocopherols inhibit cell proliferation, regulate expression of ERα, PPARγ, and Nrf2, and decrease serum inflammatory markers during the development of mammary hyperplasia. Mol. Carcinog., 52, 514–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang P.H., et al. (2013)Vitamin E facilitates the inactivation of the kinase Akt by the phosphatase PHLPP1. Sci. Signal., 6, ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smolarek A.K., et al. (2012)Dietary administration of δ- and γ-tocopherol inhibits tumorigenesis in the animal model of estrogen receptor-positive, but not HER-2 breast cancer. Cancer Prev. Res. (Phila), 5, 1310–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang C.S., et al. (2012)Does vitamin E prevent or promote cancer?Cancer Prev. Res. (Phila), 5, 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ding Z., et al. (2012)Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell, 148, 896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Grabowska M.M., et al. (2014)Mouse models of prostate cancer: picking the best model for the question. Cancer Metastasis Rev., 33, 377–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shiao S.L., et al. (2016)Regulation of prostate cancer progression by the tumor microenvironment. Cancer Lett., 380, 340–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Malloy V.L., et al. (1997)Interaction between a semisynthetic diet and indole-3-carbinol on mammary tumor incidence in Balb/cfC3H mice. Anticancer Res., 17, 4333–4337. [PubMed] [Google Scholar]
- 55. Taylor B.S., et al. (2010)Integrative genomic profiling of human prostate cancer. Cancer Cell, 18, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Robinson D., et al. (2015)Integrative clinical genomics of advanced prostate cancer. Cell, 161, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.