

Abstract
Lung exposure to radiation induces an injury response that includes the release of cytokines and chemotactic mediators; these signals recruit immune cells to execute inflammatory and wound-healing processes. However, radiation alters the pulmonary microenvironment, dysregulating the immune responses and preventing a return to homeostasis. Importantly, dysregulation is observed as a chronic inflammation, which can progress into pneumonitis and promote pulmonary fibrosis; inflammatory monocytes, which are bone marrow derived and express CCR2, have been shown to migrate into the lung after radiation exposure. Although the extent to which recruited inflammatory monocytes contribute to radiation-induced pulmonary fibrosis has not been fully investigated, we hypothesize that its pathogenesis is reliant on this population. The CC chemokine ligand, CCL2, is a chemotactic mediator responsible for trafficking of CCR2+ inflammatory cells into the lung. Therefore, the contribution of this mediator to fibrosis development was analyzed. Interleukin (IL)-1β, a potent pro-inflammatory cytokine expressed during the radiation response, and its receptor, IL-1R1, were also evaluated. To this end, CCR2−/−, IL-1β−/− and IL-1R1−/− chimeric mice were generated and exposed to 12.5 Gy thoracic radiation, and their response was compared to wild-type (C57BL/6) syngeneic controls. Fibrotic foci were observed in the periphery of the lungs of C57 syngeneic mice and CCR2−/− recipient mice that received C57 bone marrow (C57 > CCR2−/−) by 16 and 12 weeks after irradiation, respectively. In contrast, in the mice that had received bone marrow lacking CCR2 (CCR2−/−> C57 and CCR2−/− syngeneic mice), no pulmonary fibrosis was observed at 22 weeks postirradiation. This observation correlated with decreased numbers of infiltrating and interstitial macrophages compared to controls, as well as reduced proportions of pro-inflammatory Ly6C+ macrophages observed at 12–18 weeks postirradiation, suggesting that CCR2+ macrophages contribute to radiation-induced pulmonary fibrosis. Interestingly, reduced proportions of CD206+ lung macrophages were also present at these time points in CCR2−/− chimeric mice, regardless of donor bone marrow type, suggesting that the phenotype of resident subsets may be influenced by CCR2. Furthermore, chimeras, in which either IL-1β was ablated from infiltrating cells or IL-1R1 from lung tissues, were also protected from fibrosis development, correlating with attenuated CCL2 production; these data suggest that IL-1β may influence chemotactic signaling after irradiation. Overall, our data suggest that CCR2+ infiltrating monocyte-derived macrophages may play a critical role in the development of radiation-induced pulmonary fibrosis.
INTRODUCTION
Although radiation therapy for cancer treatment is common, with as many as 50% of patients receiving this therapy (1), a significant number of patients experience side effects as a result of their exposure (2, 3). Given the radiosensitivity of the lung, clinicians have not utilized radiotherapy to its fullest extent, due to the risk of radiation effects on nearby normal tissue, such as radiation pneumonitis and pulmonary fibrosis (4–6). Indeed, pulmonary fibrosis is currently an irreversible disease for which effective treatment is lacking and patient prognosis remains poor (7). Factors that increase the risk for the development of radiation-induced fibrosis include the radiation parameters of dose, volume of tissue treated and the time over which the exposure occurs; the use of many chemotherapeutic agents also increases risk (8–10). However, limiting these factors may not always be possible in the clinical setting. Therefore, current tumor treatment strategies often require post-therapy approaches to manage the adverse normal tissue outcomes (11). The mitigation of these effects would, therefore, greatly improve quality of life in survivors and increase the efficacy of radiotherapy in cancer treatment (12).
Radiation exposure of the lung, as with most normal tissues, results in DNA damage and cytotoxicity; these events invoke an early injury and wound-healing response. A component of the latter is an acute inflammatory reaction involving the production and release of inflammatory cytokines and chemotactic mediators, which recruit immune cells into the lung to execute inflammation and repair processes (13). Ideally, immune responses are orchestrated in a manner that acts to restore tissue homeostasis. However, radiation can induce microenvironmental alterations in the lung that dysregulate repair mechanisms and prevent the return of the injured niche to its quiescent state. Several groups have shown that one characteristic of dysregulation is continual inflammatory signaling, leading to the accumulation and persistent activation of immune cells, perpetuating a state of chronic inflammation and contributing to disease pathogenesis (14–16). Critically, within several months to years postirradiation, a secondary delayed reaction can manifest, which culminates in the development of pulmonary fibrosis (17, 18). Improper regulation of immune responses is thought to be one contributing factor to the development of this long-term pathology due to its role in the development of chronic inflammation. Indeed, multiple studies have shown that immune responses are critical regulators of radiation-induced lung injury outcomes, including fibrosis (15, 19, 20), and have demonstrated that limiting immune cell accumulation attenuates pathogenesis (21, 22). Despite these findings, the precise role that immune cells play in the development of pulmonary radiation late effects appears complex and remains unclear at this time.
The underlying mechanisms by which radiation-induced inflammatory signaling in lung tissue promotes and supports the pathological activation of pulmonary immune cell populations has been the focus of many investigations, including our own. For example, IL-1β, a potent inflammatory cytokine whose direct induction in the lung after irradiation has been well characterized (20, 23), also has been linked to pro-inflammatory and pro-fibrotic processes in a number of injury models (24–27). Thus, it has been suggested as playing a major role in the development of radiation-induced fibrosis in multiple organs, including the lung (22, 28, 29). Mature, extracellular IL-1β binds to its receptor, IL-1R1, on numerous cell types, initiating a cascade of pro-inflammatory signaling associated with wound healing. IL-1β can mediate the stimulation of a number of chemokines, including CCL2 (30–32), as well as the recruitment of immune cells (33). Pertinent to our studies, CCL2 is a highly effective monocyte chemoattractant and is expressed by epithelial cells, monocytes, macrophages and fibroblasts in the lung (34). Released from inflamed and injured tissues, CCL2 mobilizes a subset of highly responsive bone marrow-derived circulating inflammatory monocytes expressing the receptor for CCL2, CCR2, which then traffics into the tissue. Importantly, because inflammatory monocytes are sensitive to microenvironmental signals, they subsequently differentiate into more specialized macrophage subtypes within the injured niche, which contribute to the inflammation and repair processes (35). Interestingly, published animal studies have implicated CCL2 as a mediator of pulmonary fibrosis development (36, 37). Significantly, CCL2 is also increased in the lungs after thoracic irradiation (38–40). Furthermore, CCL2 expression is increased in the lungs of patients with idiopathic pulmonary fibrosis and other interstitial lung diseases (41, 42), and the repression of CCL2-driven responses has been shown to reduce disease pathogenesis in multiple models of fibrosis (43, 44).
Our group has long proposed that the infiltrating monocyte-derived macrophages contribute to radiation-induced pulmonary fibrosis (39, 45). For the current study, we now hypothesized that it is the CCR2+ infiltrating macrophages, specifically, that are the critical element in the development of radiation-induced pulmonary fibrosis. Furthermore, we implicate IL-1β signaling in the lung, directly induced by radiation, as the initiator of CCL2 expression, ultimately leading to the accumulation of inflammatory macrophages and the subsequent late effect progression. To test this hypothesis, chimeric mice were generated from C57BL/6J (C57) mice and mice lacking CCR2, IL-1β or IL-1R1, then exposed to 12.5 Gy thoracic radiation and evaluated for alterations in pulmonary macrophage subpopulation dynamics and phenotype, as well as the development of pulmonary fibrosis.
MATERIALS AND METHODS
Animals
Female and male C57, CCR2−/− and IL-1R1−/− mice, 6–8 weeks of age, were obtained from Jackson Laboratory (Bar Harbor, ME). IL-1β−/− mice were bred in-house as a result of a gift from Dr. David Caplin, University of Alabama. Five animals per cage were housed in microisolator units under pathogen-free conditions and were acclimated for one week prior to experimentation after arrival. Animals were fed a standard laboratory diet and water ad libitum. The University Committee on Animal Resources approved all animal protocols.
Bone Marrow Isolation
Femurs were dissected from donor mice and the ends cut off either side of the bone. An 18-g needle/1-cc syringe filled with phosphate buffered saline (PBS; Gibco® Life Technologies, Grand Island, NY) inserted into the femur was used to flush the marrow from the bone into a 50-ml conical tube on ice. The marrow was then passed through a 100-μm filter using PBS. Cells were enumerated using a hemocytometer and brought up to a concentration of 50 × 106 cells/ml in PBS. Cells were kept on ice until just before use when 50 μl aliquots of the cell suspension were loaded into 27-g insulin syringes.
