

Abstract
Mass spectrometry (MS) based footprinting is an emerging approach to study protein structure. Because integral membrane proteins are difficult targets for conventional structural biology, we recently developed a MS footprinting method to probe membrane protein/drug interactions in live cells. This method can detect structural difference between apo and drug-bound states of membrane proteins, with the changes inferred from MS quantification of the cysteine modification pattern, generated by residue-specific chemical labeling. Here, we describe the experimental design, interpretation, advantages, and limitations of using cysteine footprinting by taking as an example the warfarin interaction with vitamin K epoxide reductase, a human membrane protein. Compared with other structural methods, footprinting of proteins in live cells produces structural information for the near-native state. Knowledge of cellular conformational states is a necessary complement to the high-resolution structures obtained from purified proteins in vitro. Thus, the MS footprinting method is broadly applicable in membrane protein biology. Future directions include probing flexible motions of membrane proteins and their interaction interface in live cells, which are often beyond the reach of conventional structural methods.
Graphical Abstract
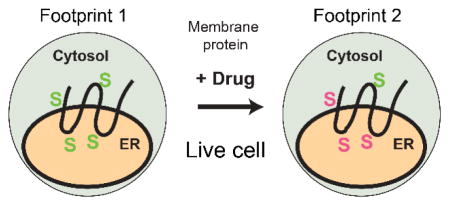
Integral membrane proteins participate in almost all physiological processes and are targets of more than half of the drugs currently in use. These proteins often undergo large conformational changes to carry out ion and molecular transport, catalysis, and signal transduction. Owing to their hydrophobic and dynamic nature, structure determination by conventional methods remains challenging, especially for membrane proteins of eukaryotic origin. To date, structures of only approximately 100 human membrane proteins have been determined. To complement the conventional high-resolution methods (x-ray crystallography, cryo-EM, and NMR), mass spectrometry (MS) is emerging as a highly useful tool to study membrane protein structures1,2.
One of the most exciting developments in MS is a probe of membrane protein structure in a cellular environment3–5. The dynamic motions of these proteins during various cellular processes are beyond the reach of traditional structural methods, which instead address purified proteins in vitro. In addition, the native structure and interaction of membrane proteins are often lost when they are removed from a membrane environment and purified in a detergent solution6. Membrane proteins are often unstable in detergent micelles, a problem that severely limits conventional structural methods. A common practice in membrane protein crystallography, for example, is to screen the stability of numerous homologous proteins or protein constructs in various detergents; most membrane proteins, however, are not pursued further because they fail this detergent test. In contrast, footprinting of membrane proteins in live cells is not subject to this requirement. Once the protein footprints are generated, detergent stability of this protein during subsequent sample preparation is no longer a concern, unlike for other structural methods.
Here we describe in detail the design, methodology, interpretation, and limitation of a footprinting method in live cells, illustrated by the use of residue-specific labeling to probe the drug interaction of a human membrane protein. This powerful tool is broadly applicable to study the motions and interactions of integral membranes proteins in live cells.
MATERIALS AND METHODS
Stable Cell lines
The 293T-REx cells7 (gift from S. Wanrooij) were derived from HEK293 and used for constructing stable cell lines. The 293T-REx cells were co-transfected by plasmids pOGG44 and pcDNA5/FRT/TO TOPO (ratio 10:1). After 48 h, the stable cell lines were selected by blasticidin and hygromycin, and the clone could be isolated after three weeks. The stable cell line was confirmed for the expression of target protein by western blot.
In-gel protease digestion and LC-MS/MS
RESULTS
Overview of MS-based footprinting of membrane proteins in live cells
MS-based footprinting is carried out in the following steps. First, different structural states, (unbound and ligand-bound states) of a membrane protein are generated in live cells. Subsequently, proteins at these states are compared by labeling with a cell-compatible chemical reagent. The modified protein is enriched in vitro by affinity purification, subjected to electrophoresis, and in-gel digested into peptide fragments for liquid chromatography and tandem MS (LC-MS/MS) analysis. Since maintaining the native protein is not necessary in these subsequent steps, the trial-and-error process (e.g., detergent optimization) required to purify a difficult membrane protein is bypassed. Consequently, sample preparation for MS becomes a routine process, and multiple protein samples can be analyzed in parallel with fast turnaround. Thus, this method can quickly detect the near native structural state of integral membrane proteins in live cells, offering an invaluable complement to high-resolution structural methods.
Experimental design
MS footprinting in live cells can provide critical structural insight for a difficult membrane protein. To illustrate, we selected a human membrane protein, vitamin K epoxide reductase (hVKOR), that is unstable in absence of lipids10. hVKOR was only identified by genetic analyses11,12, because the protein could not be isolated biochemically owing to its rapid loss of enzymatic activity during further purification in many detergents10. hVKOR is the target of warfarin, an anticoagulant used by 1% of the US population to treat and prevent thromboembolic diseases. Despite six decades of clinical experience with warfarin, there has not been an effective approach to probe the warfarin interaction with hVKOR in vivo. Thus, we have developed an MS-based footprinting method that employs isotope-coded residue-specific chemical labeling.
Our premise is that binding of warfarin to hVKOR will protect certain cysteine residues from the modification by N-ethylmaleimide (NEM), a membrane-permeable probe13,14 that irreversibly labels cysteines (Figure 1). We then immunopurified hVKOR, reduced any cysteine that was unprotected in the cells, and labeled them with deuterated NEM (NEM-d5). NEM-d5 thus serves as an internal control (via isotopic encoding) for quantitative LC-MS/MS, by which the modification level of each cysteine can be determined. The cysteine footprinting pattern has allowed us to resolve the binding topology of warfarin to hVKOR9.
Figure 1. Scheme of MS footprinting of hVKOR in live cells.

Cysteines in the native protein are footprinted by NEM (green), and all the unmodified cysteines are later modified by NEM-d5 (red). The differential isotope labeling gives a quantified footprinting pattern. See text for more explanation of each experimental step.
