

Abstract
Alcoholic hepatitis is the most severe and acute form of alcoholic liver disease. The mortality rate associated with alcoholic hepatitis is high, largely due to the lack of suitable pharmacological interventions. While there has been substantial research in the area, generating pharmacological interventions has been plagued by the lack of a robust mouse model both for testing and for understanding the underlying pathology. A number of major notable advances have been made in this area recently, with the goal of generating a mouse model of alcoholic hepatitis. The purpose of this article is to review recent advances in modeling alcoholic liver disease both in vitro and in vivo in the mouse, and place them in the context of the greater spectrum of alcoholic liver disease, with a focus on how we can translate current advances into a high-fidelity model of alcoholic hepatitis. In addition, we will review the basic mechanisms of alcoholic hepatitis as it is currently understood, focusing on recent advancements in diagnosis, prognosis and current pathophysiology, especially as it relates to the profound immune dysfunction present during alcoholic hepatitis.
Keywords: Alcoholic hepatitis, Alcohol, Liver disease, Cirrhosis, Prognosis, Keratin-18
Introduction
Alcoholic hepatitis (AH) is a type of acute-on-chronic liver failure associated with alcoholism and which has a high mortality rate.1 Corticosteroid treatment is still the gold standard pharmaceutical intervention, but has minimal efficacy.2 Orthotopic liver transplantation is the most effective cure, but there is a moral conundrum associated with giving valuable livers to patients with ongoing alcoholism, especially given the high rates of recidivism in the population.3 Moreover, the number of livers available for transplantation is limited compared to the number of AH patients in most regions, and more information is necessary to fully understand what populations respond positively to transplantation.4–6 As such, pharmacotherapy is urgently needed for this patient population.
A number of clinical trials or pilot studies have tried agents that target inflammation or inflammatory cell death, based on studies performed in the mouse model.2,7–9 There have been a few cases of positive results, and none of these agents have been translated into a legitimate clinical therapeutic candidate. This is partially due to the incredible complexity of AH pathology; however, another increasingly well understood factor is the lack of suitable models for generating therapeutic candidates for late stage alcoholic liver disease (ALD).1,10–12 As such, a major imperative in the basic science of ALD remains the generation of a high-fidelity mouse model of AH, or advanced ALD. In this review, we will attempt to bridge the gap between the mouse model and clinical AH, by defining current obstacles and highlighting innovative recent solutions to this growing dilemma.
Acute alcoholic hepatitis – Much more than just steatosis and inflammation
Understanding AH as a clinical disorder is necessary to obtain mouse models that accurately model the disease. AH patients commonly present to the hospital with one or more co-morbidities, including jaundice, esophageal varices, or fever due to co-infection.13 Underlying cirrhosis can remain silent for years prior to these precipitating events, but is present in up to 80% of AH patients.14 An overwhelming majority present with some degree of fibrosis, indicating significant chronic liver disease.14 As such, the largely silent nature of ALD in human patients is a major hurdle for early diagnosis, and a major contributor to the associated morbidity and mortality.1,15
Histologically, patients with AH commonly present with marked fibrosis, steatosis and cholestasis or bilirubinostasis in the liver.14 Patients also commonly present with elevations in plasma bilirubin, plasma creatinine and the international normalized ratio (INR), consistent with major liver and kidney dysfunction. Combinations of INR ratio, plasma creatinine and plasma bilirubin levels, make up the bulk of a number of prognostic batteries that will be discussed later in this review.16–18 As such, the major dysfunction associated with AH could be classified as an abrupt lack of liver function, that is associated with major underlying liver disease, severe inflammation and a faulty homeostatic balance of functional hepatocytes.
Surprisingly, one factor that is rarely elevated is the common marker of hepatocyte injury, plasma alanine aminotransferase (ALT). Patients with AH typically have very minor elevations in ALT, commonly less than 1.5-fold higher than the upper limit of normal. While aspartate aminotransferase (AST) values can rise as high as 200–300 IU/L, values much above this are typically considered diagnostically unlikely to be AH, and mean values in large studies are typically closer to 90–150 IU/L.2,14 AST activities are commonly elevated and AST>ALT is a common diagnostic indicator of AH versus other types of liver injury.19
Given that AST is expressed in multiple organ systems other than liver, the AST>ALT ratio may be indicative of cell death in other organ systems. These relatively low values of AST/ALT suggest only minor levels of necrosis occur in AH patients, which is supported by data that a majority of cell death during AH appears to be through apoptosis.9,20 Even still, measurements of apoptosis return very low levels of overall cell death.21 TUNEL stains on AH biopsies indicate that AH patients undergo relatively minor levels of cell death, when compared to diseases with major acute parenchymal injury.9,21 While some degree of cell death certainly occurs during AH, these data call into question whether or not cell death plays a critical role in AH progression.21
Neither AST nor ALT plasma activities are strong predictors of outcome in AH patients.21 It should be noted, though, that ALT values may be partially suppressed due to an overall reduction of ALT activity in the liver of AH cirrhotics.22 The limited degree of cell death that occurs during AH is likely not a mitigating disease factor acutely; although, the chronic cell death that occurs during the course of ALD is still a potential driver of subsequent inflammation, fibrosis and eventual cirrhosis.
Liver regeneration, which typically occurs as a subsequent response to cell death, is also highly dysregulated in AH patients. Cirrhotic patients have regenerative nodules that are universally or widely positive for proliferating cell nuclear antigen, suggesting widespread attempts at hepatocyte proliferation.23 This is suppressed in AH patients though, as these patients depose laminin instead of fibronectin, which pushes progenitor cell expansion away from mature hepatocyte differentiation, towards the bile duct epithelial cell lineage.24 The enhanced proliferation of biliary epithelial cells also seen in AH patients is also observable in the number of liver progenitor cells proliferating, which both serve to reduce the number of mature hepatocytes available to perform normal liver functions.24,25 This lack of mature hepatocytes may be a major driving factor in the lack of liver function and the subsequent liver failure. Therapeutic interventions aimed at promoting the maturation of hepatocytes and the deposition of normal extracellular matrix are an interesting therapeutic avenue for increasing liver function in alcoholic cirrhotics and AH patients.
In addition to liver dysfunction, many patients also have dysfunction of other organ systems, such as kidney and gut, which is intrinsically linked to both multiorgan system dysfunction and survival.26–29 AH patients suffer from a dysfunctional immune response as well, including elevated plasma leukocyte counts which, paradoxically, does not result in an effective immune response, as these patients are at a dramatically increased risk of systemic inflammatory response syndrome.30 This may be due to a well-characterized immune cell dysfunction in AH patients including alterations in initiation of respiratory burst in neutrophils and monocytes, and a faulty myeloperoxidase enzyme in neutrophils from cirrhotics.31–33
The lack of a normal myeloperoxidase enzyme might explain why these patients also have a paradoxical lack of chlorotyrosine adducts associated with neutrophil-mediated liver injury, despite considerable neutrophil deposition and activation.34,35 The role of neutrophils in AH is still under debate, as some evidence points towards neutrophils being drivers of AH while other studies indicate the opposite.14,36–39 Given the extended time course of AH in patients, neutrophils may play a contextual role based on current patient status. Unfortunately, very limited information is available on the time course of neutrophil activity in ALD patients, as so few patients are identified during the early portion of the disease.12 A greater understanding of neutrophil and monocyte dysfunction during AH is necessary to better understand how and why these patients become susceptible to infection.
