

Abstract
Background
One method for rejuvenating land polluted with anthropogenic contaminants is through phytoremediation, the reclamation of land through the cultivation of specific crops. The capacity for phytoremediation crops, such as Salix spp., to tolerate and even flourish in contaminated soils relies on a highly complex and predominantly cryptic interacting community of microbial life.
Methods
Here, Illumina HiSeq 2500 sequencing and de novo transcriptome assembly were used to observe gene expression in washed Salix purpurea cv. ‘Fish Creek’ roots from trees pot grown in petroleum hydrocarbon-contaminated or non-contaminated soil. All 189,849 assembled contigs were annotated without a priori assumption as to sequence origin and differential expression was assessed.
Results
The 839 contigs differentially expressed (DE) and annotated from S. purpurea revealed substantial increases in transcripts encoding abiotic stress response equipment, such as glutathione S-transferases, in roots of contaminated trees as well as the hallmarks of fungal interaction, such as SWEET2 (Sugars Will Eventually Be Exported Transporter). A total of 8252 DE transcripts were fungal in origin, with contamination conditions resulting in a community shift from Ascomycota to Basidiomycota genera. In response to contamination, 1745 Basidiomycota transcripts increased in abundance (the majority uniquely expressed in contaminated soil) including major monosaccharide transporter MST1, primary cell wall and lamella CAZy enzymes, and an ectomycorrhiza-upregulated exo-β-1,3-glucanase (GH5). Additionally, 639 DE polycistronic transcripts from an uncharacterised Enterobacteriaceae species were uniformly in higher abundance in contamination conditions and comprised a wide spectrum of genes cryptic under laboratory conditions but considered putatively involved in eukaryotic interaction, biofilm formation and dioxygenase hydrocarbon degradation.
Conclusions
Fungal gene expression, representing the majority of contigs assembled, suggests out-competition of white rot Ascomycota genera (dominated by Pyronema), a sometimes ectomycorrhizal (ECM) Ascomycota (Tuber) and ECM Basidiomycota (Hebeloma) by a poorly characterised putative ECM Basidiomycota due to contamination. Root and fungal expression involved transcripts encoding carbohydrate/amino acid (C/N) dialogue whereas bacterial gene expression included the apparatus necessary for biofilm interaction and direct reduction of contamination stress, a potential bacterial currency for a role in tripartite mutualism. Unmistakable within the metatranscriptome is the degree to which the landscape of rhizospheric biology, particularly the important but predominantly uncharacterised fungal genetics, is yet to be discovered.
Electronic supplementary material
The online version of this article (10.1186/s40168-018-0432-5) contains supplementary material, which is available to authorized users.
Keywords: Metatranscriptomics, Microbiome, Salix, Rhizosphere, Phytoremediation
Background
The observation of gene expression across multiple interacting organisms has the potential to better reflect the complex reality of biology than the observation of organisms in isolation [1]. By separating the assembly of RNA sequence data from annotation (identification) of assembled contigs, de novo metatranscriptome assembly allows for such observation without a prerequisite for, and therefore bias from, reference genome sequences from organisms expected to be present within any biological system [2, 3]. A metatranscriptomic approach designed without constraint to any a priori defined organism, but open to annotation from any sequenced strata of life, should be powerful in biological systems already recognised as highly complex, such as the human digestive tract or rhizosphere microbiome (although such microbiome complexity could arguably be defined by the current extent of study in a biological field). Here, the rhizospheric microbiome of Salix purpurea cv. ‘Fish Creek’ was challenged using hydrocarbon-contaminated soil and differential gene expression observed.
Pervasive organic pollutants, such as polycyclic aromatic hydrocarbons (PAHs), polychlorinated biphenyls (PCBs) and C10-C50 petroleum hydrocarbons, represent serious risk to human health and the environment [4]. There are thought to be greater than 400,000 contaminated sites across North America [5, 6] and estimates are as high as 2.5 million sites across the EU [7]. Currently, rehabilitation of such sites is constrained by the high costs (> 2 M$/ha, [8]) of standard restoration strategies such as excavation and transport (dig-and-dump), with the consequence that these sites are rarely restored. In recent decades, a consensus has grown that environmentally sustainable and economically viable land restoration methods should be developed; phytoremediation is one such green technology alternative [9, 10]. Phytoremediation relies on the interaction between plants and their associated microorganisms to absorb, immobilise, volatilise, degrade, translocate or transform organic and inorganic contaminants [11]. In trials across Canada, a wide range of fast growing short rotation coppice willow cultivars (Salix spp.) have been shown as highly tolerant to PAH, PCBs and other organic petroleum hydrocarbon contaminants [12], as well as inorganic contaminants [13]. While the societal, environmental and economic benefits of rehabilitating these sites can be extensive (estimated within Canadian metropolitan areas at 4.6–7 billion dollars annually [14]), an additional benefit of the use of crops such as willow is that the biomass yielded per hectare can also be utilised for several valuable end-uses, including lignocellulosic biofuels [15], renewable electricity and heat generation [16], as well as green phytochemical production. Cultivation of biomass is often seen as the most substantial hurdle for economically feasible bioenergy production [17–19]. By aligning feedstock production strategy with land decontamination, cultivation can serve as a positive value-stream, in terms of the financial profit as well as a clear local environmental benefit [14].
To achieve effective success as a phytoremediation crop, willow is thought to maintain and exploit intimate symbiotic relationships with fungi. Roughly 6000 species of fungi from Glomeromycota, Ascomycota, and Basidiomycota have been categorised to date as mycorrhizal [20]. Recent research has explored the symbiotic interaction of arbuscular mycorrhizal fungi (AMF, endomycorrhizal fungi currently exclusively categorised within Glomeromycota) with willow [13, 21, 22] although trees and shrubs such as willow are characteristically known for interacting with ectomycorrhizal (ECM) fungi when mature [20, 23]. Such interactions are thought to be predicated on the exchange of nutrients from the fungi to the plant, in particular phosphate and nitrogen attained by the fungi from soils, and a highly controlled amount of sugars exchanged from the plant to fungi. However, the comprehensive identification of fungi from an extra-laboratory environment, let alone delineating their roles in a complex biological system, is confounded by culturing difficulty and that, of the estimated 1.5 million fungal species, less than 600 have been currently (2016) sequenced and annotated (JGI MycoCosm [24]).
The peril of confounding bacterial community assessment by culturing methodology alone is also widely acknowledged, but the progression of contemporary sequencing techniques and extensive research advancement driven by fields relating to the human microbiome has led to an understanding of the ubiquitous, exceedingly diverse, presence and involvement of bacteria in eukaryotic biology. The importance of bacteria to mycorrhizal fungi and/or plant health has been established even in conditions less challenging than anthropogenic soil contamination [25–28]. Bacteria whose presence and function is beneficial to mycorrhizal fungi and/or plants are often termed mycorrhizal helper bacteria MHB [29, 30] or plant growth-promoting bacteria (PGPB), such as the recently identified Enterobacter sp. 638, recognised as improving poplar growth on challenging marginal soils by up to 40% [31]. In relation to the rhizosphere, the highly complex environment has been very well reviewed by Bonfante et al. [20] as comprising ‘tripartite’ interactions between plants, mycorrhizal fungi and bacteria. The distinction between the potential roles of bacteria within the rhizosphere, in terms of the level of host interaction (rhizospheric, extracellular interacting or intracellular) and the spectrum of interaction type (pathogeny, symbiosis or commensalism), is problematic; however, metagenomics and metatranscriptomics can help to unravel this complexity by allowing gene function to be observed.
Here, all the RNA assembled de novo from roots of 12 willow trees, pot-grown in either contaminated or non-contaminated soil from a former petroleum refinery, was annotated and differential gene expression from any and all organisms identified was explored to see if the functionality of a successful phytoremediation system can be elucidated.
Methods
Contamination composition, experimental design and sampling
Both contaminated and non-contaminated soils were gathered from the site of a former petrochemical refinery at Varennes, Canada. Contaminated soil had an average C10-C50 petroleum hydrocarbon concentration of 912 mg kg− 1 (non-contaminated soil was below detection limit: < 100 mg kg− 1). Salix purpurea cv. ‘Fish Creek’ cuttings were established for 8 weeks in conventional potting media before being transferred to 20-l pots containing treatment soil within a larger experiment consisting of six blocks where randomised contamination effect was investigated (further soil and experimental information available from Yergeau et al. [27]). Growth conditions were 16 h 20 °C day and 8 h 18 °C night with excess watering and individual plant pot saucers to reduce leeching. After 6 months of growth, roots were harvested from six replicate trees per growth condition; soil was removed manually and roots samples were flash frozen in liquid nitrogen within 5 min of the initial perturbation.
RNA extraction and Illumina sequencing
A modified CTAB protocol [32, 33] was used to extract RNA from roots with quality and quantity assessed using a BioAnalyser (Agilent, Mississauga, ON, Canada). Roots from 12 trees were sequenced: 6 from trees cultivated in contaminated soil and 6 from non-contaminated soil. Polyadenylated mRNA was amplified using Ambion’s MessageAmp™ II aRNA Amplification Kit. Amplified RNA was tested for genomic DNA content by PCR, using 18S rRNA gene primers and conditions described in Stewart et al. [34]. Indexing of cDNA samples for sequencing was performed in accordance with Meyer and Kircher [35]. The samples were sequenced (four separate runs) using an Illumina HiSeq 2500 platform.
De novo metatranscriptome assembly and differential expression
Trimmomatic [36] was used to trim nucleotides of poor quality and reads < 40 bp were removed. Reads from all 12 biological samples were assembled into a de novo transcriptome using Trinity software set to default parameters [3]. Assembled contigs shorter than 200 bp were discarded. Bowtie2 [37, 38] was used to align the RNA-seq reads back to the de novo transcriptome with -a -X 600 parameters on top of default parameters. Low count contigs, below 29 total counts across all libraries, were filtered out (corresponding to those contigs below the 80th percentile [39]). Read back-mapping rates were an average of 64.91% across all 12 root samples. Raw and normalised transcript abundance (tpm) was calculated using eXpress [40] with default parameters. Differential expression (DE) was estimated using EBSeq based on median normalisation [41] and on an empirical Bayes model framework [42, 43], keeping contigs with a posterior probability of differential expression (PPDE) ≥ 0.95 (with target false discovery rate controlled at 5%). EdgeR [44] was used to generate MA plots based on trimmed mean of M-values normalisation method [45]. Extended quality control, assembly and normalisation information is provided in Additional file 1, and scripts are provided in Additional file 2.
Annotation
The metatranscriptomic (unconstrained) annotation strategy, which queries a broad range of protein sequence repositories, was performed as outlined by Gonzalez et al. [46]. Briefly, the de novo assembled contigs were annotated using three major protein databases (nr NCBI non-redundant protein database, SwissProt and TrEMBL) as well as the S. purpurea 94006 reference genome. During the informed annotation step, Populus trichocarpa reference genome was also added. UniProt Archive (UniParc) database was used to protein blast differentially expressed (DE) contigs that did not have a hit in any of the databases. NCBI nucleotide database was used to nucleotide blast all DE contigs. E values < 10−4 (protein blast) and < 10−6 (nucleotide blast) were used as cutoffs. A previously reported method for selecting annotation (based on the percentage of maximum potential bitscore) from blastx returns was used to help select the primary annotation given multiple high scoring alignments for a single sequence, all statistically characterised as non-random [2]. BLAST hits that were not selected but have a high comparable percentage of maximum potential bitscore (within 10%) were retained for each contig as (alternative) secondary annotation. Custom scripting (in Python, R, Shell, Javascript) and Krona [47] were used to generate images and figures (Additional file 2).
Transdecoder
To find coding regions within bacterial polycistronic sequences, we used TransDecoder software (https://transdecoder.github.io/). Protocol was followed according to the manual with default parameters. Precedence of transcriptional unit structure (putative operons) was verified in all cases against the database of prokaryotic operons (DOOR [48]) unless otherwise stated. A final hand curation step was included.
Interrogation of unknown contigs
Further annotation was undergone for contigs with no hit on any database. Nucleotide blast was carried out using non-coding RNA database (NONCODE2016 from www.noncode.org), Salix purpurea 94006 genome, and NCBI ESTs database.
Results and discussion
Assembly, mapping and annotation
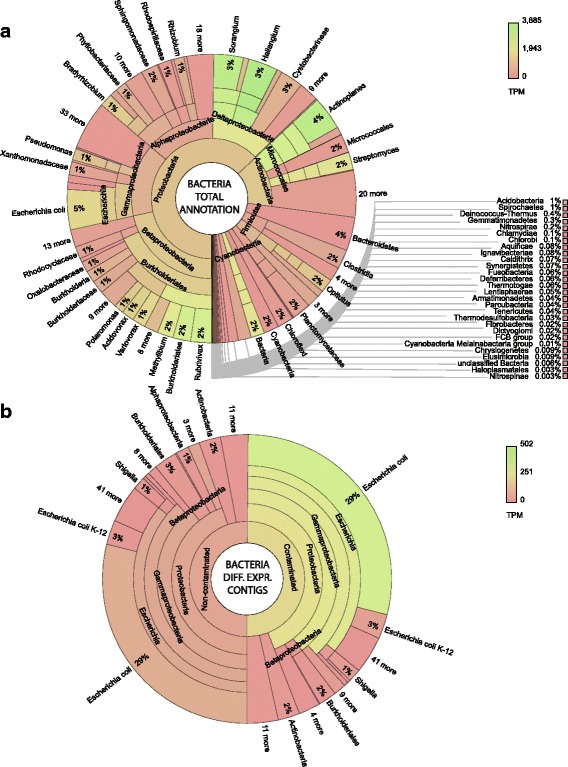
A total of 189,849 confident contigs were confidently assembled de novo from 456,182,049 sequence reads. Overall, 125,151 contigs (66%) were confidently annotated as potential protein encoding transcripts, but 64,698 contigs (34%) had no protein homologue in SwissProt, Trembl or nr databases and are referred to as unknown henceforth (a substantial number of these were DE and investigated in greater detail in Additional files 1 and 3). The annotated transcripts included 91,053 Eukaryota (48%), 33,222 Bacteria (18%), 187 Archaea (0.01%) and 139 viral sequences (0.01%; including the common Illumina spike) (Fig. 1). Transcripts best annotated from eukaryotes were, as could be expected, mostly derived from Viridiplantae (46,817), although a close second was Fungi (40,352), with Metazoa (1417), Amoebozoa (918), Stramenopiles (617), Euglenozoa (390), Alveolata (198) and others (such as metagenomic or environmental samples) comprising only minor proportions of the transcript diversity (324).