Experimental protocol
The footprinting experiments are designed and carried out in the following order (Figure 1). 1) The use of the stable cell lines allows the hVKOR protein to be expressed in a large quantity, which is important to enhance the signal of modified peptides in mass spectra. 2) The cells are either untreated or pretreated with different ligands to allow their binding to hVKOR. 3) The cells are labeled by NEM and lysed. 4) The NEM reaction is quenched by DTT. 5) Both NEM and DTT are removed from the cell lysate by a desalting column. 6) The hVKOR protein is affinity purified. 7) The purified protein is further enriched by trichloroacetic acid (TCA) precipitation. 8) The protein is subjected to electrophoresis (Figure 2A). 9) The protein is reduced and in-gel labeled with isotopically encoded NEM-d5. 10) The protein is in-gel digested into peptide fragments. 11) The extracted peptides are separated and analyzed by liquid chromatography tandem MS (LC-MS/MS). 12) The MS data are computationally analyzed to identify the peptides and locate the isotope-encoded Cys-containing peptides (Figure 2B–D).
Figure 2. Quantification analysis of cysteine modification level.

A. Reducing SDS-PAGE serves as an additional purification step after the hVKOR protein is affinity purified. The sliced gel bands (boxes) are used for in-gel isotope labeling and digestion. B. LC/MS/MS analysis of a peptide containing Cys43, which is modified by NEM (green spheres) and NEM-d5 (red). C. Peak areas of the modified peptides are used for calculating percentage of NEM-d5 modification. D. Analysis results are displayed as a bar graph.
1. NEM labeling
Each MS experiment requires cells from five 15-cm plates. Pass the cells into five plates and grow in complete DMEM medium for 24 h. Add 200 ng/mL doxycycline to induce protein expression for additional 24 h.
Add warfarin to the growth medium to give a final concentration of 5 μM and incubate for 4 h at 37 °C in a CO2 incubator. In a parallel experiment, add 4-hydroxycoumarin (4-HC), another hVKOR inhibitor, to give a final concentration of 1 mM. (In control experiments, the cells are either untreated or incubated with 10 mM DTT (final concentration) for 20 min.)
Remove the growth medium. Wash the cells on plate with 20 mL ice-cold phosphate-buffered saline (PBS) buffer.
Label the cells with NEM. To each 15-cm plate, add 1.8 mL buffer13,15 containing 20 mM NEM, 1% Triton X-100, 50 mM Tris-HCl pH 7.5, 150 mM NaCl, and protease inhibitor cocktail (Roche). Shake the plates until the cells are fully lysed. Transfer the lysate to two 1.5 mL Eppendorf tubes and incubate on ice for 20 min to allow the reaction with NEM to go to completion.
Add 100 μL 1 M DTT (final concentration ~ 100 μM) to quench the NEM reaction.
Special note 1
We tested an alternative protocol in which NEM labeling is performed before solubilizing the cell membrane by using triton X-100. The steps 4–5 described above are changed to the following steps. In addition, removing the NEM and DTT becomes unnecessary; one can skip the desalting step (next section) and directly continue to affinity purification.
Label live cells with NEM. Add 15 mL PBS buffer containing 20 mM NEM (at room temperature) to the plates. Incubate the plates for 10 min in a 37°C incubator.
Remove the labeling buffer, and add 20 mL ice-cold PBS. Detach the cells by scraping. Transfer to a 50 mL Falcon tube. Collect the cells by centrifuging at 200 g for 5 min.
Wash the cells twice with the PBS buffer. During each wash step, resuspend the cell pellet in 20 mL ice-cold PBS and centrifuge at 200 g for 5 min.
Remove the PBS buffer. To one plate of cells, add 1.8 mL lysis buffer containing 1% triton X-100, 50 mM Tris-HCl pH 7.5, 150 mM NaCl, and protease inhibitor cocktail. Incubate the lysis mixture for 20 min.
Divide the cell lysate into 1.5 mL tubes. Centrifuge at 14,000 g for 15 min. Collect the supernatant into a new 15 mL Falcon tube.
Efficient NEM labeling of membrane-buried cysteines requires membrane solubilization by detergent
The following data are interpreted based on the crystal structure of a bacterial VKOR homolog, which indicate that human VKOR forms a bundle of four transmembrane helices (TM) with a luminal loop/helix domain between TM1 and TM2 (HL1-2). Cysteines buried in the membrane are hard to deprotonate and, therefore, are less reactive to NEM13,14. Therefore, we carried out the NEM labeling with or without membrane solubilization by triton X-100 at the same time. We find that the more exposed cysteines in hVKOR, including Cys43/Cys51 and Cys96 in loop regions and Cys132/Cys135 at the end of a transmembrane helix show similar modification levels (Figure 3). In contrast, we see large differences in Cys16 and Cys85, two residues that are deeply buried in the membrane. Without membrane solubilization by triton X-100, these cysteines are not efficiently labeled by NEM. In later steps, these free cysteines are exposed after protein denaturation and labeled by the NEM-d5, increasing the modification levels of Cys16 and Cys85. Thus, we chose to carry out NEM labeling in presence of triton X-100 because this method better represents the live-cell state of the two membrane-buried cysteines, and it does not affect the state of the other five cysteines. With the increase of NEM modification efficiency, this method also affords a better contrast to detect a warfarin-induced change.
Figure 3. Control experiments to identify membrane-buried and disulfide-bonded cysteines.