Diagnosis, prognosis and presumed mechanisms
AH occasionally represents a diagnostic dilemma for clinicians. The use of transjugular biopsy (TJB) is the recommended means of differentiating between diagnoses, as it can definitively provide an answer; although, a number of detractors accurately point out that biopsy comes with inherent risk in patients with bleeding susceptibility, and may only be of limited availability in rural settings.1,2,40 As such, The American Association for the Study of Liver Disease has not yet fully endorsed TJB.40 A number of plasma biomarkers have been investigated for when TJB is either unavailable or too risky, though.
Plasma values of keratin-18 are dramatically elevated in AH patients, and may serve as a means for diagnosing the AH population.41 While plasma keratin-18 values are typically measured in the context of cell death, AH patients have dramatic elevations without the typically associated increases in plasma transaminases, or lactate dehydrogenase (Fig. 1).21 Why this occurs remains a mystery, although it is currently under investigation.21 Transient elastography can also identify alcoholic cirrhosis (AC) with some specificity and sensitivity, although its use in AH is likely limited.42 Alcoholic steatohepatitis (ASH) test is a novel multiparameter test with good specificity, and specificity that may be useful in some patient populations or in centers with limited potential for biopsy.43 A highly sensitive and specific test for diagnosis of AH would be incredibly valuable, as it would preclude the need for TJB in highly at-risk patients, or in hospital settings where TJB is not immediately available.
Fig. 1. Ratio of K18 (M65) to plasma alanine aminotransferase (ALT) activity compiled from multiple studies.

K18:ALT was calculated in a series of studies on different clinically relevant liver diseases. Acetaminophen (APAP) data are derived from Antoine et al.99; obstructive cholestasis data are derived Woolbright et al.56; alcoholic cirrhosis (AC) and alcoholic hepatitis data are derived from Woolbright et al.21
Prognosis of AH has been more widely investigated, as a diagnosis of AH has a 28-day mortality rate of 25–40%.1 A number of metrics have been evaluated for their ability to predict mortality in the population. Perhaps the most common is Maddrey’s discriminatory function score based on prothrombin time and bilirubin levels.44 This is a modified metric, where scores above 32 indicate severe dysfunction and recommend the use of corticosteroids.44 The model for end-stage liver disease score was also adapted for use in AH and was reported to predict outcome more effectively than Maddrey’s, presumably due to taking into account kidney function, which is linked to survival.17,26 The Lille score is a dynamic scoring system and not only predicts outcome in patients but also serves to determine whether or not patients are responders or non-responders to corticosteroid treatment using multiple measurements over 7 days.18
Recently, combinations of these scores were shown to be even more effective.45 The ABIC scoring system is also widely reported in the literature and essentially is as effective as Lille or Maddrey’s.16 Recently, our own group and others have reported that plasma keratin-18 ratios may also be highly predictive of mortality, despite the fact that plasma keratin-18 levels in AH appear to be disproportional to traditional measures of liver cell death in plasma.21,46 The use of prognostic scores has been a major source of understanding of clinical AH. As direct intervention is cumbersome and risky in human patients, identifying markers associated with survival or non-survival is critical to understanding the disease.
A number of different factors associated with the immune response have been correlated with survival and non-survival. Interestingly, some of these values can be viewed as contradictory in nature (i.e. plasma IL-8 values are elevated in non-surviving AH patients), yet neutrophil infiltration seems to be important for survival.14,36,39 Hepatic expression of CXC chemokines, though, is also linked to poorer survival outcomes, suggesting hepatic inflammation is detrimental to outcome.37 The lack of clarity in these data have led to some dichotomy between the basic science fields and the clinical science fields with regards to the role of neutrophils in AH.
Neutrophil infiltration has long been a desired aspect of mouse models, due to their noted presence and presumed pathological significance in AH patients, and recent advancements were rightfully hailed for their incorporation of legitimate neutrophil infiltration into the model, achieved solely with the use of alcohol.47,48 The Gao-binge model is dependent on the presence of neutrophils for cytotoxicity, as prevention of neutrophil extravasation by depletion of E-selectin is strongly protective.48 Thus, a number of recent studies using this model have capitalized on this finding and demonstrated a reduction in neutrophil recruitment or activity, and suggested the immunomodulatory activity as a potential therapeutic for AH, despite the fact that histological evidence and clinical evidence indicates that global inflammatory suppression might be highly detrimental in some patients.7,39,49–51 As noted above, neutrophils very likely play multiple roles during the course of the disease and should be investigated contextually in the future.
Even so, other markers of survival act to reinforce each other. In the instance of cholestasis, a majority of research indicates that concurrent cholestasis in patients or murine models with alcoholic liver injury dramatically worsen outcomes. In murine models, ethanol feeding dramatically exacerbates cholestatic liver injury induced by bile duct ligation (Woolbright and Jaeschke, unpublished data). Plasma bile acid values are elevated in patients with AH.52 Total plasma bile acid levels, cholic acid levels and chenodeoxycholic acid levels also correlate with severity of liver disease in ALD patients.52 Concurrent cholestasis, plasma bilirubin levels, and low deoxycholate/glycodeoxycholate levels, indicative of intrahepatic or extrahepatic bile duct blockage, are all strongly associated with mortality; however, it is unknown whether this is a primary cause or a secondary effect of the lack of liver function.37,44,52,53 Bile acids are thought to be hepatotoxic in large concentrations, especially when hepatocytes are exposed to leakage from the biliary tracts.54–56 Bile acids themselves are also pro-inflammatory and may contribute to the inflammation seen in AH patients when there is advanced jaundice and cholestasis.56,57 More research is needed using patient samples to establish whether or not some of these effects are potentially causative or a byproduct of the loss of liver function.
Alcoholic liver disease in the mouse – Limited pathology
Mouse model – Mechanisms
A number of authoritative reviews have been written on mechanisms of alcoholic steatosis, alcoholic gut dysfunction, and the subsequent effects in mouse models of ALD.58–60 This review is an attempt to focus on translation of these findings into actionable targets, and thus these mechanisms will only be touched upon briefly.
Alcohol is widely considered to be unpalatable to most rodent species, and, thus, ad libitum administration to rodents rarely results in blood alcohol levels commensurate with human exposure or human disease.61 The limited increase in blood alcohol concentrations may also be due to faster metabolism of alcohol by rodents, and is likely a component of the reduced pathology observed in murine models. Regardless, alcohol is rapidly metabolized by the mammalian body through numerous pathways, but primarily through alcohol dehydrogenase and cytochrome 2E1, and in small quantities through glucuronidation.62–64 Ethanol is converted to acetaldehyde by alcohol dehydrogenase, and to acetic acid by aldehyde dehydrogenase.62 Some additional detoxification occurs through cytochrome P4502E1 (CYP2E1), through a similar pathway as alcohol dehydrogenase.63 Acetaldehyde is far more reactive than either ethanol or acetic acid and, thus, is a likely source of cellular stress in ALD patients.65
Long-term alcohol consumption alters NADH to NAD+ ratios through the alcohol dehydrogenase pathway, as this pathway uses NAD+ as a co-factor.66 This shift alters the activity of histone deacetylase sirtuin-1, which affects numerous signaling pathways related to lipid metabolism. This includes activation of sterol response element binding protein-1, which enhances lipogenesis, and a reduction in peroxisome proliferator activated receptor co-activator gamma signaling leading to reduced fatty acid oxidation and accumulation of short chain hydrocarbons.67,68 The effect of this simultaneous dysregulation of metabolism is considerable accumulation of triglycerides in hepatocytes, leading to the characteristic steatosis present in alcohol-treated animals or cells.66–68
Most current research is focused on these processes, and our understanding of this process is far greater than the subsequent events in the mouse. Unfortunately, while simple steatosis is present in a majority of AC and AH patients, the pathology associated with simple steatosis alone is minimal, and most patients present at a point considerably past steatosis (Fig. 2).69 Moreover, steatosis is rapidly resolved by abstinence, which remains a mainline therapy for ALD and AH, and, thus, the likelihood that addressing liver steatosis as an effective therapeutic target for AH is minimal, as it is self-resolving in compliant patients.70,71
Fig. 2. Disease severity in patients versus alcohol model progression.
