

Abstract
Multiple myeloma (MM) is a disease of copy number variants (CNVs), chromosomal translocations, and single-nucleotide variants (SNVs). To enable integrative studies across these diverse mutation types, we developed a capture-based sequencing platform to detect their occurrence in 465 genes altered in MM and used it to sequence 95 primary tumor-normal pairs to a mean depth of 104×. We detected cases of hyperdiploidy (23%), deletions of 1p (8%), 6q (21%), 8p (17%), 14q (16%), 16q (22%), and 17p (4%), and amplification of 1q (19%). We also detected IGH and MYC translocations near expected frequencies and non-silent SNVs in NRAS (24%), KRAS (21%), FAM46C (17%), TP53 (9%), DIS3 (9%), and BRAF (3%). We discovered frequent mutations in IGLL5 (18%) that were mutually exclusive of RAS mutations and associated with increased risk of disease progression (p = 0.03), suggesting that IGLL5 may be a stratifying biomarker. We identified novel IGLL5/IGH translocations in two samples. We subjected 15 of the pairs to ultra-deep sequencing (1259×) and found that although depth correlated with number of mutations detected (p = 0.001), depth past ~300× added little. The platform provides cost-effective genomic analysis for research and may be useful in individualizing treatment decisions in clinical settings.
Introduction
Multiple myeloma (MM) is a fatal malignancy of mature plasma B cells. Overt MM is preceded by a premalignant phase, monoclonal gammopathy of undetermined significance (MGUS), which can progress to smoldering MM and ultimately to fatal myeloma. Genetic alterations detected in premalignant MGUS cells are likely initiating events. These may be divided into two primary subtypes that are most often non-overlapping1: hyperdiploid (HRD) myeloma is characterized by trisomies of most odd-numbered chromosomes2, while non-HRD myeloma frequently involves immunoglobulin heavy chain (IGH) translocations. These upregulate target oncogenes by placing them under the control of one or both of the powerful, B-cell-specific IGH enhancer regions; canonical IGH partner genes include WHSC1/FGFR3, CCND3, CCND1, MAF, and MAFB in translocations t(4;14), t(6;14), t(11;14), t(14;16), and t(14;20), respectively1. Secondary genetic events are detected in MM, but not its precursor phases, and are thought to drive disease progression. The most prevalent secondary events include MYC translocations (juxtaposing IGH and other loci), single-nucleotide variants (SNVs) involving KRAS, NRAS, and DIS3, and copy number variants (CNVs) that amplify chromosome arm 1q or delete 1p, 6q, 13, 14q, or 16q2.
This diversity of genetic lesions has recently been leveraged in a prognostic model that integrates the International Staging System (ISS)3 with incidence of CNVs, SNVs, and translocations4. This ISS-MUT model increases precision over ISS alone in detecting early mortality and progression. Other studies have highlighted the context-dependent prognostic significance of variants across the spectrum of mutation types1. For example, trisomies of chromosomes 3 or 5 have been found to abrogate the poor overall survival associated with t(4;14) translocations5. Collectively, these results highlight the prognostic impact of the interplay between CNVs, SNVs, and translocations—the potential for co-occurring mutations to mitigate an otherwise poor outcome implies that testing a patient for all three types of mutations may help target chemotherapy regimens more accurately to specific patient subgroups in the future2.
Detecting myeloma-relevant mutations may be done via exome sequencing, as in the ISS-MUT study4. However, approaches targeting a subset of disease-associated genes should reduce computational analysis, facilitate quicker return of clinical results, and enable deeper sequencing at a fixed budget6. Indeed, targeted, clinical sequencing is performed with increasing frequency both commercially7 and through cancer centers8–10. In the specific context of MM, an amplicon-based, 77-gene panel detects both CNVs and SNVs11. This extends an earlier panel6 used to track mutation evolution across 47 genes12. Other efforts have focused on IGH rearrangements and translocations: amplicon-based sequencing of the locus effectively detects minimal residual disease13, while capture-based approaches have been used to discover IGH and MYC translocations14,15.