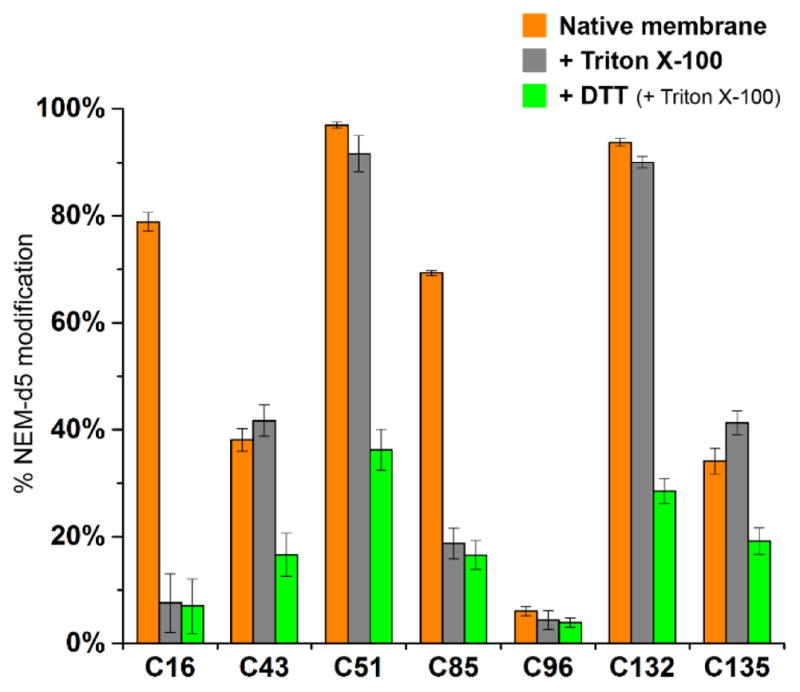
Cys16 and Cys85 are membrane buried. Cys43, Cys51, Cys132, and Cys135 can form disulfide bonds9. Native membrane: NEM labeling was carried out with intact cell membrane, as described in Special Note 1. Triton X-100: membrane solubilization and NEM labeling were carried out at the same time, as described in the Experimental Protocol. DTT: the cysteines in hVKOR were reduced in cells before the NEM labeling.
Special note 2
Because hVKOR rapidly loses activity during purification in detergents, the effect of using triton X-100 on the protein structure is a concern. There are several pieces of evidence showing that the protein conformation is largely maintained during the NEM labeling in triton X-100. First, among the seven cysteines used for footprinting, five of the exposed cysteines are not affected by triton X-100 treatment (Figure 3). In contrast, the NEM accessibilities of Cys43 (HL1-2) and Cys135 (TM4) are changed upon warfarin binding (see Figure 5 below). If triton X-100 had denatured the hVKOR structure, more drastic changes would occur to the cysteine states than with warfarin; such changes, however, are not observed. Furthermore, the denaturation should occur progressively, but using longer labeling time (40 min and 60 min) did not change significantly the cysteine modification extent9. Second, NEM footprinting with triton X-100 allowed the topology of warfarin binding to be deduced (Figure 5), affording a topology that fits well with the relative inhibitory activity of resistant mutants9. Third, with the same labeling protocol, the process of active VKOR catalysis and active electron transfer can be captured in live cells9. And fourth, adding triton X-100 and NEM can be viewed as a quenching step, similar to that in a standard enzymatic assay, because the VKOR catalysis or warfarin binding has already occurred in live cells. Taken together, the NEM labeling in triton X-100 appears to probe instantaneously the state of cysteines before the protein structure is disrupted.
Figure 5. Determination of the warfarin-binding topology.
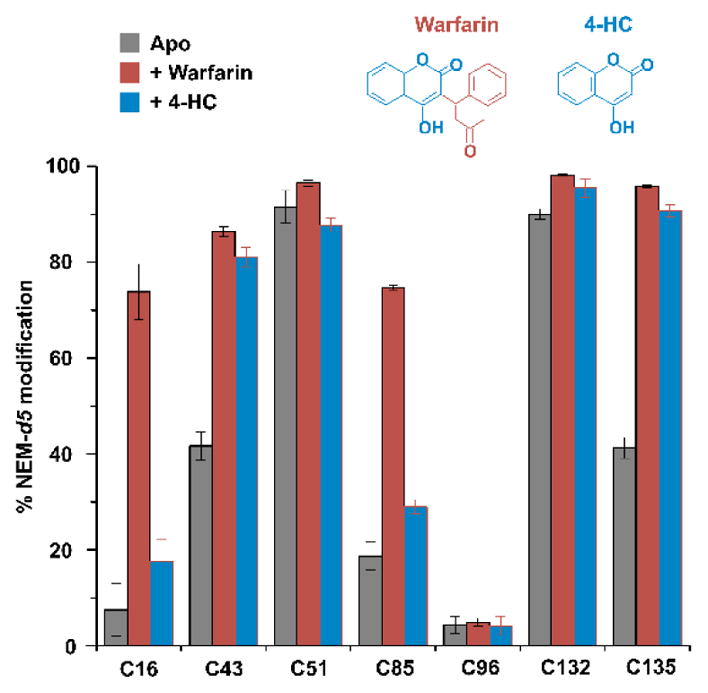
Cell treatment with warfarin and 4-hydroxycoumarin (4-HC) before the NEM labeling generates different cysteine modification patterns. Inset, chemical structures of warfarin and 4-HC.
2. Sample cleanup
The cell lysate after the NEM labeling and DTT quenching needs to be cleaned up by a desalting column, because NEM and DTT interfere with the subsequent affinity purification of hVKOR. The following steps are conducted at 4 °C or on ice to prevent protein degradation.
Solvate 40 g Sephadex G-25 resin (Sigma). De-gas and pack into a large desalting column (2 cm X 45 cm). Equilibrate the column in buffer A (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, and 1 mM PMSF) at a flow rate of 1.5 mL/min for at least 2 h.
Centrifuge the cell lysate for 15 min at 12,000 g. Transfer the supernatant (~ 11 mL) to a new 15 mL Falcon tube. Add phenol red to a final concentration of 0.1 μg/mL as an indicator to the desalting process.
Load the supernatant onto the G-25 desalting column. Elute with buffer A at a flow rate of 1.5 mL/min. After 40 mL flow through, collect 2 mL fractions for the next 40 mL of elution. Wash the column with an additional 180 mL buffer A to re-equilibrate.