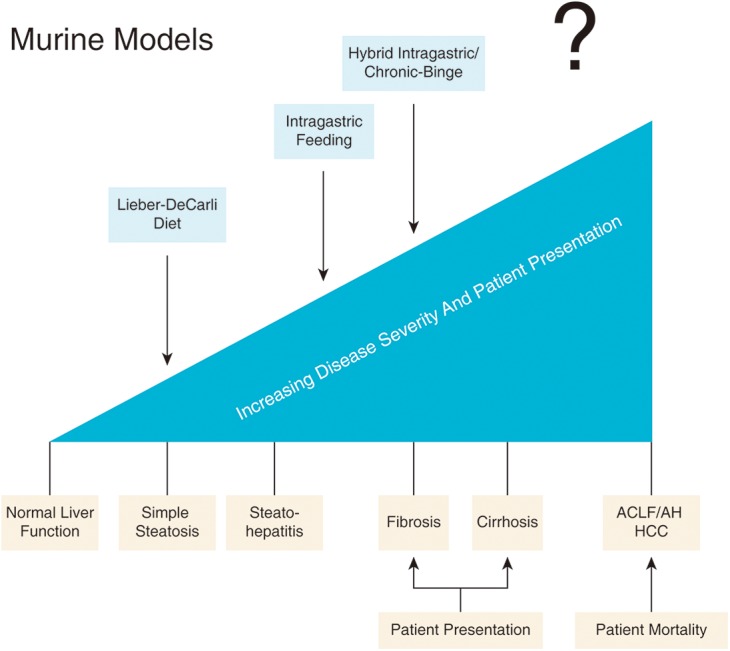
A majority of models fail to recapitulate the disease severity observed in AH patients.. Abbreviations: ACLF, acute-on-chronic liver failure; AH, alcoholic hepatitis; HCC, hepatocellular carcinoma.
Considerable oxidative stress builds rapidly upon chronic alcohol exposure. This is likely from multiple sources, including Kupffer cell-mediated oxidative stress, mitochondrial dysfunction in hepatocytes and the production of reactive metabolites.72–74 It has been hypothesized that CYP2E1 can be a relevant source of reactive oxygen species (ROS) during alcohol exposure; however, others have questioned this hypothesis and suggested a role of NADPH oxidase in Kupffer cells as the main source of alcohol-induced oxidant stress.75 Regardless, a number of different products from ROS-mediated damage are measurable after alcohol exposure, including 4-hydroxynonenal and 3-nitrotyrosine.76 These species likely contribute to cell death after alcohol consumption, but also activate measures such as autophagy.77–78
As autophagy removes both damaged mitochondria and lipid droplets, this is likely a protective measure against ROS-damaged species in alcohol-fed mice.77 Selective mitophagy, or the removal of damaged mitochondria, through the protein parkin is protective against alcohol-induced liver injury, as knockout of parkin dramatically exacerbates injury in alcohol-fed mice.78 This is due to preservation of normal mitochondrial function and removal of mitochondria damaged by excessive ROS production and alcohol metabolism.79 It is currently unknown as to whether selective or non-selective autophagy also occurs in human patients; although, this might be another potential therapeutic avenue for reducing cell death and enhancing hepatocyte function.
Simultaneous to liver dysfunction, alcohol causes severe gut dysfunction.80 The “leaky gut hypothesis” was an initial explanation as to why lipopolysaccharide (LPS) levels were elevated in alcoholics and alcohol-exposed mice.81 Chronic alcohol exposure causes intestinal overgrowth of bacteria and tight junction dysfunction in the gut.82 The combination of these two factors allows for bacteria to spill out from the gut, which increases plasma LPS levels.80 In human cirrhotics, this effect can be ameliorated by gut sterilization with high doses of rifaximin, designed to prevent the generation of ammonia and reduce hepatic encephalopathy.60,83
Elevated LPS levels in the mouse model activate toll-like receptor-4 (TLR-4) on Kupffer cells.84 This results in production of tumor necrosis factor-α (TNF-α) and release of ROS mediated by NADPH, which induces cell death in hepatocytes and provokes inflammation.84,85 Ablation of TLR-4 expression is protective against injury in the mouse model.84 The role of TLR-4 has been confirmed repeatedly in subsequent studies by multiple groups.85 This initially led to TNF-α blockers being assessed in patients; however, the clinical results were disappointing, as the blockade of inflammation led to sepsis in the patient population and higher mortality.7 Surprisingly, in most models of pure alcohol administration, the TNF-α release does not result in overt neutrophil accumulation, fibrosis or cirrhosis.12 Administration of other agents is typically required, and represents a major hurdle for generating a high-fidelity mouse model.
The mouse model of ALD – What it is and what it is not
The rodent model of ALD has evolved since the original experiments from DeCarli and Lieber identifying a liquid diet that was capable of inducing steatosis in the mouse.86 While DeCarli and Lieber used rats, the existence of a remarkable number of tools in the mouse has led to its surge as the primary animal model of use in the laboratory. Four- to six-week feeding with the liquid diet induces steatosis and mild elevations in plasma transaminase activity consistent with very early stage ALD. Extended time frames, the addition of different pathogenic molecules (such as LPS to enhance inflammation or carbon tetrachloride (CCl4) to induce fibrosis), the use of binge dosing on top of subacute dosing, direct intragastic infusion, and hybrid alcohol/high-fat diets has led to a much more robust pathology.48,87–92
The above-listed mechanisms of alcohol metabolism, hepatic steatosis and alcohol-induced inflammation in the mouse were all largely discovered in one of these associated models, validating the usefulness of these models in understanding the effects of alcohol on mammalian physiology. Advantages and disadvantages of each model are summarized in Table 1. However, the plurality of the common findings associated with AH either have not been demonstrated or are more tightly associated with the adjuvant treatment, such as in the case of liver fibrosis in mice treated with alcohol and CCl4, in mouse models of ALD. This includes: major elevations in plasma bilirubin or INR, indicating loss of liver function, multiorgan dysfunction syndrome, concomitant cholestasis, bridging fibrosis, cirrhosis, regenerative nodules of hepatocytes, progression to hepatocellular carcinoma, and acute-on-chronic type liver failure. Unfortunately, increasingly well acknowledged flaws exist that require remediation if the mouse is going to serve as the major laboratory model for ALD. Translating the above findings will require innovation from the field as a whole, and advancement of the underlying pathology present in clinical patients.
Table 1. Comparison of clinical parameters and histological findings in widely used mouse models of alcohol exposure and human AH patients.
| L-C diet | Gao-binge | “Hybrid” gastric infusion | Human AH patients | |
| AST, U/L | Slight elevation | Elevated 100–300 U/L | Elevated 100–200 U/L | Elevated 80–120 U/L |
| ALT, U/L | Slight elevation | Elevated 100–300 U/L | Elevated 100–200 U/L | Mild elevation |
| Bilirubin | No increase | No increase | No increase | Elevated 3–30+ mg/dL |
| INR | No increase | No increase | No increase | Commonly elevated |
| Bilirubinostasis | None | None | None | Common |
| PMN infiltration | No increase | Significant | Significant | Significant |
| Fibrosis | None | None | None | Up to 98% of patients |
| Cirrhosis | None | None | None | Up to 80% of patients |
| Liver failure | None | Unknown | Unknown | ACLF |
Abbreviations: ACLF, acute-on-chronic liver failure; AH, alcoholic hepatitis; ALT, alanine aminotransferase; AST, aspartate aminotransferase; INR, international normalized ratio; L-C Diet, liquid diet; PMN, polymorphonuclear cell.