Fig. 1.
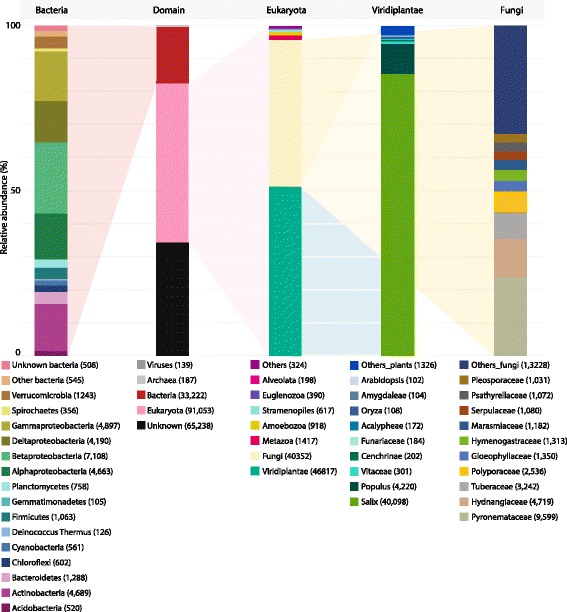
Total annotation. Annotation of the entire transcriptome assembly (including non-differentially expressed contigs). Bars representing Bacteria, Domain, Eukaryota, Viridiplantae and Fungi are selected as a useful overview of the diversity within the transcriptome. While bars represent data normalised to 100%, only ~ 65% of the sequenced reads were successfully mapped to the assembled transcriptome (so are overlooked here) and 34% of assembled contigs had no similarity to known sequences (so are again overlooked). Full annotation is provided in Additional file 4 and an interactive Krona of total annotation is available at: https://github.com/gonzalezem/Tripartite_Metatranscriptomics_article
Total community makeup
Comparative multi-omics analysis by Hultman [49] and Tveit et al. [50] has revealed some shortfalls in community assessment using internal transcribed spacer (ITS) and 16S ribosomal RNA (16S rRNA), even at the phylum level (discussed with additional Basidiomycota identification investigations in Additional file 1). Others have expressed even stronger scepticism, such as Bent et al. [51], who discussed the difficulties in providing reliable diversity indices from microbial fingerprinting methodology [52–57]. An alternative, top-down approach is employed here which accepts uncertainty of the microbiome community, assembling as many contigs as possible within the system and performing differential expression analysis on all retained contigs without any necessity for a priori sequence information before independent annotation using the world’s major protein repositories.
Transcripts clearly identified as deriving from Salix (the mass majority from the Salix purpurea 94006 genome) made up 85.6% (40,098) of the contigs from plant species. While this was expected, it is perhaps more informative to recognise that 14.4% of plant transcripts were best annotated outside of Salix, mostly from the very close relative Populus (9.0%) but also from other genera (Vitaceae 0.6%; Cenchrinae 0.4%; Funariaceae 0.4; Acalypheae 0.4%; Oryza 0.2%; Amygdaleae 0.2%; Arabidopsis 0.2% and others 2.8%). This is roughly in line with previous research suggesting between 6 and 10% of Salix purpurea genes can be better identified by querying the broader protein repositories [2, 46] and that, importantly, over 10% of likely Salix gene expression can be obscured by only mapping to a reference genome, even when species specific.
Extraordinary fungal diversity was found within the transcriptome. Although RNA was extracted from washed roots, this diversity is not surprising as the majority of plant species are thought to employ (widely diverse) below-ground fungal association in order to survive and compete in the biosphere [12, 58]. De novo sequencing provides a unique opportunity to observe such opaque below-ground genetics (Fig. 1, only fungal genera with over 1000 hits are presented, comprehensive annotation is provided in Additional file 4 and interactive Krona graphs). The fungal community comprised 40,352 distinct contigs by primary annotation, the majority were Pyronemataceae (23.8%), then Hydnangiaceae (11.7%), Tuberaceae (8.0%), Polyporaceae (6.3%), Gloeophyllaceae (3.3%), Hymenogastraceae (3.3%), Marasmiaceae (2.9%), Serpulaceae (2.7%), Psathyrellaceae (2.7%) and Pleosporaceae (2.6%) contigs as well as a very extensive quantity of others (13,228 contigs or 32.8%).
Bacterial diversity was extremely high, even at class level, with the 33,222 transcripts originating from Betaproteobacteria (21.4%), Gammaproteobacteria (14.7%), Actinobacteria (14.1%), Alphaproteobacteria (14.0%), Deltaproteobacteria (12.6%), Bacteroidetes (3.9%), Verrucomicrobia (3.7%) and Firmicutes (3.2%) as well as from other genera (Planctomycetes 2.3%; Chloroflexi 1.8%; Cyanobacteria 1.7%; Acidobacteria 1.6%; uncharacterised bacteria 1.5%; Spirochaetes 1.1%; Deinococcus-Thermus 0.4%; Gemmatimonadetes 0.3%; others 1.6%). These bacterial contigs assembled from RNA extracted from roots very closely reflected the community reported in a previous experiment performed by Yergeau et al. [59] using 16S rRNA sequencing, where rhizospheric bacteria (from soil attached to roots), in association with the same willow cultivar grown in the same contamination, was also dominated by Betaproteobacteria (~ 39%) and Gammaproteobacteria (~ 38%). The authors used 16S rRNA sequencing to positively identify a wide spectrum of bacteria at the class level; while the methodology is not quantitatively comparable to that used here (Additional file 1), the proportions of the community makeup were roughly similar at a phylum and class level.
Differentially expressed contigs
Metaorganism patterning in gene expression
A total of 12,576 contigs were identified as DE between non-contaminated and contaminated soil conditions (Fig. 2). DE contigs (putative transcripts when annotated as protein encoding) may be responding directly to hydrocarbon toxicity or to environmental alterations due to the high soil concentrations of petroleum hydrocarbons, such as nutrient or water availability. Beyond this, differential expression could also be due to secondary responses representing the shift in the microbiome community or, indeed, expression of other organisms independent of the shift in community within the metaorganism. No sequences were discarded based on annotation, especially since previous studies have demonstrated the potential for the removal of sequences (unexpected a priori) to biologically [60] and technically [2] confound data integrity and interpretation. The necessity for altering the paradigm of experimental approaches in light of a modern understanding of metaorganismal complexity has recently been recognised as essential in mammals with respect to phenotypic assessment incorporating the microbiome [1]. In particular, bacterial DE sequences were retained for analysis (despite polyA enrichment prior to RNA-seq).
Fig. 2.
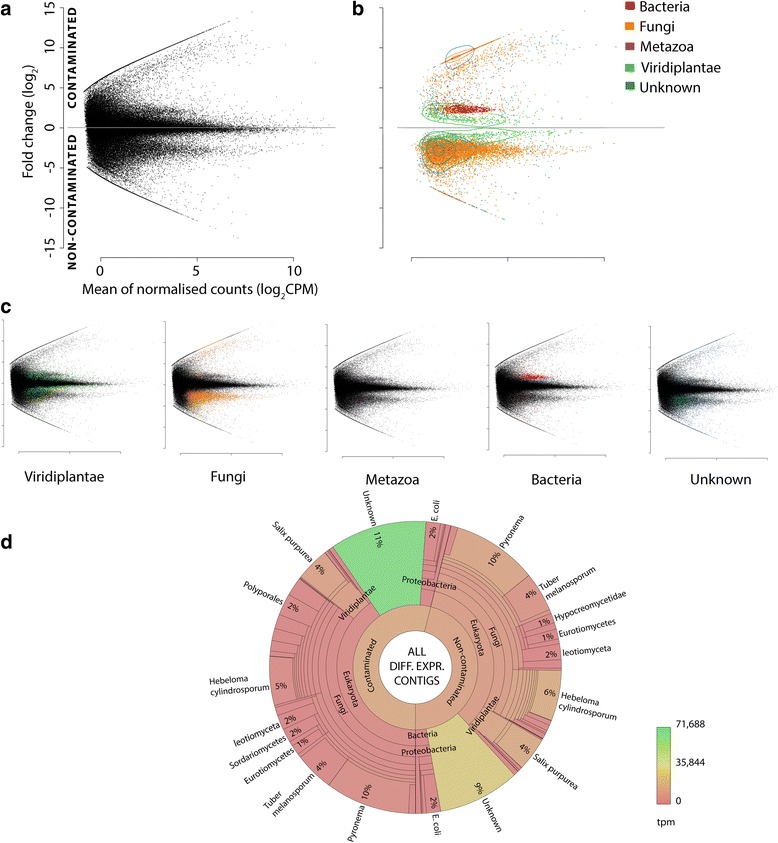
Origin of differentially expressed contigs. MA plots (a–c) of de novo assembled transcriptome; y-axis represents fold change (FC, log2) between contaminated (+ive) to non-contaminated conditions (−ive), and the x-axis represents mean normalised (EdgeR) counts per million (log2 CPM). Plot a all contigs (including non-DE) and b DE contigs only; coloured by annotation including contours to represent contig density relative within each group. c Individual MA plots of differentially expressed (DE) contigs annotated from Viridiplantae, Fungi, Metazoa, Bacteria and Unknown (no known similar sequences) are included for clarity. Data patterning from contamination-driven shifts in the community are observable (a) prior to any annotation. An epsilon factor is added in place of zero abundance where contigs are present in only one condition to allow visualisation and abundance comparison (as fold change would be infinite); the presence or absence of contigs (due to contamination) is biologically informative. d All DE contigs represented within a Krona graph [47]; the proportion of each taxonomic grouping is defined by the number of distinct contigs, whereas the colour represents the relative abundance (transcripts per million, tpm) of transcripts in each taxon. An interactive Krona graph to assist navigation of DE contig annotation origin is available at: https://github.com/gonzalezem/Tripartite_Metatranscriptomics_article. A full contig list including expression information, annotation (1° and 2°) and gene ontology is provided in Additional file 4 whereas DE only contigs are provided in Additional file 9
A substantial proportion of all the contigs assembled (including non-DE) were unknown (34%), bearing no confident homologous sequence in the major protein repositories. Although unknown sequences are often discarded, the strategy reported here very deliberately maintains data where possible due to the potential for unknown DE sequences to represent novel organisms and functionality. Over 20% of the total DE sequences were indeed unknown (Additional file 3); extended investigation into these sequences is discussed in Additional file 1.
Pooling of highly complex gene expression data from non-model plants into ontology groups can give the impression of an overview of the biological system (GO terms are provided for query Additional file 4); however, an attempt to explore DE function at a transcript level across all the organisms within the samples is presented here.
Salix purpurea
A total of 839 DE contigs were annotated as Salix purpurea, representing 6.7% of all DE contigs from roots. The number of contigs up (45%) or down-regulated (55%) was similar in response to contamination (Figs. 2 and 3). Despite the strain/cultivar studied here (S. purpurea ‘Fish Creek’) being the same species as that of the sequenced reference genome, S. purpurea 94006, 131 contigs (which we presume originate from Salix) were better annotated from other plant species. This is consistent with another study suggesting (top-down) de novo strategies can capture more sequence variation than mapping to a single non-clonal reference genome alone [2]. Although the diversity of Salix DE contigs was relatively low within the system, with the exception of the unknown, they had the highest relative abundance (analysed as transcripts per million, TPM); perhaps unsurprisingly, given RNA was sampled from washed Salix roots.
Fig. 3.

Salix purpurea differential expression (DE) transcript distribution and abundance (transcripts per million, tpm) weighted fold change (log2). Top: fold change (FC log2) distribution of DE genes contaminated (black) and non-contaminated (gold). Bottom: mean transcript counts (tpm) difference between conditions against fold change per DE contig. The highly abundant transcripts discussed within the text are labelled. A full DE transcript list including expression data, functional description (if available), gene ontology terms (if available) and secondary annotation (if available) is provided in Additional file 5. PIP plasma membrane intrinsic protein (aquaporin), GST glutathione S-transferase
Direct detoxification/stress responses
The expectations for plant gene expression within a high hydrocarbon-contaminated environment would include an increase in cytochrome P450 monooxygenases. Out of 26 DE transcripts identified as encoding cytochrome P450 family proteins, 18 were in higher abundance within control roots (cultivated in non-contaminated soil) (Additional file 5). Only two were annotated as putative monooxygenases (c585325_g1_i1 and c601406_g5_i2), with one upregulated and one downregulated with respect to contamination. Another expected set of stress-induced detoxification equipment, known to be transcript abundance dependent, are glutathione S-transferases (GSTs), which catalyse glutathione conjugation to a broad range of xenobiotics in order to facilitate vacuolar loading (compartmentalisation of cellular pollutants). Only a single GST was downregulated in roots from contaminated soil, with 13 upregulated (8 tau class, including GSTU19, 37, 48 and 50) at consistently high relative abundance levels (tpm) (Fig. 2 and Additional file 5).
UDP-glucose:flavonoid 7-O-glucosyltransferase was the most abundant upregulated Salix gene in contaminated roots (c600230_g5_i1; 216.94 tpm), and two other putative isoforms were also highly upregulated (c600230_g5_i2, 90.71 tpm; c600230_g6_i1, 52.53 tpm; Fig. 2).
Such increases in flavonoid glucosyltransferase expression represent a common conjugation mechanism in plant detoxification metabolism [61]. Alternatively, UDP-glucose:flavonoid 7-O-glucosyltransferase, which allows symbiotic interaction with microorganisms in Glycine max [62], can be limiting for vacuolar loading of the isoflavone conjugates, and the corresponding conjugate-hydrolysing β-glucosidase is localised to the plant root apoplast, intriguing in terms of the extensive interaction with microbes evidently ongoing within the roots.