Generation of Chimeras
Unanaesthetized recipient mice were placed in Plexiglas® holders and were total-body irradiated (2 × 5.5 Gy, spaced 4 h apart), using a 137Cs γ-ray source (JL Shepherd & Associates, San Fernando, CA) operating at a dose rate of approximately 1.7 Gy/min. Directly after the second exposure, mice were anesthetized with isoflurane and received 2.5 × 106 bone marrow cells via retro-orbital injection. Mice recovered for 8 weeks to allow bone marrow engraftment. For experiments investigating the effects of CCR2 on the radiation response, C57 recipient mice received bone marrow from either C57 or CC2−/− donor mice to generate C57 > C57 and CCR2−/− > C57 chimeras, respectively. Additionally, CCR2−/− recipient mice received bone marrow from either C57 or CC2−/− donor mice to generate C57 > CCR2−/− and CCR2−/− > CCR2−/− chimeras, respectively. Engraftment was verified via flow analysis 8 weeks later, by identifying CD45.1 donor-derived cells in blood samples collected from CD45.2 recipient mice (Supplementary Fig. S1; http://dx.doi.org/10.1667/RR14874.1.S1). For experiments investigating the effects of IL-β and IL-1R1−/− on the radiation response, the same procedure was performed using C57 and IL-β−/− mice to generate C57 > C57, IL-β−/− > C57, C57 > IL-β−/− and IL-β−/− > IL-β−/− chimeras or using C57 and IL-1R1−/− mice to create C57 > C57, IL-1R1−/− > C57, C57 > IL-1R1−/− and IL-1R1−/− > IL-1R1−/− chimeras.
Thoracic Irradiation
Animals, restrained in Plexiglas jigs, were exposed to 12.5 Gy thoracic radiation from a 137Cs γ-ray source operating at a dose rate of approximately 1.5 Gy/min. Age- and sex-matched control chimeras were sham irradiated with identical handling.
Sample Collection, Magnetic Cell Sorting and Flow Cytometry
Lungs were collected from CCR2−/− chimeras at 12–18 weeks postirradiation (corresponding to the pneumonitic period) for flow cytometry analysis. Lungs were digested by instillation with 1.8 units/ml dispase (Gibco Life Technologies) in Dulbecco’s modified Eagle medium (DMEM; Gibco Life Technologies) and incubated at room temperature for 45 min. Lungs were next disrupted by mincing, and the cell solution filtered through 100-, 40- and 25-μm cell strainers (Fisher Scientific™, Waltham, MA) using DMEM plus 0.01% DNAse-1 (Sigma-Aldrich® LLC, St. Louis, MO) as a wash buffer, then transferred to DMEM plus 10% fetal bovine serum (FBS; BD Biosciences, San Jose, CA).
CD45+ myeloid cells were enriched in lung digests using magnetic activated cell sorting (MACS) by incubating cells with a biotin anti-CD45 antibody (BD Pharmingen™, San Jose, CA) followed by binding to BD IMag™ Strepavidin Particles (BD Biosciences) and placement on a Magna grIP™ magnet (Millipore, Billerica, MA). Unbound CD45− cells were then removed and DMEM plus 10% FBS was used to resuspend the CD45+ cells that were retained on the magnet. CD45+ cells were then counted using a hemocytometer and 1 × 106 cells were stained for flow cytometric analysis. Cells were transferred to staining buffer (PBS plus 10% FBS) and were incubated with anti-mouse CD16/CD32 Fc block (BD Pharmingen) diluted 1:500 in staining buffer for 10 min at 4°C to block nonspecific antibody binding. Surface staining was then performed for 30 min at 4°C using PerCP-Cy5.5-conjugated rat anti-mouse CD-11b (BD Pharmingen), Alexa Fluor® 647-conjugated anti-mouse Ly6G, Brilliant Violet™ 605-conjugated anti-mouse CD206, Brilliant Violet 711-conjugated anti-mouse Ly6C, PE-conjugated anti-mouse CD45 (BioLegend® Inc., San Diego, CA), eFluor 450-conjugated anti-mouse CD11c, and PE/Cy7-conjugated anti-mouse F4/80 (eBioscience Inc., San Diego, CA). Cells were washed and then stained with LIVE/DEAD fixable aqua dead cell stain kit (Life Technologies) for 15 min at 4°C, then washed and resuspended in 2.5% phosphate buffered formalin (Fisher Scientific). Flow cytometry was performed on an 18-paramater LSRII flow cytometer (BD Biosciences). Simply Cellular® anti-mouse compensation standard (Bangs Laboratories Inc., Fishers, IN) was incubated with 1 μl of each detection antibody and used for single color positive controls. Data were analyzed using FlowJo software (Ashland, OR).
Histology
Lungs were collected for histological analysis from CCR2−/− chimeras at 12–22 weeks postirradiation (corresponding to the pneumonitic and early fibrotic phases, respectively), and were collected from IL-1β−/− and IL-1R1−/− chimeras at 32 weeks postirradiation [corresponding to the late fibrotic phase (46)]. The left lobe of the lung was inflation-fixed in 10% zinc buffered formalin (Anatech, Battle Creek, MI) and paraffin embedded. Tissue sections (6 μm) were prepared and stained with Gomori trichrome and examined by light microscopy. Images were acquired on an Olympust BX51 microscope (Olympus America, Center Valley, PA).
Ribonuclease Protection Assay
Total RNA was isolated from frozen lung tissue (50–100 mg) using 1 ml TRIzol® Reagent (Life Technologies, Grand Island, NY) according to the manufacturer’s instructions. Each final RNA pellet was resuspended in 50 μl of diethylpyrocarbonate-treated water and the RNA concentration and purity was quantified using the GeneQuant™ RNA/DNA Calculator (Pharmacia Biotech Inc., Piscataway, NJ). Quantitation of steady-state cytokine mRNA levels was performed using a multi-cytokine ribonuclease protection assay. RNase protection assays were performed as described elsewhere (47) with custom riboprobe templates for IL-1β, CCL2 and GAPDH. The protected radiolabeled RNA fragments were electrophoresed on a 5% acrylamide/8 M urea sequencing gel, and the dried gel was used to expose X-AR film (Eastman Kodak, Rochester, NY) at −80°C with intensifying screens (Quanta III; Dupont, Wilmington, DE). For quantitation, the dried gels were placed against PhosphorImager™ screens (Molecular Dynamics Inc., Sunnyvale, CA). The intensity of each specific chemokine and cytokine band was measured using a computer-linked PhosphorImager with ImageQuant software (Molecular Dynamics). Each intensity score was normalized to the intensity of hybridization for the constitutively expressed housekeeping gene, GAPDH, to correct for differences in loading.
Statistical Analysis
Data, expressed as mean ± SEM, were analyzed by two-way ANOVA, followed by Tukey’s multiple comparisons test. Differences were considered significant at P < 0.05.
RESULTS
Response of CCR2−/− Chimeras to Thoracic Irradiation
The role of CCR2+ infiltrating monocyte-derived macrophages in the development of radiation-induced pulmonary fibrosis was investigated in CCR2−/− chimeric mice exposed to 12.5 Gy thoracic radiation. Histologically, fibrotic foci could be observed in the periphery of the lungs of chimeric mice receiving C57 bone marrow [both C57 > CCR2−/− and C57 > C57 (syngeneic) chimeras] (Fig. 1A and B). The foci were apparent by 16 weeks postirradiation in C57 syngeneic mice, but were well established by 12 weeks postirradiation in C57 > CCR2−/− mice; inflammatory cell accumulation was associated with the fibrotic areas. In contrast, in mice receiving CCR2−/− bone marrow, fibrotic areas were not observed through 22 weeks postirradiation, indicating that an inhibition or delay in fibrosis development occurred in mice lacking CCR2-driven chemotaxis (Fig. 1C). A diffuse immune cell accumulation was observed throughout the tissue in CCR2−/− syngeneic mice, which was not seen in CCR2−/− > C57 chimeras. For all chimeric groups, in control chimeras that were not exposed to thoracic radiation, there were no visible pathological changes to the lung (Supplementary Fig. S2; http://dx.doi.org/10.1667/RR14874.1.S1).
FIG. 1.