Lost in translation – How do we turn basic science findings into clinical results in the alcohol field?
With the limited pathology available in mice, how do you attempt to translate these findings into a more complete understanding of clinical manifestations of ALD? A number of groups have aggressively pursued better models, especially in recent years.48,88–92 While the obvious clinical need is an effective therapeutic, the most critical need for the basic science field is not therapeutic candidates but models of ALD that recapitulate all aspects of the human pathophysiology. Unfortunately, this task has proven difficult for the field, for a number of the reasons listed in this review. New models are being generated every year at this point as the field recognizes that current models are inefficient and unlikely to translate to the clinic. In the interim, additional methods are constantly being developed to help bridge the gap between mice and humans. A number of notable studies with innovative attempts at bridging this gap have recently been published or presented and will be discussed in the subsequent sections.
It is clear that patients with AH present with three critical factors that are either missing or are poorly established in mouse models: 1) the presence of major underlying liver dysfunction (i.e. fibrosis or cirrhosis) before the onset of liver failure (acute-on-chronic liver failure); 2) severe multiorgan dysfunction; and 3) dysfunctional innate immune responses. This is a critical point of understanding: mouse models fail to recapitulate any of these underlying factors, with the possible exception of point 3. Even the most advanced models typically only recapitulate findings up to the point of steatohepatitis (Fig. 2).48,91 This state can occur undetected for decades before the onset of full-blown cirrhosis or concurrent AH, and is not a single event precipitated by a single binge but more likely the result of cumulative binges in both mice and humans.91,92 Moreover, inflammation in the setting of steatosis is benign compared to the advanced pathology associated with cirrhosis or AH.
While human patients likely encounter a similar progression of pathology at some point, there is no diagnosis of ALD for the overwhelming number of patients during this period. AC and AH patients commonly present at or near the end of their disease.1 This is clearly evidenced by the high mortality rate associated with AH.1 Thus, interventions that attempt to target this period would require a dramatic paradigm shift in the diagnosis of ALD. As such, interventional studies should be re-evaluated in the context of more advanced ALD.
Experimental therapeutic intervention in murine models should be initiated after the major onset of pathology and not before pathology begins. These studies can coincide with prophylactic studies that address the role of the protein in progression of the injury. Knockout mouse models have become the gold standard for the assessment of protein function during disease. While the authors of this review concur with their use in assessment of protein function during disease, they do not always make excellent therapeutic models. This is especially true in the case of chronic disease states, where an intervention is only possible in the later stages of the disease.
A number of different mechanisms for selective protein knockdown have become more widely available in recent years, including knockdown via the hepatotropic adeno-associated virus-8, inducible genetic knockdown, and cell-specific knockdown therapies.93,94 Fortunately, therapeutic efficacy is still demonstrable even when administered late in ALD in mice. Administration of heat-shock protein 90 inhibitor 17-DMAG prevented alcohol-induced liver injury when given directly before a binge of ethanol, but after 10 days of chronic alcohol treatment.95 By giving the drug at a point after chronic alcohol administration, the authors insured that the drug did not affect the onset of steatosis, but instead directly affected the immune dysfunction associated with binge on chronic alcohol treatment.95 Similar results with both prophylactic schemes and treatment schemes were obtained in a separate study investing the effect of pepducins on neutrophil recruitment in a model of moderate ASH.51 More studies testing late-stage therapeutics in addition to prophylactic treatment are recommended as they allow for the differentiation between effects on disease progression and effects on disease state.
A number of studies have recently begun validating their murine findings by following up in patients with ALD of varying degrees. Impressive results were recently obtained when investigators noticed that a protein called fat-specific protein 27 was up-regulated in microarray analysis of both human AH patients and mice fed an ASH-inducing diet.92 Up-regulation of the protein was confirmed in AH patients, and correlated with mortality and disease severity; however, knockout of the protein was protective in the mouse model, indicating direct involvement in the injury.92 As such, the study bridged the gap between murine ASH and human AH. Similarly, a study indicating expression of osteopontin in AH patients correlated with outcome and knockout of osteopontin was also protective in the mouse; although, contrary results were obtained in other alcohol models.49,90
These strategies may apply to diagnostic markers as well, as microRNA-192 was up-regulated in plasma samples of both mouse and humans exposed to alcohol, and was proposed as a potential diagnostic marker.96 “Humanizing” mice in an attempt to get a more human-like response from mice has also yielded interesting results. Fecal transfer between AH patients and germ-free or conventional mice resulted in a dramatic increase in susceptibility to ALD, indicating some microbiota may be inherently protective against ALD in patients that do not undergo progression to advanced steatohepatitis and subsequent disease states.97
Other studies have used treatment of primary human hepatocytes to determine how ethanol affects hepatocytes acutely in man. Treatment of primary human hepatocytes with ethanol resulted in posttranslational modifications of forkhead box protein Class O 3 (FOXO3) as seen with in vivo treatment of ethanol.98 Studies focused on validating murine results with direct human impact are integral to both our understanding of what the murine model represents and potential therapeutic targets in patients. Furthermore, these studies might elucidate novel information that assists in the development of high fidelity mouse models.
Induction of cirrhosis followed by administration of alcohol may also yield a better understanding of the effects of alcohol in populations with underlying liver disease. Cirrhosis of all origins, likely has more in common with AC than benign forms of murine ALD that feature consumption of alcohol. In support of this, patients with AC that remain abstinent for 6 months have limited steatosis, but still suffer from a number of associated complications and still have substantial neutrophil infiltrate in the liver.21 New models of cirrhosis that incorporate known effects of alcohol need to be developed.
While the effects of alcohol on naïve livers is well understood from decades of research, very few studies have examined the effects of alcohol on the cirrhotic liver. Administration of alcohol in addition to agents that can cause cirrhosis, such as repeated CCL4 injection, accelerates progression of disease by enhancing fibrosis.88 Chiang et al.12 hypothesized this was due to enhancement of the same pathways present in CCl4-treated mice; thus, agents that can dramatically enhance normal alcohol pathology may be useful. A number of knockout mouse models also experience fibrosis after chronic ethanol feeding. Given that only a minority of chronic alcohol drinkers undergo the conversion from steatosis to steatohepatitis, a genetic component may be involved, although little is understood about this area. Nevertheless, knockout mouse models with exacerbated effects should be validated in human patients when possible by assessing protein levels in patients at varying stages of ALD.
ALD – What does the future hold?
The number of studies on AH published has doubled in the years between 2006 and 2015, according to NCBI, and is continuing to increase at a rapid pace. As alcohol has widely been understudied relative to its impact on liver health, the field is poised for rapid expansion. During this period, it will be imperative to focus on development of legitimate models of late-stage ALD and AH if progress is to be made clinically on these fronts. Continued attempts at translating our understanding of the mouse to human patients using novel and innovative means will provide the greatest return, and will hopefully generate real therapeutics in the immediate future.
Acknowledgements
The work in the authors’ laboratory was supported in part by the National Institutes of Health grant R01 AA12916, the Robert Hanlon Trust, and grants from the National Institute of General Medical Sciences (P20 GM103549 and P30 GM118247) of the National Institutes of Health. Additional support came from the “Training Program in Environmental Toxicology” T32 ES007079-26A2 (to B.L.W.) from the National Institute of Environmental Health Sciences.