Similar enrichment technologies could be used to simultaneously detect CNVs, SNVs, and translocations. Indeed, one such platform was recently described—the approach involved targeted sequencing of the IGH locus and 246 genes implicated in MM and/or other cancers16. The platform was used to profile 14 MM cell lines and five primary samples. Maturing such approaches will require extensive validation and tuning of the associated bioinformatics methods. Towards this end, we developed a related MM-specific targeted sequencing platform and validated it against 95 primary tumor samples, 44 of which were previously subjected to exome sequencing and 22 of which were previously assayed by fluorescence in situ hybridization (FISH). We demonstrate high concordance of the platform with these exome sequencing and FISH results. Additionally, we describe novel approaches to tuning computational CNV and translocation calling methods. These optimized methods facilitated integrative analysis across mutation types, which revealed patterns of mutual exclusivity and co-occurrence involving CNVs, SNVs, and translocations. We discovered novel translocations juxtaposing IGLL5 with IGH and detected high-frequency mutations in IGLL5. These IGLL5 mutations were mutually exclusive of RAS mutations and associated with disease progression, suggesting that IGLL5 may be involved in disease pathogenesis and/or serve as a biomarker of high-risk MM.
Methods
We designed a Nimblegen probe set (Roche) targeting 3.3 Mb of space that includes 465 genes and the IGH region. Sequencing library pools were prepared, hybridized to the probes, and sequenced on the HiSeq2000 (2 × 100 reads for initial sequencing of 95 tumor-normal pairs) or the HiSeq2500 (2 × 125 reads for deep sequencing of a subset of 15 pairs). Reads were aligned against human reference genome GRCh37-lite using BWA17. Acquired SNVs were called in each tumor sample relative to its paired normal sample using samtools18, SomaticSniper19, MuTect20, Strelka21, and VarScan2 (ref. 22). Translocations were called using LUMPY23, with results filtered by a machine learning approach optimized to achieve high precision relative to available FISH results. CNVs were called using CopyCAT2 (https://github.com/abelhj/cc2/; ref.24) parameterized to detect copy number alterations exceeding the level of noise estimated from diploid regions using a gaussian mixture model (https://github.com/genome/bmm). Additional information is provided in Supplementary Methods.
Results
We developed a MM-specific custom capture sequencing platform capable of detecting CNVs, SNVs, and translocations. We designed oligonucleotide probes covering 3.3 Mb of genomic space and complementary to the exons, untranslated regions, and splice sites of 465 genes (Tables S1 and S2) expressed in MM that: (1) are annotated as cancer genes (in COSMIC25 or MutSig26), (2) function in DNA repair or B-cell biology, (3) are mutated at a frequency of >3% in published studies27,28, or (4) have mutations that cluster in hotspots. To detect IGH translocations, we also designed probes tiled in an unbiased manner across the locus, including within the variable (IGHV), diversity (IGHD), joining (IGHJ), and constant/switch regions. We also designed probes targeting the exonic regions of canonical IGH translocation partners (CCND1, CCND3, FGFR3, MAF, MAFB, WHSC1, and WWOX). To capture secondary MYC translocations, we tiled probes across exonic and intronic regions of the MYC locus.
We used the platform to sequence DNA isolated from 95 tumor (CD138-purified cells isolated from bone marrow aspirates) and paired normal (blood) samples. These samples were specifically selected to validate our platform and to tune our computational methods as a subset of them were previously subjected to exome sequencing (44 samples) and/or FISH (22 samples) analysis of IGH translocations. We achieved a mean sequencing depth of 104× (min = 33×, max = 140×; Supplementary Figure S1A) across the tumor samples and of 107× (min = 43×, max = 168×; Supplementary Figure S1B) across the normal samples.
Targeted capture sequencing identifies copy number alterations with prognostic significance
The broad coverage of the platform [across chromosome arms (Table S3) and 465 genes] facilitated detection of chromosome-level, arm-level, and focal CNVs. We identified these events computationally from the per-probe ratios of tumor to normal sequencing depth using CopyCAT2 (ref.24). To reduce false positives, we developed an approach that filtered CNV calls with ratios below a noise level estimated from diploid regions (Supplementary Figure S2A–C). Our approach identified the full range of CNVs, from genome-scale hyperdiploid events (Fig. 1a; Table S4) to focal events, including a homozygous deletion that encompassed BRCA2 (Fig. 1b). Detected arm-level events included those associated with poor prognosis such as amp(1q), del(1p), del(13q), and del(17p) (reviewed in ref.1; Supplementary Figure S2D).
Fig. 1. Targeted sequencing identifies chromosome-level, arm-level, and focal CNVs.