Take 5 μL aliquots from each fraction and pipet onto filter paper. Dry briefly and stain with Coomassie blue solution (1 mg/mL Coomassie brilliant blue R250, 50% v/v methanol, 10% v/v acetic acid) for 1 min. De-stain the filter paper by flushing with water. The fractions containing the protein show blue color. Combine these fractions and divide into two 15 mL Falcon tubes.
3. Sample enrichment by affinity purification and TCA precipitation
Equilibrate the anti-flag M2 resin (Sigma A2220). Pipet 100 μL anti-flag M2 resin into a 1.5 mL tube, and prepare two tubes for each MS sample. Add 1 mL buffer A to wash the resin. Invert the tubes 4–6 times and centrifuge at 6,000 g for 1 min. Discard the supernatant. Repeat the wash three times.
Add 100 μL equilibrated resin to each 15 mL tube containing the protein fractions.
Incubate the mixture at 4 °C overnight (12–16 h) with rotation of the tubes.
Collect the resin by centrifugation at 8,000 g for 1 min. Carefully aspirate and discard the supernatant.
Wash the resin with 1 mL buffer A three times.
Elute the hVKOR protein. Add 350 μL elution buffer (0.1 M glycine-HCl, pH 2.8) per tube. Incubate for 5 min at room temperature with gentle rotation. Centrifuge at 8,000 g for 1 min at room temperature. Transfer the supernatant to a new 1.5 mL tube for TCA precipitation.
Add 70 μL of 1 M Tris-HCl, pH 7.5 to the 350 μL supernatant.
Add 350 μL 30% TCA (w/v, sigma T9159). Mix thoroughly and incubate on ice for 30 min to precipitate the protein.
Centrifuge 20,000 g for 15 min at 4 °C. Remove the supernatant.
Wash the pellet with 1 mL cold acetone. Centrifuge 20,000 g for 10 min at 4 °C. Remove the supernatant. Repeat the wash. Optional: the experiment can be temporarily stopped at this step by storing the protein pellet in 1 mL cold acetone at −20 °C.
Air dry the pellet for 5–10 min at room temperature.
Re-suspend the pellet in 40 μL buffer containing 100 mM Tris-HCl pH 8.5, 6 M urea, 1% SDS. Vortex rigorously for 5 min and centrifuge 20,000 g for 1 min. Repeat 6 times until the pellet is fully dissolved.
4. In-gel isotope labeling
Add 5 μL SDS-loading buffer and 5 μL 1 M DTT to each tube and mix well. Load the sample on a 15% lab-made SDS-PAGE gel. Run the electrophoresis at 120 V for 1.5 h.
After the electrophoresis, transfer the gel to a clean box, stain the gel for 2 min by Coomassie blue solution at room temperature. Note: do not boil the staining solution.
Destain the gel by deionized water at room temperature with shaking. Change water every 30 min, and repeat five times. After the sixth time, put the gel in deionized water and shake overnight at room temperature.
Slice the gel band of hVKOR protein into 0.5 mm cubic pieces. Transfer to a 0.5 mL LoBind microcentrifuge tube (Eppendorf). (Optional: the gel pieces can be stored in 100 μL deionized water at 4 °C for two weeks.)
Aspirate the deionized water.
Add 350 μL of neat acenonitrile (MS grade). Incubate tubes at room temperature for 10 min until gel pieces shrink and appear white. Centrifuge at 14,000 g for 1 min and remove the liquid. Repeat acenonitrile wash one more time. Air dry for 10 min.
Add 100 μL TCEP solution (5 mM TCEP , 50 mM Tris-HCl pH 7.5) to the tube. Centrifuge at 14,000 g for 1 min. Incubate the tube at 56 °C for 30 min. Note: the dry gel pieces swell and absorb liquid. If the 100 μl solution is fully absorbed, add more TCEP solution.
Cool the tube to room temperature. Aspirate the extra TCEP solution.
Repeat step 6.
Add 100 μL NEM-d5 solution (10 mM NEM-d5, 50 mM Tris-HCl pH 7.5) to label the TCEP-reduced cysteines. Centrifuge at 14,000 g for 1 min. Incubate for 1 h at room temperature to allow the reaction to go to completion.
Aspirate the deuterated-NEM solution,
Repeat step 6.
Digest in-gel and conduct LC-MS/MS analysis9. Optional: the gel pieces can be stored at −20 °C for a few weeks prior to analysis.
Special note 3
Cysteines in reduced form can react with residual acrylamide in the gel, producing propionamide derivatized cysteines. To address this complication, we have analyzed the data of the apo- and warfarin-bound hVKOR by including in the search (MASCOT search engine) propionamide-modified cysteine residues. The search results show that the highest extent of this modification is on Cys43 and Cys51. The modification extents on the other five cysteines are much lower in the ions score and in the number of hits of the modified peptides. Therefore, we compared the Cys43- or Cys51-containing peptides with the highest propionamide modification to the sum of the same peptides modified by NEM and NEM-d5 (Figure 4). The propionamide modification is ~1.1%–2.9% of the NEM+NEM-d5 modification. This low level of modification does not affect the overall conclusion of the footprinting experiments because the binding of warfarin increases the NEM-d5 modification level of Cys43 from 42% to 86% (Figures 4 and 5).
Figure 4. Propionamide modification of cysteines represents a minor fraction of NEM and NEM-d5 modification.
