

Abstract
Background
The heterogeneity of genetic effects on Major Depressive Disorder (MDD) may be partly attributable to moderation of genetic effects by environment, such as exposure to childhood trauma (CT). Indeed, previous findings in two independent cohorts showed evidence for interaction between polygenic risk scores (PRS) and CT, albeit in opposing directions. This study aims to meta-analyze MDD-PRSxCT interaction results across these two and other cohorts, while applying more accurate PRS based on a larger discovery sample.
Methods and Materials
Data were combined from 3,024 MDD cases and 2,741 controls from nine cohorts contributing to the MDD Working Group of the Psychiatric Genomics Consortium. MDD-PRS were based on a discovery sample of approximately 110,000 independent individuals. CT was assessed as exposure to sexual or physical abuse during childhood. In a subset of 1957 cases and 2002 controls, a more detailed 5-domain measure additionally included emotional abuse, physical neglect and emotional neglect.
Results
MDD was associated with the MDD-PRS (OR=1.24, p=3.6e-5, R2=1.18%) and with CT (OR=2.63, p=3.5e-18 and OR=2.62, p=1.4e-5 for the 2- and 5-domain measures respectively). No interaction was found between MDD-PRS and the 2-domain and 5-domain CT measure (OR=1.00, p=0.89 and OR=1.05, p=0.66).
Conclusions
No meta-analytic evidence for interaction between MDD-PRS and CT was found. This suggests that the previously reported interaction effects, although both statistically significant, can best be interpreted as chance findings. Further research is required, but this study suggests that the genetic heterogeneity of MDD is not attributable to genome-wide moderation of genetic effects by CT.
Keywords: Depression, polygenic risk, childhood trauma, interaction, meta-analysis, genetics
INTRODUCTION
Recent studies have found the first associated genetic variants for Major Depressive Disorder (MDD) and depressive complaints (1–3), but research on MDD still hasn’t met the success of research on schizophrenia, for which 108 genetic variants were found in 2014 (4). This discrepancy is attributable to several factors, including the higher population prevalence of MDD (so that the difference in liability between cases and controls is smaller than in schizophrenia) (5, 6), the lower heritability of MDD (assuming the same degree of polygenicity in terms of number of risk loci) (5), and the greater genetic and phenotypic heterogeneity of MDD (7). To illustrate the possible consequence of heterogeneity, Wray and Maier showed that the power to detect a causal SNP decreases dramatically when a disorder is caused by two distinct pathways (8), while Milaneschi et al found that genetic effects in those with typical MDD might partially differ from genetic effects in those with atypical MDD (9, 10).
Another source of genetic heterogeneity may arise from gene-by-environment (GxE) interaction: the moderation of genetic effects on MDD by specific environmental factors. Much research concerning GxE-interaction has been conducted with candidate genes, in particular the interaction between the serotonin transporter gene (5-HTTLPR) and childhood trauma (11), but this research has produced contradictory findings (12–15) that have been attributed, at least in part, to publication bias (16). Recently, Culverhouse et al published results from a collaborative meta-analysis showing no evidence for interaction between 5-HTTLPR and childhood trauma (17) based on a previously published protocol for analyses (18). Nevertheless, in the last couple of years, methods have been developed to assess the combined impact of all genotyped SNPs, such as polygenic risk score (PRS) analyses (19). Kendler proposed that a confirmed main effect is a desirable condition for GxE-interaction testing (20). This suggests that PRS may be preferable over candidate genes to test for GxE-interaction, because PRS have a confirmed significant effect on MDD (21, 22) contrasting the non-replicated and non-consistent effects of candidate genes (23, 24).
In GxE interaction research numerous environmental factors can be tested, which may have catalyzed publication bias in the candidate gene literature (16) and may also present as a challenge for GxE interaction tests with PRS. Nevertheless, a plausible environmental factor to test in the context of GxE-interaction is childhood trauma, which is one of the strongest risk factors with a lifelong impact on MDD risk (25), and may perhaps be more uniformly defined than stress later in life. Moreover, exposure to childhood trauma has been hypothesized to distinguish a clinically and neurobiologically distinct subtype of MDD, because MDD patients exposed to childhood trauma have an earlier onset, more chronic course, higher severity with more neurovegetative and psychotic symptoms, more comorbidities, more suicide attempts and poorer treatment outcome than MDD patients that did not experience childhood trauma (26).
Following this reasoning, Peyrot et al. tested for GxE interaction between PRS and CT in the Netherlands Study of Depression and Anxiety (NESDA) and found a significantly stronger impact of PRS on MDD risk in individuals exposed to childhood trauma compared to individuals not exposed to childhood trauma (27). In a replication study, Mullins et al found a significant but opposing interaction effect in the RADIANT UK sample with a stronger impact of PRS on MDD risk in those unexposed to childhood trauma (28). These opposing findings, that were both significant, are not well understood, and it remains unclear whether these reflect actual differences between cultures, between recruitment of participants into cohorts, or chance-findings. The aim of the current study is (i) to re-analyze NESDA and RADIANT UK with more accurate PRS based on discovery results from approximately 110,000 individuals (compared to ~15,000 applied previously), and (ii) to place the NESDA and RADIANT UK findings in a broader perspective by meta-analyzing their results with seven additional cohorts from the Psychiatric Genomics Consortium (PGC) MDD wave 2 (29). Secondary analyses used PRS calculated from discovery GWAS results for schizophrenia and bipolar disorder, as these are genetically related to MDD (7, 30).
METHODS
Subjects
Subjects were recruited from the Psychiatric Genomics Consortium (PGC) wave 2, which combines genotype and phenotype data of individuals of European ancestry in 29 different cohorts (29). The combined samples include data of 16,823 MDD cases and 25,632 controls. Of these 29 cohorts, nine cohorts included a measure of childhood trauma: Cognition and Function in Mood Disorders Study (COFAMS) from Australia (31), Depression Gene Network (DGN) from the USA (32), the Netherlands Study of Depression and Anxiety (NESDA) (33), the Queensland Institute of Medical Research (QIMR in three different cohorts defined by genotyping platform) from Australia (23), RADIANT UK (34), and Study of Health in Pomerania (SHIP-0, and SHIP-TREND) from Germany (see Table S1 for more detailed information) (35). Briefly, SHIP-O, SHIP-T and QIMR are community studies with MDD cases and screened controls defined from responses to self-report questionnaires, whilst the other studies recruit MDD cases from in- or out-patient clinics and recruit screened controls with both cases and controls completing the same childhood trauma questionnaires. The definition of MDD in all studies was based on structured psychiatric interviews following DSM-criteria.
Childhood Trauma Questionnaire
The Childhood Trauma Questionnaire (CTQ) was applied to assess childhood trauma, defined as trauma before the age of 16, in five of the nine cohorts (COFAMS, NESDA/NTR, RADIANT UK, SHIP-0, and SHIP-TREND). The CTQ covers the five domains of sexual abuse (SA), physical abuse (PA), emotional abuse (EA), emotional neglect (EN), and physical neglect (PN). Each domain is assessed by five questions (scored 1 to 5) resulting in a domain score ranging from 5 to 25, and an overall CTQ continuous score ranging from 25 to 125 (36). Per domain, cutoffs were applied to define a narrow definition of childhood trauma separating no or mild trauma from moderate or severe trauma (Supplemental Methods). From this, an overall dichotomous CTQ indicator was constructed to separate trauma in any of the five domains (indicator=1) from trauma in none of the domains (indicator=0). The analyses were based on the continuous and dichotomous 5-domain CT scores. The five domains were highly correlated: all pairwise correlation coefficients were larger than 0.4 except for sexual abuse which was slightly less connected (Table S2) as has previously also been reported by Spinhoven et al (37).