a Hyperdiploid and (b; arrow) focal copy number events detected by CopyCat2 (blue; p < 0.05) from log2 ratios of tumor to paired normal sequencing depth (y-axis) across chromosomes (x-axis). (Clonal) single-copy gains occur at a log2 ratio of log2(3/2) =0.58, whereas (clonal) heterozygous/single-copy losses occur at a log2 ratio of log2(1/2) = −1
Targeted capture sequencing identifies IGH translocations
We detected IGH translocations using LUMPY and again developed a filtering strategy to reduce likely false positives. We filtered putative translocations based on thresholds on the number of supporting split reads and discordant paired-end reads. We tuned the thresholds to maximize precision using a machine learning approach and available FISH data, resulting in a precision of 100% and a recall of 64% (Supplementary Figure S3; Supplementary Table S5). Canonical IGH translocations were then detected by the platform near expected frequencies (Table 1; Fig. 2a) and occurred predominantly within the IGH constant region, but also telomeric of the IGHM switch region and occasionally within the D and J regions (Fig. 2c). Notably, one of the t(11;14) translocations occurred within the constant region, but outside all constant and switch segments. These results are consistent with a recent study15, which found IGH translocations occurring within each of these regions. No translocations within the V region passed the filtering step.
Table 1.
Frequency of Translocations
| Transloaction | Frequency (%) |
|---|---|
| t(11;14) | 14 |
| t(4;14) | 13 |
| t(8;14) | 7 |
| t(6;14) | 2 |
| t(14;20) | 2 |
Fig. 2. Targeted sequencing detects IGH and MYC translocations.

Circos plots of (a) IGH and (b) MYC translocations. Chromosomes involved in translocations are magnified to highlight regions and genes near breakpoints. c Breakpoints (vertical lines) of canonical IGH translocations within IGH locus. E3A2 and E3A1: 3’ enhancer elements downstream of IGHA2 and IGHA1 genes, respectively. Eμ: μ enhancer. Purple boxes: switch regions. Figure is to scale
IGLL5 is translocated and co-incident with overexpression of DERL3 in multiple myeloma
To prioritize novel IGH translocations as potential driver mutations, we identified cancer-associated genes within 1 Mb of each chromosomal breakpoint (Supplementary Table S6). The two annotated translocations with largest total evidence (sum of number of supporting split reads and number of discordant paired-end reads) were analyzed further. The first was a complex translocation involving chromosomes 11, 13, and 14. The putative breakpoint on chromosome 13 was nearby FLT3 (<0.5 Mb; Supplementary Figure S4); we validated that chromosome 13q12.2 was indeed translocated to IGH on chromosome 14 using PCR.
Breakpoints of the second highly-supported translocation, t(14,22)(q32.33;q11.22), were located within IGH and IGLL5, which is spanned by the immunoglobulin lambda light chain locus (Fig. 3a). To validate this translocation, we performed PCR amplification of the putative breakpoint on DNA isolated from the patient in which it was detected. A PCR product of the expected size was detected in CD138+ tumor cells but not in the peripheral blood mononuclear control [Fig. 3b (top)]. Re-sequencing and mapping of the tumor-specific PCR product confirmed the reciprocal translocation spanned chromosomes 14 and 22. Small regions were deleted on both derivative chromosomes and thus could be used to selectively amplify the corresponding wild-type (WT) chromosomes. We designed primers within these deleted regions and used them to perform PCR amplification, which confirmed the retention of one copy of each of the WT chromosomes in the tumor sample [Fig. 3b (bottom)].
Fig. 3. IGLL5 is translocated and DERL3 is overexpressed in multiple myeloma.

a Schematic of validated t(14;22) translocation. (Left) WT chromosomes 14 and 22 with horizontal lines indicating location of breakpoints within the IGH and IGLL5 loci, respectively. (Right) Two derivative (der) chromosomes, each retaining a portion of its respective IGH or IGLL5 gene. Cancer-associated genes within 1 Mb of breakpoint on der(14) are shown. b PCR validation of t(14,22) translocation. Oligos specific to each breakpoint used in PCR reactions (top). Oligos specific to the small regions deleted on der(14) and der(22) were designed to detect non-translocated allele (bottom). T tumor, N germline (peripheral blood) control. cDERL3 expression across 84 MM patients. Red circle indicates sample in which putative (non-validated) t(14;22) translocation was detected. FPKM Fragments Per Kilobase of transcript per Million mapped reads
To search for additional IGLL5 translocations, we relaxed our filtering constraints and found a second sample predicted by LUMPY to harbor a t(14;22) translocation, though no DNA was available for validation. The validated t(14;22) translocation was predicted to juxtapose the μ and 3’ enhancers (chr14:106032614–106167601) with DERL3. Hence, we looked for evidence of overexpression of DERL3 and other cancer-associated genes within 1 Mb of the predicted breakpoint on chromosome 22 (IGLL5, BCR, and SMARCB1) by examining RNA-seq expression data from a partially overlapping set of 84 MM patients. We found outlying expression of DERL3 in six of these samples (exceeding 1.5× the FPKM interquartile range), including the second sample with a putative t(14;22) translocation (Fig. 3c; no expression data were available for the sample harboring the validated translocation). Additionally, we found that DERL3 was overexpressed in MM relative to other cancer types within the Cancer Cell Line Encyclopedia (CCLE)29 (Supplementary Figure S5), with high expression also reported in other B-cell malignancies. We also examined expression of IGLL5 both within our cohort and CCLE. IGLL5 expression was in the 82nd percentile in the putatively translocated sample for which we had RNA-seq data, though it is not noticeably elevated with respect to the mean expression (data not shown). IGLL5 expression in MM cell lines within the CCLE is second only to Burkitt’s Lymphoma cell lines; more generally, and as with DERL3, IGLL5 expression is higher in B-cell malignancies than in other cancer types. Taken together, these data suggest that DERL3 may be dysregulated in MM via IGH translocation or another unknown mechanism.