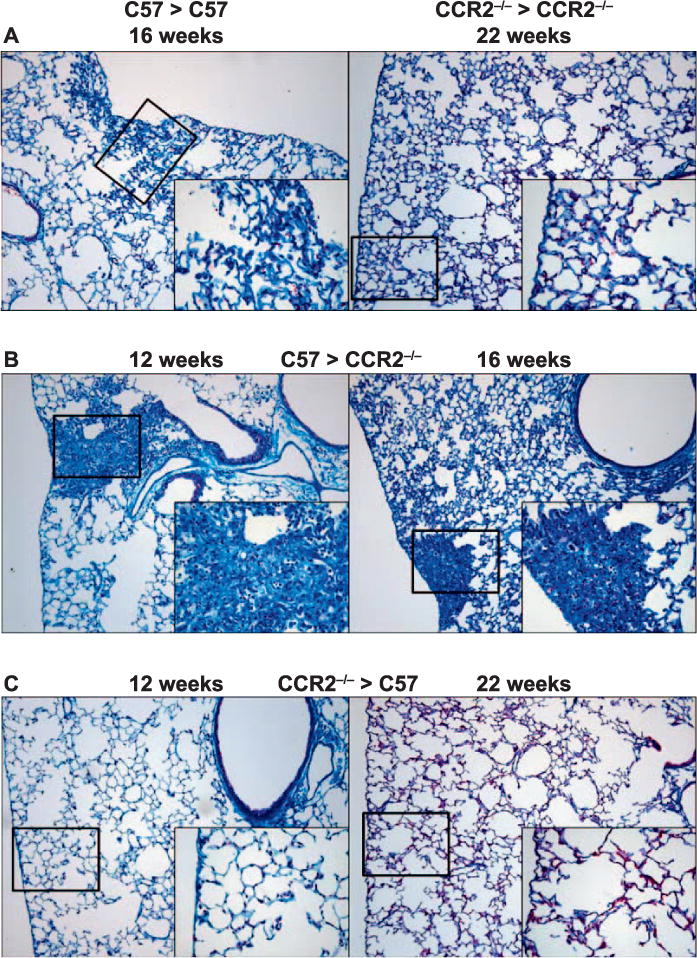
Chimeras lacking CCR2 from donor derived cells are protected from pulmonary fibrosis. Gomori trichrome-stained lung sections of syngeneic (panel A), C57 > CCR2−/− (panel B) and CCR2−/− > C57 (panel C) chimeras were prepared between 12 and 22 weeks after 0 or 12.5 Gy thoracic irradiation (n = 3–4 mice/treatment group). Original magnification 200× (inset 400×).
To investigate the influence of the CCR2+ infiltrating macrophage subset on resident macrophage population dynamics, pulmonary macrophage subpopulations were analyzed in CD45+ immune cell-enriched lung digests collected from CCR2−/− chimeras. Samples were collected from most groups at 18 weeks after thoracic irradiation, time points corresponding to the pneumonitic/fibrotic phase, but before significant morbidity occurred. A 12-week time sample was used for the C57 > CCR2−/− chimeras due to the earlier onset of fibrosis. As previously reported (48), alveolar, interstitial and infiltrating macrophage subsets were identified in the CD45+ population (Fig. 2A; the gating scheme is provided in Supplementary Fig. S3; http://dx.doi.org/10.1667/RR14874.1.S1). In the C57 syngeneic mice, the numbers of each macrophage subpopulation were similar to control chimeras that had not received thoracic irradiation. In contrast, in chimeras that had received CCR2−/− bone marrow (both CCR2−/− > C57 and CCR2−/− syngeneic chimeras), numbers of infiltrating macrophages were significantly decreased compared to nonirradiated controls, suggesting that CCR2+ cells may be the primary contributors to the infiltrating macrophage subpopulation (Fig. 2B). A similar effect was also noted in the interstitial populations, with differences reaching significance in CCR2−/− > C57 chimeras (Fig. 2C). Numbers of alveolar macrophages were increased in CCR2−/− syngeneic chimeras after irradiation, however, no significant differences in this subset were observed in either of the congenic chimeras (Fig. 2D).
FIG. 2.
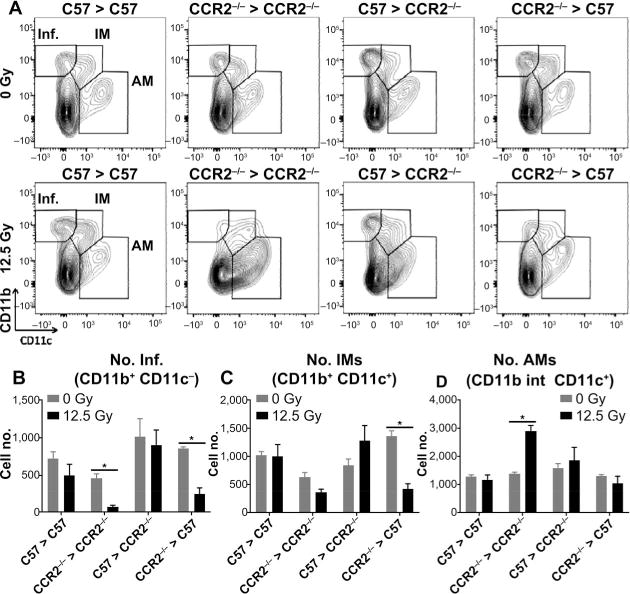
Chimeras lacking CCR2 from donor-derived cells have altered pulmonary macrophage subpopulation dynamics. In CCR2−/− chimeric mice, between 12 and 18 weeks after 0 or 12.5 Gy thoracic irradiation, CD45+ cells were enriched from lung digests using MACS, and alveolar (AM), interstitial (IM) and infiltrating (Inf) macrophages were analyzed by flow cytometry. Contour plots show a representative sample (panel A). Each subpopulation is calculated as the number of cells in the CD45+ per whole lung (panels B–D). Each bar represents mean ± SEM (n = 3–4 mice/treatment group). *Significantly different (P > 0.05) from nonirradiated chimera-matched controls.
CD11c is expressed in pulmonary macrophages that are resident to the lung, and is upregulated when infiltrating monocytes differentiate into lung macrophages (49). Therefore, the effects of radiation-induced alterations in macrophage phenotype were further analyzed in CD11c+ resident populations. The proportions of CD11c+ cells that expressed the inflammatory cell marker Ly6C (50) were reduced after irradiation in chimeras that had received CCR2−/− bone marrow (both CCR2−/− > C57 and CCR2−/− syngeneic chimeras) when compared to the nonirradiated controls, indicating that infiltrating CCR2+ cells likely contribute to this phenotype (Fig. 3A). In C57 syngeneic and C57 > CCR2−/− chimeras, proportions of Ly6C+ CD11c+ cells were not significantly different from irradiated controls at this time point. In contrast, the proportions of CD11c+ macrophages expressing the alternative activation marker CD206 (51), were reduced after irradiation in chimeras in which the lung tissue was lacking CCR2 (both C57 > CCR2−/− and CCR2−/− syngeneic chimeras), regardless of bone marrow donor type, suggesting this resident phenotype is influenced by local CCR2 expression (Fig. 3B). No significant differences were noted in the proportions of CD206+ CD11c+ macrophages after irradiation in chimeras that retained lung CCR2 expression (C57 syngeneic and CCR2−/− > C57 chimeras). In all chimeric groups, the percentage of CD11c+ resident macrophages that expressed F4/80, a marker present on mature macrophage and monocyte populations (52, 53), was decreased after thoracic irradiation (Fig. 3C).
FIG. 3.
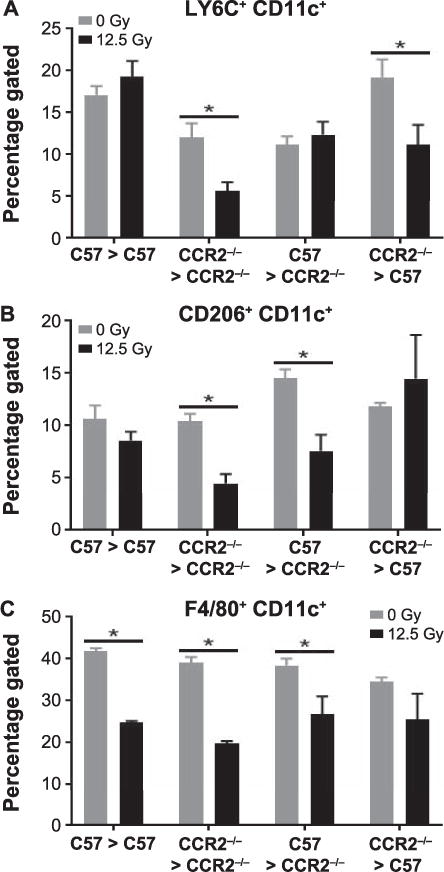
Chimeras lacking CCR2 from donor-derived cells have altered pulmonary macrophage subpopulation phenotypes. In CCR2−/− chimeric mice, between 12 and 18 weeks after 0 or 12.5 Gy thoracic irradiation, CD45+ cells were enriched from lung digests using MACS and analyzed by flow cytometry. CD11c+ resident macrophages were gated on and expression of Ly6C, CD206, and F4/80 was assessed. Each bar represents mean ± SEM (n = 3–4 mice/treatment group). *Significantly different (P > 0.05) from nonirradiated chimera-matched controls.