Apart from the UDP-glucose:flavonoid 7-O-glucosyltransferase and glutathione transferases, the most highly expressed DE transcripts in contaminated roots were aquaporins and dehydrin. This could be expected given petroleum hydrocarbon contamination is likely to reduce access to water and thus induce osmotic stress in roots. Three PIP1.1 transcripts (c483320_g1_i2, 125.11 tpm; c589371_g5_i4, 88.10 tpm; c589371_g5_i8, 30.63 tpm), three PIP2.1 (c599604_g6_i1, 79.73 tpm; c576656_g7_i3, 70.73 tpm; c599604_g8_i6, 4.23) and PIP2.7 (c602203_g2_i9, 21.70 tpm) were all highly abundant in contaminated conditions. Beyond a direct plant response to soil conditions, it has long been known that root-associated fungi, particularly ECM fungi, can improve not only nutrient uptake in trees, but also water [63–65]. It is interesting to note, in light of subsequent fungal gene expression, that extreme PIP upregulation has previously been observed as induced by the ECM fungi Amanita muscaria in fine ectomycorrhized roots of poplar (a close relative of Salix, sharing macrosynteny) [66] as well as in Salix by the AMF Rhizophagus irregularis [22].
Nitrogen and carbohydrate transport
Five amino acid transporters were DE, two in higher abundance in non-contaminated trees and three in contaminated trees. Two were distinct cationic amino acid transporters, one in higher abundance in non-contaminated conditions (c598534_g2_i3, 6.65 tpm) and one higher in contaminated conditions (c584679_g1, 18.25 tpm). Another of the transcripts higher in contaminated conditions (c594518_g1_i1) was most similar to AAP6 in poplar (87% identity, blastn e-value = 7e-29), and the two remaining transporters (one in higher abundance in each condition) were both similar to vacuolar amino acid transporter 1. AAP6, an acidic and neutral amino acid transporter known to be expressed in roots, is distinctive in having high substrate affinity (so could potentially be relevant to low amino acid concentration uptake) as well as having an affinity for aspartate [67]. Seven nitrate/peptide transporters where DE with six in higher abundance in contaminated conditions, sharing close homology with poplar NPF (NTR/PTR family) 5.4, 3.1 and 5.2 proteins. Alongside PIP expression, these are hallmarks of root symbiosis [68] in terms of differential expression and are thought to represent oligopeptide import of AM- and ECM-packaged nitrogen, the principal fungi to plant facet of resource exchange.
In terms of potential concomitant plant to fungi exchange of resources, differential expression of a putative sucrose transporter (c582330_g6_i4) with high abundance in roots from contaminated soil was identified, which would be expected if increased ECM interaction were underway in these conditions. Three distinct trehalose-6-phosphate synthase transcripts were DE (c598745_g5_i1, c536396_g3_i1, c599008_g3_i2), all in higher abundance in roots cultivated in contaminate soil. While the role of trehalose synthesis in plants is somewhat obscure [69], trehalose is the dominant storage carbohydrate in AMF hyphae and implicated in ECM abiotic stress tolerance [70, 71], so expression here is intriguing in light of concomitant fungal DE. Additionally, the sugar transporter SWEET2 (c555872_g1_i5) was in higher abundance in roots from contaminated soil. SWEETs (Sugars Will Eventually Be Exported Transporter) have only been identified relatively recently [72] but have already been strongly implicated in plant-fungal interactions [73], including Solanum tuberosum roots colonised by AMF [74]. In Arabidopsis, SWEET2 has been shown to accumulate in root hairs, cap and epidermis, and in cells in close contact with the rhizosphere [75]. Interestingly, the authors suggest that SWEET2 functions as a bidirectional glucose transporter that could control glucose secretion through limitation/vacuolar loading as part of highly complex and co-ordinated interactions with rhizospheric microbes.
Community association
The most abundant downregulated contigs in Salix roots from non-contaminated soil encoded two distinct organ-specific S2 proteins (Fig. 2).
While these proteins are of unknown function, they have been found to be downregulated in a Medicago truncatula 1-deoxy-d-xylulose 5-phosphate synthase 2 (catalysing the beginning of the MEP pathway) knockdown, which also reduced AMF colonisation of roots [76]. An acidic endochitinase (EC 3.2.1.14, c583738_g7_i2) was upregulated in contamination-treated roots. While this would intuitively be associated with plant defence against pathogenic fungi, a number of studies have found that this endochitinase can encourage symbiotic association with ECM fungi, such as Hebeloma, Suillus, Wilcoxina, Pisolithus, Paxillus and Amanita with spruce, birch and Eucalyptus; the suggested mode of action being that non-fungi-damaging apoplastic chitinase modifies ectomycorrhizal elicitors to facilitate symbiotic interaction [77–80]. When considered alongside the preceding data, Salix root DE reflects plant recognition of foreign molecular patterns [81], the non-recognition impact of effectors [82] as well as alterations in nutrient exchange and water availability known to be drastically altered by fungal interactions [65].
Fungi
Division by taxonomy
Fungi represented the kingdom with the most diverse genetic response to contamination within the root samples, being the annotation source of the 8252 distinct DE contigs. The paradigm of direct up vs down-regulation collapses when multiple organisms and natural biological complexity are acknowledged in sequencing studies (when the dynamic nature of the metagenome is considered [2]), as the presence of an organism can vary as well as gene expression within organisms. Therefore, DE contigs can represent direct or indirect responses to treatment within a given organism of stable presence (with respect to treatment), including responses to highly diverse, changeable and potentially hostile biological environment, but also represent baseline transcription and metabolism of newly present, absent, growing or diminishing organisms within the system. The contigs annotated as fungal had very distinct patterning in relation to contamination; of the contigs present in both conditions, 88.8% (6184 contigs) had higher abundance in non-contaminated trees while, in contrast to this, of the contigs only present in trees of one condition, 96.3% (1239 contigs) were in contaminated conditions (Figs. 2b, c; 4a and 5). These expression patterns, even before investigating gene function, can be interpreted as a general downregulation of constitutive fungal expression due to contamination and the arrival of a distinct, contamination specific, set of expressing genes potentially representing a shift in community makeup. By comparison, only seven Salix DE contigs were expressed in only trees of a single condition.
Fig. 4.
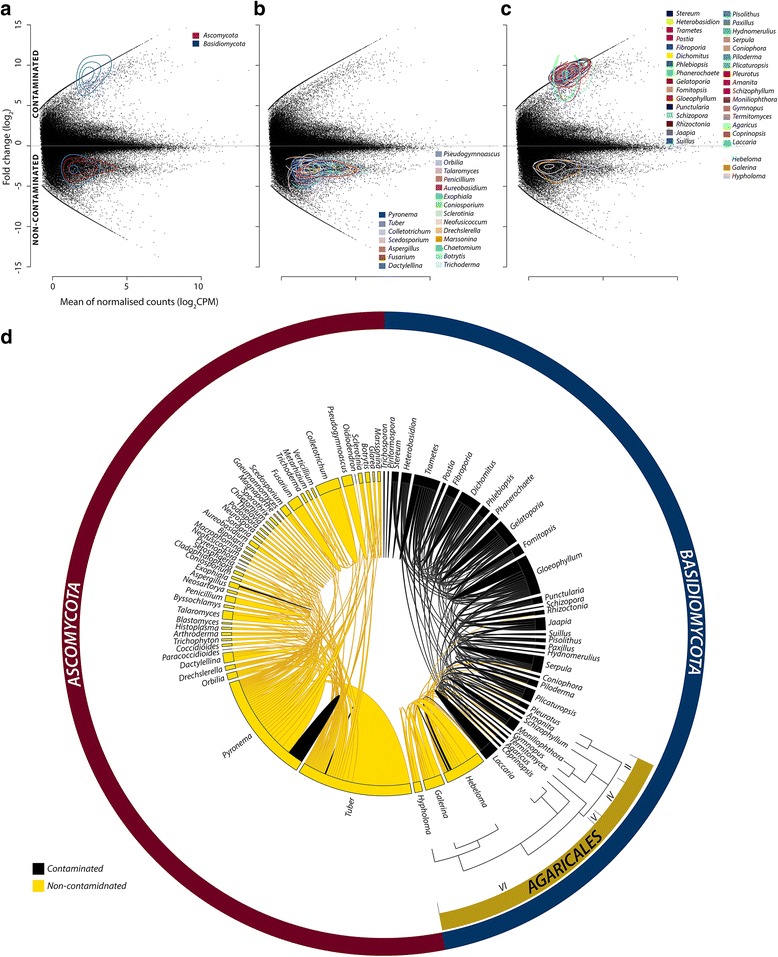
Taxonomy of fungal differential expression and secondary annotation. MA plots of de novo assembled transcriptome; y-axis represents fold change (FC, log2) between contaminated (+ive) to non-contaminated conditions (−ive), and the x-axis represents mean normalised (EdgeR) counts per million (log2 CPM). Contours representing relative DE contig density. a DE contigs annotated from fungi with Ascomycota (red) and Basidiomycota (blue), b DE Ascomycota contigs with genera annotating > 20 contigs highlighted and c DE Basidiomycota contigs with genera annotating > 20 contigs highlighted. d Secondary annotation of each DE fungal contig illustrating alternative, equally valid annotation [2] from other species (presented as genera for clarity). Genera with correspondences > 20 are presented and coloured by DE direction (more abundant in contaminated roots = black; more abundant in non-contaminated roots = gold). Agaricles phylogeny (an order of Agaricomycetes) is provided to visualise expression profiles against relatedness, with clade II (Pluteoid), IV (Marasmoid), V (Tricholomatoid) and VI (Agaricoid) structure (taken from Matheny et al. [86]). An interactive chord diagram and Krona graph to assist more comprehensive navigation of taxonomy and fungal secondary annotation are available at: https://github.com/gonzalezem/Tripartite_Metatranscriptomics_article. A full fungal DE contig list including expression information, annotation (1° and 2°) and gene ontology is provided in Additional file 6 whereas a full list of Basidiomycota DE contigs upregulated in roots of contaminated trees is provided in Additional file 7
Fig. 5.
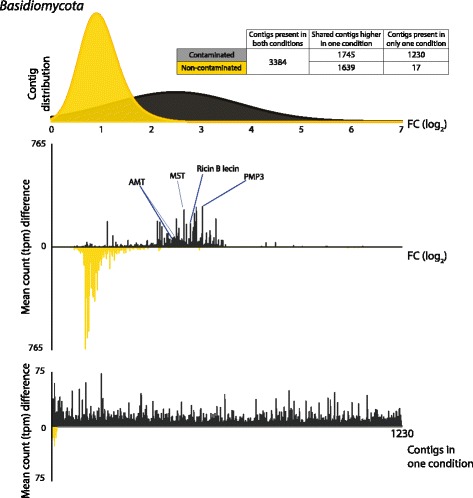
Basidiomycota differential expression (DE) transcript distribution, abundance (transcripts per million, tpm) weighted fold change (log2) and contigs present in only one condition. Top: fold change (FC log2) distribution of DE genes contaminated (black) and non-contaminated (gold). Middle: mean counts (tpm) difference between conditions against fold change per DE contig. The highly abundant transcripts discussed within the text are labelled. A full DE transcript list including expression data, functional description (if available), gene ontology terms (if available) and secondary annotation (if available) is provided in Additional file 7. MST monosaccharide transporter, AMT ammonium transporter, PMP3 plasma membrane proteolipid 3. Bottom: contigs present in only one condition (termed infinity genes in Additional files)
The two Ascomycota species most represented within DE transcripts were the closely related Pyronema omphalodes (a saprophyte) and Tuber melanosporum (an ECM fungi) (Fig. 4b and Additional file 6). These species both dominated fungal differential expression and had high enough levels of primary annotated contigs (2303 contigs and 1077 contigs) to merit very confident identification of these species, or of close relatives, within the system. Transcript levels were almost all more abundant within roots of non-contaminated trees (94.1 and 98.4% for P. omphalodes and T. melanosporum, respectively). While an alteration in gene expression in response to hydrocarbon conditions is possible, the extremity of polar expression might also suggest a lower abundance for these organisms due to the impact of contamination stress and/or increased competition from more contamination-tolerant life forms. Tuber has been previously identified as associated with Salix [83, 84]; non-constitutively expressed ECM high-abundance markers were DE in Tuber and were indeed in higher abundance in non-contaminated conditions, such as RAS protein (c594475_g3_i1) [85], implying successful Tuber ECM interaction was underway in non-contaminated conditions but was suppressed by hydrocarbon contamination.
Unlike Ascomycota, Basidiomycota expression was dynamic with respect to contamination, as 1639 contigs annotated from three closely related species (from the Agaricoid family’s Hymenogastraceae and Strophariaceae [86]) Heboloma cylindrosporum (an ECM fungi), Galerina marginata (predominantly white rot) and Hypholoma sublateritium (white rot) were downregulated, while 1745 contigs, with a highly distinctive expression pattern, were upregulated but broadly annotated from 61 genera (Fig. 4c and Additional files 6 and 7). These upregulated contigs were distinctive in the extreme diversity of annotation origin and as the majority (71%) were present only in roots from contaminated soil. To investigate the origin of these phytoremediation responsive contigs, and provide additional evidence towards their species of origin, we explored the species diversity of equally good hits using ‘secondary annotation’ [2] (Fig. 4d; Additional file 1) as well as using nucleotide blastn (a strict blastn reduces observation of fungal data by 99.5%, Additional file 8).
Secondary annotation
By utilising secondary annotation, the retention of equally (statistically) ‘good’ annotation hits, all 12,576 DE contigs can be more confidently assigned to an organism of origin by elucidating any ambiguity in annotation which is often overlooked. The set of 1745 Basidiomycota genes most often had secondary annotation from known ECM (such as Scleroderma citrinum and Paxillus involutus) and saprophytes (such as Pleurotus ostreatus and Trametes versicolor), in agreement with the current awareness that the continuum of mutualism is complex [87]. Very little crossover was observed between Basidomycota and Ascomycota in terms of differential expression due to contamination response, as Basidomycota extensively dominated the increased expression in response to contamination conditions (Fig. 4a–d). Secondary annotation provides an additional benefit in terms of mining useful functional description from across the world’s major data repositories (Additional files 2, 3, 4, 5, 6, 7, 8 and 9), but it is also interesting to note that retention of secondary annotation independently reflects classical Basidiomycota phylogeny ([86]; Fig. 4d) (albeit with a crude thresholds limiting data retention to only the very closest homologues), particularly interesting is if this idea is considered in the context of the 189,839 total annotated contigs that include 491,505 secondary annotation (Additional file 4).