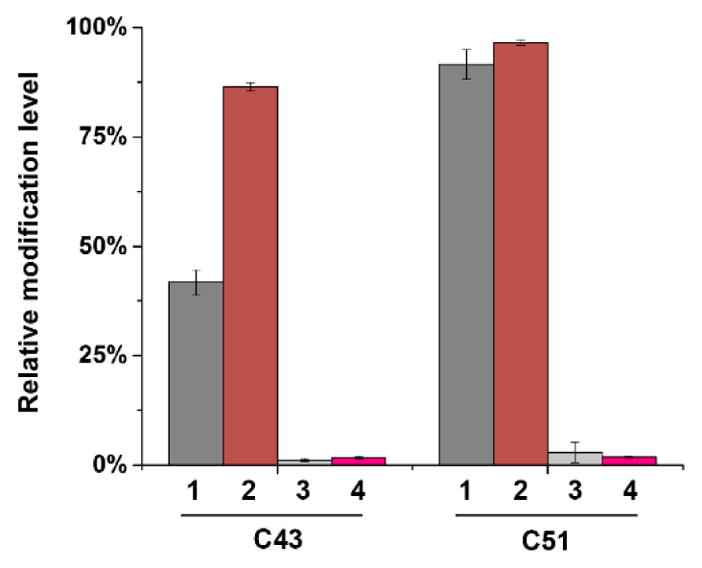
The data shown for Cys43 and Cys51 are: 1, NEM-d5 modification extent of apo-hVKOR protein, i.e., NEM-d5/(NEM+NEM-d5); 2, NEM-d5 modification extent of warfarin-treated hVKOR; 3, propionamide modification extent of apo-hVKOR; and 4, propionamide modification extent of warfarin-treated hVKOR.
Interpretation of ligand-binding interactions from cysteine footprints
The cysteines in the hVKOR protein are sequentially labeled in two steps by the isotopically encoded NEM to give “light” and “heavy” samples. The first NEM modification is on the native protein (light sample). Subsequently, the protein is denatured and reduced for NEM-d5 labeling to capture all the cysteines not modified by NEM (heavy sample). The relative modification levels of NEM and NEM-d5 on each cysteine are calculated as the percentage of NEM-d5 modification (i.e., NEM-d5/(NEM + NEM-d5) where the “NEM” terms refer to integrated peak intensities. In a previous paper9, we defined this percentage as the apparent oxidation fraction, which is essentially a reverse measure of the NEM modification extent of a cysteine at the first labeling step. The NEM-d5 percentages obtained for each cysteine, with higher percentage indicating lower cysteine reactivity in the native protein, collectively give the footprinting data.
The cysteine reactivity in the apo- and warfarin-bound protein is affected by several factors. The following sections describe these factors and the control experiments required to interpret properly the cysteine footprinting results.
1. Factors affecting the cysteine modification extent in absence of warfarin
Because hVKOR is an ER membrane protein, cysteines facing the ER lumen undergo a redox equilibrium. A cellular fraction of each of these cysteine is in the disulfide-bonded form, which does not react with NEM. To address this complication, we reduced the native hVKOR protein with DTT before the NEM labeling. This control experiment identifies the disulfide-bonded cysteines that become reactive to NEM after being reduced. Comparison of the DTT-treated and untreated samples (Figure 3) shows that, in hVKOR, Cys43, Cys51, Cys132, and Cys135 form disulfide bonds, whereas Cys16, Cys85, and Cys96 stay in reduced form because these three cysteines are either in the cytosol or are membrane-buried.
The cysteines buried in the ER membrane are difficult to deprotonate to give a thiolate (i.e., SH → S−) that is reactive to NEM13,14. Therefore, we compared the cysteine modification levels with and without membrane solubilization by triton X-100 (Figure 3). We find that, only with triton X-100 treatment, Cys16 and Cys85 become exposed to NEM labeling, indicating that these cysteines are buried in the membrane.
Despite DTT reduction or triton X-100 solubilization, we find a relatively high fraction of Cys16 and Cys85 that cannot be modified by NEM (Figure 3). This is because these cysteines are not only membrane-buried but also are partially buried in the protein structure, which prevents further NEM modification. Alternatively, these cysteines can be in a flexible region that switches between exposed and buried states. These two possibilities, however, cannot be distinguished by any control experiments we have considered.
2. Deduce the warfarin-binding orientation from the comparison of cysteine modification patterns
Warfarin treatment significantly changes the modification levels of Cys16, Cys43, Cys85 and Cys135 in hVKOR, but not those of Cys51, Cys96, Cys135 (Figure 5). These changes in cysteine modification are due to at least three different factors: 1) warfarin either directly interacts with a cysteine residue or sterically protects it from NEM labeling, 2) the warfarin binding induces a conformation change so that the cysteine becomes more buried (or less flexible) in the membrane or in the protein structure, and 3) warfarin induces the formation of disulfide bond(s).
To distinguish these possibilities, we treated the cells with warfarin and then reduced with DTT before the NEM labeling. Remarkably, we found that the DTT treatment in the warfarin-binding state barely changes the labeling efficiency of all cysteines, including Cys51 and Cys132, which are known to form disulfide bonds in the apo-protein (Figure 6). Although it is known that the Cys51-Cys132 disulfide is preferred for warfarin binding9, this disulfide bond should exist, but it cannot be reduced by DTT. Therefore, warfarin has induced a very tight conformation for VKOR, one that protects all the reducible cysteines (Cys43, Cys51, Cys132, and Cys135; Figures 3 and 6) from reacting with DTT.
Figure 6. Warfarin binding protects cysteines from reacting to DTT.
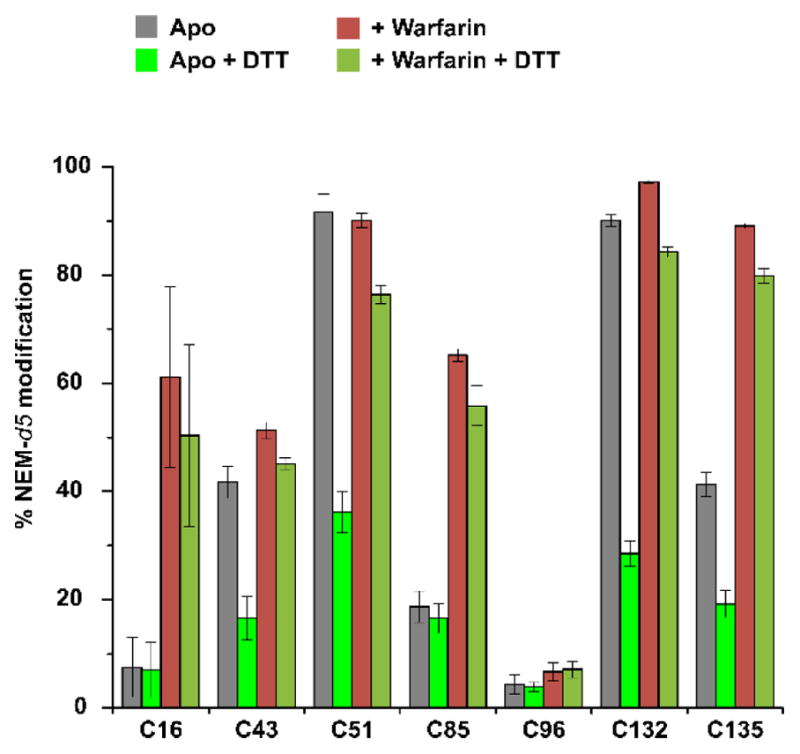
The cells are either untreated or treated with warfarin, and subsequently reduced with DTT.