Response of IL-1β−/− and IL-1R1−/− Chimeras to Thoracic Irradiation
Immediate induction of IL-1β by thoracic irradiation has been well characterized. However, in this study, we endeavored to determine whether the signaling initiated by this cytokine is the major contributor to inflammatory cell infiltration and development of fibrosis. To investigate this, chimeras were generated from C57 and IL-1β−/− mice or C57 and IL-1R1−/− mice. Chimeras were then exposed to 12.5 Gy thoracic radiation. At 32 weeks postirradiation, a time corresponding to the late fibrotic phase of the radiation response (46), trichrome staining revealed focal areas of fibrosis in lung peripheries of both C57 syngeneic and C57 > IL-1β−/− chimeras, in which IL-1β expression in bone marrow-derived cells remained intact (Fig. 4A and C). Small fibrotic areas were also detected in syngeneic IL-1β−/− mice (Fig. 4D). In contrast, fibrosis did not develop in IL-1β−/− > C57 chimeras, in which IL-1β expression was lost from bone marrow-derived cells (Fig. 4B). Protection from fibrosis was also apparent in chimeras for which IL-1R1 was missing from lung tissues (C57 > IL-1R1−/− and IL-1R1−/− syngeneic chimeras; Fig. 4F and G), but did develop in IL-1R1−/− > C57 mice in which the receptor was present in resident lung cells, but not in bone marrow-derived cells (Fig. 4E), indicating that IL-1β/IL-1R1 signaling also contributes to the fibrotic response.
FIG. 4.
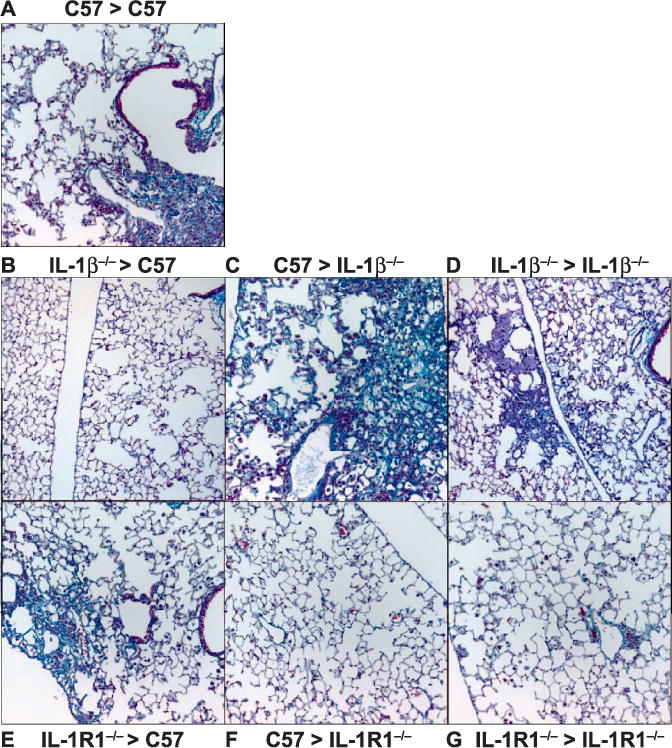
Development of radiation-induced pulmonary fibrosis is altered in IL-1β−/− and IL-1R1−/− chimeric mice. Gomori trichrome-stained lung sections were prepared 32 weeks after 0 or 12.5 Gy thoracic irradiation (n > 5 mice/treatment group). Original magnification 200×.
Because loss of IL-1β from bone marrow-derived cells, but not lung tissues, protected from radiation-induced pulmonary fibrosis development, the relative contributions of the tissue and infiltrating immune compartments to the total expression of IL-1β in the lung was examined in whole-lung lysates from IL-1β−/− and IL-1R1−/− chimeric mice at 32 weeks postirradiation or in nonirradiated control chimeras. Loss of IL-1β from the bone marrow (both IL-1β−/− > C57 and IL-1β−/− syngeneic chimeras) significantly reduced the expression of IL-1β (Fig. 5A). In contrast, in C57 > IL-1β−/− mice that retained IL-1β activity in their bone marrow-derived cells, whole-lung IL-1β expression was not significantly different from C57 syngeneic chimeras, suggesting that it is the infiltrating cells that are a major source of this cytokine in the lung. Loss of IL-1R1 did not affect expression of IL-1β. In both IL-1R1−/− syngeneic and congenic chimeras, IL-1β expression did not significantly differ from C57 syngeneic chimeras, and was not altered by thoracic irradiation.
FIG. 5.
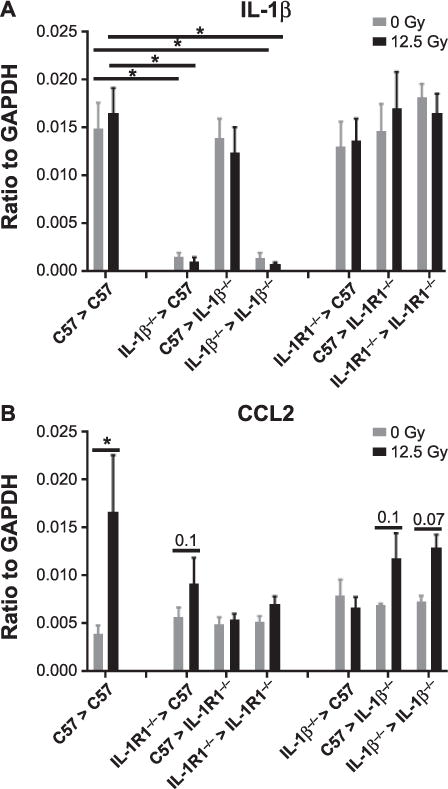
Lung expression of IL-1β and CCL2 mRNA is altered in IL-1β−/− and IL-1R1−/− chimeric mice. Whole-lung lysates were prepared 32 weeks after 0 or 12.5 Gy thoracic irradiation. mRNA abundance was measured using RNase Protection assay. Each bar represents mean ± SEM (n > 5 mice/treatment group). *Significantly different (P > 0.05) from nonirradiated chimera-matched controls.
Since the development of radiation-induced pulmonary fibrosis was abrogated in chimeras in which either expression of IL-1β was lost from bone marrow-derived cells or its receptor, IL-1R1, was not expressed in lung tissue, the contribution of IL-1β to the production of chemotactic factors was investigated. This was performed by analyzing lung expression of CCL2, responsible for recruiting inflammatory CCR2+ monocytes into the lung, in chimeras lacking IL-1β and IL-1R1. Thoracic irradiation significantly increased CCL2 expression in C57 syngeneic chimeras (Fig. 5B). This trend was also noted in the chimeras in which fibrosis occurred, in C57 > IL-1β−/− and IL-1β−/− syngeneic chimeras, as well as in IL-1R1−/− > C57 chimeras. However, in chimeras in which fibrosis was not observed, in IL-1β−/− > C57, C57 > IL-1R1−/− and IL-1R1−/− syngeneic chimeras, increased CCL2 expression was not observed, suggesting a strong and interactive relationship between IL-1β signaling, CCL2 production and fibrogenesis.
DISCUSSION
The lung’s response to radiation injury progresses over an extended period of time and often involves the development of pulmonary fibrosis (12, 18, 54). Consistent with all biological damage, the initial pulmonary injury stems from the direct exposure of DNA and other cellular components to ionizing radiation events, resulting in cytotoxicity and the initiation of an acute inflammation and wound-healing response (13, 16). However, after high doses, the irradiated lung microenvironment can fail to return to homeostasis (12), and recurrent periods of DNA damage as well as chronic inflammation ensues in a response that develops over the following weeks and months. Dysregulation within the niche is observed as the production and release of pro-inflammatory cytokines, chemokines and adhesion molecules, as well as an accumulation of activated macrophages and lymphocytes (19, 55, 56). Our earlier published studies, using a similar thoracic radiation model to that used in these studies, demonstrated that, after irradiation, there is early induction of pro-inflammatory and pro-fibrotic cytokines, including IL-1β (20, 23). In fibrosis-sensitive mouse strains, this was followed by production of chemokines, including CCL2, the recruitment of inflammatory cells and the development of pulmonary fibrosis (40). Interactions between the lung parenchyma and activated immune cells have been implicated in fibrogenesis (57), and suggest that immune cell recruitment and activation in the irradiated lung likely contribute to a pro-fibrotic environment. We have previously observed that lung macrophage subpopulations are phenotypically altered after thoracic irradiation, and that these dysregulated phenotypes persist throughout the radiation response, through fibrosis progression (48).
Infiltrating monocyte-derived macrophages have been found to contribute to pulmonary fibrosis development in multiple experimental models (43, 58, 59), and although published studies on radiation-induced lung injury and fibrosis have demonstrated that exposure promotes inflammatory cell infiltration and accumulation (21, 22), we are unaware as to whether the contributions of the infiltrating macrophage subset to fibrogenesis have been assessed. Earlier findings reported by our group have shown that, after thoracic irradiation, CCR2 expression in the lung was increased at late end points in a mouse strain that is deemed radiation-fibrosis susceptible, but not in a strain that does not develop radiation fibrosis (39, 40). In the current studies, our findings demonstrated that loss of bone marrow-derived CCR2, and thus CCR2-mediated immune cell infiltration, reduced the number of infiltrating monocyte-derived macrophages in the lungs of irradiated mice, confirming that trafficking of this population into the lung is dependent on CCR2. Furthermore, while pulmonary fibrosis did not occur when CCR2-mediated chemotaxis into the lung was ablated, its development was noted in chimeras lacking CCR2 on resident lung cells. This provides further support of a pro-fibrotic role for CCR2+ infiltrating monocyte-derived macrophages in this process, in line with results from other models of pulmonary fibrosis. For example, CCR2−/− mice were shown to be protected from fibrosis development in both FITC- and bleomycin-induced pulmonary fibrosis models (43, 60, 61), as well as in genetically susceptible Hermansky-Pudlak syndrome mice that were crossed with CCR2−/− mice and challenged with bleomycin (62).