Targeted capture sequencing identifies intra- and inter-chromosomal MYC translocations
FISH validation data of MYC translocations were not available to tune LUMPY parameters and, as a result, intra- and (non-IGH) inter-chromosomal MYC translocations were called at a high false-positive rate (in every tumor and normal sample, Supplementary Figure S6). To accurately detect somatic MYC translocations, we developed a machine learning-based approach tuned to filter putative MYC translocations called in normal samples. Applying this method to tumor samples resulted in five intra-chromosomal and two non-IGH inter-chromosomal MYC translocations, with one sample having one intra- and one inter-chromosomal translocation (6 of 95, 6%, Fig. 2b; Supplementary Table S7). The intra-chromosomal translocations involved neighboring genes PVT1 and POU5F1B, as previously reported14.
Targeted capture sequencing identifies non-silent single-nucleotide variants in all tumor samples
All tumor samples harbored at least one somatic (missense, nonsense, or frame-shift) mutation, with each sample having a mean of 20 mutations (Supplementary Table S8). A total of 443 genes had a non-synonymous (frame-shift insertion or deletion, missense, or nonsense) mutation in one or more samples; 581 genes had a mutation of any kind in one or more samples. Ninety-four of 95 tumor samples had a mutation predicted to be deleterious by Poly-Phen230 or SIFT31, with each sample having a mean of twelve deleterious mutations. In 24 instances, we observed a gene harboring multiple mutations previously associated with cancer (via COSMIC). This occurred in thirteen samples across seventeen genes, including KRAS and RB1; both were among the most frequently observed (in three samples).
Increased sequencing depth yields few additional variants
To determine whether MM is characterized by deeply subclonal variants of biological significance, we performed additional sequencing of 15 tumor (mean depth = 1,259×, min = 506×, max = 1,660×) and paired normal (mean = 1,326×, min = 763×, max = 1,727×) samples. We then compared the allele frequencies of variants discovered during the original and/or subsequent deep sequencing (Fig. 4). To focus on high-confidence events likely to be of biological relevance, we removed silent variants, those in intronic, intergenic, or flanking regions, those in IGH (and, hence, likely arising due to somatic hypermutation), or those that were flagged as likely germline variants by at least one caller in at least one study. This resulted in 57 variants in the original sequencing study (mean depth = 92×) and 67 variants in the subsequent study (mean depth = 1,169×). Variant allele frequencies (VAFs) of mutations shared across the two studies were highly correlated (R2 = 0.80; p < 2.2e-16). As expected, the vast majority of variants unique to either study had low VAFs: one of the four variants unique to the original study had a VAF <10%, though all had an alternate allele count of three or fewer supporting reads, while 12 of the 14 variants unique to the subsequent study had a VAF <10%. Though relatively few new variants were discovered by the additional sequencing, these did include several annotated in COSMIC in genes KRAS, HECW1, and ZFHX4.
Fig. 4. Greater sequencing depth yields few additional variants.

VAF of variants discovered during initial targeted sequencing (x-axis) and/or with subsequent deeper sequencing (y-axis)
These results were recapitulated in a comparison of the variants discovered in 44 samples subjected to capture-based sequencing (mean depth of 1,562 capture-based variants = 123×) and previously to exome sequencing (mean depth of 3,563 exome variants = 136×; Supplementary Figure S7). As in our above comparison, the 1,450 mutations discovered in both studies had highly correlated VAFs (R2 = 0.85; p < 2.2e-16). Again, the majority of variants unique to one study had low VAF: 79 of 112 variants unique to the capture-based study (Supplementary Fig. S7C) and 2,066 of the 2,113 variants unique to the exome-based study (Supplementary Figure S7B) had VAFs <10%.