To identify which cysteines are directly involved in warfarin binding, we systematically mutated these cysteines. We found that, compared to the wild-type hVKOR, the Cys51Ala, Cys132Ala and Cys135Ala mutations changed the modification pattern after warfarin treatment, whereas other cysteine mutations did not have this effect (Figure 7). Thus, these three cysteines seem to be directly involved in warfarin binding; Cys135 may form an hydrogen bond with warfarin9, and the Cys51-Cys132 disulfide may interact with warfarin through van der Waals forces. On the other hand, the warfarin-induced protection of other cysteines (Cys43, Cys16, and Cys85) from NEM labeling seems to result from a conformational or flexibility change in the hVKOR structure when warfarin is bound.
Figure 7. Identification of warfarin-interacting cysteines by mutagenesis.

The different modification patterns between warfarin-treated and untreated samples are shown for wild-type hVKOR and for each cysteine mutant. Mutations of Cys51Ala, Cys132Ala, and Cys135Ala change the warfarin-induced footprinting pattern compared to wild-type hVKOR (*P < 0.001). In contrast, Cys43Ala, Cys16Ala, Cys85Ala, and Cys96Ala mutations produce similar warfarin-induced footprinting pattern as the wild type hVKOR.
To determine the topology of warfarin binding, we compared warfarin with 4-hydroxycoumarin (4-HC), a compound that retains the 4-HC ring of warfarin but lacks its phenyl butanone side group (Figure 5). When cells were treated by 4-HC, Cys16 and Cys85 in the transmembrane region of hVKOR are not affected (Figure 5). In contrast, Cys43 and Cys135 at the membrane interface are affected by both compounds. Thus, warfarin should bind in an orientation with its 4-HC group facing the membrane surface and its side group facing the transmembrane region (Figure 8). This binding orientation is supported by biochemistry experiments that measure hVKOR’s relative resistance to these two drugs, and by molecular dynamics simulation based on the structure of a bacterial VKOR homolog9,18,19.
Figure 8. Model of warfarin binding deduced from MS footprinting.
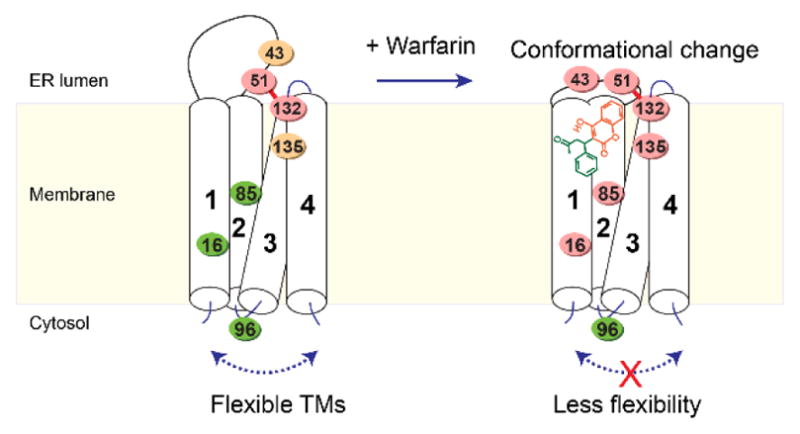
The color changes on the cysteines (spheres) indicate the changes in their NEM accessibility (green: high accessibility; orange: medium; red: low) upon warfarin binding. The 4-HC group of warfarin is shown in red, and its side group in green.
Taken together, the MS footprinting data suggest that warfarin binds close to Cys51, Cys132, and Cys135 with its 4-HC ring facing up (Figure 8). The binding induces a conformational change in the ER luminal domain containing Cys43, thereby burying Cys43 and protecting it from NEM modification. The tightly bound warfarin also reduces the high flexibility of the TM region, rendering Cys16 in TM1 and Cys85 in TM2 less accessible to NEM.
3. General guidelines to the design and interpretation of MS footprinting
Most of factors and control experiments described above are generally applicable to the interpretation of MS footprinting data. 1) The ligand-binding topology can be deduced by comparing the footprinting patterns of ligands with different chemical groups, such as warfarin and 4-HC (Fig. 4). 2) Target residues directly involved in the ligand binding can be identified by mutation, which should prevent the binding and change the entire modification pattern (Fig. 6). If not, the apparent change in a residue’s modification level may be due to a protein conformational change induced by the ligand binding. 3) The apparent modification level of the target residue can be affected by its interaction with another protein residue, either through a covalent or noncovalent bond (e.g., a disulfide bond or a salt bridge). This complication can be resolved by changing experimental conditions to disrupt such a bond (e.g., reducing the disulfide bond; Fig. 5), or by substituting the pairing residue by mutation if it can be identified from a high-resolution structure. 4) Membrane-buried residues can be identified by comparing the modification content with or without detergent solubilization of the membrane. 5) The footprinting results can be integrated with in silico modeling9, such as homology modeling, ligand docking, and molecular dynamics simulation, to generate a more thorough structural understanding.