Abbreviations
- AC
alcoholic cirrhosis
- AH
alcoholic hepatitis
- ALD
alcoholic liver disease
- ALT
alanine aminotransferase
- ASH
alcoholic steatohepatitis
- AST
aspartate aminotransferase
- CCL4
carbon tetrachloride
- INR
international normalized ratio
- LPS
lipopolysaccharide
- ROS
reactive oxygen species
- TJB
transjugular biopsy
- TLR-4
toll-like receptor-4
- TNF-α
tumor necrosis factor-α
References
- 1.Mathurin P, Bataller R. Trends in the management and burden of alcoholic liver disease. J Hepatol. 2015;62:S38–S46. doi: 10.1016/j.jhep.2015.03.006. doi: 10.1016/j.jhep.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thursz MR, Richardson P, Allison M, Austin A, Bowers M, Day CP, et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med. 2015;372:1619–1628. doi: 10.1056/NEJMoa1412278. doi: 10.1056/NEJMoa1412278. [DOI] [PubMed] [Google Scholar]
- 3.Jesudian AB, Brown RS., Jr Acute alcoholic hepatitis as indication for liver transplantation. Curr Opin Organ Transplant. 2016;21:107–110. doi: 10.1097/MOT.0000000000000285. doi: 10.1097/MOT.0000000000000285. [DOI] [PubMed] [Google Scholar]
- 4.Singal AK, Duchini A. Liver transplantation in acute alcoholic hepatitis: Current status and future development. World J Hepatol. 2011;3:215–218. doi: 10.4254/wjh.v3.i8.215. doi: 10.4254/wjh.v3.i8.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singal AK, Bashar H, Anand BS, Jampana SC, Singal V, Kuo YF. Outcomes after liver transplantation for alcoholic hepatitis are similar to alcoholic cirrhosis: exploratory analysis from the UNOS database. Hepatology. 2012;55:1398–1405. doi: 10.1002/hep.25544. doi: 10.1002/hep.25544. [DOI] [PubMed] [Google Scholar]
- 6.Russ KB, Chen NW, Kamath PS, Shah VH, Kuo YF, Singal AK. Alcohol use after liver transplantation is independent of liver disease etiology. Alcohol Alcohol. 2016;51:698–701. doi: 10.1093/alcalc/agw032. doi: 10.1093/alcalc/agw032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naveau S, Chollet-Martin S, Dharancy S, Mathurin P, Jouet P, Piquet MA, et al. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology. 2004;39:1390–1397. doi: 10.1002/hep.20206. doi: 10.1002/hep.20206. [DOI] [PubMed] [Google Scholar]
- 8.Enomoto N, Ikejima K, Yamashina S, Hirose M, Shimizu H, Kitamura T, et al. Kupffer cell sensitization by alcohol involves increased permeability to gut-derived endotoxin. Alcohol Clin Exp Res. 2001;25:51S–54S. doi: 10.1097/00000374-200106001-00012. doi: 10.1111/j.1530-0277.2001.tb02418.x. [DOI] [PubMed] [Google Scholar]
- 9.Natori S, Rust C, Stadheim LM, Srinivasan A, Burgart LJ, Gores GJ. Hepatocyte apoptosis is a pathologic feature of human alcoholic hepatitis. J Hepatol. 2001;34:248–253. doi: 10.1016/s0168-8278(00)00089-1. doi: 10.1016/S0168-8278(00)00089-1. [DOI] [PubMed] [Google Scholar]
- 10.Schnabl B. Liver capsule: Mechanisms of alcoholic hepatitis. Hepatology. 2016;64:276. doi: 10.1002/hep.28488. doi: 10.1002/hep.28488. [DOI] [PubMed] [Google Scholar]
- 11.Mathews S, Xu M, Wang H, Bertola A, Gao B. Animals models of gastrointestinal and liver diseases. Animal models of alcohol-induced liver disease: pathophysiology, translational relevance, and challenges. Am J Physiol Gastrointest Liver Physiol. 2014;306:G819–G823. doi: 10.1152/ajpgi.00041.2014. doi: 10.1152/ajpgi.00041.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilkin RJ, Lalor PF, Parker R, Newsome PN. Murine models of acute alcoholic hepatitis and their relevance to human disease. Am J Pathol. 2016;186:748–760. doi: 10.1016/j.ajpath.2015.12.003. doi: 10.1016/j.ajpath.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Kim W, Kim DJ. Severe alcoholic hepatitis-current concepts, diagnosis and treatment options. World J Hepatol. 2014;6:688–695. doi: 10.4254/wjh.v6.i10.688. doi: 10.4254/wjh.v6.i10.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altamirano J, Miquel R, Katoonizadeh A, Abraldes JG, Duarte-Rojo A, Louvet A, et al. A histologic scoring system for prognosis of patients with alcoholic hepatitis. Gastroenterology. 2014;146:1231–1239.e6. doi: 10.1053/j.gastro.2014.01.018. doi: 10.1053/j.gastro.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol. 2015;12:231–242. doi: 10.1038/nrgastro.2015.35. doi: 10.1038/nrgastro.2015.35. [DOI] [PubMed] [Google Scholar]
- 16.Dominguez M, Rincón D, Abraldes JG, Miquel R, Colmenero J, Bellot P, et al. A new scoring system for prognostic stratification of patients with alcoholic hepatitis. Am J Gastroenterol. 2008;103:2747–2756. doi: 10.1111/j.1572-0241.2008.02104.x. doi: 10.1111/j.1572-0241.2008.02104.x. [DOI] [PubMed] [Google Scholar]
- 17.Dunn W, Jamil LH, Brown LS, Wiesner RH, Kim WR, Menon KV, et al. MELD accurately predicts mortality in patients with alcoholic hepatitis. Hepatology. 2005;41:353–358. doi: 10.1002/hep.20503. doi: 10.1002/hep.20503. [DOI] [PubMed] [Google Scholar]
- 18.Louvet A, Naveau S, Abdelnour M, Ramond MJ, Diaz E, Fartoux L, et al. The Lille model: a new tool for therapeutic strategy in patients with severe alcoholic hepatitis treated with steroids. Hepatology. 2007;45:1348–1354. doi: 10.1002/hep.21607. doi: 10.1002/hep.21607. [DOI] [PubMed] [Google Scholar]
- 19.Botros M, Sikaris KA. The de ritis ratio: the test of time. Clin Biochem Rev. 2013;34:117–130. [PMC free article] [PubMed] [Google Scholar]
- 20.Ziol M, Tepper M, Lohez M, Arcangeli G, Ganne N, Christidis C, et al. Clinical and biological relevance of hepatocyte apoptosis in alcoholic hepatitis. J Hepatol. 2001;34:254–260. doi: 10.1016/s0168-8278(00)00047-7. doi: 10.1016/S0168-8278(00)00047-7. [DOI] [PubMed] [Google Scholar]
- 21.Woolbright BL, Bridges BW, Dunn W, Olson JC, Weinman SA, Jaeschke H. Cell death and prognosis of mortality in alcoholic hepatitis patients using plasma keratin-18. Gene Expr. 2017;17:301–312. doi: 10.3727/105221617X15016197658871. doi: 10.3727/105221617X15016197658871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matloff DS, Selinger MJ, Kaplan MM. Hepatic transaminase activity in alocholic liver disease. Gastroenterology. 1980;78:1389–1392. [PubMed] [Google Scholar]
- 23.Delhaye M, Louis H, Degraef C, Le Moine O, Devière J, Gulbis B, et al. Relationship between hepatocyte proliferative activity and liver functional reserve in human cirrhosis. Hepatology. 1996;23:1003–1011. doi: 10.1053/jhep.1996.v23.pm0008621125. doi: 10.1053/jhep.1996.v23.pm0008621125. [DOI] [PubMed] [Google Scholar]