Other childhood trauma instruments
In addition to the five cohorts that assessed childhood trauma with the CTQ instrument, four additional PGC cohorts (DGN and the three sub-cohorts of QIMR) assessed childhood trauma with other instruments (before the age of 18 in QIMR). To obtain the largest possible dataset, childhood trauma information was matched across all nine cohorts for sexual abuse and physical abuse (Supplemental Methods). A broad definition (no abuse versus mild, moderate or severe abuse) was applied to create a childhood trauma indicator separating those with trauma (exposed to sexual and/or physical abuse) from those not exposed to childhood trauma (neither exposed to sexual nor physical abuse). The correlation (Spearman’s rho) between the 2-domain dichotomous CT indicator and the 5-domain continuous CT score equaled 0.50 (p<2.e-16).
Genotyping, quality control and imputation
The cohorts were genotyped following their local protocols, after which quality control and imputation against the 1000 genomes reference panel (38) were performed centrally in the PGC per cohort (29). The SNP probabilities were converted to best guess data with a genotype call probability cut-off of 0.8, after which individuals were removed with missing-rate >2%. A total of 1,171,526 HapMap 3 SNPs passed post-imputation QC in at least 2 of 9 batches (missing-rate <2%, minor allele frequency >0.01, and imputation INFO-score >0.6). These 1,171,526 SNPs were used to calculate the genetic relatedness matrix (GRM) with PLINK2 (39), which was thus based on a different set of SNPs for individuals from each cohort and between each pair of cohorts (Table S3), in this way providing genome-wide coverage of well described HapMap 3 SNPs. From the GRM, unrelated individuals were selected with relatedness <0.05, and ancestry informative principal components were calculated with GCTA (40).
Polygenic risk scores
Polygenic risk scores for MDD (MDD-PRS) were based on meta-analysis of the GWAS results from the twenty PGC MDD wave 2 cohorts with no childhood trauma information available (10,409 cases, 18,640 controls) (29), deCODE (1,980 cases, 9,536 controls) (29), GenScotland (997 cases, 6,358 controls) (41, 42), GERA (7,162 cases, 38,307 controls) (43), iPsych (16,242 cases, 15,847 controls) (29) and UK Biobank (8,248 cases, 16,089 controls) (44, 45). This discovery sample comprised 45,038 cases and 104,777 controls yielding a power similar to a sample of 56,134 cases and 56,134 controls (Neffective = 56,134 + 56,134 = 112,268). Additional PRS were based on GWAS results from schizophrenia (SCZ-PRS) (4) and bipolar disorder (BIP-PRS) (46), because these disorders are genetically related to MDD (7, 30). PRS were calculated using 463,215 SNPs shared between the discovery sample results and passing QC in all cohorts (missing-rate <2%, minor allele frequency >0.01, and imputation INFO-score >0.6). Thus, PRS were based on the same set of SNPs in all analyses to increase comparability of results across cohorts. These SNPs were clumped with PLINK (–clump-p1 1 –clump-p2 1 –clump-r2 0.25 –clump-kb 500), and provided 73,576 lowly correlated SNPs for MDD, 73,559 for SCZ, and 73,656 for BIP. The MDD-PRS were based on five different thresholds of GWAS significance for SNP inclusion (p-value smaller than 0.01, 0.05, 0.1, 0.5 and 1 respectively). The SCZ-PRS was based on a threshold of p<0.05, which provided optimal predictive power on SCZ (4). The BIP-PRS was based on a threshold of p<0.5 with best predictive performance on BIP (46). The PRS were calculated by summing the number of risk alleles weighted by their effect size (–score command in PLINK) (39).
Statistical analyses
The prevalences at the population level of the 5-domain and 2-domain dichotomous CT indicators were approximated from this study assuming a population lifetime risk of MDD of 15%, with a lifetime risk of 20% in women and 10% in men (5, 47). The impact of the PRS, CT and PRSxCT was first estimated in the individual cohorts, and the effects in the total sample were subsequently assessed with random-effect meta-analysis. Within each cohort, the impact of CT on MDD was assessed with logistic regression including sex as covariate. The tests for the main effects of the PRS on MDD included sex and the first three ancestry informative principal components as covariates. Interaction analyses were conducted with the 5-domain continuous CT measure and with the 2-domain dichotomous CT indicator. Interaction analyses of PRSxCT were corrected for sex, three principal components, PRS, CT, and the interaction-terms of PRS and CT with sex and the principal components in line with Keller’s recommendation (48). With logistic regression, interaction is tested as departure from multiplicativity (combined impact different from the product of the individual effects), but it has been argued that interaction as departure from additivity (combined impact different from the sum of the individual effects) is more meaningful biologically (49). For testing interaction as departure from additivity, the relative excess risks due to interaction (RERI) were estimated with the coefficients from logistic regression as , and their 95% confidence intervals by means of bootstrapping with 10,000 iterations. The impact of the PRS on MDD was further expressed as variation explained on the liability scale, R2 (50). The PRS and continuous 5-domain CT measure were standardized (i.e. mean of 0 and variance of 1), and the presented ORs can thus be interpreted as increased MDD risk per standard deviation increase in PRS or CT. The analyses were conducted in R (51).
Genetic Relationship Matrix (GRM)-based analyses
The variance in MDD liability and CT explained by genotyped SNPs (SNP heritability) was assessed with cross product Haseman-Elston regression (52). These analyses were corrected for covariates by calculating the residuals of linear regression of MDD and CT on sex, genotyping batch and 20 ancestry informative principal components (PCs). We included 20 PCs, because GRM-based analyses are more sensitive to population stratification than PRS analyses (7). To test for interaction between CT and genome-wide genetic effects in MDD, the genetic correlation between MDD in unexposed individuals and MDD in exposed individuals can give information about differences in genetic effects (53). Unfortunately, the current data did not allow for the latter analyses because of limited sample size (e.g. only 389 exposed controls) while analyses had to be corrected for 9 cohorts.
RESULTS
Phenotypic association between MDD and CT
The 5-domain continuous and dichotomous CT measures were available for 1957 cases and 2002 controls, and the 2-domain dichotomous indicator was available for 3024 cases and 2741 controls. The prevalence of CT was estimated at 0.25 based on the 5-domain indicator (Table 1), and at 0.17 for the 2-domain indicator (Table 3). As expected, the prevalence was considerably larger in cases than controls (0.50 vs 0.21 for the 5-domain measure and 0.35 vs 0.14 for the 2-domain measure). This was reflected in an OR for MDD of 3.80 (p=3.0e-6) for the 5-domain dichotomous measure, and an OR of 2.63 (p=3.5e-18) for the 2-domain measure. For the 5-domain continuous CT measure, an OR for MDD of 2.62 (p=1.4e-5) per standard deviation increase in CT was found (Table 1 & Figure 1). The impact of CT on MDD was comparable in men and women, with ORs of 2.18 (males, p=1.1e-4) and 2.74 (females, p=3.6e-5) per standard deviation increase in the continuous 5-domain CT measures (Table 1). CT had an impact on MDD risk in all cohorts (Table 1), and the five CTQ domains all had an impact on MDD risk (Table S4).
Table 1.