Key advantages of MS footprinting in live cells and comparison to previous approaches
The dynamic structural motions of membrane proteins usually determine their function. Different structural states of the proteins generally coexist as an equilibrium mixture in live cells. MS footprinting can detect this mixed cellular state and the equilibrium shift induced by ligand binding or protein interaction. In contrast, high-resolution crystal or cryo-EM structures only capture still images of the individual conformational states. The high-resolution structures, however, still provide the best basis to interpret the MS footprints, although footprinting can be done in the absence of a high-resolution structure.
MS footprinting infers structural information from the accessibility of protein residues probed by a chemical reagent. The resolution is at the peptide or even the amino-acid residue level, a middle ground between high-resolution methods and traditional biochemical and spectroscopic methods. Owing to the complications mentioned above, MS data are less certain in the absence of a three-dimensional structure. On the other hand, conventional structural biology methods are unable to study the membrane proteins in live cells. Even maintaining the lipid condition is a large obstacle because the lipid vesicles hinder crystal packing and interfere with NMR owing to increases in overall size and tumbling time. Thus, integration of MS footprinting with the high-resolution structural methods is the best approach.
Nevertheless, determining the ligand-binding structures of a membrane protein by crystallography or cryo-EM is often not straightforward. The ligand can be instead directly applied to live cells for protein footprinting, thereby overcoming an obstacle of conventional structural studies. Combined with in silico modeling, MS footprinting should find useful applications in drug development.
A large advantage of footprinting in live cells is that purification of membrane proteins in detergent solution can be avoided. Footprinting, thus, is much simpler and more direct than high-resolution methods, which, even with a detergent-stable protein, are impractical to apply when many structures of homologs and functional variants are needed. The requirement for purification in detergent also hinders the application of fluorescence, NMR, single-molecule spectroscopy, and electron paramagnetic resonance (EPR) to follow structural motions of membrane proteins. Compared to most of these methods, an additional advantage of MS footprinting is its capability of analyzing multiple chemical modification sites in parallel, with accurate quantification and high sensitivity to map structural motions.
Residue-specific labeling (e.g., NEM to cysteines) generally employs irreversible modification that is not disturbed during the protein enrichment from a cell mixture. In contrast, footprinting with labile or reversible labeling, such as hydrogen-deuterium (HD) exchange, may not survive the sample preparation prior to MS analysis. HD exchange, however, offers a broad-based modification scheme involving the protein backbone, providing higher coverage compared to residue-specific labeling. A compromise that includes the broad-coverage feature of HD exchange is to use irreversible broad-based labeling, which will certainly rely on free radicals or carbenes20 that react with a range of amino acids. Protein oxidation by the OH• radical has been applied both to purified membrane proteins21 and to footprinting in E. coli cells5. Compared to the use of free radicals, residue-specific labeling has the advantage of ready application of isotope encoding, a powerful method that confirms the peak identification in a complex peptide mixture and, with almost identical ionization efficiency of the light and heavy peptide, provides more accurate quantification of the modification level. With isotope encoding, we observed excellent reproducibility in the NEM labeling experiments.
Limitations
The assumption for all chemical labeling experiments is that the structural integrity of the target protein is preserved during the chemical modification. Because this assumption may not always hold true, additional biochemical or structural analyses are required to support the MS footprinting results. In addition, the chemical labeling should not disturb cell pathways affecting the conformation of the target membrane protein (e.g., a protein involved in a signal transduction pathway). Another complication is the use of detergent during the chemical labeling, as discussed earlier. Nevertheless, if in-cell labeling occurs sufficiently fast, an instantaneous state of the protein conformation may be captured (to give a “snapshot” of the protein) preempting any adverse effects in protein conformation owing to the modification.
A significant technical challenge to the MS footprinting of membrane proteins is obtaining high sequence coverage of the hydrophobic protein and the digest. Clearly, to obtain a footprint, a residue must be within the sequence coverage. A 20 30% sequence coverage in the transmembrane region, however, is typically realized according to the literature22–24, and, although adequate in proteomics, is inadequate in footprinting. To overcome this problem, we recently developed an optimized in-gel protease digestion method to fragment effectively and preserve hydrophobic peptides9. This method allows quantitative analysis of all the cysteines in hVKOR, four of which are located on transmembrane helices, and generated more than 90% sequence coverage including over the transmembrane region.
The extent or resolution of residue-specific footprinting labeling (e.g., NEM, GEE) depends on the amino acid composition of a particular membrane protein. For example, NEM labeling requires a protein to contain several cysteines. The seven cysteines in hVKOR are sufficient to provide a coarse-grained footprint to deduce the warfarin-binding topology. Better footprinting coverage, however, is obviously desired for this approach to be broadly applicable. Therefore, alternative cell-compatible methods, such as specific labeling on charged residues25, should be tested in the future. In addition, new chemical probes should be developed to provide broader footprinting coverage20,26, especially at the membrane-buried region of the protein. A few of these complementary labeling methods can be combined to provide a good coverage to study the protein structural changes.
Perspectives
Many chemical reagents have been used in MS structural proteomics, offering a rich source to identify those compatible with footprinting in cells. The chemical reaction should be selected to occur under mild conditions (e.g., at near neutral pH and without organic solvent) to maintain the target protein in its native cellular state. The chemical reaction should also be highly efficient and the incubation time short (within minutes or preferably less) during which the probe must diffuse into the cells and react with the target protein. Thus, cell permeability of the probe needs to be considered. NEM, a lipid-soluble reagent, and H2O2, which is a source of the OH• radical in FPOP, can efficiently enter cells9,27. Larger chemical probes carrying multiple charges, however, may be less permeable, but this problem may be overcome by including cell-poration reagents that are used in cell biology28,29.
MS footprinting of membrane proteins in live cells can answer a broad range of significant structural questions. With this new method, we recently deduced the native folding topology of hVKOR and the binding orientation of warfarin, and observed an active electron-transfer process that maintains hVKOR activity9. Other important applications of footprinting can be envisioned. One is to investigate the motions of a highly flexible protein region, which often appears “disordered” in crystal or cryo-EM structures owing to an averaging effect. The second is to probe the interface between membrane proteins that are transiently interacting or forming a stable complex or oligomer in cell membranes; such protein-protein interactions, however, are frequently lost in a detergent solution. With further technical developments to provide higher footprinting coverage, MS-based footprinting in live cells may provide unprecedented insights into membrane structural biology, particularly with the medically important human proteins where the both the need and the difficulty are great.