- 24.Dubuquoy L, Louvet A, Lassailly G, Truant S, Boleslawski E, Artru F, et al. Progenitor cell expansion and impaired hepatocyte regeneration in explanted livers from alcoholic hepatitis. Gut. 2015;64:1949–1960. doi: 10.1136/gutjnl-2014-308410. doi: 10.1136/gutjnl-2014-308410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sancho-Bru P, Altamirano J, Rodrigo-Torres D, Coll M, Millán C, José Lozano J, et al. Liver progenitor cell markers correlate with liver damage and predict short-term mortality in patients with alcoholic hepatitis. Hepatology. 2012;55:1931–1941. doi: 10.1002/hep.25614. doi: 10.1002/hep.25614. [DOI] [PubMed] [Google Scholar]
- 26.Altamirano J, Fagundes C, Dominguez M, García E, Michelena J, Cárdenas A, et al. Acute kidney injury is an early predictor of mortality for patients with alcoholic hepatitis. Clin Gastroenterol Hepatol. 2012;10:65–71.e3. doi: 10.1016/j.cgh.2011.09.011. doi: 10.1016/j.cgh.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 27.Seki E, Schnabl B. Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. J Physiol. 2012;590:447–458. doi: 10.1113/jphysiol.2011.219691. doi: 10.1113/jphysiol.2011.219691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maiwall R, Chandel SS, Wani Z, Kumar S, Sarin SK. SIRS at admission is a predictor of AKI development and mortality in hospitalized patients with severe alcoholic hepatitis. Dig Dis Sci. 2016;61:920–929. doi: 10.1007/s10620-015-3921-4. doi: 10.1007/s10620-015-3921-4. [DOI] [PubMed] [Google Scholar]
- 29.Michelena J, Altamirano J, Abraldes JG4, Affò S, Morales-Ibanez O, Sancho-Bru P, et al. Systemic inflammatory response and serum lipopolysaccharide levels predict multiple organ failure and death in alcoholic hepatitis. Hepatology. 2015;62:762–772. doi: 10.1002/hep.27779. doi: 10.1002/hep.27779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar K, Mohindra S, Raj M, Choudhuri G. Procalcitonin as a marker of sepsis in alcoholic hepatitis. Hepatol Int. 2014;8:436–442. doi: 10.1007/s12072-014-9540-x. doi: 10.1007/s12072-014-9540-x. [DOI] [PubMed] [Google Scholar]
- 31.Mookerjee RP, Stadlbauer V, Lidder S, Wright GA, Hodges SJ, Davies NA, et al. Neutrophil dysfunction in alcoholic hepatitis superimposed on cirrhosis is reversible and predicts the outcome. Hepatology. 2007;46:831–840. doi: 10.1002/hep.21737. doi: 10.1002/hep.21737. [DOI] [PubMed] [Google Scholar]
- 32.Vergis N, Khamri W, Beale K, Sadiq F, Aletrari MO, Moore C, et al. Defective monocyte oxidative burst predicts infection in alcoholic hepatitis and is associated with reduced expression of NADPH oxidase. Gut. 2017;66:519–529. doi: 10.1136/gutjnl-2015-310378. doi: 10.1136/gutjnl-2015-310378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boussif A, Rolas L, Weiss E, Bouriche H, Moreau R, Périanin A. Impaired intracellular signaling, myeloperoxidase release and bactericidal activity of neutrophils from patients with alcoholic cirrhosis. J Hepatol. 2016;64:1041–1048. doi: 10.1016/j.jhep.2015.12.005. doi: 10.1016/j.jhep.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 34.Woolbright B, Dunn W, Malle E, Jaeschke H. Biomarkers of neutrophil-mediated liver injury in patients with alcoholic liver disease. FASEB J. 2016;30:516.9.. [Google Scholar]
- 35.Gujral JS, Liu J, Farhood A, Jaeschke H. Reduced oncotic necrosis in Fas receptor-deficient C57BL/6J-lpr mice after bile duct ligation. Hepatology. 2004;40:998–1007. doi: 10.1002/hep.20380. doi: 10.1002/hep.20380. [DOI] [PubMed] [Google Scholar]
- 36.Hill DB, Marsano LS, McClain CJ. Increased plasma interleukin-8 concentrations in alcoholic hepatitis. Hepatology. 1993;18:576–580. doi: 10.1002/hep.1840180316. [PubMed] [Google Scholar]
- 37.Dominguez M, Miquel R, Colmenero J, Moreno M, García-Pagán JC, Bosch J, et al. Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology. 2009;136:1639–1650. doi: 10.1053/j.gastro.2009.01.056. doi: 10.1053/j.gastro.2009.01.056. [DOI] [PubMed] [Google Scholar]
- 38.Degré D, Lemmers A, Gustot T, Ouziel R, Trépo E, Demetter P, et al. Hepatic expression of CCL2 in alcoholic liver disease is associated with disease severity and neutrophil infiltrates. Clin Exp Immunol. 2012;169:302–310. doi: 10.1111/j.1365-2249.2012.04609.x. doi: 10.1111/j.1365-2249.2012.04609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mathurin P, Duchatelle V, Ramond MJ, Degott C, Bedossa P, Erlinger S, et al. Survival and prognostic factors in patients with severe alcoholic hepatitis treated with prednisolone. Gastroenterology. 1996;110:1847–1853. doi: 10.1053/gast.1996.v110.pm8964410. doi: 10.1053/gast.1996.v110.pm8964410. [DOI] [PubMed] [Google Scholar]
- 40.O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Am J Gastroenterol. 2010;105:14–32. doi: 10.1038/ajg.2009.593. doi: 10.1038/ajg.2009.593. [DOI] [PubMed] [Google Scholar]
- 41.Bissonnette J, Altamirano J, Devue C, Roux O, Payancé A, Lebrec D, et al. A prospective study of the utility of plasma biomarkers to diagnose alcoholic hepatitis. Hepatology. 2017;66:555–563. doi: 10.1002/hep.29080. doi: 10.1002/hep.29080. [DOI] [PubMed] [Google Scholar]
- 42.Mueller S, Seitz HK, Rausch V. Non-invasive diagnosis of alcoholic liver disease. World J Gastroenterol. 2014;20:14626–14641. doi: 10.3748/wjg.v20.i40.14626. doi: 10.3748/wjg.v20.i40.14626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rudler M, Mouri S, Charlotte F, Cluzel P, Ngo Y, Munteanu M, et al. Validation of AshTest as a non-invasive alternative to transjugular liver biopsy in patients with suspected severe acute alcoholic hepatitis. PLoS One. 2015;10:e0134302. doi: 10.1371/journal.pone.0134302. doi: 10.1371/journal.pone.0134302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maddrey WC, Boitnott JK, Bedine MS, Weber FL, Jr, Mezey E, White RI., Jr Corticosteroid therapy of alcoholic hepatitis. Gastroenterology. 1978;75:193–199. [PubMed] [Google Scholar]
- 45.Louvet A, Labreuche J, Artru F, Boursier J, Kim DJ, O’Grady J, et al. Combining data from liver disease scoring systems better predicts outcomes of patients with alcoholic hepatitis. Gastroenterology. 2015;149:398–406.e8. doi: 10.1053/j.gastro.2015.04.044. doi: 10.1053/j.gastro.2015.04.044. [DOI] [PubMed] [Google Scholar]
- 46.Mueller S, Nahon P, Rausch V, Peccerella T, Silva I, Yagmur E, et al. Caspase-cleaved keratin-18 fragments increase during alcohol withdrawal and predict liver-related death in patients with alcoholic liver disease. Hepatology. 2017;66:96–107. doi: 10.1002/hep.29099. doi: 10.1002/hep.29099. [DOI] [PubMed] [Google Scholar]