Information is displayed for the cohorts that assessed childhood trauma with the Childhood Trauma Questionnaire (CTQ) covering the 5 domains of sexual abuse, physical abuse, emotional abuse, physical neglect and emotional neglect in a dichotomous 5-domain indicator (exposed versus unexposed) and continuous measure (ranging from 25–125). For the dichotomous CT measure, the proportion of exposed individuals is presented in cases, controls, and in terms of the full population (Pop) assuming a population prevalence of MDD of 15% with twice the prevalence in females (20%) as in males (10%), as well as the odds ratio (OR) of exposed versus unexposed to develop MDD. For the continuous CT measure, the means are displayed in the original scale, and the odds ratio for MDD was assessed for the CTQ measure scaled to variance 1, and can thus be interpreted as increased odds per standard deviation (SD) increase in childhood trauma. The ORs were estimated with logistic regression including sex as covariate. The ORs in the Total sample were estimated with random effect meta-analysis.
| Dichotomous CT indicator
|
Continuous CT measure
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| N
|
Proportion of CT
|
Mean (SD)
|
|||||||
| Cohort | Case | Control | Case | Control | Pop | OR (p-value) | Case | Control | OR (p-value) |
| Male and female
| |||||||||
| COFAMS | 56 | 22 | 0.70 | 0.23 | 0.30 | 7.22 (8.6e-04) | 54.7 (21.4) | 33.2 (11.6) | 5.60 (1.2e-03) |
| NESDA | 1143 | 272 | 0.53 | 0.21 | 0.26 | 4.18 (6.9e-19) | 43.0 (14.6) | 33.6 (9.1) | 3.29 (3.4e-21) |
| RADIANT UK | 269 | 267 | 0.62 | 0.18 | 0.24 | 7.60 (1.1e-22) | 46.4 (16.2) | 32.7 (8.8) | 4.08 (7.4e-21) |
| SHIP-0 | 340 | 993 | 0.36 | 0.23 | 0.25 | 1.94 (1.1e-06) | 37.4 (12.3) | 33.0 (8.4) | 1.52 (7.4e-11) |
| SHIP-TREND | 149 | 448 | 0.28 | 0.15 | 0.17 | 2.43 (1.5e-04) | 36.9 (14.2) | 31.6 (7.3) | 1.72 (2.4e-07) |
|
| |||||||||
| Total | 1957 | 2002 | 0.50 | 0.21 | 0.25 | 3.80 (3.0e-06) | 42.4 (15.1) | 32.7 (8.4) | 2.62 (1.4e-05) |
|
Male only | |||||||||
| COFAMS | 20 | 12 | 0.55 | 0.25 | 0.28 | 3.67 (1.1e-01) | 50.2 (19.9) | 34.8 (14.5) | 2.94 (4.4e-02) |
| NESDA | 357 | 111 | 0.53 | 0.19 | 0.22 | 4.70 (5.4e-09) | 42.0 (13.5) | 33.4 (9.1) | 3.17 (3.4e-09) |
| RADIANT UK | 73 | 109 | 0.62 | 0.18 | 0.23 | 7.42 (7.8e-09) | 45.5 (14.5) | 33.2 (9.1) | 3.43 (4.4e-08) |
| SHIP-0 | 112 | 562 | 0.39 | 0.25 | 0.26 | 1.95 (1.8e-03) | 37.0 (9.1) | 33.2 (7.8) | 1.48 (1.8e-05) |
| SHIP-TREND | 44 | 246 | 0.27 | 0.18 | 0.19 | 1.71 (1.5e-01) | 35.7 (10.9) | 32.3 (7.5) | 1.42 (1.3e-02) |
|
| |||||||||
| Total | 606 | 1040 | 0.49 | 0.22 | 0.25 | 3.30 (8.7e-05) | 41.3 (13.4) | 33.0(8.2) | 2.18 (l.le-04) |
|
Female only | |||||||||
| COFAMS | 36 | 10 | 0.78 | 0.20 | 0.32 | 14.0 (2.9e-03) | 57.2 (22.0) | 31.4 (7.0) | 18.44 (2.2e-02) |
| NESDA | 786 | 161 | 0.53 | 0.23 | 0.29 | 3.90 (2.1e-11) | 43.5 (15.1) | 33.7 (9.0) | 3.30 (1.5e-13) |
| RADIANT UK | 196 | 158 | 0.61 | 0.17 | 0.26 | 7.70 (2.4e-15) | 46.8 (16.8) | 32.3 (8.6) | 4.41 (3.0e-14) |
| SHIP-0 | 228 | 431 | 0.35 | 0.22 | 0.24 | 1.94 (1.7e-04) | 37.5 (13.6) | 32.6 (9.0) | 1.57 (5.5e-07) |
| SHIP-TREND | 105 | 202 | 0.29 | 0.11 | 0.15 | 3.10 (2.6e-04) | 37.4 (15.4) | 30.7 (6.9) | 2.04 (1.2e-05) |
|
| |||||||||
| Total | 1351 | 962 | 0.50 | 0.19 | 0.25 | 4.03 (2.5e-06) | 42.8 (15.8) | 32.3 (8.6) | 2.74 (3.6e-05) |
Table 3.
The impact on major depressive disorder (MDD) is displayed for polygenic risk scores (PRS) and their interaction with the childhood trauma (CT) dichotomous indicator covering sexual abuse and physical abuse (broad definition). The prevalence of CT is presented in MDD cases, controls, and in terms of the full population (Pop) assuming a population prevalence of MDD of 15% with twice the prevalence in females (20%) as in males (10%). The impact of the PRS and CT is presented as the odds ratio (OR) from logistic regression corrected for sex and three principal components, as well as with the variance explained by the PRS on the liability scale. Interaction of PRS with CT (PRSxCT) was assessed as departure from multiplicativity with logistic regression while additionally correcting for the main effects of PRS and CT. The PRS were based on discovery GWAS results from MDD including all SNPs, i.e. with significance threshold p<1.
| Impact on MDD
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N
|
Proportion exposed to CT
|
CT
|
PRS
|
PRSxCT
|
||||||||
| Cohorts | Case | Control | Case | Control | Pop | OR | P | OR | P | R2 (SE, %) | OR | P |
| COFAMS | 56 | 22 | 0.43 | 0.27 | 0.30 | 1.85 | 0.268 | 1.41 (0.82:2.49) | 0.212 | 3.13 (4.61) | 0.51 (0.21:1.05) | 0.088 |
| DGN | 461 | 458 | 0.40 | 0.20 | 0.22 | 2.49 | 1.9e-09 | 1.30 (1.13:1.50) | 2.5e-04 | 1.77 (0.94) | 1.06 (0.91:1.22) | 0.465 |
| NESDA | 1133 | 271 | 0.32 | 0.11 | 0.14 | 3.83 | 8.3e-11 | 1.24 (1.09:1.43) | 0.002 | 1.36 (0.85) | 1.06 (0.87:1.28) | 0.587 |
| QIMR_3 | 186 | 55 | 0.44 | 0.18 | 0.22 | 3.66 | 7.0e-04 | 1.07 (0.79:1.46) | 0.670 | 0.13 (0.60) | 0.82 (0.52:1.25) | 0.355 |
| QIMR_3_M7 | 126 | 29 | 0.48 | 0.31 | 0.34 | 2.10 | 0.092 | 1.16 (0.75:1.80) | 0.494 | 0.66 (1.80) | 0.83 (0.49:1.40) | 0.496 |
| QIMR_6 | 121 | 107 | 0.38 | 0.23 | 0.29 | 2.05 | 0.016 | 0.90 (0.67:1.19) | 0.452 | 0.30 (0.78) | 0.87 (0.61:1.22) | 0.418 |
| QIMR_C | 180 | 46 | 0.40 | 0.33 | 0.33 | 1.36 | 0.387 | 0.83 (0.58:1.17) | 0.297 | 0.92 (1.70) | 0.89 (0.60:1.30) | 0.564 |
| RADIANT UK | 262 | 263 | 0.42 | 0.15 | 0.19 | 4.33 | 1.5e-11 | 1.61 (1.33:1.97) | 2.1e-06 | 5.46 (2.14) | 1.04 (0.83:1.30) | 0.761 |
| SHIP_0 | 352 | 1042 | 0.22 | 0.12 | 0.14 | 2.10 | 6.0e-06 | 1.31 (1.15:1.49) | 4.2e-05 | 1.95 (0.93) | 0.97 (0.86:1.10) | 0.606 |
| SHIP-TREND | 147 | 448 | 0.20 | 0.08 | 0.10 | 2.77 | 2.0e-04 | 1.34(1.09:1.64) | 0.005 | 2.14 (1.50) | 1.08 (0.88:1.35) | 0.460 |
|
| ||||||||||||
| Total | 3024 | 2741 | 0.35 | 0.14 | 0.17 | 2.63 | 3.5e-18 | 1.24(1.12:1.37) | 3.6e-05 | 1.18 (0.31) | 1.00 (0.93:1.07) | 0.894 |
Figure 1.