Acknowledgments
G. S. is supported by National Natural Science Foundation of China (81770140). M.L.G. is supported by the NIH NIGMS (P41 GM103422), which supported the MS measurements. W.L. is supported by the NIH National Heart, Lung, and Blood Institute (R01 HL121718).
References
- 1.Barrera NP, Di Bartolo N, Booth PJ, Robinson CV. Science. 2008;321:243–246. doi: 10.1126/science.1159292. [DOI] [PubMed] [Google Scholar]
- 2.Hopper JTS, Yu YTC, Li D, Raymond A, Bostock M, Liko I, Mikhailov V, Laganowsky A, Benesch JLP, Caffrey M, Nietlispach D, Robinson CV. Nat Methods. 2013;10:1206–1208. doi: 10.1038/nmeth.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou M, Robinson CV. Curr Opin Struct Biol. 2014;28:122–130. doi: 10.1016/j.sbi.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Pan Y, Konermann L. Analyst. 2010;135:1191. doi: 10.1039/b924805f. [DOI] [PubMed] [Google Scholar]
- 5.Zhu Y, Guo T, Park JE, Li X, Meng W, Datta A, Bern M, Lim SK, Sze SK. Mol Cell Proteomics. 2009;8:1999–2010. doi: 10.1074/mcp.M900081-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seddon AM, Curnow P, Booth PJ. Biochim Biophys Acta - Biomembr. 2004;1666:105–117. doi: 10.1016/j.bbamem.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 7.Wanrooij S, Goffart S, Pohjoismäki JLO, Yasukawa T, Spelbrink JN. Nucleic Acids Res. 2007;35:3238–3251. doi: 10.1093/nar/gkm215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shevchenko A, Tomas H, Havlis̆ J, Olsen JV, Mann M. Nat Protoc 1 VN-re. 2007:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 9.Shen G, Cui W, Zhang H, Zhou F, Huang W, Liu Q, Yang Y, Li S, Bowman GR, Sadler JE, Gross ML, Li W. Nat Struct Mol Biol. 2017;24:69–76. doi: 10.1038/nsmb.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu PH, Huang TY, Williams J, Stafford DW. Proc Natl Acad Sci U S A. 2006;103:19308–19313. doi: 10.1073/pnas.0609401103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li T, Chang CYY, Jin DYY, Lin PJJ, Khvorova A, Stafford DW. Nature. 2004;427:541–544. doi: 10.1038/nature02254. [DOI] [PubMed] [Google Scholar]
- 12.Rost S, Fregin A, Ivaskevicius V, Conzelmann E, Hortnagel K. Nature. 2004;427:537–541. doi: 10.1038/nature02214. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Toei M, Forgac M. J Biol Chem. 2008;283:20696–20702. doi: 10.1074/jbc.M803258200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamura N, Konishi S, Iwaki S, Kimura-Someya T, Nada S, Yamaguchi A. J Biol Chem. 2001;276:20330–20339. doi: 10.1074/jbc.M007993200. [DOI] [PubMed] [Google Scholar]
- 15.Jessop CE, Watkins RH, Simmons JJ, Tasab M, Bulleid NJ. J Cell Sci. 2009;122:4287–4295. doi: 10.1242/jcs.059154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wallin R, Martin LF. J Clin Invest. 1985;76:1879–1884. doi: 10.1172/JCI112182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodroge A, Longin-Sauvageon C, Fourel I, Benoit E, Lattard V. Arch Biochem Biophys. 2011;515:14–20. doi: 10.1016/j.abb.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 18.Liu S, Cheng W, Fowle Grider R, Shen G, Li W. Nat Commun. 2014;5:3110. doi: 10.1038/ncomms4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li W, Schulman S, Dutton RJ, Boyd D, Beckwith J, Rapoport TA. Nature. 2010;463:507–512. doi: 10.1038/nature08720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manzi L, Barrow AS, Scott D, Layfield R, Wright TG, Moses JE, Oldham NJ. Nat Commun. 2016;7:13288. doi: 10.1038/ncomms13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Y, Zhang H, Niedzwiedzki DM, Jiang J, Blankenship RE, Gross ML. Anal Chem. 2016;88:8827–8834. doi: 10.1021/acs.analchem.6b01945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schey KL, Grey AC, Nicklay JJ. Biochemistry. 2013;52:3807–3817. doi: 10.1021/bi301604j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X, Chien EYT, Chalmers MJ, Pascal BD, Gatchalian J, Stevens RC, Griffin PR. Anal Chem. 2010;82:1100–1108. doi: 10.1021/ac902484p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung KY, Rasmussen SGF, Liu T, Li S, DeVree BT, Chae PS, Calinski D, Kobilka BK, Woods VL, Sunahara RK. Nature. 2011;477:611–615. doi: 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mendoza VL, Vachet RW. Mass Spectrom Rev. 2009;28:785–815. doi: 10.1002/mas.20203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng M, Zhang B, Cui W, Gross ML. Angew Chemie Int Ed. 2017;56:14007–14010. doi: 10.1002/anie.201706697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Espino JA, Mali VS, Jones LM. Anal Chem. 2015;87:7971–7978. doi: 10.1021/acs.analchem.5b01888. [DOI] [PubMed] [Google Scholar]
- 28.Holden P, Horton WA. BMC Res Notes. 2009;2:243. doi: 10.1186/1756-0500-2-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyamoto K, Yamashita T, Tsukiyama T, Kitamura N, Minami N, Yamada M, Imai H. Cloning Stem Cells. 2008;10:535–542. doi: 10.1089/clo.2008.0020. [DOI] [PubMed] [Google Scholar]