- 47.Tanner AR, Powell LW. Corticosteroids in liver disease: possible mechanisms of action, pharmacology, and rational use. Gut. 1979;20:1109–1124. doi: 10.1136/gut.20.12.1109. doi: 10.1136/gut.20.12.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E-selectin. Hepatology. 2013;58:1814–1823. doi: 10.1002/hep.26419. doi: 10.1002/hep.26419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morales-Ibanez O, Domínguez M, Ki SH, Marcos M, Chaves JF, Nguyen-Khac E, et al. Human and experimental evidence supporting a role for osteopontin in alcoholic hepatitis. Hepatology. 2013;58:1742–1756. doi: 10.1002/hep.26521. doi: 10.1002/hep.26521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wieser V, Tymoszuk P, Adolph TE, Grander C, Grabherr F, Enrich B, et al. Lipocalin 2 drives neutrophilic inflammation in alcoholic liver disease. J Hepatol. 2016;64:872–880. doi: 10.1016/j.jhep.2015.11.037. doi: 10.1016/j.jhep.2015.11.037. [DOI] [PubMed] [Google Scholar]
- 51.Wieser V, Adolph TE, Enrich B, Kuliopulos A, Kaser A, Tilg H, et al. Reversal of murine alcoholic steatohepatitis by pepducin-based functional blockade of interleukin-8 receptors. Gut. 2017;66:930–938. doi: 10.1136/gutjnl-2015-310344. doi: 10.1136/gutjnl-2015-310344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trinchet JC, Gerhardt MF, Balkau B, Munz C, Poupon RE. Serum bile acids and cholestasis in alcoholic hepatitis. Relationship with usual liver tests and histological features. J Hepatol. 1994;21:235–240. doi: 10.1016/s0168-8278(05)80401-5. doi: 10.1016/S0168-8278(05)80401-5. [DOI] [PubMed] [Google Scholar]
- 53.Rachakonda V, Gabbert C, Raina A, Bell LN, Cooper S, Malik S, et al. Serum metabolomic profiling in acute alcoholic hepatitis identifies multiple dysregulated pathways. PLoS One. 2014;9:e113860. doi: 10.1371/journal.pone.0113860. doi: 10.1371/journal.pone.0113860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spivey JR, Bronk SF, Gores GJ. Glycochenodeoxycholate-induced lethal hepatocellular injury in rat hepatocytes. Role of ATP depletion and cytosolic free calcium. J Clin Invest. 1993;92:17–24. doi: 10.1172/JCI116546. doi: 10.1172/JCI116546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Woolbright BL, Li F, Xie Y, Farhood A, Fickert P, Trauner M, et al. Lithocholic acid feeding results in direct hepato-toxicity independent of neutrophil function in mice. Toxicol Lett. 2014;228:56–66. doi: 10.1016/j.toxlet.2014.04.001. doi: 10.1016/j.toxlet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Woolbright BL, Dorko K, Antoine DJ, Clarke JI, Gholami P, Li F, et al. Bile acid-induced necrosis in primary human hepatocytes and in patients with obstructive cholestasis. Toxicol Appl Pharmacol. 2015;283:168–177. doi: 10.1016/j.taap.2015.01.015. doi: 10.1016/j.taap.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol. 2011;178:175–186. doi: 10.1016/j.ajpath.2010.11.026. doi: 10.1016/j.ajpath.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beier JI, McClain CJ. Mechanisms and cell signaling in alcoholic liver disease. Biol Chem. 2010;391:1249–1264. doi: 10.1515/BC.2010.137. doi: 10.1515/BC.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barnes MA, Roychowdhury S, Nagy LE. Innate immunity and cell death in alcoholic liver disease: role of cytochrome P4502E1. Redox Biol. 2014;2:929–935. doi: 10.1016/j.redox.2014.07.007. doi: 10.1016/j.redox.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szabo G. Gut-liver axis in alcoholic liver disease. Gastroenterology. 2015;148:30–36. doi: 10.1053/j.gastro.2014.10.042. doi: 10.1053/j.gastro.2014.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chester JA, Blose AM, Froehlich JC. Further evidence of an inverse genetic relationship between innate differences in alcohol preference and alcohol withdrawal magnitude in multiple selectively bred rat lines. Alcohol Clin Exp Res. 2003;27:377–387. doi: 10.1097/01.ALC.0000056619.98553.50. doi: 10.1097/01.ALC.0000056619.98553.50. [DOI] [PubMed] [Google Scholar]
- 62.Cederbaum AI. Alcohol metabolism. Clin Liver Dis. 2012;16:667–685. doi: 10.1016/j.cld.2012.08.002. doi: 10.1016/j.cld.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cederbaum A. Nrf2 and antioxidant defense against CYP2E1 toxicity. Expert Opin Drug Metab Toxicol. 2009;5:1223–1244. doi: 10.1517/17425250903143769. doi: 10.1517/17425250903143769. [DOI] [PubMed] [Google Scholar]
- 64.Skopp G, Schmitt G, Pötsch L, Drönner P, Aderjan R, Mattern R. Ethyl glucuronide in human hair. Alcohol Alcohol. 2000;35:283–285. doi: 10.1093/alcalc/35.3.283. doi: 10.1093/alcalc/35.3.283. [DOI] [PubMed] [Google Scholar]
- 65.Yu HS, Oyama T, Isse T, Kitagawa K, Pham TT, Tanaka M, et al. Formation of acetaldehyde-derived DNA adducts due to alcohol exposure. Chem Biol Interact. 2010;188:367–375. doi: 10.1016/j.cbi.2010.08.005. doi: 10.1016/j.cbi.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 66.You M, Jogasuria A, Taylor C, Wu J. Sirtuin 1 signaling and alcoholic fatty liver disease. Hepatobiliary Surg Nutr. 2015;4:88–100. doi: 10.3978/j.issn.2304-3881.2014.12.06. doi: 10.3978/j.issn.2304-3881.2014.12.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP) J Biol Chem. 2002;277:29342–29347. doi: 10.1074/jbc.M202411200. doi: 10.1074/jbc.M202411200. [DOI] [PubMed] [Google Scholar]
- 68.Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. doi: 10.1128/MCB.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shoreibah M, Raff E, Bloomer J, Kakati D, Rasheed K, Kuo YF, et al. Alcoholic liver disease presents at advanced stage and progresses faster compared to non-alcoholic fatty liver diseas. Ann Hepatol. 2016;15:183–189. doi: 10.5604/16652681.1193707. doi: 10.5604/16652681.1193707. [DOI] [PubMed] [Google Scholar]
- 70.Lefkowitch JH. Morphology of alcoholic liver disease. Clin Liver Dis. 2005;9:37–53. doi: 10.1016/j.cld.2004.11.001. doi: 10.1016/j.cld.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 71.Farooq MO, Bataller R. Pathogenesis and management of alcoholic liver disease. Dig Dis. 2016;34:347–355. doi: 10.1159/000444545. doi: 10.1159/000444545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kono H, Rusyn I, Yin M, Gäbele E, Yamashina S, Dikalova A, et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest. 2000;106:867–872. doi: 10.1172/JCI9020. doi: 10.1172/JCI9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wheeler MD, Nakagami M, Bradford BU, Uesugi T, Mason RP, Connor HD, et al. Overexpression of manganese superoxide dismutase prevents alcohol-induced liver injury in the rat. J Biol Chem. 2001;276:36664–36672. doi: 10.1074/jbc.M105352200. doi: 10.1074/jbc.M105352200. [DOI] [PubMed] [Google Scholar]