An important take home message here, when querying unknown sequences using BLAST, is the benefit of not taking the single top returns as fact. Pertsemlidis and Fondon [88] have detailed how differentiating proteins of high homology from BLAST scoring system should be performed with caution and others have outlined the extensive risk of confounding biological interpretation by not acknowledging the uncertainty of annotation [2]. Secondary annotation of Basidiomycota was spread widely across Agaricomycotina for upregulated contigs whereas downregulated contigs (> 95%) derived from a specific Agaricoid clade comprising the three closely related genera (Hypholoma, Galerina and Hebeloma) (Fig. 4c) which, understandably, shared the majority of secondary annotation with each other (Fig. 4d). For the distinct, upregulated Basidiomycota, secondary annotation revealed no dominant source of annotation (homology) to our contigs within Agaricomycotina (more detailed sequence origin investigation is presented in Additional files 1 and 8).
Upregulated Basidiomycota function
Given that the intricacy, or perhaps futility, in differentiating between saprophytes and ECM fungi has been well discussed [89, 90], determining the fungal mode of action from expression study is problematic [91] and is further confounded by the generally accepted belief of multiple ECM evolutionary events in Basidiomycota ([92]). The scale of mycorrhizospheric complexity is very high; once DE genes have been sub-selected as best annotated from fungi (keeping in mind that 2575 DE contigs were unknown, having no known sequence similarity), further sub-selected based on Basidiomycota annotation and even further sub-selected based on response to contamination (just those contigs in higher abundance in contaminated conditions), 1745 DE-annotated transcripts remain to describe potential functionality (driven by positive expression) within the system. Of these 1745 DE transcripts, 70% (1227 contigs) had unknown function and were annotated as hypothetical, predicted, putative or uncharacterised proteins from Basidiomycota species. These poor functional description terms are selected against during annotation if an equally good hit is available in secondary annotation, in practice re-mining any confident functional descriptions available within the major protein data repositories.
In terms of recognisable gene function, the most abundant fungal contig expressed in contaminated conditions was the relatively cryptic plasma membrane proteolipid 3 (Pmp3), best annotated from Moniliophthora, with 301.42 tpm (c553133_g5_i1, 2128.20 FC) (Additional file 7). The small hydrophobic Pmp3 (dissimilar to recognised hydrophobin structure) has been characterised as highly conserved in fungi, is environmental stress induced (cryptic in yeast under standard laboratory conditions), involved in cytotoxic cation tolerance [93] and sphingolipid synthesis [94] so could be related to membrane integrity maintenance given the challenge of contamination conditions. Generally, the most highly expressed contigs within the group of distinctive Basidiomycota (Figs. 4c and 5) were not putatively related to hydrocarbon degradation but, instead, were related to classical association with plant roots and/or the highly upregulated bacteria (Enterobacteriaceae sp.), namely carbohydrate import, nitrogen (nitrate, ammonia and amino acid) metabolism and transport, and bacterial interaction. This could be expected in light of the hydrocarbon degradation mechanisms DE in the Enterobacteriaceae sp. (outlined below) if the interaction did indeed involve some degree of tripartite mutualism.
Putative hydrocarbon degradation
Surprisingly none of the expected hydrocarbon degrading monooxygenases were identified within the 1745 DE Basidiomycota contigs which increased in abundance due to contamination (Fig. 5, Additional file 7). Only three dioxygenase encoding contigs were identified as DE, all of which had relatively low abundance levels. Two were poorly characterised dioxygenase family proteins (c596412_g1_i1, 8.78 tpm and c593762_g1_i1, 5.93 tpm), but one was a putative extradiol aromatic ring-opening dioxygenase (c598058_g2_i2, 4.23 tpm), functionality well recognised in bacterial PAH degradation studies [95–97] but less familiar in fungi. No lignin peroxidases, manganase-dependent peroxidases (class II peroxidases) or laccases (with potential degradation functionality [26]) were identified as DE, expected if a ligninolytic (white rot) mode of action was underway, particularly as the associated enzyme secretion is highly dependent on transcript levels [91]. Two DE contigs, however, did encode glutathione peroxidase-like proteins (c591991_g1_i5, 8.78 tpm; c591991_g1_i10, 2.14 tpm), and one encoded a thioredoxin-dependent peroxidase (c565655_g1_i1, 48.94 tpm) were upregulated, although this is a common response to contamination-induced oxidative stress [98].
Carbohydrate transport and CAZy
One of most intuitive approaches to distinguish between saprophitic and ECM ecological strategies (the most likely in Basidiomycota here) would be to compare the expression of genes for plant cell wall degradation and carbohydrate import mechanisms, although this is a non-trivial task [91]. For instance, the molecular mechanisms underpinning much of the carbon transfer to ECM fungi from plants is unclear; while there is strong evidence that up to 30% of the plant’s total photoassimilates can be transferred to ECM fungi [99], few hexose transporters have been experimentally validated. The monosaccharide transporter MST1 in both Amanita muscaria (fourfold upregulation during symbiosis [100]) and Laccaria bicolor [101] being the exception to this.
The third most abundant (275.43 tpm, Table 1) Basidiomycota DE contig positively responding to contamination was a probable monosaccharide transporter (c601571_g1_i1; equally well annotated from either Serendipita vermifera or Piriformospora indica, Additional file 7), as well as two similarly annotated putative isoforms (25.71 tpm and 18.19 tpm) (Fig. 5 and Table 1). This is especially noteworthy as Hynson et al. [102] detected potential ECM lineages within Serendipitaceae and because P. indica has been shown to stimulate plant growth, but the mechanics of the symbiosis is somewhat cryptic (and can even cause plant cell death [103]). Interestingly, Zuccaro et al. [104] also found that this monosaccharide transporter was highly upregulated in Piriformospora indica and identified expression as clearly associated with barley root biotrophism (as opposed to saprotropism). The Saccharomyces cerevisiae homologue (e-value = 5e−17, 67% identity) of this MST, the extracellular glucose sensor rgt2, is well studied as also having sensor functionality [99, 105]; however, given the extremely high relative abundance, we would speculate high glucose import function as more likely to drive differential expression (due to increased transcript dependency of function). This probable monosaccharide transporter (c601571_g1_i1) is also similar to the abovementioned Laccaria bicolor MST1.3 (monosaccharide importer CAQ53118.1; lacbi1:301992, blastx e-value = 2e−21, 48% identity), whose crucial role in ECM glucose import is further supported by 14C-labelled glucose trials which demonstrated not only strongly upregulation during ECM formation when compared to extraradical mycelium expression but also strong evidence of substantial glucose import functionality [101]. Interestingly, in Laccaria bicolor, glucose uptake by MST1.3 was only very slightly affected by the presence of fructose (whereas uptake was inhibited by fructose in others, allowing for the possibility of sucrose hydrolysis at the rhizospheric interface). Alongside potential plant cell wall binding machinery, glucose import represents a highly expressed, recognisable function within the Basidomycota transcriptionally responding to contamination conditions and seems a credible candidate describing the currency of plant to fungi symbiosis with Salix roots.
Table 1.
Fungal carbohydrate metabolism and CAZy. EBSeq [42, 43] was used to estimate posterior probability of differential expression (PPDE) ≥ 0.95. A full DE transcript list including expression data, functional description (if available), gene ontology terms (if available) and secondary annotation (if available) is provided in Additional file 6
| Monosaccharide transporters | |||||
|---|---|---|---|---|---|
| Query id | Cont mean tpm | Treat mean tpm | FC | 1° annotation id | Subject description |
| c601571_g1_i1 | 0.33 | 275.43 | 830.24 | gi|751683823|gb|KIM33975.1| | Monosaccharide transporter (MST) |
| c601571_g1_i3 | 0.00 | 25.71 | High | gi|751683823|gb|KIM33975.1| | Monosaccharide transporter (MST) |
| c601571_g1_i7 | 0.00 | 18.19 | inf | gi|749760949|gb|KII85851.1| | Monosaccharide transporter (MST) |
| CAZy | |||||
| c601768_g1_i4 | 0.01 | 12.09 | 891.69 | tr|A8N526|A8N526_COPC7 | Exo-beta-1,3-glucanase (GH5) |
| c601246_g1_i2 | 0.00 | 9.29 | inf | tr|S7QIL0|S7QIL0_GLOTA | Glycoside hydrolase (GH13) |
| c601246_g1_i3 | 0.00 | 8.75 | inf | tr|S7QIL0|S7QIL0_GLOTA | Glycoside hydrolase (GH13) |
| c601246_g1_i1 | 0.00 | 3.40 | inf | tr|S7QIL0|S7QIL0_GLOTA | Glycoside hydrolase (GH13) |
| c598537_g2_i2 | 0.00 | 20.09 | inf | gi|695542974|ref.|XP_009543687.1| | Glycoside hydrolase family 13 protein |
| c600093_g2_i1 | 0.00 | 2.50 | inf | gi|646308860|gb|KDQ30003.1| | Glycoside hydrolase family 13 protein |
| c598962_g2_i1 | 0.00 | 4.02 | inf | gi|691791220|emb|CDO73299.1| | Glycoside Hydrolase Family 13 protein |
| c546152_g1_i1 | 0.00 | 7.98 | inf | tr|F8PDS7|F8PDS7_SERL9 | Glycoside hydrolase family 13/GT5 protein |
| c594633_g1_i2 | 0.11 | 50.90 | 449.23 | gi|751693072|gb|KIM43056.1| | Glycoside hydrolase family 131 protein |
| c569099_g1_i1 | 0.00 | 6.22 | inf | gi|595767974|ref.|XP_007262530.1| | Glycoside hydrolase family 16 protein |
| c601155_g2_i1 | 0.00 | 3.25 | inf | gi|751697951|gb|KIM47928.1| | Glycoside hydrolase family 16 protein |
| c601155_g2_i2 | 0.00 | 9.44 | inf | gi|751718111|gb|KIM67108.1| | Glycoside hydrolase family 16 protein |
| c601339_g1_i1 | 0.00 | 8.66 | inf | tr|M2QB49|M2QB49_CERS8 | Glycoside hydrolase family 3 protein |
| c594608_g1_i1 | 0.00 | 4.13 | inf | tr|S7Q7X9|S7Q7X9_GLOTA | Glycoside hydrolase family 38 protein |
| c601768_g1_i2 | 0.00 | 4.18 | High | gi|749771114|gb|KII95444.1| | Glycoside hydrolase family 5 protein |
| c601768_g1_i3 | 0.22 | 2.91 | 13.51 | gi|749771114|gb|KII95444.1| | Glycoside hydrolase family 5 protein |
| c598880_g1_i5 | 0.04 | 104.28 | 2617.19 | gi|660966484|gb|KEP50967.1| | Barwin-like endoglucanase GH45 |
| c600699_g2_i2 | 0.00 | 5.19 | inf | gi|761954826|gb|KIY74457.1| | Glycoside hydrolase family 45 protein |
| c600240_g5_i1 | 0.04 | 37.05 | 862.09 | gi|597903192|ref|XP_007298895.1| | Glycoside hydrolase family 72 protein |
| c597573_g1_i2 | 0.00 | 4.93 | inf | tr|M2RHY3|M2RHY3_CERS8 | Glycosyltransferase family 1 protein |
| c601034_g1_i3 | 0.00 | 5.05 | inf | gi|751699172|gb|KIM49104.1| | Glycosyltransferase family 15 protein |
| c601818_g1_i1 | 0.00 | 6.08 | inf | tr|F8NSE4|F8NSE4_SERL9 | Glycosyltransferase family 2 protein |
| c601727_g2_i2 | 0.00 | 4.46 | inf | gi|646307867|gb|KDQ29011.1| | Glycosyltransferase family 2 protein |
| c602102_g1_i1 | 0.00 | 1.47 | inf | gi|749763786|gb|KII88520.1| | Glycosyltransferase family 2 protein |
| c602018_g4_i1 | 0.00 | 21.01 | inf | gi|761950386|gb|KIY70060.1| | Glycosyltransferase family 20 protein |
| c593342_g2_i1 | 0.00 | 33.59 | inf | gi|754372083|gb|KIP04804.1| | Glycosyltransferase family 4 protein |
| c586703_g1_i3 | 0.00 | 7.27 | inf | gi|751716241|gb|KIM65247.1| | Glycosyltransferase family 48 protein |
| c593828_g1_i1 | 0.00 | 4.05 | inf | tr|G4T6Z6|G4T6Z6_PIRID | Pectate lyase 1 |
| c576776_g1_i2 | 0.05 | 2.53 | 50.56 | tr|R7SST1|R7SST1_DICSQ | Galactose oxidase (AA5) |
| c594647_g2_i1 | 0.20 | 25.38 | 127.24 | gi|752370879|gb|KIO28638.1| | Carbohydrate esterase family 12 protein |
| c592601_g1_i1 | 0.03 | 13.15 | 414.76 | gi|749760957|gb|KII85859.1| | Carbohydrate esterase family 4 protein |
| c600395_g1_i1 | 0.00 | 3.82 | inf | gi|691792079|emb|CDO72533.1| | Carbohydrate-binding module family 1/GH18 |