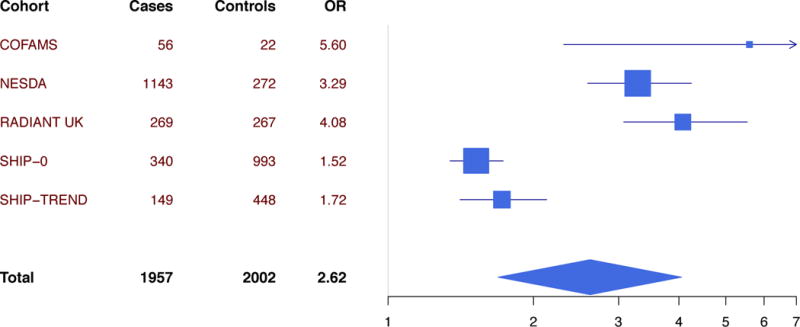
Forest plot of impact on major depressive disorder of the continuous childhood trauma (CT) score covering the 5 domains of sexual abuse, physical abuse, emotional abuse, emotional neglect, and physical neglect. The odds ratio (OR) represents one standard deviation increased in CT.
Polygenic risk score analyses
The MDD-PRS based on all SNPs (inclusion threshold of p<1) had the greatest predictive power, with an OR of 1.34 (p=5.1e-11, R2=1.71%) in the 1957 cases and 2002 controls with availability of the 5-domain CT measures (Table 2). The SCZ-PRS and BIP-PRS also predicted MDD but to a lesser extent than the MDD-PRS (Table 2), reflecting the well-described genetic correlation between MDD, BIP and SCZ (7). Because GE-correlation can lead to spurious GxE-results (54), we tested for an association between the MDD-PRS and CT. The MDD-PRS did predict the 5-domain continuous CT measure (beta=0.76, p=0.004 in linear regression), but this was approximated to only reflect a small correlation in terms of the full population of ~0.04 (Table S5). No interaction between the PRS and the 5-domain continuous CTQ measure was found, with an impact of MDD-PRSxCT on MDD of OR=1.05 (p=0.52; Table 2). In addition, no evidence was found for interaction as departure from additivity (RERI=0.83, 95%CI= −0.62 to 18.03). The BIP-PRS and SCZ-PRS showed no evidence for interaction with the 5-domain CT measure.
Table 2.
The impact on major depressive disorder (MDD) is displayed for polygenic risk scores (PRS) and their interaction with the 5-domain continuous childhood trauma (CT) measure including sexual abuse, physical abuse, emotional abuse, physical neglect and emotional neglect. The impact of the PRS is presented as the odds ratio (OR) from logistic regression corrected for sex and three principal components, as well as with the variance explained by the PRS on the liability scale. Interaction of PRS with CT (PRSxCT) was assessed as departure from multiplicativity with logistic regression while additionally correcting for the main effects of PRS and CT. Interaction as departure from additivity was expressed as the relative excess risks due to interaction (RERI) estimated as described in the main text, and their 95% confidence intervals (CI) were estimated with bootstrapping with 10,000 iterations. The PRS were based on discovery GWAS results from MDD, schizophrenia (SCZ) and bipolar disorder (BIP). Results in the Total sample were based on random-effect meta-analysis of the effects in the individual cohorts.
| Impact on MDD
|
||||||||
|---|---|---|---|---|---|---|---|---|
| N
|
PRS
|
PRSxCT
|
||||||
| Discovery | Case | Control | OR | P | R2 (SE, %) | OR | P | RERI (95% CI) |
| COFAMS
| ||||||||
| MDD p<1 | 56 | 22 | 1.41 (0.82:2.49) | 0.212 | 3.13 (4.61) | 0.38 (0.08:1.74) | 0.201 | −2.07 (NA-NA) |
| SCZ p<0.05 | 56 | 22 | 1.18 (0.59:2.33) | 0.623 | 0.54 (1.95) | 0.01 (0.00:0.37) | 0.030 | −62.80 (NA-NA) |
| BIP p<0.5 | 56 | 22 | 0.85 (0.44:1.58) | 0.612 | 0.44 (1.77) | 0.13 (0.01:0.96) | 0.076 | −2.46 (NA-NA) |
|
NESDA | ||||||||
| MDD p<1 | 1143 | 272 | 1.24 (1.08:1.42) | 0.002 | 1.33 (0.84) | 1.08 (0.83:1.39) | 0.556 | 1.06 (−1.07:10.48) |
| SCZ p<0.05 | 1143 | 272 | 1.25 (1.07:1.46) | 0.006 | 1.02 (0.74) | 0.91 (0.68:1.22) | 0.510 | 0.39 (−1.18:8.78) |
| BIP p<0.5 | 1143 | 272 | 1.14 (1.00:1.31) | 0.049 | 0.53 (0.53) | 1.19 (0.92:1.52) | 0.182 | 1.97 (−0.28:17.61) |
|
RADIANT UK | ||||||||
| MDD p<1 | 269 | 267 | 1.64 (1.35:2.00) | 6.8e-07 | 5.90 (2.19) | 0.93 (0.66:1.31) | 0.670 | 4.42 (−1.78:178.22) |
| SCZ p<0.05 | 269 | 267 | 1.61 (1.31:2.01) | 1.3e-05 | 4.44 (1.92) | 0.90 (0.62:1.30) | 0.581 | 9.87 (−0.43:275.79) |
| BIP p<0.5 | 269 | 267 | 1.19 (1.00:1.43) | 0.053 | 0.85 (0.86) | 1.02 (0.75:1.38) | 0.920 | 4.25 (−0.95:137.22) |
|
SHIP-0 | ||||||||
| MDD p<1 | 340 | 993 | 1.30 (1.14:1.48) | 1.0e-04 | 1.81 (0.91) | 1.02 (0.89:1.18) | 0.737 | 0.52 (−0.18:2.86) |
| SCZ p<0.05 | 340 | 993 | 1.05 (0.91:1.22) | 0.470 | 0.06 (0.17) | 0.95 (0.83:1.10) | 0.497 | −0.22 (−0.97:0.60) |
| BIP p<0.5 | 340 | 993 | 0.95 (0.84:1.09) | 0.477 | 0.06 (0.16) | 0.92 (0.81:1.05) | 0.230 | −0.12 (−0.89:0.96) |
|
SHIP-TREND | ||||||||
| MDD p<1 | 149 | 448 | 1.33 (1.09:1.63) | 0.005 | 2.10 (1.47) | 1.28 (0.96:1.72) | 0.103 | 0.22 (−0.50:1.43) |
| SCZ p<0.05 | 149 | 448 | 1.10 (0.89:1.37) | 0.379 | 0.20 (0.46) | 0.90 (0.71:1.15) | 0.404 | −0.09 (−1.09:1.62) |
| BIP p<0.5 | 149 | 448 | 1.20 (0.99:1.46) | 0.071 | 0.86 (0.95) | 1.05 (0.85:1.32) | 0.659 | 0.07 (−0.75:1.51) |
|
Total | ||||||||
| MDD p<0.01 | 1957 | 2002 | 1.22 (1.08:1.37) | 0.001 | 0.58 (0.26) | 1.02 (0.89:1.17) | 0.790 | −0.17 (−2.86:10.25) |
| MDD p<0.05 | 1957 | 2002 | 1.29 (1.14:1.45) | 4.0e-05 | 1.08 (0.36) | 0.98 (0.79:1.22) | 0.846 | 0.27 (−2.46:15.37) |
| MDD p<0.1 | 1957 | 2002 | 1.34 (1.18:1.53) | 1.0e-05 | 1.49 (0.42) | 1.01 (0.84:1.22) | 0.910 | 0.51 (−2.02:15.72) |
| MDD p<0.5 | 1957 | 2002 | 1.35 (1.22:1.48) | 2.2e-09 | 1.70 (0.45) | 1.03 (0.86:1.23) | 0.755 | 0.84 (−0.52:22.18) |
| MDD p<l | 1957 | 2002 | 1.34(1.23:1.47) | 5.1e-11 | 1.71(0.45) | 1.05 (0.91:1.20) | 0.519 | 0.83 (−0.62:18.03) |
|
| ||||||||
| SCZ p<0.05 | 1957 | 2002 | 1.22(1.04:1.43) | 0.013 | 0.57 (0.26) | 0.91 (0.79:1.04) | 0.172 | −0.15 (−2.87:11.06) |
| BIP p<0.5 | 1957 | 2002 | 1.10 (0.98:1.23) | 0.114 | 0.16 (0.14) | 1.00 (0.85:1.18) | 0.997 | 0.39 (−1.13:20.78) |
Applying the 2-domain dichotomous CT indicator of sexual or physical abuse allowed inclusion of four additional cohorts in the analyses (Table 3): DGN and 3 QIMR cohorts (one of the QIMR cohorts was split in two to acknowledge different instruments applied to assess childhood trauma). The total sample size thus increased to 3024 cases and 2741 controls, in which the MDD-PRS had an impact on MDD with an OR of 1.24 (p=3.6e-5, R2=1.18%). The polygenic risk scores did predict MDD in DGN, but not in all QIMR cohorts, which is attributable to the relatively small number of QIMR subjects with CT information available compared to the full QIMR sample (in which PRS predict MDD as expected). No interaction was found between the PRS and 2-domain dichotomous CT indicator (Table 3).