Macrophages accumulate in the lungs throughout the development of the pneumonitic and fibrotic stages of the radiation response. Since these cells are highly responsive to signals they encounter in their microenvironment, their phenotypes also evolve and can, in turn, act as mediators in disease outcomes (63). Our previously published studies have demonstrated that, in the first few weeks after exposure of the lung to radiation, resident macrophage populations are depleted and replenished in a subpopulation-dependent manner, culminating in phenotypes that differ from those originally contained within the lung (48). Hashimoto et al. have also demonstrated that, after genotoxic injury to the lung, the ability of depleted resident macrophage populations to maintain macrophage pools through local proliferation is impaired and, instead, infiltrating monocytes from the circulation are retained to maintain an adequate resident population (64). In line with this, our results support the possibility that CCR2+ infiltrating monocytes are recruited into the lung to replenish reduced interstitial subpopulations, since numbers of both infiltrating and interstitial macrophages were significantly decreased after thoracic irradiation in chimeras in which CCR2-mediated monocyte infiltration into the lung was abolished (in CCR2−/− > C57 mice). Because the phenotypes of resident macrophage populations in the lung can be affected by the infiltrating macrophage population, either through their production of phenotypic mediators or through differentiation into activated macrophages (35), it is possible that the retention of CCR2+ infiltrating cells in the irradiated lung could stimulate the development of a pro-inflammatory phenotype in resident macrophage populations. Our finding, that the expression of pro-inflammatory Ly6C was decreased on CD11c+ resident macrophages in those chimeras in which CCR2-driven monocyte chemotaxis into the lung was eliminated (in CCR2−/− > C57 mice), provides support for this contention. Furthermore, these findings appeared to be specific to the bone marrow-derived infiltrating cells, rather than resident immune or epithelial populations, since this result was not observed in C57 > CCR2−/− chimeric mice where CCR2 expression was lost from the parenchyma.
Effects due to the loss of lung parenchymal expression of CCR2 in C57 > CCR2−/− mice were also noted, but were in contrast to the more protective effects that were observed with loss of bone marrow-derived CCR2. Since alveolar macrophages themselves do not express CCR2 and do not migrate in response to CCL2, our findings are not likely the result of intrinsic differences between the alveolar macrophages of CCR2−/− mice and wild-type (65). However, lung alveolar epithelial cells do express CCR2, and bind CCL2 to migrate within the lung in response to epithelial cell injury, a part of the canonical healing response (66). A loss of epithelial migration within the lung during the acute wound-response period may explain the earlier onset of fibrosis after irradiation noted in C57 > CCR2−/− chimeric mice when compared to C57 syngeneic chimeras. While alveolar macrophages are CCR2 expression-independent, they appear to be responsive to the altered pulmonary microenvironment that develops with loss of local CCR2 expression. This was demonstrated by the fact that proportions of CD206+ resident macrophages, indicative of alternatively activated resident phenotype (51), are decreased after thoracic irradiation in chimeric mice lacking lung CCR2 expression, but not in those in which CCR2 was present in the lung, regardless of the CCR2 status of the bone marrow.
Given the findings that infiltrating macrophages contribute to radiation-induced pulmonary fibrosis, the conditions under which the irradiated lung promotes the accumulation of inflammatory cells was further investigated. It has been well characterized that a multitude of cytokines are induced and cyclically expressed throughout the radiation response, mediating the acute and chronic outcomes (20, 23, 39, 40, 67). Much of this work has suggested that IL-1β is critical to the development of pulmonary and other normal tissue irradiation outcomes (22, 23, 28, 29, 68). IL-1β, a potent pro-inflammatory cytokine, is associated with the induction and propagation of inflammation, as well as wound-healing processes (69, 70). IL-1β can also act as a mediator of fibrogenesis and plays a role in lung tissue remodeling (27), in part due to its ability to regulate the balance between matrix deposition and degradation (71, 72). This is evidenced in mouse studies showing that transient overexpression of IL-1β was associated with persistent pulmonary fibrosis (26), and by findings of increased levels of this cytokine in bronchoalveolar lavage fluid from mice with bleomycin-induced pulmonary fibrosis (27, 73). The IL-1β receptor, IL-1R1, has also been found to play a crucial role in disease pathogenesis in multiple models of pulmonary fibrosis. For example, in bleomycin and silica models, pulmonary fibrosis was attenuated in IL-1R1−/− mice or with treatment with an IL-1 receptor antagonist (27, 74), and mice lacking IL-1R1 were protected from developing skin fibrosis after irradiation (28).
Our study demonstrated that fibrosis still occurs in chimeras in which only bone marrow-derived cells retain the ability to produce IL-1β, but it was not detected in IL-1β−/− > C57 chimeras lacking the cytokine in donor-derived inflammatory cells. Immune cells appear to be the major source of this cytokine since lung levels of IL-1β were maintained in chimeras in which only infiltrating cells retained expression of this cytokine, but were significantly decreased in those chimeras in which IL-1β production was lost from the infiltrating cells. Considering these findings, it was therefore unexpected that small fibrotic areas were present in IL-1β−/− syngeneic chimeras. A potential explanation for this discrepancy may be that the additional radiation dose used for bone marrow ablation in chimera generation may have exceeded the lung threshold for injury repair in these mice, such that the total dose was sufficient to initiate fibrosis, which is a multifactorial process. This theory is supported by work from our laboratory (unpublished data) using the same thoracic radiation model, which revealed that fibrosis was attenuated in IL-1β−/− mice (Supplementary Fig. S4; http://dx.doi.org/10.1667/RR14874.1.S1). Additionally, based on our previously reported findings in C57 mice that have not undergone bone marrow ablation and reconstitution, in which fibrosis becomes established past 20 weeks postirradiation, the fibrotic lesions observed in C57 syngeneic chimeras at 16 weeks postirradiation indicate that an early onset of fibrosis occurred in these mice, suggesting that compared with our normal time line of fibrosis development using a 12.5 Gy dose, the contribution from the earlier ablation dose may have shifted the induction curve (67, 75).
Although production of IL-1β by infiltrating cells appears to be a significant contributor to fibrogenesis, loss of IL-1R1 from resident lung populations, but not the bone marrow-derived cells, abrogated the fibrotic response. This suggests that downstream signaling by the resident lung cell populations is an essential component of the process. Again, this observation is not likely due to differences in IL-1β production in the IL-1R1−/− chimeric mice, since loss of the receptor from either compartment did not affect the production of IL-1β at the observed time points. Importantly, the findings in both IL-1β−/− and IL-1R1−/− chimeric mice showed that loss of IL-1β-mediated production of CCL2 correlated with an apparent protection from fibrosis. Importantly, radiation-associated increases in CCL2 expression were lost when IL-1β was lacking from bone marrow-derived infiltrating cells, as well as when the receptor was absent from resident lung populations. This suggests that after irradiation, IL-1β-induced signaling directly stimulates CCL2 production, and thus, inflammatory cell recruitment. Furthermore, although mechanistic information is currently limited, these findings provide support for a relationship between IL-1R1 and CCL2, and are in line with other studies demonstrating this link. For example, CCL2 expression was abolished in IL-1R1 conditional knock-out mice (30), while elevated levels of CCL2, induced by cardiac infarcts in nonmyeloid cells, have been shown to be abrogated in IL-1R1−/− mice (31). We therefore suggest that inflammatory cell production of IL-1β stimulates IL-1R1 in resident lung populations, initiating the downstream signaling that results in CCL2 production. This in turn recruits circulating CCR2+ inflammatory monocytes into the lung, which are capable of further IL-1β production, and can promote macrophage activation to a pro-fibrotic phenotype and the development of a pro-fibrotic environment (Fig. 6). We believe that these findings indicate that it is the pro-inflammatory signaling, instigated by infiltrating immune cell populations and their response to the dysregulated microenvironment, that is critical to the development of radiation-induced pulmonary fibrosis.
FIG. 6.