| c600797_g1_i1 | 0.00 | 21.07 | inf | tr|V2WEI0|V2WEI0_MONRO | Carbohydrate-binding module family 12 protein |
| c600797_g1_i4 | 0.00 | 6.68 | inf | gi|695558353|ref.|XP_009547634.1| | Carbohydrate-binding module family 12 protein |
| c590812_g2_i2 | 0.00 | 7.48 | inf | gi|751000067|gb|KIK42253.1| | Carbohydrate-binding module family 50 protein |
| EM pathway | |||||
| c598496_g1_i1 | 0.00 | 15.04 | inf | tr|E2M098|E2M098_MONPE | Glucose-6-phosphate isomerase |
| c596025_g1_i2 | 0.00 | 7.96 | inf | tr|S7Q9Q0|S7Q9Q0_GLOTA | Glucokinase regulator |
| c594601_g1_i2 | 0.00 | 13.43 | inf | gi|751180168|gb|KIL66119.1| | Hexokinase |
| c585806_g1_i1 | 0.02 | 18.89 | 1107.17 | gi|646397316|gb|KDQ61522.1| | Phosphoglucomutase |
| c602057_g2_i2 | 0.00 | 11.14 | High | gi|646396300|gb|KDQ60552.1| | Phosphoglycerate kinase |
| c602057_g2_i3 | 0.07 | 11.10 | 151.91 | tr|V2XST0|V2XST0_MONRO | Phosphoglycerate kinase |
| c598088_g2_i2 | 0.00 | 5.55 | inf | gi|749895786|gb|KIJ63485.1| | Phosphoglycerate mutase-like protein |
| c600469_g2_i1 | 0.00 | 10.71 | inf | gi|749764415|gb|KII89090.1| | Phosphoglycerate mutase-like protein |
| c572773_g2_i1 | 0.00 | 51.66 | inf | gi|695531349|ref.|XP_009540596.1| | Enolase |
| c596540_g1_i2 | 0.00 | 4.59 | inf | gi|646309757|gb|KDQ30900.1| | Pyruvate kinase |
| c600228_g1_i1 | 0.00 | 15.88 | inf | A0A0C9X171|A0A0C9X171_9AGAR | Fructose-1,6-bisphosphatase |
| c600228_g2_i1 | 0.00 | 3.03 | inf | gi|807746795|ref|XP_012178192.1| | Fructose-1,6-bisphosphatase |
| Trehalose and glycogen metabolism | |||||
| c593922_g1_i3 | 0.01 | 2.87 | 238.65 | tr|A8NBB7|A8NBB7_COPC7 | Trehalose-6-phosphate phosphatase |
| c593922_g1_i2 | 0.01 | 1.75 | 233.45 | tr|A8NBB7|A8NBB7_COPC7 | Trehalose-6-phosphate phosphatase |
| c590104_g1_i2 | 0.00 | 5.27 | inf | gi|576992104|gb|EUC64784.1| | Alpha,alpha-trehalose-phosphate synthase |
| c601514_g2_i2 | 0.00 | 8.92 | inf | gi|636619097|ref|XP_008040683.1| | Glycogen phosphorylase |
| c601447_g1_i3 | 0.00 | 24.27 | High | tr|S7PZV3|S7PZV3_GLOTA | Glycogen synthase |
| c601447_g1_i4 | 0.02 | 5.34 | 267.52 | tr|R7S2M9|R7S2M9_PUNST | Glycogen synthase |
| Pentose phosphate pathway | |||||
| c599283_g2_i1 | 0.06 | 23.65 | 403.23 | tr|S7QHH6|S7QHH6_GLOTA | Glucose-6-phosphate 1-dehydrogenase |
| c593556_g1_i1 | 0.00 | 4.43 | inf | tr|F8PCX3|F8PCX3_SERL9 | 6-Phosphogluconolactonase |
| c579299_g1_i2 | 0.00 | 4.57 | inf | gi|695543481|ref|XP_009543815.1| | 6-Phosphogluconate dehydrogenase |
| c596260_g2_i1 | 0.00 | 6.07 | inf | tr|S7QN86|S7QN86_GLOTA | 6-Phosphogluconate dehydrogenase, decarboxylating |
| c599261_g6_i1 | 0.40 | 29.50 | 73.50 | laccaria|B0DN26|B0DN26_LACBS | Transketolase |
| c586684_g1_i2 | 0.00 | 9.94 | inf | gi|807755903|ref|XP_012182746.1| | Transaldolase |
| Ethanol | |||||
| c595474_g1_i1 | 0.00 | 16.64 | inf | gi|754377558|gb|KIP10174.1| | Pyruvate decarboxylase |
| TCA cycle | |||||
| c578376_g1_i1 | 0.00 | 20.05 | inf | tr|A8N9T7|A8N9T7_COPC7 | Pyruvate carboxylase |
| c590892_g1_i1 | 0.00 | 16.91 | inf | gi|924125151|emb|CUA68456.1| | Pyruvate dehydrogenase E1 component subunit alpha |
| c602021_g6_i1 | 0.00 | 20.10 | inf | tr|D8PKQ0|D8PKQ0_SCHCM | Acetyltransferase component of pyruvate dehydrogenase |
| c569980_g1_i1 | 0.00 | 12.30 | High | tr|S8EDA9|S8EDA9_FOMPI | ATP-utilising phosphoenolpyruvate carboxykinase |
| c601865_g3_i1 | 0.00 | 21.12 | inf | tr|R7STC2|R7STC2_DICSQ | ATP-utilising phosphoenolpyruvate carboxykinase |
| c601865_g3_i2 | 0.00 | 9.71 | inf | tr|R7STC2|R7STC2_DICSQ | ATP-utilising phosphoenolpyruvate carboxykinase |
| c569980_g1_i2 | 0.09 | 16.00 | 168.90 | gi|749767591|gb|KII92023.1| | ATP-utilising phosphoenolpyruvate carboxykinase |
| c596905_g1_i4 | 0.00 | 12.95 | inf | tr|F8ND69|F8ND69_SERL9 | Malate dehydrogenase |
| c596978_g1_i2 | 0.01 | 18.99 | 1489.83 | gi|599119246|ref.|XP_007387111.1| | NAD-malate dehydrogenase |
| c596978_g1_i1 | 0.01 | 13.56 | 1360.27 | gi|599119246|ref.|XP_007387111.1| | NAD-malate dehydrogenase |
| c601738_g1_i3 | 0.00 | 9.89 | inf | gi|751717279|gb|KIM66280.1| | Succinyl-coa synthetase alpha chain, gdp-forming |
| c600881_g1_i4 | 0.01 | 1.61 | 299.16 | tr|M2RB43|M2RB43_CERS8 | Succinyl-CoA synthetase beta chain SSC-beta |
| c600756_g1_i1 | 0.00 | 11.35 | inf | gi|646392723|gb|KDQ57237.1| | Homocitrate synthase |
| c601939_g2_i2 | 0.05 | 13.42 | 261.20 | gi|754372092|gb|KIP04812.1| | Isocitrate dehydrogenase |
| c583773_g1_i1 | 0.00 | 5.65 | inf | gi|636616019|ref|XP_008039144.1| | Peroxysomal citrate synthase |
| c585571_g1_i2 | 0.03 | 2.64 | 81.61 | gi|751696020|gb|KIM45999.1| | Aconitase |
| c591611_g1_i1 | 0.00 | 10.96 | inf | gi|636618909|ref|XP_008040589.1| | Aconitate hydratase |
| c575147_g1_i1 | 0.00 | 5.73 | inf | gi|597981973|ref|XP_007363646.1| | 2-Oxoglutarate dehydrogenase mitochondrial precursor |
| c597062_g1_i2 | 0.00 | 16.03 | inf | gi|749834860|gb|KIJ12495.1| | Succinate dehydrogenase |
| c602048_g2_i2 | 0.00 | 5.52 | inf | gi|749830076|gb|KIJ08477.1| | Succinate dehydrogenase |
| c600915_g2_i1 | 0.07 | 13.16 | 190.79 | tr|M2PCH1|M2PCH1_CERS8 | Succinate dehydrogenase |
| c600002_g1_i4 | 0.00 | 4.55 | inf | tr|D8Q9I2|D8Q9I2_SCHCM | Fumarate reductase |
| c600002_g1_i1 | 0.00 | 17.05 | inf | gi|752345450|gb|KIO04521.1| | Fumarate reductase |
| Leading to AAs | |||||
| c601513_g2_i5 | 0.00 | 6.72 | inf | tr|M2PP80|M2PP80_CERS8 | Glutamine sythetase |
| c600930_g1_i1 | 0.00 | 9.88 | inf | tr|V2X687|V2X687_MONRO | Glutamate synthase |
Basidomycota carbohydrate active enzymes were identified as upregulated in response to contamination conditions, with the majority only expressed in contaminated trees (only 9 contigs were also expressed in control trees). Of these, 20 contigs belonged to one of nine glycosyl hydrolase (GH) families, eight contigs to one of six glycosyl transferase (GT) families and one contig was a pectin/pectate lyase (PL; pectate lyase 1). The most highly represented group was GH13, with seven contigs (5 distinct enzymes including 3 likely isoforms) including a secreted alpha-amylase. It is possible that these may represent extracellular invertase activity (sucrose hydrolysis) as opposed to native fungal α-glucan metabolism.
The most abundant CAZy contig (104.28 tpm) was a GH45 Barwin-like endoglucanase (expansin family protein) (Table 1). Expansin domain containing proteins have previously been recognised in Laccaria bicolor as expressed only in ECM tissue [106], which is fascinating in light of the functional similarity to cell wall loosening in plants [107] and if considered in the context of non-necrosis inducing Salix root tissue remodelling and potential interaction with middle lamella and primary cell walls. In agreement with this, a carbohydrate esterase family 12 protein (putative rhamnogalacturonan acetylesterase, pectin-related) [108] annotated only from the orchid symbiote Tulasnella calospora was DE. Further to this, a pectin/pectate lyase was also DE, recently identified as one of the few cell wall-degrading enzymes retained by the ECM fungus Tuber melanosporum for cell wall remodelling of Corylus avellana as part of their symbiotic interaction [109]. The second most abundant CAZy was GH131 (best annotated from Hebeloma but with equally good hits in Plicaturopsis, Laccaria, Jaapia, Tulasnella and Gelatoporia), including a cellulose binding module (CBM1) with broad β-glucanase activity. While this activity allows for the potential for saprophytic action, remodelling of the plant cell wall and progression through lamella is in agreement with ECM colonisation of plant roots towards the effective formation of the hartig net [110] and, interestingly, GH131 has previously been identified as expressed in Jaapia argillacea as a potential adaption from saprotrophic ancestors [92]. Three GH5 contigs (exo-beta-1,3-glucanase) were also highly expressed including an ectomycorrhiza-upregulated exo-beta-1,3-glucanase [106, 111]. In disagreement with this is the lack of identifiable lignases or maganase-dependent peroxidases necessary for deconstruction of heavily lignified middle lamella as well as the differential expression of galactose oxidase (AA5), a secreted extracellular catalyse which has twice as much activity on galactomannan (mannan backbone with galactose sidechains) compared to galactose [112], important given this very common hemicellulose component is more indicative of plant secondary cell walls [113].
In terms of the potential direct interface between the plant and putative ECM Basidiomycota, two Stereum hirsutum ricin B-like lectins of the carbohydrate-binding module family 13 (CMB13) contigs were very highly abundant in treated conditions at 144.48 tpm (14th most abundant, 1350 FC) and 98.05 tpm (22nd most abundant, 350 FC). Interchangeable homologous hits were also found using secondary annotation within Heterobasidion, Fomitiporia and Jaapia. Lectin carbohydrate-binding is well documented as one of the means of intense binding at the interface between the ECM hartig net and the plant host cell wall [114, 115]. CMB13 has been shown to have specificity for backbone xylan (such as the majority of hemicellulose within willow root 2° root cell wall), although is also found in a diverse array of non-xylanase glycosyl hydrolases [116].
The metabolic machinery necessary as a downstream consequence of monosaccharide import (in light of very highly expressed MST) includes the Embden-Meyerhof (EM) pathway, pentose phosphate pathway and tricarboxylic acid cycle (Additional file 7). Trehalose and glycogen metabolism within the EM pathway, clearly represented within DE contigs, are glycolytic pathways expected in ECM fungi [117]. Alpha,alpha-trehalose-phosphate synthase (TPS1), two trehalose-6-phosphate phosphatases (TPS2), two glycogen synthases, glycogen phosphorylase, glucokinase regulator, hexokinase, phosphoglucomutase, glucose-6-phosphate isomerase, two phosphoglycerate kinases, two phosphoglycerate mutase-like proteins, enolase, pyruvate kinase and two fructose-1,6-bisphosphatases were identified as DE and in higher abundance in root from contaminated soil. Within the pentose phosphate pathway: glucose-6-phosphate 1-dehydrogenase, 6-phosphogluconolactonase, two 6-phosphogluconate dehydrogenases, transketolase and transaldolase were also DE. While carbon was fated towards ethanol fermentation (pyruvate carboxylase was DE), the citric acid cycle was also comprehensively represented as upregulated: pyruvate dehydrogenase (E1 component subunit alpha; acetyltransferase component), four phosphoenolpyruvate carboxykinases, pyruvate carboxylase, three succinate dehydrogenases, two fumarate reductases, succinyl-coa synthetase (alpha chain and beta chain), three malate dehydrogenases, 2-oxoglutarate dehydrogenase, two homocitrate synthases, two aconitate hydratases and isocitrate dehydrogenase, supporting carbon scaffold assembly towards amino acid production.
Nitrogen management
This extensive representation of the citric acid cycle upregulated in Basidiomycota from contaminated conditions is relevant in the context of potential amino acid export to Salix roots, given very similar ECM expression profiles in ECM Laccaria and Tuber [118, 119], and considering the differential expression of contigs encoding glutamate synthase and glutamine synthetase (Table 1). Contigs encoding N-related transport machinery were highly expressed (Table 2), including two ammonium transporters, which were some of the most highly abundant Basidiomycota contigs present in the samples (AmtB, tpm 66.39; Amt1, tpm 64.18) as well as a very highly expressed contig encoding a putative FUN34-transmembrane protein involved in ammonia production (tpm 47.03). Additionally, a peptide/nitrate transporter, five amino acid transporters, a purine transporter, a glutathione transporter, two oligopeptide transporters (OPTs), two plasma membrane h+ symports, two mitochondrial carriers as well as urease, amine oxidase, D-aspartate oxidase and two carbon-nitrogen hydrolases were upregulated, all enzymes involved in the reduction of organic nitrogen compounds and ammonia production (Additional file 7). This wide ranging selection of N-related compound transporters identified as DE very closely matched those previously reported in the ECM fungi Paxillus involutus [120] and Laccaria bicolor [121].
Table 2.