- 74.Niemelä O, Parkkila S, Koll M, Preedy VR. Generation of protein adducts with malondialdehyde and acetaldehyde in muscles with predominantly type I or type II fibers in rats exposed to ethanol and the acetaldehyde dehydrogenase inhibitor cyanamide. Am J Clin Nutr. 2002;76:668–674. doi: 10.1093/ajcn/76.3.668. [DOI] [PubMed] [Google Scholar]
- 75.Arteel GE. Alcohol-induced oxidative stress in the liver: in vivo measurements. Methods Mol Biol. 2008;447:185–197. doi: 10.1007/978-1-59745-242-7_14. doi: 10.1007/978-1-59745-242-7_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med. 2008;44:723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Williams JA, Ni HM, Ding Y, Ding WX. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am J Physiol Gastrointest Liver Physiol. 2015;309:G324–G340. doi: 10.1152/ajpgi.00108.2015. doi: 10.1152/ajpgi.00108.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Williams JA, Ding WX. A mechanistic review of mitophagy and its role in protection against alcoholic liver disease. Biomolecules. 2015;5:2619–2642. doi: 10.3390/biom5042619. doi: 10.3390/biom5042619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thurman RG, Bradford BU, Iimuro Y, Knecht KT, Arteel GE, Yin M, et al. The role of gut-derived bacterial toxins and free radicals in alcohol-induced liver injury. J Gastroenterol Hepatol. 1998;13:S39–S50. doi: 10.1111/jgh.1998.13.s1.39. doi: 10.1111/jgh.1998.13.s1.39. [DOI] [PubMed] [Google Scholar]
- 81.Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology. 2000;32:1008–1017. doi: 10.1053/jhep.2000.19621. doi: 10.1053/jhep.2000.19621. [DOI] [PubMed] [Google Scholar]
- 82.Yan AW, Fouts DE, Brandl J, Stärkel P, Torralba M, Schott E, et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology. 2011;53:96–105. doi: 10.1002/hep.24018. doi: 10.1002/hep.24018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Testa R, Eftimiadi C, Sukkar GS, De Leo C, Rovida S, Schito GC, et al. A non-absorbable rifamycin for treatment of hepatic encephalopathy. Drugs Exp Clin Res. 1985;11:387–392. [PubMed] [Google Scholar]
- 84.Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology. 2001;34:101–108. doi: 10.1053/jhep.2001.25350. doi: 10.1053/jhep.2001.25350. [DOI] [PubMed] [Google Scholar]
- 85.Nagy LE. The role of innate immunity in alcoholic liver disease. Alcohol Res. 2015;37:237–250. doi: 10.35946/arcr.v37.2.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.DeCarli LM, Lieber CS. Fatty liver in the rat after prolonged intake of ethanol with a nutritionally adequate new liquid diet. J Nutr. 1967;91:331–336. doi: 10.1093/jn/91.3_Suppl.331. [DOI] [PubMed] [Google Scholar]
- 87.Enomoto N, Yamashina S, Kono H, Schemmer P, Rivera CA, Enomoto A, et al. Development of a new, simple rat model of early alcohol-induced liver injury based on sensitization of Kupffer cells. Hepatology. 1999;29:1680–1689. doi: 10.1002/hep.510290633. doi: 10.1002/hep.510290633. [DOI] [PubMed] [Google Scholar]
- 88.Chiang DJ, Roychowdhury S, Bush K, McMullen MR, Pisano S, Niese K, et al. Adenosine 2A receptor antagonist prevented and reversed liver fibrosis in a mouse model of ethanol-exacerbated liver fibrosis. PLoS One. 2013;8:e69114. doi: 10.1371/journal.pone.0069114. doi: 10.1371/journal.pone.0069114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tsukamoto H, French SW, Benson N, Delgado G, Rao GA, Larkin EC, et al. Severe and progressive steatosis and focal necrosis in rat liver induced by continuous intragastric infusion of ethanol and low fat diet. Hepatology. 1985;5:224–232. doi: 10.1002/hep.1840050212. doi: 10.1002/hep.1840050212. [DOI] [PubMed] [Google Scholar]
- 90.Lazaro R, Wu R, Lee S, Zhu NL, Chen CL, French SW, et al. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology. 2015;61:129–140. doi: 10.1002/hep.27383. doi: 10.1002/hep.27383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xu J, Liu X, Gao B, Karin M, Tsukamoto H, Brenner D, et al. New approaches for studying alcoholic liver disease. Curr Pathobiol Rep. 2014;2:171–183. doi: 10.1007/s40139-014-0053-z. doi: 10.1007/s40139-014-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xu MJ, Cai Y, Wang H, Altamirano J, Chang B, Bertola A, et al. Fat-specific protein 27/CIDEC promotes development of alcoholic steatohepatitis in mice and humans. Gastroenterology. 2015;149:1030–1041.e6. doi: 10.1053/j.gastro.2015.06.009. doi: 10.1053/j.gastro.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ho KJ, Bass CE, Kroemer AH, Ma C, Terwilliger E, Karp SJ. Optimized adeno-associated virus 8 produces hepatocyte-specific Cre-mediated recombination without toxicity or affecting liver regeneration. Am J Physiol Gastrointest Liver Physiol. 2008;295:G412–G419. doi: 10.1152/ajpgi.00590.2007. doi: 10.1152/ajpgi.00590.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Friedel RH, Wurst W, Wefers B, Kühn R. Generating conditional knockout mice. Methods Mol Biol. 2011;693:205–231. doi: 10.1007/978-1-60761-974-1_12. doi: 10.1007/978-1-60761-974-1_12. [DOI] [PubMed] [Google Scholar]
- 95.Ambade A, Catalano D, Lim A, Kopoyan A, Shaffer SA, Mandrekar P. Inhibition of heat shock protein 90 alleviates steatosis and macrophage activation in murine alcoholic liver injury. J Hepatol. 2014;61:903–911. doi: 10.1016/j.jhep.2014.05.024. doi: 10.1016/j.jhep.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Momen-Heravi F, Bala S, Kodys K, Szabo G. Exosomes derived from alcohol-treated hepatocytes horizontally transfer liver specific miRNA-122 and sensitize monocytes to LPS. Sci Rep. 2015;5:9991. doi: 10.1038/srep09991. doi: 10.1038/srep09991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Llopis M, Cassard AM, Wrzosek L, Boschat L, Bruneau A, Ferrere G, et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut. 2016;65:830–839. doi: 10.1136/gutjnl-2015-310585. doi: 10.1136/gutjnl-2015-310585. [DOI] [PubMed] [Google Scholar]
- 98.Li Z, Zhao J, Tikhanovich I, Kuravi S, Helzberg J, Dorko K, et al. Serine 574 phosphorylation alters transcriptional programming of FOXO3 by selectively enhancing apoptotic gene expression. Cell Death Differ. 2016;23:583–595. doi: 10.1038/cdd.2015.125. doi: 10.1038/cdd.2015.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Antoine DJ, Jenkins RE, Dear JW, Williams DP, McGill MR, Sharpe MR, et al. Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J Hepatol. 2012;56:1070–1079. doi: 10.1016/j.jhep.2011.12.019. doi: 10.1016/j.jhep.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]