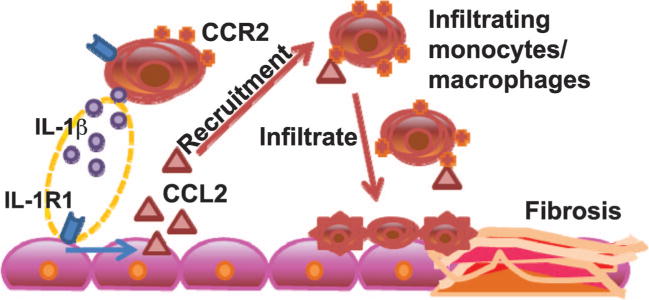
IL-1β signaling and CCR2-dependent inflammatory monocyte recruitment in the development of radiation-induced pulmonary fibrosis. IL-1β produced by immune cells induces local signaling through IL-1R1 that promotes CCL2 production, and thus, inflammatory cell recruitment and the development of a pro-inflammatory microenvironment.
Irradiation of normal lung tissue is an inevitable consequence of the use of radiation therapy when treating thoracic neoplasms, despite the technological advances that have reduced the exposure of healthy tissues. Dependent on treatment characteristics, such as dose and volume, as well as patient characteristics, such as age, smoking habits, etc., the use of radiation as a treatment modality carries with it the risk for developing devastating side effects, such as radiation pneumonitis and pulmonary fibrosis. Effective therapies to treat or prevent these outcomes are currently limited, especially with respect to fibrosis. The identification of new therapeutic targets will benefit the clinician’s ability to improve treatment options. Research is needed to more precisely elucidate how the processes regulating the inflammatory and wound-healing responses induced by radiation fail to return normal tissues, such as the lung, to homeostasis, and how such conditions lead to the development of an environment that promotes fibrogenesis. It is well accepted that fibrotic remodeling is a complex process, regulated by multiple cytokines and chemokines, and involving the actions of multiple cell types that both release and are responsive to these signals. We believe that our findings provide more detailed and, indeed, novel information describing the contributions of the infiltrating and resident immune cell compartments to the development of this long-term pathology, corroborating the findings of multiple experimental models of pulmonary fibrosis, but more importantly, establishing these effects in a radiation setting. The actions of an accumulation of a specific population of infiltrating macrophages and the production of the chemoattractant, CCL2, by resident lung populations are now highlighted as drivers of disease pathogenesis and warrant further investigation into the interaction between the lung parenchymal and immune compartments in the context of pulmonary fibrosis mitigation. Interestingly, our hypothesis that fibrogenesis may, in fact, be initiated by the direct induction of IL-1β expression, which is observed immediately after irradiation, is supported by the current use of stereotactic body radiation therapy (SBRT), i.e., delivery of a limited number of high-dose fractions. Published clinical studies have shown little to no change in the risk of treatment-related lung fibrosis with the use of SBRT for lung cancer (4, 76). This may be a consequence of improved conformal and image-guided beam delivery. However, if radiation biology dogma holds true, such results also may suggest that, despite their delayed appearance, pneumonitis and fibrosis are dependent on one or more acutely-responding cell populations, supporting our focus on the immune cell response.
Supplementary Material
Fig. S1. Bone marrow engraftment validation in CCR2−/− chimeric mice. Engraftment was verified via flow analysis 8 weeks after generation of chimeras by identifying CD45.1 donor-derived cells in blood samples collected from CD45.2 recipient mice.
Fig. S2. Lung histology from CCR2−/− chimeric mice. Gomori trichrome-stained lung sections were prepared between 12 and 22 weeks after 0 or 12.5 Gy thoracic irradiation (n = 3–4 mice/treatment group). Original magnification 200× (inset 400×).
Fig. S3. Gating scheme for characterization of pulmonary macrophage subsets. In CCR2−/− chimeric mice, between 12 and 18 weeks after 0 or 12.5 Gy thoracic irradiation, CD45+ cells were enriched from lung digests using MACS and analyzed by flow cytometry. Following doublet and dead cell discrimination, CD45+, Ly6G- cells were gated. CD11b and CD11c were used to discern alveolar (AMs; CD11b intermediate, CD11c+), interstitial (IMs; CD11b+, CD11c+) and infiltrating (Inf; CD11b+, CD11c-) macrophages for analysis of population dynamics. To analyze resident macrophages, CD11c+ cells were gated, and the percentages of this population expressing Ly6C, CD206 and F4/80 were determined.
Fig. S4. Lung histology from C57Bl/6 and IL-1β−/− mice. Gomori trichrome-stained lung sections were prepared 32 weeks after 0 or 12.5 Gy thoracic irradiation. Original magnification 200×.
Acknowledgments
This work is supported by the National Institutes of Health, NIH nos. R01 AI101732-01, U19AI091036, P30 ES-01247 and ES T32 07026.
Footnotes
Editor’s note. The online version of this article (DOI: 10.1667/RR14874.1) contains supplementary information that is available to all authorized users.
References
- 1.Baskar R, Lee KA, Yeo R, Yeoh KW. Cancer and radiation therapy: current advances and future directions. Int J Med Sci. 2012;9:193–9. doi: 10.7150/ijms.3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carver JR, Shapiro CL, Ng A, Jacobs L, Schwartz C, Virgo KS, et al. American Society of Clinical Oncology clinical evidence review on the ongoing care of adult cancer survivors: cardiac and pulmonary late effects. J Clin Oncol. 2007;25:3991–4008. doi: 10.1200/JCO.2007.10.9777. [DOI] [PubMed] [Google Scholar]
- 3.Ghafoori P, Marks LB, Vujaskovic Z, Kelsey CR. Radiation-induced lung injury. Assessment, management, and prevention Oncology (Williston Park) 2008;22:37–47. discussion 52–3. [PubMed] [Google Scholar]
- 4.Trovo M, Linda A, El Naqa I, Javidan-Nejad C, Bradley J. Early and late lung radiographic injury following stereotactic body radiation therapy (SBRT) Lung Cancer. 2010;69:77–85. doi: 10.1016/j.lungcan.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Peulen H, Karlsson K, Lindberg K, Tullgren O, Baumann P, Lax I, et al. Toxicity after reirradiation of pulmonary tumours with stereotactic body radiotherapy. Radiother Oncol. 2011;101:260–6. doi: 10.1016/j.radonc.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 6.Gustavsson A, Eskilsson J, Landberg T, Larusdottir H, Svahn-Tapper G, White T, et al. Long-term effects on pulmonary function of mantle radiotherapy in patients with Hodgkin’s disease. Ann Oncol. 1992;3:455–61. doi: 10.1093/oxfordjournals.annonc.a058234. [DOI] [PubMed] [Google Scholar]
- 7.Kong FM, Ten Haken R, Eisbruch A, Lawrence TS. Non-small cell lung cancer therapy-related pulmonary toxicity: an update on radiation pneumonitis and fibrosis. Semin Oncol. 2005;32:S42–54. doi: 10.1053/j.seminoncol.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 8.Sleijfer S. Bleomycin-induced pneumonitis. Chest. 2001;120:617–24. doi: 10.1378/chest.120.2.617. [DOI] [PubMed] [Google Scholar]
- 9.Skinner R, Kaplan R, Nathan PC. Renal and pulmonary late effects of cancer therapy. Semin Oncol. 2013;40:757–73. doi: 10.1053/j.seminoncol.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 10.Kirkbride P, Hatton M, Lorigan P, Joyce P, Fisher P. Fatal pulmonary fibrosis associated with induction chemotherapy with carboplatin and vinorelbine followed by CHART radiotherapy for locally advanced non-small cell lung cancer. Clin Oncol (R Coll Radiol) 2002;14:361–6. doi: 10.1053/clon.2002.0119. [DOI] [PubMed] [Google Scholar]
- 11.Straub JM, New J, Hamilton CD, Lominska C, Shnayder Y, Thomas SM. Radiation-induced fibrosis: mechanisms and implications for therapy. J Cancer Res Clin Oncol. 2015;141:1985–94. doi: 10.1007/s00432-015-1974-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams JP, Calvi L, Chakkalakal JV, Finkelstein JN, O’Banion MK, Puzas E. Addressing the symptoms or fixing the problem? Developing countermeasures against normal tissue radiation injury. Radiat Res. 2016;186:1–16. doi: 10.1667/RR14473.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ward JF. Some biochemical consequences of the spatial distribution of ionizing radiation-produced free radicals. Radiat Res. 1981;86:185–95. [PubMed] [Google Scholar]
- 14.Sprung CN, Forrester HB, Siva S, Martin OA. Immunological markers that predict radiation toxicity. Cancer Lett. 2015;368:191–7. doi: 10.1016/j.canlet.2015.01.045. [DOI] [PubMed] [Google Scholar]