An alternative method sometimes applied to test for interaction as departure from additivity is linear regression with the disease trait as outcome (28). We suggest for caution in interpreting findings from this approach, because this method has, to the best of our knowledge, not been formally described. Nevertheless, for reasons of completeness, this approach was applied and also showed no evidence for interaction with the 5-domain CT measure (beta=-0.004, p=0.67) and the 2-domain CT measure (beta=−0.005, p=0.45).
GRM based analyses
The SNP heritability of MDD was estimated at 0.14 (SE=0.03; p=3.7e-8) based on the 6,348 cases and 6,751 controls across the nine cohorts (Table S1; these analyses included additional individuals with no CT information available). The SNP heritability of CT was estimated at 0.00 (SE=0.07; p=1; N=3,959) for the 5-domaine continuous measure, and at 0.09 (SE=0.08; p=0.27; N=5,765) for the 2-domain dichotomous indicator.
DISCUSSION
This study was conducted to test for interaction between polygenic risk for MDD and childhood trauma (CT) in 5,765 individuals from nine cohorts contributing to the Psychiatric Genomics Consortium that had a childhood trauma assessment available. CT occurred in 25% of individuals based on an indicator of 5-domains (sexual abuse, physical abuse, emotional abuse, emotional neglect, and physical neglect), and in 17% based on broad definition of 2-domains (sexual and/or physical abuse). As expected, the prevalence was considerably higher in cases than controls (0.50 vs 0.21 for the 5-domain measure and 0.35 vs 0.14 for the 2-domain measure). The 5-domain measure was more detailed and uniformly assessed in 1957 cases and 2002 controls; the 2-domain indicator was assessed heterogeneous across cohorts, but available for a larger sample comprising of 3024 cases and 2741 controls. The polygenic risk scores (PRS) explained 1.18% to 1.71% of variation in MDD risk. No evidence for interaction between PRS and childhood trauma was found with 5-domain CT measure (Table 2) and the 2-domain CT indicator (Table 3). Secondary analyses also showed no evidence for interaction in analyses with PRS based on discovery results from schizophrenia and bipolar disorders, in tests for interaction as departure from additivity, in analyses in males and females separately (Table S6), and in analysis in the five separate domains of CT (Table S7; significance threshold 0.01=0.05/5). Analyses excluding NESDA and RADIANT UK showed no evidence for interaction between the MDD-PRS (p-value threshold 1) and 5-domain CT measure (OR=1.06, p=0.67) and 2-domain CT measure (OR=0.98, p= 0.61) in the remainder of the cohorts.
Remarkably, no interaction-effects were found in NESDA (OR=1.08, 95%CI=0.83–1.39, p=0.56) and RADIANT UK (OR=0.93, 95%CI=0.66–1.31, p=0.67) with the 5-domain CT measure (Table 2), which contrasts previous findings in these respective cohorts by Peyrot et al (OR=1.12, p=0.018, discovery sample Neffective=15,295) (27) and Mullins et al (OR=0.96 based on differently scaled PRS and CT, p=0.002, discovery sample Neffective=15,540) (28). Aiming to clarify these discrepancies, we analyzed PRS based on discovery results from PGC MDD wave 2 with an effective sample size of approximately 37,000 (Table S8) and confirmed the previously reported interaction-effects in NESDA (OR=1.38, 95%CI=1.07–1.76, p=0.011) and RADIANT UK (OR=0.67, 95%CI=0.51–0.90, p=0.006). Therefore, it appears that the OR of the interaction-effects are reduced by adding deCODE (29), GenScotland (41, 42), GERA (43), iPsych (29) and UK Biobank (44, 45) to the PRS discovery sample. These discrepancies in interaction results may reflect different study designs in the discovery datasets with application of self-reported depression status in UKB and clinical records in iPsych and GERA, contrasting the semi-structured interviews (such as the SCID, CIDI and MINI) applied in most PGC cohorts (29). However, these discrepancies may also reflect random variation in effects with discovery sample size increasing from ~37,000 to ~110,000. The latter possibility seems more likely since: (1) we observe an increase in the variance explained by the PRS from 0.66% (p=2.8e-5) to 1.71% (p=5.1e-11) (Table S8), which corresponds with the increase predicted from theory given the increased sample size (55); (2) a genetic correlation of 0.91–0.96 between the PGC wave 2 discovery results and the extended discovery results as estimated with LD-score regression (30); and (3) an overlap of the 95% CI of the interaction-effects based on the PGC discovery sample and the larger discovery sample applied in this paper (Table S8). In other words, our results suggest that the additional discovery cohorts (deCODE, GenScotland, GERA, iPsych, and UK Biobank) capture the same genetic information as the PGC cohorts. Therefore, we hypothesize that the previously reported interaction results in NESDA (27) and RADIANT UK (28) were both chance findings. The fact that these findings were both significant in an opposite direction may reflect the statistical vulnerability of interaction testing (48, 54, 56).
A source of spurious interaction effects can be found in gene-environment (GE) correlation as explained for twin analyses by Purcell (54). Notably, the PRS based on the PGC wave 2 discovery results were slightly more correlated with childhood trauma in the full population (with approximately −0.09 in NESDA and 0.13 in RADIANT UK) than the PRS based on the extended sample (~0.02 and ~0.06 respectively). A simulation study suggested that the type I error rate can indeed be inflated in the context of GE-correlation, but to a modest extent of 0.075 (with alpha set at 0.05) for a strong correlation of 0.3 between G and E (Supplemental Methods). It is, therefore, unlikely that the GxE-interactions previously found would be attributable to GE-correlation.