To further explore the effects of sequencing depth, we downsampled the sequencing reads from the 15 deeply-sequenced samples, called variants on the downsampled reads, and plotted the total number of variants following filtering (as above) in the downsampled and full data set (Supplementary Figure S8). As expected, the number of variants was correlated with sequencing depth (R2 = 0.52; p = 10−3). However, beyond ~25% of the final sequencing depth (or a mean depth of ~300×), the increase in number of discovered variants is marginal. Similar performance was observed at an even lower depth of 92× in the aforementioned case of the 15 original capture results later re-sequenced. Together, these results indicate that depths as low as 100× can capture the majority of variants of interest and that coverage beyond 300× will lead to sharply diminishing returns.
Targeted capture sequencing facilitates integrative analysis across mutation types
Integrated analysis of CNVs, SNVs, and translocations highlights patterns of mutual exclusivity and co-occurrence both within and across mutation types (Fig. 5). We tested for significance of these patterns after excluding the apparent hypermutator sample (leftmost column; Fig. 5a) to improve statistical power. This revealed mutation co-occurrence (blue; Fig. 6; Table S9) within CNVs [i.e., of del(6q) with del(16q) and amp(1q) and of del(13q) with amp(1q) and del(14q)] and involving CNVs and translocations [i.e., of del(14q) with t(4;14)]. As expected, we detected mutual exclusivity (red; Fig. 6; Table S9) between hyperdiploidy and t(11;14). We also detected cross-mutation type exclusivity between CNVs and SNVs [i.e., both RAS mutations (i.e., KRAS or NRAS) and FAM46C are mutually exclusive with del(6q)]. IGLL5 was the third most frequently mutated gene in our data set (Fig. 5b; 18%), with (silent and non-synonymous) IGLL5 mutations enriched for a c-AID signature (i.e., C to T/G mutation at WRCY motifs; p = 2.7 × 10−6; binomial test). Thirty-eight of the 40 detected IGLL5 SNVs occurred in amino acid positions one to 100, outside of any annotated protein domains. The remaining two (K166N and K189R) occurred in the immunoglobulin C1-set domain. Mutations in IGLL5 were mutually exclusive of RAS mutations (p = 0.006), with trends toward mutual exclusivity with KRAS (p = 0.054), NRAS (p = 0.111), and FAM46C (p = 0.113), independently (Figs 5 and 6). IGLL5 mutations in diploid loci had a median VAF of 58% and a first quartile VAF of 39%, suggesting that the majority are likely clonal (Supplementary Figure S9). Finally, we found that IGLL5 SNVs are associated with disease progression [Fig. 7; hazard ratio = 1.46 (95% confidence interval: 1.03–2.08); p = 0.03 (log-rank test)].
Fig. 5. Targeted sequencing identifies CNVs, SNVs, and translocations.

a Mutations per Mb, b SNVs, and c CNVs and translocations detected across 95 samples (columns). MYC-ITX: intra-chromosomal MYC translocations; MYC-CTX: inter-chromosomal MYC translocations; Non MYC-IGH CTX: inter-chromosomal IGH translocations, excluding those involving MYC
Fig. 6. Targeted sequencing identifies co-occurrence and mutual exclusivity across mutation types.
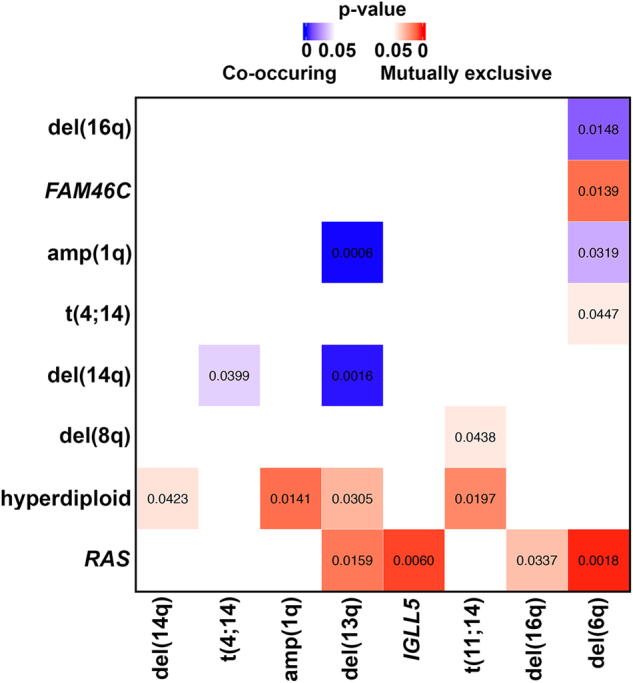
Co-occurring (blue) and mutually exclusive (red) mutations (p < 0.05). Numbers indicate p-values
Fig. 7. IGLL5 mutations are associated with increased risk of disease progression.