Fungal nitrogen-related DE genes from upregulated Basidiomycota. EBSeq [42, 43] was used to estimate posterior probability of differential expression (PPDE) ≥ 0.95. A full DE transcript list including expression data, functional description (if available), gene ontology terms (if available) and secondary annotation (if available) is provided in Additional file 6
| Query id | Cont mean tpm | Treat mean tpm | FC | 1° annotation id | Subject description |
|---|---|---|---|---|---|
| c596378_g2_i3 | 0.14 | 66.39 | 477.06 | gi|749759516|gb|KII84515.1| | Ammonium transporter |
| c596378_g2_i2 | 0.14 | 64.18 | 458.91 | tr|Q8NKD5|Q8NKD5_HEBCY | Ammonium transporter |
| c599446_g2_i1 | 0.14 | 57.28 | 405.50 | gi|660970957|gb|KEP54657.1| | Rab GTPase family protein |
| c555426_g1_i1 | 0.04 | 47.83 | 1162.00 | gi|646294952|gb|KDQ16118.1| | Small GTPase-binding protein |
| c590384_g1_i3 | 0.36 | 47.03 | 130.17 | tr|R7RZR8|R7RZR8_PUNST | fun34-transmembrane protein of ammonia production |
| c597663_g1_i3 | 0.02 | 31.86 | 1277.49 | tr|R7S8J0|R7S8J0_TRAVS | Small GTPase-binding protein |
| c576075_g1_i1 | 0.02 | 22.26 | 1356.92 | gi|691790442|emb|CDO74199.1| | Amino acid transporter |
| c597663_g1_i1 | 0.01 | 20.67 | 2581.27 | tr|V2X8E2|V2X8E2_MONRO | Small GTPase-binding protein |
| c596122_g2_i1 | 0.00 | 18.08 | 4389.38 | tr|V2WTU3|V2WTU3_MONRO | Plasma membrane h(+)-atpase 1 |
| c592122_g2_i1 | 0.00 | 18.00 | inf | tr|S7PWC9|S7PWC9_GLOTA | Mitochondrial carrier |
| c598258_g3_i1 | 0.00 | 16.68 | inf | gi|597977025|ref.|XP_007362384.1| | Urease |
| c597755_g1_i1 | 0.00 | 16.37 | inf | gi|754376368|gb|KIP08998.1| | Subtilisin-like serine protease pepC |
| c601027_g2_i1 | 0.00 | 15.18 | inf | gi|660966237|gb|KEP50782.1| | Serine carboxypeptidase |
| c590409_g1_i1 | 0.00 | 13.93 | inf | tr|E2LF88|E2LF88_MONPE | Oligopeptide transporter |
| c602018_g3_i1 | 0.01 | 12.29 | 949.61 | tr|R7S2N4|R7S2N4_PUNST | Endopeptidase |
| c601958_g7_i1 | 0.00 | 11.68 | inf | tr|M2R776|M2R776_CERS8 | Aminopeptidase 2 |
| c597846_g2_i1 | 0.01 | 11.17 | 1682.64 | gi|695572973|ref|XP_009551472.1| | Purine transporter |
| c598346_g1_i1 | 0.00 | 10.15 | inf | gi|807753175|ref.|XP_012181382.1| | ATP-dependent metallopeptidase Hfl |
| c596122_g2_i3 | 0.05 | 9.94 | 216.32 | laccaria|A0A0C9XV60_9AGAR | Plasma membrane ATPase |
| c600930_g1_i1 | 0.00 | 9.88 | inf | tr|V2X687|V2X687_MONRO | Glutamate synthase |
| c575280_g1_i3 | 0.00 | 9.72 | inf | tr|F8NRP9|F8NRP9_SERL9 | Oligopeptide transporter |
| c586208_g2_i4 | 0.00 | 9.42 | inf | gi|595766652|ref|XP_007261869.1| | Small GTPase |
| c599446_g3_i1 | 0.00 | 8.68 | inf | tr|B0CRR7|B0CRR7_LACBS | Ras-related protein Rab-5B |
| c597351_g5_i3 | 0.05 | 8.59 | 185.55 | tr|D8PW94|D8PW94_SCHCM | Carbon-nitrogen hydrolase |
| c600850_g1_i2 | 0.00 | 8.38 | inf | gi|751184086|gb|KIL70023.1| | Aspartic peptidase A1 |
| c591514_g1_i1 | 0.00 | 8.25 | inf | gi|597967903|ref.|XP_007360041.1| | Ras protein |
| c600898_g1_i3 | 0.02 | 7.43 | 316.59 | tr|M2RCY8|M2RCY8_CERS8 | Zinc carboxypeptidase |
| c590849_g1_i2 | 0.00 | 7.23 | inf | gi|751185175|gb|KIL71111.1| | Rab-type small GTP-binding protein |
| c594481_g1_i1 | 0.00 | 7.10 | inf | uniparc|UPI000444A56C | Amino acid transporter |
| c595033_g1_i1 | 0.00 | 7.06 | inf | tr|V2WPM3|V2WPM3_MONRO | Ras GTPase-activating protein |
| c596094_g1_i2 | 0.00 | 6.76 | inf | gi|695535578|ref|XP_009541732.1| | Metallo peptidase M16B |
| c601513_g2_i5 | 0.00 | 6.72 | inf | tr|M2PP80|M2PP80_CERS8 | Glutamine synthetase |
| c587799_g1_i1 | 0.00 | 5.74 | inf | laccaria|B0CVJ7|B0CVJ7_LACBS | Aspartic peptidase A1 |
| c583462_g2_i3 | 0.00 | 5.67 | inf | tr|A8N171|A8N171_COPC7 | Glutathione transporter |
| c570564_g1_i1 | 0.00 | 5.19 | inf | gi|827762056|gb|KLO16666.1| | Zn-dependent exopeptidase |
| c594381_g2_i2 | 0.00 | 5.13 | inf | gi|599097713|ref.|XP_007380030.1| | Zn-dependent exopeptidase |
| c600158_g1_i5 | 0.03 | 5.12 | 203.31 | tr|F8NFV4|F8NFV4_SERL9 | rab GDP-dissociation inhibitor |
| c583177_g1_i2 | 0.00 | 4.84 | inf | gi|751693001|gb|KIM42985.1| | Transmembrane GTPase fzo1 |
| c600177_g1_i1 | 0.00 | 4.71 | inf | gi|914260206|gb|KNZ75358.1| | Peptide/nitrate transporter |
| c601958_g3_i1 | 0.00 | 4.68 | inf | tr|M2R776|M2R776_CERS8 | Aminopeptidase 2 |
| c594481_g1_i2 | 0.00 | 4.50 | inf | gi|618814194|ref.|XP_007309736.1| | Amino acid transporter |
| c567674_g1_i1 | 0.00 | 4.48 | inf | gi|599098740|ref.|XP_007380368.1| | Carbon-nitrogen hydrolase |
| c597783_g1_i3 | 0.00 | 4.45 | inf | tr|F8QHR7|F8QHR7_SERL3 | Glutamate carboxypeptidase |
| c594170_g1_i2 | 0.00 | 3.78 | inf | gi|751737188|gb|KIM85475.1| | Mitochondrial carrier |
| c601872_g2_i1 | 0.00 | 3.76 | inf | gi|628824529|ref|XP_007762568.1| | ATP-dependent metallopeptidase Hfl |
| c599577_g1_i2 | 0.00 | 3.73 | inf | gi|597902114|ref.|XP_007298356.1| | Peptidase M24A methionine aminopeptidase |
| c602154_g2_i2 | 0.01 | 3.51 | 328.31 | gi|761927343|gb|KIY48481.1| | D-aspartate oxidase |
| c600522_g1_i1 | 0.00 | 3.41 | inf | gi|695555686|ref.|XP_009546958.1| | Bleomycin hydrolases/aminopeptidases (cys family) |
| c598679_g1_i2 | 0.00 | 3.40 | 852.70 | tr|M2QPL6|M2QPL6_CERS8 | Aspartic-type endopeptidase |
| c601070_g1_i3 | 0.00 | 3.26 | inf | gi|599098141|ref|XP_007380172.1| | Amine oxidase |
| c595365_g1_i2 | 0.00 | 2.95 | inf | tr|E2LXQ1|E2LXQ1_MONPE | Amino acid transporter |
| c601230_g1_i3 | 0.01 | 2.92 | 561.26 | gi|695571495|ref|XP_009551148.1| | Metallo peptidase M24B |
| c600602_g1_i1 | 0.00 | 2.84 | inf | gi|695544112|ref.|XP_009543978.1| | Small monomeric GTPase |
| c601825_g4_i5 | 0.00 | 2.32 | inf | gi|754370636|gb|KIP03412.1| | Vacuolar amino acid transporter |
A broad suite of proteases was also DE including four endopeptidases (including subtilisin and bleomycin), two Zn-dependent exopeptidases, three carboxypeptidases (glutamate, serine and zinc), four metallopeptidases, a methionine aminopeptidase, two putative aminopeptidase isoforms and two distinct aspartic peptidase A1 (Table 2). Secreted suites of proteases are thought to be similar between Basidiomycota saprotrophs and EMC [90]. However, the extracellular protein degradation pathways identified here distinctly match those expressed in the EMC P. involutus [120]. One of the markers identified from Laccaria biocolor but recognised across ECM are Ras/Ras-like proteins [85, 122, 123], small GTPases involved in cargo sorting in coated vesicules [124] (vesicular turnover is thought to be high in ECM due nutrient and signal exchange with hosts [125]) that have been shown to increase in transcript abundance after successful establishment of symbiosis [126]. Eleven Ras-like proteins were DE and in high abundance in the phytoremediation responsive Basidiomycota, importantly including likely Ras, or Ras interacting, proteins known as host interaction-specific ECM markers identified in Laccaria [85]. These comprised three highly abundant uncharacterised small GTPase-binding proteins (47.83 tpm, 31.86 tpm, 9.42 tpm), Rho1 (20.67 tpm), Ras-related protein Rab-5B, Ras protein, Rab-type small GTPase, Ras GTPase-activating protein, Rab GDP-dissociation inhibitor, GTPase foz1, Sar1-like protein member of Ras-family as well as an extremely highly expressed Rab5 (ypt5) GTPase family protein (57.28 tpm), involved in endocytotic vascular trafficking [127].
While a clear depth of functional detail can be revealed by metatranscriptomics, the majority of putative fungal proteins were uncharacterised (having no functionally characterised homologue), a useful reminder that the vast majority of the natural world remains to be explored.
Bacteria
Polyadenylation in bacteria
Polyadenylation of RNA has long been known to occur in bacteria [128, 129]. Some studies have established the potential involvement of polyadenylation in mRNA degradation [130, 131] and RNA quality control [132, 133] in a limited number of bacterial species, although the contemporary picture of polyadenylation in bacteria suggests complexity beyond this, as recently described by Kushner [134]. Using pulse-labelling, levels of polyadenylated RNA have been measured at up to 15% of RNA in Escherichia coli [135] (in contrast to those of up to 25% in gram-positive Bacillus brevis [136]) and more recent research using microarray analysis revealed, in an E. coli K-12 wild-type transcriptome, that 90% of transcribed ORFs underwent some degree of polyadenlyation [137].
Within the plant transcriptomic research community, bacterial mRNA is routinely discarded during early quality control of common bioinformatics pipelines as distinct from the target organism of study or discarded intrinsically when mapping to a reference genome. Gonzalez et al. [46] recently reported how the sub-selection of only transcriptomic sequences expected a priori can confound biological results, the example leading to S. purpurea biotic stress genes being misidentified as abiotic stress responsive genes due to a strong treatment-specific interaction of a plant herbivore (Tetranycus urticae). Similarly, Brereton et al. [2] demonstrated the potential for mistaken mapping (mis-mapping) RNA-seq reads from unexpected foreign organisms to technically confound results by mapping known foreign sequences to the S. purpurea genome. Given these potential pitfalls, it would seem prudent to acknowledge the complexity of extra-laboratory biological systems involving higher eukaryotes by investigating all mRNA molecules present within any sample even when strong expectations exist within the experimental design. The approach is limited here as an unknown absolute proportion (and community) of bacterial mRNA could be lost during polyA enrichment.
Despite polyA enrichment, an extremely broad diversity of bacteria was observed within the system (including contigs that were not DE; Fig. 6a)). The majority of these contigs derived from Proteobacteria, including Alphaproteobacteria (14%), Betaproteobacteria (34%), Deltaproteobacteria (13%) and Gammaproteobacteria (23%). Actinobacteria (14%), Bacteroidetes (4%) and Firmicutes (4%) were also highly represented. This community makeup is similar to that previously reported in metagenomic studies of contaminated soils [25].
Fig. 6.
Bacterial contigs, total and differentially expressed (DE) transcript origin. Krona graphs [47] represent a total annotation of bacterial transcripts (including non-DE) and b annotation of DE bacterial transcripts. The proportion of each taxonomic grouping is defined by the number of unique transcripts, whereas the colour represents the relative abundance (transcripts per million tpm) of transcripts in each taxon. A full contig list including expression data, functional description (if available), gene ontology terms (if available) and secondary annotation (if available) is provided in Additional file 4. A list of bacterial DE transcripts (including protein coding sequences within polycistronic contigs annotated with transdecoder is provided in Additional file 10. Interactive versions of these Krona graphs available at: https://github.com/gonzalezem/Tripartite_Metatranscriptomics_article
A different picture was discerned in DE contigs (Fig. 6b); of 638 DE contigs, 86% were in higher abundance in contaminated trees. Enterobacteriaceae species were the most represented, annotating 72% of all the bacteria contigs. Importantly, 100% of these Enterobacteriaceae contigs were of higher abundance in contaminated trees (Fig. 2b, c), suggesting strong biological association with contamination and/or the other organisms. Therefore, their functionality was explored, albeit with caution. Of potential relevance to this, growth under more challenging conditions than standard rich media, or repression of growth using transformation, can increase the level of polyadenylated transcripts within E. coli [138–140].
Decoding polycistronic transcriptional units
Determining transcriptional units (putative operons) in bacteria is computationally problematic in terms of prediction from genomic sequence [141, 142]. While prediction of E. coli K12 operon structure is as advanced as in any bacteria, little confidence can be established in the transferability of such predictions given operon variation across organisms and very high responsiveness to environmental change. The unique nature of any extra-laboratory environment, such as that explored here, as well as the high likelihood of numerous novel organisms present (not the least being the bacteria themselves present within the system) further complicates transcriptional unit prediction. That being said, top-down de novo assembly provides very high confidence in the presence of observed sequences within the system, of particular technical value is the high requirement for equal mapping coverage applied within Trinity’s contig assembly [3]. Of the 639 DE and potentially polycistronic contigs annotated as bacterial in origin, 3134 protein coding regions were identified using Trinity’s transdecoder. In total, 489 of these contigs were identified as deriving from species within the family Enterobacteriaceae (100% in higher abundance in contaminated trees) comprised 2834 proteins, although a substantial proportion were duplicates with only minor sequence variation (Additional file 10). These could represent common expression by distinct organisms or multiple operons from a single organism (being somewhat reminiscent of eukaryotic splice variants at a sequence level). The majority of the 2834 Enterobacteriaceae putative proteins were annotated from E. coli (68%, across a broad spectrum of strains, Additional file 10) or the genus Shigella (29%). A recent identification and genome sequence of a poplar growth promoting endophyte (Enterobacter sp. 638) by Taghavi et al. [31] shared similar levels of homology as those found here but at a genome level, with 69% of predicted CDS being similar to E. coli.