The current study has both strengths and limitations. First, this study is the largest to date to test for interaction between polygenic risk scores and CT in MDD risk. Second, polygenic risk scores were based on a powerful discovery GWAS with approximately 110,000 individuals. Third, diagnoses were DSM-based aiming to select clinically relevant cases of MDD. A limitation of our study is that CT was not assessed uniformly across cohorts for the 2-domain measure, but analyses restricted to cohorts assessed uniformly with the 5-domain CTQ-instrument showed similar results. Although this study is the largest to date, power to detect an interaction-effect between PRS and CT was still limited (power≥0.8 for interaction effects with OR≤0.83 or OR≥1.21 for analyses with the 2-domain CT measure in 5,765 individuals based on power analyses with the QUANTO software) (57). Of note, tests of interaction with PRS do not rule out interaction with individual SNPs; the PRS were based on many SNPs, some, but not all of which may be involved in interaction. The current study tested for interaction with childhood trauma, because childhood trauma has been hypothesized to define a distinct type of MDD,(26) but other environmental factors could have also been tested. Nevertheless, testing too many environmental conditions assessed with a variety of instruments may increase risk of publication bias when significant findings would be published selectively (16, 58).
Lastly, we would like to emphasize the complex nature of interaction testing with PRS based on genome-wide SNPs. For analyses with twin data, Purcell described the distinction between qualitative interaction (different genes have an effect across different environments) and quantitative interactions (the same genes have an effect but they explain a different proportion of variance) (54). In an attempt to elucidate some of the characteristics of interaction testing with PRS, we conducted a second simulation study constructing PRS from simulated SNP-level data for different underlying genetic architectures (Supplemental Methods and Table S9). First, we note that the discovery results are typically based on a discovery sample with an unknown mixture of individuals unexposed (CT=0) and individuals exposed to childhood trauma (CT=1). When assuming qualitative genome-wide interaction with different directions of SNP effects in exposed and unexposed individuals (explaining the same proportion of variance in both groups), the discovery GWAS would mainly tag the effects in unexposed individuals that form the majority of the discovery sample. Consequently, negative interaction between PRS and CT would be detected under this scenario. Second and contrary, for quantitative interaction a positive interaction effect may be expected when SNPs would explain more variance in exposed individuals.
To conclude, no overall evidence was found for interaction between PRS and CT. Previously found interaction effects (27, 28) were no longer significant when applying more powerful discovery results. This study provides a cautionary tale for interaction analyses with PRS: it emphasizes the need to meta-analyze results across different cohorts to obtain external validity. The quest continues to clarify the nature of the heterogeneity of MDD, but the present study has shown that the heterogeneity is unlikely to be attributable to moderation of genome-wide genetic effects by CT. Future research may focus on interaction effects between CT and individual SNPs. We hereby call for large GWAS cohorts to assess CT in a uniform manner to facilitate such research in the years the come.
Supplementary Material
Acknowledgments
NRW was funded by the Australian National Health and Medical Research Council 1078901, 1087889 and EMB was supported by fellowhip 1053639. The Netherlands Study of Depression and Anxiety (NESDA) was funded by the Netherlands Organization for Scientific Research (MagW/ZonMW Grants 904-61-090, 985-10-002, 904-61-193, 480-04-004, 400-05-717, 912-100-20; Spinozapremie 56-464-14192; Geestkracht program Grant 10-000-1002); the Center for Medical Systems Biology (NWO Genomics), Biobanking and Biomolecular Resources Research Infrastructure, VU University’s Institutes for Health and Care Research and Neuroscience Campus Amsterdam, NBIC/BioAssist/RK (2008.024); the European Science Foundation (EU/QLRT-2001-01254); the European Community’s Seventh Framework Program (FP7/2007-2013); ENGAGE (HEALTH-F4-2007-201413); and the European Science Council (ERC, 230374). Genotyping was funded in part by the Genetic Association Information Network (GAIN) of the Foundation for the US National Institutes of Health, and analysis was supported by grants from GAIN and the NIMH (MH081802). CoFaMS was supported by a grant from the National Health and Medical Research Council (NHMRC APP 1060524 to BTB). SHIP is part of the Community Medicine Research net of the University of Greifswald, Germany, which is funded by the Federal Ministry of Education and Research (grants no. 01ZZ9603, 01ZZ0103, and 01ZZ0403), the Ministry of Cultural Affairs and the Social Ministry of the Federal State of Mecklenburg-West Pomerania. Genome-wide data analyses in SHIP have been supported by a joint grant from Siemens Healthineers, Erlangen, Germany and the Federal State of Mecklenburg-West Pomerania. Genome-wide genotyping in SHIP-TREND-0 was supported by the Federal Ministry of Education and Research (grant no. 03ZIK012). This work was also funded by the German Research Foundation (DFG: GR 1912/5-1). In addition, this work was supported by the German Federal Ministry of Education and Research (BMBF) within the framework of the e:Med research and funding concept (Integrament; grant no. 01ZX1314E). Royal Netherlands Academy of Science Professor Award (PAH/6635) to DIB. MR received funding from the German Federal Ministry of Education and Research (BMBF) within the context of the Integrated Network IntegraMent (Integrated Understanding of Causes and Mechanisms in Mental Disorders; grant 01ZX1314G). MR and SHW received funding from the German Research Foundation (DFG) within the context of FOR2107 (DFG-Forschergruppe 2107; grant RI908/11-1 to M.R.; grant WI 3439/3-1 to SHW). This report represents independent research part-funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health. The RADIANT studies were funded by a joint grant from the UK Medical Research Council (G0701420), GlaxoSmithKline and by the National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health at South London and Maudsley NHS Foundation Trust and Institute of Psychiatry, Psychology and Neuroscience, King’s College London. N.M. and C.M.L. have received funding from the European Community’s Seventh Framework Programme under the Marie Curie Industry-Academia Partnership and Pathways (grant 286213). E.C.D. is supported by the National Institute of Mental Health (NIMH; 1K01MH102403). H.L.F. is supported by an MQ Fellows Award (MQ14F40). We thank all individuals who participated in the RADIANT study and all involved with data collection and management.
The Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium is a collaborative co-author on this paper. The individual authors are (affiliations are listed in the Supplement): Naomi R Wray, Stephan Ripke, Manuel Mattheisen, Maciej Trzaskowski, Enda M Byrne, Abdel Abdellaoui, Mark J Adams, Esben Agerbo, Tracy M Air, Till F M Andlauer, Silviu-Alin Bacanu, Marie Bækvad-Hansen, Aartjan T F Beekman, Tim B Bigdeli, Elisabeth B Binder, Douglas H R Blackwood, Julien Bryois, Henriette N Buttenschøn, Jonas Bybjerg-Grauholm, Na Cai, Enrique Castelao, Jane Hvarregaard Christensen, Toni-Kim Clarke, Jonathan R I Coleman, Lucía Colodro-Conde, Baptiste Couvy-Duchesne, Nick Craddock, Gregory E Crawford, Gail Davies, Ian J Deary, Franziska Degenhardt, Eske M Derks, Nese Direk, Conor V Dolan, Erin C Dunn, Thalia C Eley, Valentina Escott-Price, Farnush, Farhadi Hassan Kiadeh, Hilary K Finucane, Andreas J Forstner, Josef Frank, Héléna A Gaspar, Michael Gill, Fernando S Goes, Scott D Gordon, Jakob Grove, Lynsey S Hall, Christine Søholm Hansen, Thomas F Hansen, Stefan Herms, Ian B Hicki, Per Hoffmann, Georg Homuth, Carsten Horn, Jouke-Jan Hottenga, David M Hougaard, Marcus Ising, Rick Jansen, Eric Jorgenson, James A Knowles, Isaac S Kohane, Julia Kraft, Warren W. Kretzschmar, Jesper Krogh, Zoltán Kutalik, Yihan Li, Penelope A Lind, Donald J MacIntyre, Dean F MacKinnon, Robert M Maier, Wolfgang Maier, Jonathan Marchini, Hamdi Mbarek, Patrick McGrath, Peter McGuffin, Sarah E Medland, Divya Mehta, Christel M Middeldorp, Evelin Mihailov, Yuri Milaneschi, Lili Milani, Francis M Mondimore, Grant W Montgomery, Sara Mostafavi, Niamh Mullins, Matthias Nauck, Bernard Ng, Michel G Nivard, Dale R Nyholt, Paul F O’Reilly, Hogni Oskarsson, Michael J Owen, Jodie N Painter, Carsten Bøcker Pedersen, Marianne Giørtz Pedersen, Roseann E. Peterson, Erik Pettersson, Wouter J Peyrot, Giorgio Pistis, Danielle Posthuma, Jorge A Quiroz, Per Qvist, John P Rice, Brien P. Riley, Margarita Rivera, Saira Saeed Mirza, Robert Schoevers, Eva C Schulte, Ling Shen, Jianxin Shi, Stanley I Shyn, Engilbert Sigurdsson, Grant C B Sinnamon, Johannes H Smit, Daniel J Smith, Hreinn Stefansson, Stacy Steinberg, Fabian Streit, Jana Strohmaier, Katherine E Tansey, Henning Teismann, Alexander Teumer, Wesley Thompson, Pippa A Thomson, Thorgeir E Thorgeirsson, Matthew Traylor, Jens Treutlein, Vassily Trubetskoy, André G Uitterlinden, Daniel Umbricht, Sandra Van der Auwera, Albert M van Hemert, Alexander Viktorin, Peter M Visscher, Yunpeng Wang, Bradley T. Webb, Shantel Marie Weinsheimer, Jürgen Wellmann, Gonneke Willemsen, Stephanie H Witt, Yang Wu, Hualin S Xi, Jian Yang, Futao Zhang, Volker Arolt, Bernhard T Baune, Klaus Berger, Dorret I Boomsma, Sven Cichon, Udo Dannlowski, EJC de Geus, J Raymond DePaulo, Enrico Domenici, Katharina Domschke, Tõnu Esko, Hans J Grabe, Steven P Hamilton, Caroline Hayward, Andrew C Heath, Kenneth S Kendler, Stefan Kloiber, Glyn Lewis, Qingqin S Li, Susanne Lucae, Pamela AF Madden, Patrik K Magnusson, Nicholas G Martin, Andrew M McIntosh, Andres Metspalu, Ole Mors, Preben Bo Mortensen, Bertram Müller-Myhsok, Merete Nordentoft, Markus M Nöthen, Michael C O’Donovan, Sara A Paciga, Nancy L Pedersen, Brenda WJH Penninx, Roy H Perlis, David J Porteous, James B Potash, Martin Preisig, Marcella Rietschel, Catherine Schaefer, Thomas G Schulze, Jordan W Smoller, Kari Stefansson, Henning Tiemeier, Rudolf Uher, Henry Völzke, Myrna M Weissman, Thomas Werge, Cathryn M Lewis, Douglas F Levinson, Gerome Breen, Anders D Børglum, Patrick F Sullivan
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST
All authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Cai N, Bigdeli TB, Kretzschmar W, Li Y, Liang J, Song L, et al. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature. 2015;523:588–91. doi: 10.1038/nature14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okbay A, Baselmans BML, De Neve J-E, Turley P, Nivard MG, Fontana MA, et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat Genet. 2016 doi: 10.1038/ng.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet. 2016 doi: 10.1038/ng.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, Holmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sullivan PF, Daly MJ, O’Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet. 2012;13:537–51. doi: 10.1038/nrg3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peyrot WJ, Boomsma DI, Penninx BWJH, Wray NR. Disease and Polygenic Architecture: Avoid Trio Design and Appropriately Account for Unscreened Control Subjects for Common Disease. Am J Hum Genet. 2016;98:382–391. doi: 10.1016/j.ajhg.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–94. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wray NR, Maier R. Genetic Basis of Complex Genetic Disease: The Contribution of Disease Heterogeneity to Missing Heritability. Curr Epidemiol Reports. 2014;1:220–227. [Google Scholar]
- 9.Milaneschi Y, Lamers F, Mbarek H, Hottenga J-J, Boomsma DI, Penninx BWJH. The effect of FTO rs9939609 on major depression differs across MDD subtypes. Mol Psychiatry. 2014;19:960–2. doi: 10.1038/mp.2014.4. [DOI] [PubMed] [Google Scholar]
- 10.Milaneschi Y, Lamers F, Peyrot WJ, Abdellaoui A, Willemsen G, Hottenga J-J, et al. Polygenic dissection of major depression clinical heterogeneity. Mol Psychiatry. 2015;21:516–22. doi: 10.1038/mp.2015.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–9. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 12.Fergusson DM, Horwood LJ, Miller AL, Kennedy Ma. Life stress, 5-HTTLPR and mental disorder: findings from a 30-year longitudinal study. Br J Psychiatry. 2011;198:129–35. doi: 10.1192/bjp.bp.110.085993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Munafò MR, Durrant C, Lewis G, Flint J. Gene X environment interactions at the serotonin transporter locus. Biol Psychiatry. 2009;65:211–9. doi: 10.1016/j.biopsych.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 14.Karg K, Burmeister M, Shedden K, Sen S. The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Arch Gen Psychiatry. 2011;68:444–54. doi: 10.1001/archgenpsychiatry.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Risch N, Herrell R, Lehner T, Liang K-Y, Eaves L, Hoh J, et al. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA. 2009;301:2462–71. doi: 10.1001/jama.2009.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duncan LE, Keller MC. A critical review of the first 10 years of candidate gene-by-environment interaction research in psychiatry. Am J Psychiatry. 2011;168:1041–9. doi: 10.1176/appi.ajp.2011.11020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Culverhouse RC, Saccone NL, Horton AC, Ma Y, Anstey KJ, Banaschewski T, et al. Collaborative meta-analysis finds no evidence of a strong interaction between stress and 5-HTTLPR genotype contributing to the development of depression. Mol Psychiatry. 2017 doi: 10.1038/mp.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Culverhouse RC, Bowes L, Breslau N, Nurnberger JI, Burmeister M, Fergusson DM, et al. Protocol for a collaborative meta-analysis of 5-HTTLPR, stress, and depression. BMC Psychiatry. 2013;13:304. doi: 10.1186/1471-244X-13-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kendler KS, Gardner CO. Interpretation of interactions: guide for the perplexed. Br J Psychiatry. 2010;197:170–1. doi: 10.1192/bjp.bp.110.081331. [DOI] [PubMed] [Google Scholar]
- 21.Demirkan A, Penninx BWJH, Hek K, Wray NR, Amin N, Aulchenko YS, et al. Genetic risk profiles for depression and anxiety in adult and elderly cohorts. Mol Psychiatry. 2011;16:773–83. doi: 10.1038/mp.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peyrot WJ, Lee SH, Milaneschi Y, Abdellaoui A, Byrne EM, Esko T, et al. The association between lower educational attainment and depression owing to shared genetic effects? Results in ~25 000 subjects. Mol Psychiatry. 2015 doi: 10.1038/mp.2015.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wray NR, Pergadia ML, Blackwood DHR, Penninx BWJH, Gordon SD, Nyholt DR, et al. Genome-wide association study of major depressive disorder: new results, meta-analysis, and lessons learned. Mol Psychiatry. 2012;17:36–48. doi: 10.1038/mp.2010.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clarke H, Flint J, Attwood AS, Munafò MR. Association of the 5-HTTLPR genotype and unipolar depression: a meta-analysis. Psychol Med. 2010;40:1767–78. doi: 10.1017/S0033291710000516. [DOI] [PubMed] [Google Scholar]