Kaplan-Meier curves of IGLL5 mutant samples (with non-synonymous SNVs and/or indels) versus IGLL5 WT samples
Discussion
We developed a MM-specific, capture-based sequencing platform targeting 465 genes that detects CNVs, SNVs, and translocations. We used the platform to sequence 95 tumor/normal pairs and validated its ability to detect SNVs and translocations using exome sequencing and FISH data. After developing custom computational approaches for reducing false positives, we detected CNVs (Fig. 1) and known IGH translocations at expected frequencies (Fig. 2; Table 1) and discovered rare novel chromosomal translocations (Fig. 3). By deeply sequencing 15 pairs, we showed that there is a marginal increase in number of non-silent variants discovered with sequencing depth (Fig. 4). Hence, many scientific and some clinical goals may be achievable with the more modest sequencing depth (~100×) used for the complete cohort of 95 pairs.
We detected (intra- and inter-chromosomal) MYC translocations at a frequency of 13% [95% CI (7%-20%); 12 of 95; Fig. 2; Tables S5 and S7]. This is generally consistent with prior studies, though a considerable range of frequencies has been published. Such discrepancies are likely due in part to differences in sample size, disease stage, assay, and assay optimization/tuning. An early study using FISH reported complex MYC abnormalities in 50% of primary tumors—however, the analyzed samples were few [7 of 14; 95% CI (26%-74%)] and advanced (stage III)32. MYC translocation frequencies more similar to those we observed have been detected in newly diagnosed MM patients using both FISH—15% [79 of 529; 95% CI (12%-18%)]33 and 23% [62 of 274; 95% CI (18%-28%)]34,35—and a capture-based next-generation sequencing approach similar to that presented here—20% [21 of 104; 95% CI (13%-28%)]14.
Our data revealed that IGLL5 is mutated via both point substitutions and translocations in MM (Figs 2, 3 and 5). The SNVs within the locus likely result from somatic hypermutation of the immunoglobulin lambda light chain locus, which spans IGLL5. Indeed, we saw an enrichment for AID-induced mutations within IGLL5, as has been previously observed in chronic lymphocytic leukemia (CLL), where the gene is frequently mutated36. We found that non-synonymous IGLL5 mutations are both mutually exclusive with RAS mutations (Fig. 6) and associated with disease progression (Fig. 7) and, with a median VAF of 58%, are likely present in the founding clone. Together, these findings suggest that IGLL5 mutation may contribute to myeloma pathogenesis, but without additional data their functional significance remains unknown. The fact that mutations and translocations do not recurrently affect specific residues in the IGLL5 protein supports a loss of function model, but an alternative hypothesis is that IGLL5 mutations are simply a biomarker for high-risk disease.
We observed that IGLL5 was translocated to the IGH locus in two patient samples (Figs. 2 and 3). Translocations between IGH and either of the two light chain loci have been reported in B cells and their associated disorders: IGL/IGH translocations have been reported in non-Hodgkin lymphoma37 and in activated B cells38 where it was suggested that the translocations resulted from deficiencies in non-homologous end-joining that induced both V(D)J-recombination-associated breaks at the IGL locus and class switch recombination-associated IGH breaks. The authors speculated that, since the translocations conferred no obvious selective advantage, these translocations simply reflected the mechanistic opportunity presented by two frequently-broken and spatially-proximal loci38. IGH translocations partnered instead with IGK have been detected in B-cell lymphomas39 and in a patient-derived B lymphoblastoid cell line40 with the latter plausibly attributable to the patient’s prior, long-term treatment with a DNA-damaging alkylating agent. The possibility remains that the translocations we detected play a functional role by co-opting a large super-enhancer downstream of IGLL5, which was found to be active within the MM1.S MM cell line41. Its position between IGLL5 and BCR place it on the der(14) chromosome of the t(14;22) translocation we validated. Hence, this super-enhancer may amplify the effect of the IGH enhancers in upregulating the expression of nearby genes translocated to chromosome 14. Indeed, we observed upregulation of one such gene, DERL3, across MM tumors relative to other cancer types, generally (Supplementary Figure S5), and in the sample with the second putative t(14;22) translocation, specifically (Fig. 3). Overexpression of DERL3 could play a role in MM by increasing endoplasmic reticulum (ER)-associated degradation within the proteasome. DERL3 forms an export channel in the ER through which misfolded proteins fated for degradation reach the proteasome42. The proteasome inhibitor bortezomib is highly efficacious in MM cells. Hence, were it to instead promote, rather than inhibit, activity of the proteasome, DERL3 overexpression might confer a selective advantage to MM cells. Future experiments are required to decipher the importance of IGLL5 translocations and the significance of DERL3 overexpression.