- 15.Schaue D, McBride WH. Links between innate immunity and normal tissue radiobiology. Radiat Res. 2010;173:406–17. doi: 10.1667/RR1931.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schaue D, Micewicz ED, Ratikan JA, Xie MW, Cheng GH, McBride WH. Radiation and inflammation. Semin Radiat Oncol. 2015;25:4–10. doi: 10.1016/j.semradonc.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams JP, Brown SL, Georges GE, Hauer-Jensen M, Hill RP, Huser AK, et al. Animal models for medical countermeasures to radiation exposure. Radiat Res. 2010;173:557–78. doi: 10.1667/RR1880.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trott KR, Herrmann T, Kasper M. Target cells in radiation pneumopathy. Int J Radiat Oncol Biol Phys. 2004;58:463–9. doi: 10.1016/j.ijrobp.2003.09.045. [DOI] [PubMed] [Google Scholar]
- 19.McBride WH, Chiang CS, Olson JL, Wang CC, Hong JH, Pajonk F, et al. A sense of danger from radiation. Radiat Res. 2004;162:1–19. doi: 10.1667/rr3196. [DOI] [PubMed] [Google Scholar]
- 20.Rubin P, Johnston CJ, Williams JP, McDonald S, Finkelstein JN. A perpetual cascade of cytokines postirradiation leads to pulmonary fibrosis. Int J Radiat Oncol Biol Phys. 1995;33:99–109. doi: 10.1016/0360-3016(95)00095-G. [DOI] [PubMed] [Google Scholar]
- 21.Hallahan DE, Virudachalam S. Intercellular adhesion molecule 1 knockout abrogates radiation induced pulmonary inflammation. Proc Natl Acad Sci U S A. 1997;94:6432–7. doi: 10.1073/pnas.94.12.6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung EJ, Sowers A, Thetford A, McKay-Corkum G, Chung SI, Mitchell JB, et al. Mammalian target of rapamycin inhibition with rapamycin mitigates radiation-induced pulmonary fibrosis in a murine model. Int J Radiat Oncol Biol Phys. 2016;96:857–66. doi: 10.1016/j.ijrobp.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnston CJ, Piedboeuf B, Rubin P, Williams JP, Baggs R, Finkelstein JN. Early and persistent alterations in the expression of interleukin-1 alpha, interleukin-1 beta and tumor necrosis factor alpha mRNA levels in fibrosis-resistant and sensitive mice after thoracic irradiation. Radiat Res. 1996;145:762–7. [PubMed] [Google Scholar]
- 24.Borthwick LA. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin Immunopathol. 2016;38:517–34. doi: 10.1007/s00281-016-0559-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindroos PM, Coin PG, Badgett A, Morgan DL, Bonner JC. Alveolar macrophages stimulated with titanium dioxide, chrysotile asbestos, and residual oil fly ash upregulate the PDGF receptor-alpha on lung fibroblasts through an IL-1beta-dependent mechanism. Am J Respir Cell Mol Biol. 1997;16:283–92. doi: 10.1165/ajrcmb.16.3.9070613. [DOI] [PubMed] [Google Scholar]
- 26.Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest. 2001;107:1529–36. doi: 10.1172/JCI12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest. 2007;117:3786–99. doi: 10.1172/JCI32285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu W, Ding I, Chen K, Olschowka J, Xu J, Hu D, et al. Interleukin 1beta (IL1B) signaling is a critical component of radiation-induced skin fibrosis. Radiat Res. 2006;165:181–91. doi: 10.1667/rr3478.1. [DOI] [PubMed] [Google Scholar]
- 29.Chiang CS, Liu WC, Jung SM, Chen FH, Wu CR, McBride WH, et al. Compartmental responses after thoracic irradiation of mice: strain differences. Int J Radiat Oncol Biol Phys. 2005;62:862–71. doi: 10.1016/j.ijrobp.2005.02.037. [DOI] [PubMed] [Google Scholar]
- 30.Abdulaal WH, Walker CR, Costello R, Redondo-Castro E, Mufazalov IA, Papaemmanouil A, et al. Characterization of a conditional interleukin-1 receptor 1 mouse mutant using the Cre/LoxP system. Eur J Immunol. 2016;46:912–8. doi: 10.1002/eji.201546075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saxena A, Chen W, Su Y, Rai V, Uche OU, Li N, et al. IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J Immunol. 2013;191:4838–48. doi: 10.4049/jimmunol.1300725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Standiford TJ, Kunkel SL, Phan SH, Rollins BJ, Strieter RM. Alveolar macrophage-derived cytokines induce monocyte chemo-attractant protein-1 expression from human pulmonary type II-like epithelial cells. J Biol Chem. 1991;266:9912–8. [PubMed] [Google Scholar]
- 33.Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, et al. IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol. 2011;187:4835–43. doi: 10.4049/jimmunol.1102048. [DOI] [PubMed] [Google Scholar]
- 34.Rose CE, Jr, Sung SS, Fu SM. Significant involvement of CCL2 (MCP-1) in inflammatory disorders of the lung. Microcirculation. 2003;10:273–88. doi: 10.1038/sj.mn.7800193. [DOI] [PubMed] [Google Scholar]
- 35.Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional dD\ifferentiation. Front Immunol. 2014;5:514. doi: 10.3389/fimmu.2014.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mercer PF, Johns RH, Scotton CJ, Krupiczojc MA, Konigshoff M, Howell DC, et al. Pulmonary epithelium is a prominent source of proteinase-activated receptor-1-inducible CCL2 in pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:414–25. doi: 10.1164/rccm.200712-1827OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng X, Xu M, Yuan C, Yin L, Chen X, Zhou X, et al. Transcriptional regulation of increased CCL2 expression in pulmonary fibrosis involves nuclear factor-kappaB and activator protein-1. Int J Biochem Cell Biol. 2013;45:1366–76. doi: 10.1016/j.biocel.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 38.Cappuccini F, Eldh T, Bruder D, Gereke M, Jastrow H, Schulze-Osthoff K, et al. New insights into the molecular pathology of radiation-induced pneumopathy. Radiother Oncol. 2011;101:86–92. doi: 10.1016/j.radonc.2011.05.064. [DOI] [PubMed] [Google Scholar]
- 39.Johnston CJ, Williams JP, Okunieff P, Finkelstein JN. Radiation-induced pulmonary fibrosis: examination of chemokine and chemokine receptor families. Radiat Res. 2002;157:256–65. doi: 10.1667/0033-7587(2002)157[0256:ripfeo]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 40.Johnston CJ, Wright TW, Rubin P, Finkelstein JN. Alterations in the expression of chemokine mRNA levels in fibrosis-resistant and -sensitive mice after thoracic irradiation. Exp Lung Res. 1998;24:321–37. doi: 10.3109/01902149809041538. [DOI] [PubMed] [Google Scholar]
- 41.Antoniades HN, Neville-Golden J, Galanopoulos T, Kradin RL, Valente AJ, Graves DT. Expression of monocyte chemoattractant protein 1 mRNA in human idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 1992;89:5371–5. doi: 10.1073/pnas.89.12.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suga M, Iyonaga K, Ichiyasu H, Saita N, Yamasaki H, Ando M. Clinical significance of MCP-1 levels in BALF and serum in patients with interstitial lung diseases. Eur Respir J. 1999;14:376–82. doi: 10.1034/j.1399-3003.1999.14b23.x. [DOI] [PubMed] [Google Scholar]
- 43.Moore BB, Paine R, 3rd, Christensen PJ, Moore TA, Sitterding S, Ngan R, et al. Protection from pulmonary fibrosis in the absence of CCR2 signaling. J Immunol. 2001;167:4368–77. doi: 10.4049/jimmunol.167.8.4368. [DOI] [PubMed] [Google Scholar]
- 44.Inoshima I, Kuwano K, Hamada N, Hagimoto N, Yoshimi M, Maeyama T, et al. Anti-monocyte chemoattractant protein-1 gene therapy attenuates pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1038–44. doi: 10.1152/ajplung.00167.2003. [DOI] [PubMed] [Google Scholar]
- 45.Williams JP, Johnston CJ, Finkelstein JN. Treatment for radiation-induced pulmonary late effects: spoiled for choice or looking in the wrong direction? Curr Drug Targets. 2010;11:1386–94. doi: 10.2174/1389450111009011386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Travis EL. The sequence of histological changes in mouse lungs after single doses of x-rays. Int J Radiat Oncol Biol Phys. 1980;6:345–7. doi: 10.1016/0360-3016(80)90145-5. [DOI] [PubMed] [Google Scholar]
- 47.Pan LH, Ohtani H, Yamauchi K, Nagura H. Co-expression of TNF alpha and IL-1 beta in human acute pulmonary fibrotic diseases: an immunohistochemical analysis. Pathol Int. 1996;46:91–9. doi: 10.1111/j.1440-1827.1996.tb03584.x. [DOI] [PubMed] [Google Scholar]
- 48.Groves AM, Johnston CJ, Misra RS, Williams JP, Finkelstein JN. Whole-lung irradiation results in pulmonary macrophage alterations that are subpopulation and strain specific. Radiat Res. 2015;184:639–49. doi: 10.1667/RR14178.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Noti JD, Reinemann BC. The leukocyte integrin gene CD11c is transcriptionally regulated during monocyte differentiation. Mol Immunol. 1995;32:361–9. doi: 10.1016/0161-5890(94)00164-v. [DOI] [PubMed] [Google Scholar]