- 25.Hovens JGFM, Wiersma JE, Giltay EJ, van Oppen P, Spinhoven P, Penninx BWJH, Zitman FG. Childhood life events and childhood trauma in adult patients with depressive, anxiety and comorbid disorders vs. controls. Acta Psychiatr Scand. 2010;122:66–74. doi: 10.1111/j.1600-0447.2009.01491.x. [DOI] [PubMed] [Google Scholar]
- 26.Teicher MH, Samson Ja. Childhood maltreatment and psychopathology: A case for ecophenotypic variants as clinically and neurobiologically distinct subtypes. Am J Psychiatry. 2013;170:1114–33. doi: 10.1176/appi.ajp.2013.12070957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peyrot WJ, Milaneschi Y, Abdellaoui A, Sullivan PF, Hottenga JJ, Boomsma DI, Penninx BWJH. Effect of polygenic risk scores on depression in childhood trauma. Br J Psychiatry. 2014;205:113–119. doi: 10.1192/bjp.bp.113.143081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mullins N, Power RA, Fisher HL, Hanscombe KB, Euesden J, Iniesta R, et al. Polygenic interactions with environmental adversity in the aetiology of major depressive disorder. Psychol Med. 2015:1–12. doi: 10.1017/S0033291715002172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Major Depressive Disorder Working Group of the PGC. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depressive disorder. bioRxiv. 2017 doi: 10.1101/167577. [DOI] [PMC free article] [PubMed]
- 30.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh P-R, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015 doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baune BT, Air T. Clinical, Functional, and Biological Correlates of Cognitive Dimensions in Major Depressive Disorder - Rationale, Design, and Characteristics of the Cognitive Function and Mood Study (CoFaM-Study) Front psychiatry. 2016;7:150. doi: 10.3389/fpsyt.2016.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mostafavi S, Battle A, Zhu X, Potash JB, Weissman MM, Shi J, et al. Type I interferon signaling genes in recurrent major depression: increased expression detected by whole-blood RNA sequencing. Mol Psychiatry. 2014;19:1267–74. doi: 10.1038/mp.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Penninx BWJH, Beekman ATF, Smit JH, Zitman FG, Nolen WA, Spinhoven P, et al. The Netherlands Study of Depression and Anxiety (NESDA): rationale, objectives and methods. Int J Methods Psychiatr Res. 2008;17:121–40. doi: 10.1002/mpr.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis CM, Ng MY, Butler AW, Cohen-Woods S, Uher R, Pirlo K, et al. Genome-wide association study of major recurrent depression in the U.K. population. Am J Psychiatry. 2010;167:949–57. doi: 10.1176/appi.ajp.2010.09091380. [DOI] [PubMed] [Google Scholar]
- 35.Völzke H, Alte D, Schmidt CO, Radke D, Lorbeer R, Friedrich N, et al. Cohort profile: the study of health in Pomerania. Int J Epidemiol. 2011;40:294–307. doi: 10.1093/ije/dyp394. [DOI] [PubMed] [Google Scholar]
- 36.Bernstein DP, Stein Ja, Newcomb MD, Walker E, Pogge D, Ahluvalia T, et al. Development and validation of a brief screening version of the Childhood Trauma Questionnaire. Child Abuse Negl. 2003;27:169–190. doi: 10.1016/s0145-2134(02)00541-0. [DOI] [PubMed] [Google Scholar]
- 37.Spinhoven P, Penninx BW, Hickendorff M, van Hemert AM, Bernstein DP, Elzinga BM. Childhood Trauma Questionnaire: factor structure, measurement invariance, and validity across emotional disorders. Psychol Assess. 2014;26:717–29. doi: 10.1037/pas0000002. [DOI] [PubMed] [Google Scholar]
- 38.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernandez-Pujals AM, Adams MJ, Thomson P, McKechanie AG, Blackwood DHR, Smith BH, et al. Epidemiology and Heritability of Major Depressive Disorder, Stratified by Age of Onset, Sex, and Illness Course in Generation Scotland: Scottish Family Health Study (GS:SFHS) In: Ebmeier K, editor. PLoS One. Vol. 10. 2015. p. e0142197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith BH, Campbell A, Linksted P, Fitzpatrick B, Jackson C, Kerr SM, et al. Cohort Profile: Generation Scotland: Scottish Family Health Study (GS:SFHS). The study, its participants and their potential for genetic research on health and illness. Int J Epidemiol. 2013;42:689–700. doi: 10.1093/ije/dys084. [DOI] [PubMed] [Google Scholar]
- 43.Banda Y, Kvale MN, Hoffmann TJ, Hesselson SE, Ranatunga D, Tang H, et al. Characterizing Race/Ethnicity and Genetic Ancestry for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics. 2015;200:1285–95. doi: 10.1534/genetics.115.178616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith DJ, Nicholl BI, Cullen B, Martin D, Ul-Haq Z, Evans J, et al. Prevalence and characteristics of probable major depression and bipolar disorder within UK biobank: cross-sectional study of 172,751 participants. In: Potash JB, editor. PLoS One. Vol. 8. 2013. p. e75362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sklar P, Ripke S, Scott LJ, Andreassen OA, Cichon S, Craddock N, Edenberg HJ, Jr, Nurnberger JI, Rietschel M, Blackwood D, Corvin A, Flickinger M, Guan W, Mattingsdal M, McQuillen A, Kwan P, Wienker TF, Daly M, Dudbridge F, Holmans PA, Lin D, Burmeister MPS. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43:977–83. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Graaf R, ten Have M, van Gool C, van Dorsselaer S. Prevalence of mental disorders and trends from 1996 to 2009. Results from the Netherlands Mental Health Survey and Incidence Study-2. Soc Psychiatry Psychiatr Epidemiol. 2012;47:203–13. doi: 10.1007/s00127-010-0334-8. [DOI] [PubMed] [Google Scholar]
- 48.Keller MC. Gene × environment interaction studies have not properly controlled for potential confounders: the problem and the (simple) solution. Biol Psychiatry. 2014;75:18–24. doi: 10.1016/j.biopsych.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knol MJ, van der Tweel I, Grobbee DE, Numans ME, Geerlings MI. Estimating interaction on an additive scale between continuous determinants in a logistic regression model. Int J Epidemiol. 2007;36:1111–8. doi: 10.1093/ije/dym157. [DOI] [PubMed] [Google Scholar]
- 50.Lee SH, Goddard ME, Wray NR, Visscher PM. A Better Coefficient of Determination for Genetic Profile Analysis. Genet Epidemiol. 2012;36:214–224. doi: 10.1002/gepi.21614. [DOI] [PubMed] [Google Scholar]
- 51.R Core Team. R: A Language and Environment for Statistical Computing. 2015 Retrieved from http://www.r-project.org.
- 52.Golan D, Lander ES, Rosset S. Measuring missing heritability: Inferring the contribution of common variants. Proc Natl Acad Sci U S A. 2014;111:E5272–81. doi: 10.1073/pnas.1419064111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Falconer D. The problem of environment and selection. Am Nat. 1952 Retrieved April 18 2016, from http://www.jstor.org/stable/2457811.
- 54.Purcell S. Variance components models for gene-environment interaction in twin analysis. Twin Res. 2002;5:554–71. doi: 10.1375/136905202762342026. [DOI] [PubMed] [Google Scholar]
- 55.Palla L, Dudbridge F. A Fast Method that Uses Polygenic Scores to Estimate the Variance Explained by Genome-wide Marker Panels and the Proportion of Variants Affecting a Trait. Am J Hum Genet. 2015;97:250–9. doi: 10.1016/j.ajhg.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eaves LJ. Genotype x Environment interaction in psychopathology: fact or artifact? Twin Res Hum Genet. 2006;9:1–8. doi: 10.1375/183242706776403073. [DOI] [PubMed] [Google Scholar]
- 57.Kraft P, Yen Y, Stram O, Morrison J. Exploiting Gene-Environment Interaction. 2007;02115:111–119. doi: 10.1159/000099183. [DOI] [PubMed] [Google Scholar]
- 58.Sullivan PF. Spurious genetic associations. Biol Psychiatry. 2007;61:1121–6. doi: 10.1016/j.biopsych.2006.11.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.