In addition to detecting known mutations, the platform was designed to enable discovery: (1) We queried 465 genes, a much larger set than assayed by previous targeted platforms6,11,16, and (2) we tiled across the entire V, D, and J regions, as opposed to restricting probes to annotated segments within these regions15 in an attempt to detect translocations involving inter-segment regions of the locus. Additional experience will be necessary to evaluate these design decisions. For example, lack of inferred translocations involving the J region may suggest that probes used to tile across this ~700 Kb region are better invested elsewhere. In particular, future versions of the platform should provide currently lacking coverage of chromosome arm Yq, though the tandem-rich arms of acrocentric chromosomes (13p, 14p, 15p, 22p, and Yp) will remain difficult to capture. Additionally, targeting probes to common SNPs would enable detection of allele-specific ploidy at a given locus43, which would facilitate SNV-based inference of clonal evolution44.
Mutation4 and expression45 data, coupled with ISS stage, have been shown to improve prognostic accuracy over ISS stage alone. Indeed, two recent crowd-sourcing competitions [a DREAM challenge (https://www.synapse.org/#!Synapse:syn6187098/wiki/401884; refs46,47 and a Topcoder challenge (http://crowdsourcing.topcoder.com/myeloma_predictor)] aim to improve MM patient stratification using genomic, transcriptomic/expression, cytogenetic, and clinical features. The ease of employing a single platform to assay CNVs, SNVs, and translocations would facilitate these and related efforts. Significantly, the same probes could be used to perform targeted RNA sequencing, which would also furnish these studies with comparative gene expression across samples.
Electronic supplementary material
Acknowledgments
Author contribution
B.S.W., I.L., J.O., H.G., and M.H.T. designed the experiments, performed research and wrote the manuscript; C.F., E.A.B., M.F., G.J.A., M.D., and J.K. performed research; R.S.F. and H.S. designed the experiments and performed research; E.R.M., R.V., J.F.D., and J.L. designed the experiments; D.A. designed the experiments and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
These authors contributed equally: Brian S. White, Irena Lanc.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41408-018-0062-y).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Manier, S. et al. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 14, 100–103 (2016). [DOI] [PubMed]
- 2.Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat. Rev. Cancer. 2012;12:335–348. doi: 10.1038/nrc3257. [DOI] [PubMed] [Google Scholar]
- 3.Greipp PR, et al. International staging system for multiple myeloma. J. Clin. Oncol. 2005;23:3412–3420. doi: 10.1200/JCO.2005.04.242. [DOI] [PubMed] [Google Scholar]
- 4.Walker BA, et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J. Clin. Oncol. 2015;33:3911–3920. doi: 10.1200/JCO.2014.59.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chretien ML, et al. Understanding the role of hyperdiploidy in myeloma prognosis: which trisomies really matter? Blood. 2015;126:2713–2719. doi: 10.1182/blood-2015-06-650242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kortum KM, et al. Targeted sequencing using a 47 gene multiple myeloma mutation panel (M(3) P) in -17p high risk disease. Br. J. Haematol. 2015;168:507–510. doi: 10.1111/bjh.13171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frampton GM, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng DT, et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cottrell CE, et al. Validation of a next-generation sequencing assay for clinical molecular oncology. J. Mol. Diagn. 2014;16:89–105. doi: 10.1016/j.jmoldx.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garofalo A, et al. The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med. 2016;8:79. doi: 10.1186/s13073-016-0333-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kortuem KM, et al. Panel sequencing for clinically oriented variant screening and copy number detection in 142 untreated multiple myeloma patients. Blood Cancer J. 2016;6:e397. doi: 10.1038/bcj.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kortum KM, et al. Longitudinal analysis of 25 sequential sample-pairs using a custom multiple myeloma mutation sequencing panel (M(3)P) Ann. Hematol. 2015;94:1205–1211. doi: 10.1007/s00277-015-2344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez-Lopez J, et al. Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood. 2014;123:3073–3079. doi: 10.1182/blood-2014-01-550020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker BA, et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J. 2014;14:13. doi: 10.1038/bcj.2014.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walker BA, et al. Characterization of IGH locus breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pregerminal center B cells. Blood. 2013;121:3413–3419. doi: 10.1182/blood-2012-12-471888. [DOI] [PubMed] [Google Scholar]