- 50.Sunderkotter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–7. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 51.Zhang J, Tachado SD, Patel N, Zhu J, Imrich A, Manfruelli P, et al. Negative regulatory role of mannose receptors on human alveolar macrophage proinflammatory cytokine release in vitro. J Leukoc Biol. 2005;78:665–74. doi: 10.1189/jlb.1204699. [DOI] [PubMed] [Google Scholar]
- 52.Hirsch S, Austyn JM, Gordon S. Expression of the macrophage-specific antigen F4/80 during differentiation of mouse bone marrow cells in culture. J Exp Med. 1981;154:713–25. doi: 10.1084/jem.154.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leenen PJ, Jansen AM, van Ewijk W. Murine macrophage cell lines can be ordered in a linear differentiation sequence. Differentiation. 1986;32:157–64. doi: 10.1111/j.1432-0436.1986.tb00568.x. [DOI] [PubMed] [Google Scholar]
- 54.Ding NH, Li JJ, Sun LQ. Molecular mechanisms and treatment of radiation-induced lung fibrosis. Curr Drug Targets. 2013;14:1347–56. doi: 10.2174/13894501113149990198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beach TA, Johnston CJ, Groves AM, Williams JP, Finkelstein JN. Radiation induced pulmonary fibrosis as a model of progressive fibrosis: Contributions of DNA damage, inflammatory response and cellular senescence genes. Exp Lung Res. 2017:1–16. doi: 10.1080/01902148.2017.1318975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paun A, Kunwar A, Haston CK. Acute adaptive immune response correlates with late radiation-induced pulmonary fibrosis in mice. Radiat Oncol. 2015;10:45. doi: 10.1186/s13014-015-0359-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chambers RC, Mercer PF. Mechanisms of alveolar epithelial injury, repair, and fibrosis. Ann Am Thorac Soc. 2015;12:S16–20. doi: 10.1513/AnnalsATS.201410-448MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Osterholzer JJ, Olszewski MA, Murdock BJ, Chen GH, Erb-Downward JR, Subbotina N, et al. Implicating exudate macrophages and Ly-6C(high) monocytes in CCR2-dependent lung fibrosis following gene-targeted alveolar injury. J Immunol. 2013;190:3447–57. doi: 10.4049/jimmunol.1200604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, et al. Ly6C(hi) monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med. 2011;184:569–81. doi: 10.1164/rccm.201010-1719OC. [DOI] [PubMed] [Google Scholar]
- 60.Baran CP, Opalek JM, McMaken S, Newland CA, O’Brien JM, Jr, Hunter MG, et al. Important roles for macrophage colony-stimulating factor, CC chemokine ligand 2, and mononuclear phagocytes in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176:78–89. doi: 10.1164/rccm.200609-1279OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gharaee-Kermani M, McCullumsmith RE, Charo IF, Kunkel SL, Phan SH. CC-chemokine receptor 2 required for bleomycin-induced pulmonary fibrosis. Cytokine. 2003;24:266–76. doi: 10.1016/j.cyto.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 62.Young LR, Gulleman PM, Short CW, Tanjore H, Sherrill T, Qi A, et al. Epithelial-macrophage interactions determine pulmonary fibrosis susceptibility in Hermansky-Pudlak syndrome. JCI Insight. 2016;1:e88947. doi: 10.1172/jci.insight.88947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229:176–85. doi: 10.1002/path.4133. [DOI] [PubMed] [Google Scholar]
- 64.Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Opalek JM, Ali NA, Lobb JM, Hunter MG, Marsh CB. Alveolar macrophages lack CCR2 expression and do not migrate to CCL2. J Inflamm (Lond) 2007;4:19. doi: 10.1186/1476-9255-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Christensen PJ, Du M, Moore B, Morris S, Toews GB, Paine R., 3rd Expression and functional implications of CCR2 expression on murine alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L68–72. doi: 10.1152/ajplung.00079.2003. [DOI] [PubMed] [Google Scholar]
- 67.Johnston CJ, Piedboeuf B, Baggs R, Rubin P, Finkelstein JN. Differences in correlation of mRNA gene expression in mice sensitive and resistant to radiation-induced pulmonary fibrosis. Radiat Res. 1995;142:197–203. [PubMed] [Google Scholar]
- 68.Kyrkanides S, Moore AH, Olschowka JA, Daeschner JC, Williams JP, Hansen JT, et al. Cyclooxygenase-2 modulates brain inflammation-related gene expression in central nervous system radiation injury. Brain Res Mol Brain Res. 2002;104:159–69. doi: 10.1016/s0169-328x(02)00353-4. [DOI] [PubMed] [Google Scholar]
- 69.Geiser T, Atabai K, Jarreau PH, Ware LB, Pugin J, Matthay MA. Pulmonary edema fluid from patients with acute lung injury augments in vitro alveolar epithelial repair by an IL-1beta-dependent mechanism. Am J Respir Crit Care Med. 2001;163:1384–8. doi: 10.1164/ajrccm.163.6.2006131. [DOI] [PubMed] [Google Scholar]
- 70.Broekman W, Amatngalim GD, de Mooij-Eijk Y, Oostendorp J, Roelofs H, Taube C, et al. TNF-alpha and IL-1beta-activated human mesenchymal stromal cells increase airway epithelial wound healing in vitro via activation of the epidermal growth factor receptor. Respir Res. 2016;17:3. doi: 10.1186/s12931-015-0316-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamamoto T, Eckes B, Mauch C, Hartmann K, Krieg T. Monocyte chemoattractant protein-1 enhances gene expression and synthesis of matrix metalloproteinase-1 in human fibroblasts by an autocrine IL-1 alpha loop. J Immunol. 2000;164:6174–9. doi: 10.4049/jimmunol.164.12.6174. [DOI] [PubMed] [Google Scholar]
- 72.Leung LY, Tian D, Brangwynne CP, Weitz DA, Tschumperlin DJ. A new microrheometric approach reveals individual and cooperative roles for TGF-beta1 and IL-1beta in fibroblast-mediated stiffening of collagen gels. FASEB J. 2007;21:2064–73. doi: 10.1096/fj.06-7510com. [DOI] [PubMed] [Google Scholar]
- 73.Aumiller V, Balsara N, Wilhelm J, Gunther A, Konigshoff M. WNT/beta-catenin signaling induces IL-1beta expression by alveolar epithelial cells in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2013;49:96–104. doi: 10.1165/rcmb.2012-0524OC. [DOI] [PubMed] [Google Scholar]
- 74.Piguet PF, Vesin C, Grau GE, Thompson RC. Interleukin 1 receptor antagonist (IL-1ra) prevents or cures pulmonary fibrosis elicited in mice by bleomycin or silica. Cytokine. 1993;5:57–61. doi: 10.1016/1043-4666(93)90024-y. [DOI] [PubMed] [Google Scholar]
- 75.Groves AM, Johnston CJ, Misra RS, Williams JP, Finkelstein JN. Effects of IL-4 on pulmonary fibrosis and the accumulation and phenotype of macrophage subpopulations following thoracic irradiation. Int J Radiat Biol. 2016;92:754–65. doi: 10.1080/09553002.2016.1222094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Timmerman RD, Herman J, Cho LC. Emergence of stereotactic body radiation therapy and its impact on current and future clinical practice. J Clin Oncol. 2014;32:2847–54. doi: 10.1200/JCO.2014.55.4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Bone marrow engraftment validation in CCR2−/− chimeric mice. Engraftment was verified via flow analysis 8 weeks after generation of chimeras by identifying CD45.1 donor-derived cells in blood samples collected from CD45.2 recipient mice.
Fig. S2. Lung histology from CCR2−/− chimeric mice. Gomori trichrome-stained lung sections were prepared between 12 and 22 weeks after 0 or 12.5 Gy thoracic irradiation (n = 3–4 mice/treatment group). Original magnification 200× (inset 400×).
Fig. S3. Gating scheme for characterization of pulmonary macrophage subsets. In CCR2−/− chimeric mice, between 12 and 18 weeks after 0 or 12.5 Gy thoracic irradiation, CD45+ cells were enriched from lung digests using MACS and analyzed by flow cytometry. Following doublet and dead cell discrimination, CD45+, Ly6G- cells were gated. CD11b and CD11c were used to discern alveolar (AMs; CD11b intermediate, CD11c+), interstitial (IMs; CD11b+, CD11c+) and infiltrating (Inf; CD11b+, CD11c-) macrophages for analysis of population dynamics. To analyze resident macrophages, CD11c+ cells were gated, and the percentages of this population expressing Ly6C, CD206 and F4/80 were determined.
Fig. S4. Lung histology from C57Bl/6 and IL-1β−/− mice. Gomori trichrome-stained lung sections were prepared 32 weeks after 0 or 12.5 Gy thoracic irradiation. Original magnification 200×.