Upregulated bacterial gene function
A broad spectrum of contigs encoding proteins putatively involved in survival within a rhizospheric environment were identified as DE due to contamination conditions (Additional file 10), including those putatively interacting extracellularly and/or intracellularly with ECM hyphae and/or plant roots. As the vast majority of DE bacterial genes were best annotated as Enterobacteriaceae (either E. coli or Shilgella strains), E. coli K-12 MG1655 gene nomenclature was used where possible.
Hydrocarbon degradation and biosurfactant production
For PAH degradation, bacterial dioxygenase enzymes are considered the principal means of ring fission [26, 143] (although cytochrome p450 monooxygenase are common), whereas alpha-ketoglutarate-dependent dioxygenases (alkB) are one of the most recognised enzymes driving degradation of aliphatic hydrocarbons [25] (not to be confused with alkane monooxygenase, alkB). The contig c596278_g1_i5 was upregulated here (containing yojIalkBadaapbEmqo), functionally comprising an ABC transporter ATP-binding protein (yojI), alpha-ketoglutarate-dependent dioxygenase (alkB), regulatory protein (ada), thiamine biosynthesis lipoprotein (apbE) and malate:quinone oxidoreductase (mqo) (Fig. 7 and Additional file 10). Additionally to this, a number of genes well characterised as accompanying toluene tolerance [144, 145] were DE, including a putative operon which contained the toluene transporter subunits mlaCDEF and kdsCDlptACkdsCD encoding a lipopolysaccharide export system. Degradation of alkanesulphonates, previously identified as involved in crude oil degradation by metagenomic study [25], occurs through desulfonation and relies on expression of the operon ssuABCDE [146, 147] comprising an alkanesulfonate transporter subunit, a putative alkanesulfonate transporter subunit, an FMNH(2)-dependent alkanesulfonate monooxygenase, putative aliphatic sulfonate binding protein and NAD(P)H-dependent FMN reductase. Here, the entirety of the ssuABCDE operon was assembled and DE in higher abundance in contaminated conditions (two similar contigs c595976_g1_i2 and c595976_g1_i9). As would be expected alongside the ssu system, the associated tau system (comprising tauABCD genes) was also upregulated within two separate putative operons containing tauAB proteins (taurine transporter subunits A and B; c596422_g1_i2) and tauCD (taurine transporter subunit C and alpha-ketoglutarate-dependent taurine dioxygenase; c596422_g2_i1). These oxidising mechanisms, such as the well-known alkanesulfonate monooxygenase ssuD [148], suggest the bacterial species present in association with willow roots is actively expressing an enzyme suite capable of degrading the hydrocarbons present within contaminated soil and so may play an important role within any tripartite interaction of mutual benefit.
Fig. 7.

A selection of differentially expressed bacterial putative operons in higher abundance in roots of contaminated trees (and discussed in the text). Bacterial contigs were first identified within the assembly as best annotated with a single bacterial protein. To find multiple potential coding regions within bacterial polycistronic sequences, we used TransDecoder software (https://transdecoder.github.io/) [3] with default parameters. A final hand annotation step was included to remove a minor number of overlapping uncharacterised ORFs. Precedence of transcriptional unit structure (putative operons) was verified in all cases against the database of prokaryotic operons (DOOR [48]) unless otherwise stated. The in-house contig label is presented with the structure of the putative operon annotated using E. coli nomenclature. The three putative operons c60225_g2_i5, c60225_g2_i7 and c60225_g2_i8 all include the transposable element insH9, similar read coverage may falsely conjoin up- and downstream DE sequence combinations around the common insert. A full list of bacterial DE putative operons (transcriptional units) including expression data, functional description (if available), gene ontology terms (if available) and secondary annotation (if available) is provided in Additional file 10
Genes necessary for three biosurfactants were found to be most represented in metagenomic study of crude oil contaminated soil [25], (in descending order of abundance): trehalose lipids [149], polyol lipids and mono/di-rhamnolipids [150]. Interestingly, the principal enzymes involved in the trehalose degradation pathway were DE, with two putative operons, the first containing treAdhaLKMR (c602247_g2_i1), from E. coli and the second (c567422_g1_i2) containing treF (from shigella) alongside a transcriptional regulator yhjB (from E. coli). These potential operons comprise the periplasmic trehalase (treA) and PTS proteins (dhaLKM, although no transport activity is expected [151]) as well as the cytoplasmic trehalase (treF) expected for trehalase utilisation [152]. While this suggests trehalose may not be being employed as a biosurfactant, it is compelling in terms of the potential interaction with ECM Basidiomycota as the specific contamination-responsive DE contigs include trehalose biosynthesis genes. The entire rmlABCD (often termed rfb) operon (c433320_g1_i1) was in higher abundance in contaminated conditions, comprising the l-rhamnose synthesis genes essential for rhamnolipid production in E. coli [150, 153]. Additionally to this, rhamnulose-1-phosphate aldolase (rhaD), rhamnulokinase l (rhaB) and rhamnose-isomerase (rhaA) were all upregulated allowing provision of l-rhamnose from dihydroxyacetone-phosphate [154], present within two operons comprising rhaDyiiL with genes for PTS system IIA and IIB components (c599913_g4_i1, operon structure common in Firmicutes) and rhaABSRyifCLMaidB (c433320_g1_i1). While the l-rhamnose synthesis pathway is considered well characterised, the exact 3-hydroxy fatty acid precursors of rhamnolipids are less confidently known, although it has been hypothesised that both the type II fatty acid synthesis pathway [155] and β-oxidation [156] can provide lipid precursors. An operon was upregulated (c601578_g3_i4) containing numerous members of the type II fatty acid synthesis pathway (FAS-II): fabG (syn: beta-ketoacyl-ACP reductase, similar to rhlG of rhamnolipid synthesis in Pseudomonas aeruginosa [157]) fabDFHychHyceDFGthiK, the operon structure being unsurprising in E. coli. rhlAB, recognisable as driving rhamnolipid production in Pseudomonas aeruginosa [155], was not detected however, and an operon containing fabA, potentially a competitor for β-hydroxydecanoyl-ACP [158], was DE. Of β-oxidation, two fadIJsixA upregulated operons (c591107_g2_i1 and c591107_g2_i2), a common operon if E. coli K-12, were in higher abundance under contaminated conditions. fadIJ are recently discovered homologues of β-oxidation fadAB in E. coli with a suggested preference for short and medium chain fatty acid degradation [159].
Community interaction
These DE potential operons encompass a wide array of genes considered cryptic in E. coli (silent under standard laboratory conditions), for example, cryptic genes in E. coli K-12 substr. MG1655 [160] that were expressed here included ecpABCD (c589067_g3_i3), ebgAC (c601454_g4_i1), gspAB c602128_g2_i1), gspCD (c600465_g1_i1) and chiAgspO (c600465_g2_i2) (Fig. 7 and Additional file 10). The expression of genes which are silent in many E. coli species cultured under laboratory conditions is perhaps unsurprising given the mycorrhizal environment; this is further supported by DE of genes representing fungiphile metabolism [161] in the Enterobacteriaceae species responding to contamination conditions. The most convincing in this regard were genes involved in interaction with chitin (fungal cell wall) and the well-known fungal exudate oxalate, used for habitat manipulation through pH lowering and increasing availability of nutrients [162] as well as for lignocellulosic degradation in many Basidiomycota. The putative operon c591446_g1_i5 comprised the well-studied two-component (stimulus-response) regulatory system evgSA [163] with an Oxalyl-CoA decarboxylase, acetyl-CoA:oxalate CoA-transferase and transporter similar to the metabolic machinery essential for E. coli tolerance of oxalate [164]. The putative operon c599707_g2_i2 comprised 8 proteins including chiPQ (oprD family, a chitoporin for uptake of chitosugars and chitosugar-induced lipoprotein which is not constitutively expressed [165, 166]) and a phosphoglymutase, suggesting tight interaction with the chitin phosphotransferase system (PTS) system [167]. In line with this is the upregulation of a potential operon containing four proteins including chbBC (N,N′-diacetylchitobiose-specific enzyme IIB component and permease IIC component of the PTS system) alongside osmE and nadE (an uncharacterised osmotically induced lipoprotein and NH(3)-dependent NAD(+) synthase). Further supporting this, the operon chbACFGR (c599728_g5_i1) comprising the more classically recognisable chitobiose operon [168], with the exception that chbB is absent (expressed in the previous operon alongside a distinct chbC). The cryptic chitinase chiA (c600465_g2_i2) was also expressed in an operon including bfr and gspO, pertinent given the ECM fungal interaction suggested by gene expression (potentially explaining cryptic expression as the fungal environment is difficult to replicate in culture) and logical in light of associated [169] DE of (non-constitutively expressed) type II general secretion systems (gspAB, c602128_g2_i1 and gspCD, c600465_g1_i1 functionality is discussed below).
While the clear expression of chitin degrading mechanisms could suggest the interaction with ECM fungi is endocellular biotrophy or necrotrophy, the possibility of extracellular biotrophy cannot be discounted as any complex interaction could involve modification of, or macro-(biofilm)-adhesion with, the Hartig net involving chitinases. In line with this, Chittero et al. [170] report biotrophic interaction of Pseudomonas fluorescens and Bacillaceae through the use of chitinases with fruit bodies of the ECM Tuber borchii; while others found bacterial chitinase production did not inhibit ECM fungi [171, 172] and that when comparing rhizospheric and ectomycorrhizosphere bacteria, those capable of hydrolysing chitin were present only within the ectomycorrhizosphere [173].
Biofilm formation
There are five stages to biofilm formation which are generally accepted: (i) surface contact and reversible attachment, (ii) irreversible attachment, (iii) microcolony formation and early development of architecture, (iv) maturation and (v) dispersal. The accomplished mini-review by Van Houdt and Michiels [174] outlines the proposed surface determinants associated with each stage in E. coli. Flagella have been associated with the initial surface contact and reversible attachment as well as final dispersal and motility (although development and maturation stages are not dependent on flagella). Irreversible attachment coincides with production of type I fimbriae, extracellular polysaccharides and curli fimbriae. Development of biofilm architecture also involves curli and extracellular polysaccharides as well as colonic acid production and the self-recognising adhesion antigen 43 (also called flu). Maturation of the biofilm architecture also involves colonic acid production and curli as well as conjugative pili. Dispersal of bacterial cells back into the environment again involves flagella. Operons encoding proteins from each of these processes were DE, all being in higher abundance in contaminated conditions. The formation and adhesion of bacterial biofilms to fungal hyphae has been well characterised as facilitating the exchange of nutrients [28, 29] but has also been shown to allow the mobilisation of pollutant degrading bacteria through the environment for mutual benefit [175, 176].
As substantial numbers of genes involved in biofilm formation were DE, we compared gene expression here to the list generated by Tenorio et al. [177], who systematically investigated genes affecting biofilm formation in E. coli through overexpression. While the study was performed using LB media, we expect extensive overlap in machinery involved in potential biofilm formation within an ECM environment. Ten genes were identified as fundamentally altering biofilm abundance: ccmf; fdrA (syn yahF); flgFGIL, fliH (c601578_g1_i1); gspA; secY and wcaK (Fig. 7 and Additional file 10). All were DE here with the exception of secY and wcaK; however, other colonic acid production genes were upregulated (wbz wcaJLF) as well as extensive DE of the secYEG translocon (peptide export complex proteins) including, secA, secE and SecDFyajC. A total of 11 of the 35 genes whose overexpression altered biofilm architecture in LB media were DE with increased abundance in roots of contaminated soil, while 11 of the 27 genes causing filmentous morphology of the biofilm were also upregulated. The flagellin synthesis genes FliA and FliC, and flagellar biosynthesis master regulator FlhC, were identified as in high abundance in treated samples as well as fliEFGHIJKLMNOP, fliDST, flgKL and flgGHIL. Flagella can allow increased fitness when growing/forming biofilm on fungal hyphae [178], although E. coli are thought to require flagella only for the initial stages of biofilm formation [177]. Expression of these genes has been shown to be positively regulated by polyadenylation [139]; this is particularly interesting given the suggestion that this mechanism could be specifically in place to allow the bacteria to adapt and survive in challenging environmental conditions (through PAPI regulation).
The chaperone-usher pathway is a delivery system for type I pili through the outer membrane. The periplasmic transport of proteins is essential for successful pili biogenesis and the chaperone-usher pathway utilises the SecYEG translocon to achieve this, also upregulated here in contaminated conditions. Concordantly, the fimDFGH operon (c600702_g6_i3, structurally common in E. coli K-12 strains), encoding the outer membrane usher protein for type 1 fimbrial synthesis and three minor component of F-type fimbriae, was upregulated in contaminated conditions, as was sfmCD (c575401_g1_i2; fimACI was not identified as DE). Interestingly, given the likely interaction with fungal cells, sfm is a chaperone-usher operon that is silent under laboratory conditions but was shown to express when the type 1 fimbriae complex is knocked out and could promote adhesion to eukaryotic cells [160]. Exploring the idea that a substantial amount of biofilm-related expression within this microbiome environment could indeed be cryptic within laboratory environments, Korea et al. [160] identified a wide range of these chaperone-usher fimbrae as associated with distinct surface specialties in E. coli K-12.