- 16.Bolli N, et al. A DNA target-enrichment approach to detect mutations, copy number changes and immunoglobulin translocations in multiple myeloma. Blood Cancer J. 2016;6:e467. doi: 10.1038/bcj.2016.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larson DE, et al. SomaticSniper: identification of somatic point mutations in whole-genome sequencing data. Bioinformatics. 2012;28:311–317. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cibulskis K, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saunders CT, et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 22.Koboldt DC, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15:R84. doi: 10.1186/gb-2014-15-6-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sehn JK, Abel HJ, Duncavage EJ. Copy number variants in clinical next-generation sequencing data can define the relationship between simultaneous tumors in an individual patient. Exp. Mol. Pathol. 2014;97:69–73. doi: 10.1016/j.yexmp.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 25.Forbes SA, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 499, 214–218. [DOI] [PMC free article] [PubMed]
- 27.Bolli N, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014;5:2997. doi: 10.1038/ncomms3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chapman MA, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar P, Henikoff S, Fau—Ng PC, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 32.Shou Y, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc. Natl Acad. Sci. USA. 2000;97:228–233. doi: 10.1073/pnas.97.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Avet-Loiseau H, et al. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood. 2001;98:3082–3086. doi: 10.1182/blood.V98.10.3082. [DOI] [PubMed] [Google Scholar]
- 34.Weinhold N, et al. Concomitant gain of 1q21 and MYC translocation define a poor prognostic subgroup of hyperdiploid multiple myeloma. Haematologica. 2016;101:e116–e119. doi: 10.3324/haematol.2015.136929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neben K, et al. Administration of bortezomib before and after autologous stem cell transplantation improves outcome in multiple myeloma patients with deletion 17p. Blood. 2012;119:940–948. doi: 10.1182/blood-2011-09-379164. [DOI] [PubMed] [Google Scholar]
- 36.Kasar S, et al. Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat. Commun. 2015;6:8866. doi: 10.1038/ncomms9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aamot HV, Bjornslett M, Delabie J, Heim S. t(14;22)(q32; q11) in non-Hodgkin lymphoma and myeloid leukaemia: molecular cytogenetic investigations. Br. J. Haematol. 2005;130:845–851. doi: 10.1111/j.1365-2141.2005.05688.x. [DOI] [PubMed] [Google Scholar]
- 38.Wang JH, et al. Mechanisms promoting translocations in editing and switching peripheral B cells. Nature. 2009;460:231–236. doi: 10.1038/nature08159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Subramaniyam S, Geyer JT, Liu YC, Mathew S. A translocation t(2;14)(p11.2; q32) involving rearrangements of immunoglobulin heavy chain and kappa light chain genes in B-cell lymphoma. Leuk. Lymphoma. 2015;56:2992–2994. doi: 10.3109/10428194.2015.1018249. [DOI] [PubMed] [Google Scholar]
- 40.Kennedy MA. Novel chromosome translocation caused by fusion of immunoglobulin heavy and light chain V genes in a human B lymphoblastoid cell line. J. Exp. Med. 1991;173:1033–1036. doi: 10.1084/jem.173.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loven J, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oda Y, et al. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J. Cell. Biol. 2006;172:383–393. doi: 10.1083/jcb.200507057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Loo P, et al. Allele-specific copy number analysis of tumors. Proc. Natl Acad. Sci. USA. 2010;107:16910–16915. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roth A, et al. PyClone: statistical inference of clonal population structure in cancer. Nat. Methods. 2014;11:396–398. doi: 10.1038/nmeth.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuiper R, et al. Prediction of high- and low-risk multiple myeloma based on gene expression and the International Staging System. Blood. 2015;126:1996–2004. doi: 10.1182/blood-2015-05-644039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mason N, et al. Multiple Myeloma DREAM Challenge: A crowd-sourced challenge to improve identification of high-risk patients. Cancer Research. 2017;77:4725–4725. doi: 10.1158/1538-7445.AM2017-4725. [DOI] [Google Scholar]
- 47.Dervan AP, et al. Crowdsourcing a High-Risk Classifier for Multiple Myeloma Patients. Blood. 2017;130:265. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.