The contigs assembled here from root samples and upregulated in contaminated conditions did indeed represent the majority of these previously identified cryptic chaperone-usher fimbriae, including yfc (c601154_g1_i3; yfcRQOU(usher papC)SP(chaperone papD)B(prmB)aroC), sfm (c575401_g1_i2; sfmCD); fim (c600702_g6_i3; fimDFGH), yad (c601794_g1_i1; yadHG, c601794_g2_i3; yadCKLMhtrEecpD and c601794_g3_i1; yadECDI), yde (c591908_g2_i3; ydeTQSR), yqi (c601683_g14_i1; yqiGHI), yeh (c598114_g3_i1; yehABC) and yra (c600252_g5_i1; yraJ) (Fig. 7 and Additional file 10). These represent putative operons, predicted in E. coli K-12, which do not contribute to adhesion of E. coli under normal laboratory conditions but do promote biofilm formation on uncommon abiotic surfaces (‘unknown environmental niches’ [160]) and do contribute to adhesion to eukaryotic surfaces. The ELF operon (elfADCG) [179] was observed within a single contig as the seven member elfADCG-ycbUVF operon (c601320_g2_i5) predicted in E. coli K-12 with an additional protein, pyrD dihydroorotate dehydrogenase gene (quinone). Interestingly, a number of proteins expressed within these contigs are suggestive of potential functionality of association with a host: ydeTQSR included a serine/threonine-protein kinase HipA and the toxin component of a HipA family toxin/antitoxin system, and pantothenate synthetase, 3-methyl-2-oxobutanoate hydroxymethyltransferase and aspartate 1-decarboxylase (both of which participate in pantothenate and coA biosynthesis) were expressed along with yadECDI.
In addition to these extra-laboratory expressed operons associated with biofilm formation, a number of DE genes were associated with swarming in Pseudomonas aeruginosa, a process strongly related to biofilm formation. Four of the six genes explored as swarming negative mutants by Overhage et al. [180] were necessary for good biofilm formation; all were DE here and in higher abundance in contaminated conditions: rhlE (c601160_g1_i1, ATP-dependent RNA helicase), lptA (c602252_g5_i2, lipopolysaccharide transport periplasmic protein), gshB (c586692_g1_i1, glutathione synthetase) and acsA (c600318_g2_i1, acetyl-CoA synthetase). Additionally, two variants of traABCEKLNPUVWYtrbCL (c582511_g1_i6 and c582511_g1_i7) are comprised of conjugative apparatus (pili construction) common to F-like plasmids and necessary for mobility of genetic elements between bacteria (Fig. 7 and Additional file 10). None of the bacterial secretion systems are thought to be constitutively expressed but are instead triggered by recognition of host molecular pattern through adhesins [181, 182]. As discussed with relation to chitinase secretion, gspAB and gspCDO (with chiA) of the type II secretion system were upregulated in contaminated conditions. Given this pattern, it would be expected that xcpU (the general secretion pathway protein H, gspH in E. coli) would be DE; however, it was not identified as such.
The common pilus (ECP) operon is widely conserved throughout E. coli and required for early-stage biofilm development and host cell recognition [183], although usually silent in E. coli K-12 MG1655 under laboratory conditions [184]. As discussed above, an operon containing the regular common pilus machinery, ecpRABCDEykgJyagU (c589067_g3_i3; synonyms include but are not limited to matABCDEFykgJyagU) was in higher abundance under contamination conditions. Interestingly, the yagTSRQ operon (c589067_g1_i4; synonym paoABCD), common in E. coli K-12 strains, was also DE, encoding periplasmic detoxification enzymes (xanthine oxidase family [185]) including yagT, a twin arginine translocator (tat) pathway signal sequence. While the secYEG translocon can transport unfolded proteins, the tat can transport folded proteins to the periplasm (around 6% of all secreted proteins are thought to be tat dependent [186]). These secreted proteins include hydrogenases, dehydrogenase and nitrate reductases. The tat-dependent hydrogenase-2 operon (redox) (hybGFEDCBAO; c600034_g1_i12) was identified as DE, including the Tat pathway signal sequence domain protein hybA [187].
Curli fimbriae are outer membrane adhesins employed by E. coli to facilitate biofilm adhesion [177, 188, 189]. A master regulator of biofilm formation, csgD [189], is at the centre of the curli fimbriae regulatory network. Two variants of the csgD containing operon csgDEFGycdXYZ (c596842_g1_i2 and c596842_g1_i4) were DE and in higher abundance in contaminated roots (Fig. 7 and Additional file 10). csgDEFG encode four proteins necessary for curli assembly: csgG is involved in pore formation in the outer membrane, whereas csgE and csgF are periplasmic proteins which physically interact with csgG [188]. csgD is a positive regulator of csgAB (classically expressed on a distinct operon), the major curli extracellular structural subunit (csgA) and its nucleator (csgB). While csgAB was not identified as DE here, it has been recognised that this is often the case (csgAB not being expressed in cells forming curli) and that assembly can occur via interbacterial complementation, it may also be true here that subunit production is not transcript dependent or is constitutive and constant (and so not DE). Additionally to curli, the prominent surface protein flu was also upregulated (c601850_g2_i6), a self-recognising adhesin known to induce autoaggregation to promote biofilm formation [190].
Nitrogen management
Similar to the recently sequenced plant growth-promoting endophyte Enterobacter sp. 638, which was isolated from poplar stems, it seems likely that no nitrogen fixation was occurring within the Enterobacteriaceae species responding to contamination as no nif genes were identified as DE, with the exception of the pyruvate-flavodoxin oxidoreductase (nifJ/ydbK) (Fig. 7 and Additional file 10). Two variants of the fixABCX operon were also upregulated (c602208_g1_i1 and c602208_g1_i3; intricately linked with N fixation in diazotrophs [191, 192]), but in E. coli, these genes form an important reducing role in carnitine metabolism [193, 194].
Differential expression of well-characterised respiratory nitrate reduction (nar) operons were identified including narK (c598952_g5_i3) used for nitrate/nitrite extrusion in E. coli [195]; narUZ (c598132_g7_i1) where narU is also used for nitrate/nitrite extrusion while narZ is part of nitrate reduction, as well as narXLychONChaC (c598952_g3_i7; similar to 282397 (TU0650)), nitrate/nitrite sensor and regulator proteins. Two slightly divergent versions of the periplasmic nitrate reduction (nap) operon was also DE, napABCGHccmABCFE (c602069_g2_i2 and c602069_g2_i4; similar to 425132 (TU1418)) [196]. The important nitric oxide reduction (dissimilatory reduction) equipment in E. coli, via flavorubredoxin expression encoded by the NorVW operon [197], was also DE. The nitrogen assimilation control regulator nac was also present within an upregulated operon ((c598923_g2_i7; common to in structure to 580938 in E. coli BL21(DE3)) containing the cbl regulator, involved in aliphatic sulfonate utilisation, and yeeO MATE efflux transporter (involved in flavin secretion). In terms of ammonia generation, two versions of anaerobically expressed nitrite transport and reduction nir operons [31, 198] were DE: nirBtsgA (c598930_g2_i1) and nirBCDcysG (c598930_g1_i1). Amino acid (glutamine) synthesis under nitrogen demand has also been shown to be fine-tuned (ammonia/ammonium) by the nitrogen assimilatory protein glnK regulation of amtB (ammonium transporter) [199], present here within an upregulated operon containing the amtB and two multidrug efflux proteins, mdlAB, (c602197_g2_i5; all commonly expressed together). Additionally to this, nrfA, a cytochrome c-552 which catalyses nitrite to ammonia in a formate-dependent manner, was upregulated (c600318_g2_i1) as well as an operon containing yddG, an aromatic amino acid extrusion protein, in a operon (c601567_g3_i3) containing the nitrate-inducible formate dehydrogenase major subunit fdnG.
The conversion of inorganic nitrogen, through nitrate assimilation and reduction, to the biologically more useful nitrite and ammonia (for example, to construct amino acids) provides bacteria with nutrition for survival within the environment but also a potential currency for mutualism within the microbiome, or mycorrhizosphere, system. E. coli only assimilates nitrate under anaerobic conditions [200]; this broad spectrum of assimilatory and dissimilatory nitrate reduction equipment with higher abundance under contaminated conditions is perhaps unsurprising given that facultative anaerobic Enterobacteriaceae can outcompete obligate anaerobes in anoxic, organic compound-rich forest soils [201].
Bacterial functional role
So-called mycorrhizal helper bacteria MHB [30] are thought to promote the symbiotic association between plant roots and mycorrhizae, creating a tripartite relationship [29], such as the Bacillus sphaericus isolate EJP109 promoting Suillus luteus (ECM) growth in association with Pinus sylvestris as well as the Streptomyces sp. nov. 505 and S. anulatus (Beijerinck) Waksman 1003 promoting Amanita muscaria (ECM) growth in association with Picea abies [172, 202]. MHB and PGPR (plant growth-promoting rhizobacteria) strains of Pseudomonas have been visualised attaching to ECM hyphae (suggesting extracellular biotrophic mycophagy) of the genus Laccaria, in a bacterial and fungal strain-specific manner [203]. Differentiating between the potential roles of interacting bacterial species within the biological system is difficult; Leveau and Preston [204] suggested three potential modes of interaction, necrotrophy, extracellular biotrophy and endocellular biotrophy, as well as a spectrum of expected functionality associated to each mode. In general here, expression suggested extracellular biotrophy, however, not only are the other two modes of interaction possible but a more complex continuum of interaction would seem likely given the diverse environment. Before such functionality can be confidently elucidated, in addition to the necessary promotion of cross-disciplinary microbiome research, observation of gene expression without constraint to an organism/s expected within a biological system needs to become a standard requirement for transcriptomic investigations.
Conclusions
From gene expression alone, the major responses of Salix purpurea cv. ‘Fish Creek’ to soil contamination do not seem to be direct degradation, immobilisation or exclusion of contaminants but rather widespread, extraordinarily complex, alterations to interactions with microbiota. The extremely diverse fungal community, revealed using a bioinformatics approach unconstrained by the requirement of a priori nucleotide sequence, responds dynamically to contamination both with changes to gene expression but also with substantial shifts in the community makeup. Much like the host plant, fungal gene expression was dominated by alterations in pathways associated to microbiome interactions as opposed to direct hydrocarbon degradation. Surprisingly, gene expression representing increased petroleum hydrocarbon metabolism equipment came from an Enterobacteriaceae species whose total expression was uniformly more abundant in roots of contaminated trees. It seems possible that, given the successful contamination tolerance, willows may depend on symbiosis to tolerate stressful environmental conditions instead of relying solely on their own metabolism.
While polyadenylated bacterial sequences are often not quantified across experimental systems investigated with eukaryote expression in mind, the bacterial functionality observed here is thought-provoking, revealing a plausible set of expressed genes indicating tripartite mutualism. A finding which is useful in providing a convincing reminder that observing the entirety of generated data, even that which is commonly discarded, is always of potential value for exposing the unexpected. Most importantly, with regard to the attempted all-inclusive strategy of observing all expressed sequences within a biological sample, regardless of origin, is how extensively interpretation of expression could be confounded if plant, fungal or bacterial expression were investigated as responsive to treatment (here being contamination) in isolation, and thus how crucial it is to attempt observation of the entire microbiome and wider metaorganism. This metatranscriptomic approach, which marries an understanding of the uncertainty of microbiome biology with the strengths and limitations of current bioinformatics, should be broadly transferable through utility for biological samples containing expressed sequences from well-characterised, poorly characterised or entirely unknown organisms at a diversity of low or high complexity.
Additional files
Custom scripts. (DOCX 133 kb)
Unknown DE spreadsheet. (XLSX 2375 kb)
Total annotation spreadsheet. (XLSX 65570 kb)
Salix DE spreadsheet. (XLSX 2169 kb)
Fungi DE spreadsheet. (XLSX 6048 kb)
Upregulated DE Basidiomycota spreadsheet. (XLSX 1898 kb)
Upregulated DE Basidiomycota blastn spreadsheet. (XLSX 958 kb)
Total DE spreadsheet. (XLSX 7230 kb)
Bacteria DE transdecoded (annotation of polycistronic contigs) spreadsheet. (XLSX 2104 kb)
Acknowledgements
We thank the Genome Quebec Innovation Centre for the support and Calcul Quebec for the computing resources.
Funding
The project was funded by the GenoRem Project (Genome Canada and Genome Québec), NSERC Strategic grant for projects (STPGP 494702) as well as BioFuelNet Canada and NCE (Networks of 1583 Center of Excellence).
Availability of data and materials
Sequence data from this article can be found in the European Nucleotide Archive online repository (PRJEB16316).
Authors’ contributions
EY, MSA, SJ, ML and FP conceived and designed the study. JM, WGN and AP performed the plant growth trials, sample and sequencing preparation. EG and NJBB analysed the data and drafted the manuscript. All authors commented on and approved the final manuscript.
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s40168-018-0432-5) contains supplementary material, which is available to authorized users.
References
- 1.Stappenbeck TS, Virgin HW. Accounting for reciprocal host–microbiome interactions in experimental science. Nature. 2016;534:191–199. doi: 10.1038/nature18285. [DOI] [PubMed] [Google Scholar]
- 2.Brereton NJB, Gonzalez E, Marleau J, Nissim WG, Labrecque M, Joly S, Pitre FE. Comparative transcriptomic approaches exploring contamination stress tolerance in Salix sp. reveal the importance for a metaorganismal de novo assembly approach for nonmodel plants. Plant Physiol. 2016;171:3–24. doi: 10.1104/pp.16.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henner P, Schiavon M, Morel JL, Lichtfouse E. Polycyclic aromatic hydrocarbon (PAH) occurrence and remediation methods. Analusis. 1997;25:M56–M59. [Google Scholar]
- 5.De Sousa C. Contaminated sites: the Canadian situation in an international context. J Environ Manag. 2001;62:131–154. doi: 10.1006/jema.2001.0431. [DOI] [PubMed] [Google Scholar]
- 6.Hamin EM. Turning brownfields into greenbacks. J Am Plan Assoc. 1999;65:236–237. [Google Scholar]
- 7.Panagos P, Van Liedekerke M, Yigini Y, Montanarella L. Contaminated sites in Europe: review of the current situation based on data collected through a European network. J Environ Public Health. 2013;2013:158764. doi: 10.1155/2013/158764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glass D, Raskin I, Ensley B. Phytoremediation of toxic metals: using plants to clean up the environment. Phytoremediation toxic metals. Wiley; 2000. p. 304.
- 9.Pilon-Smits E. Phytoremediation. Annu Rev Plant Biol. 2005;56:15–39. doi: 10.1146/annurev.arplant.56.032604.144214. [DOI] [PubMed] [Google Scholar]
- 10.Pulford ID, Watson C. Phytoremediation of heavy metal-contaminated land by trees—a review. Environ Int. 2003;29:529–540. doi: 10.1016/S0160-4120(02)00152-6. [DOI] [PubMed] [Google Scholar]
- 11.Bell TH, Joly S, Pitre FE, Yergeau E. Increasing phytoremediation efficiency and reliability using novel omics approaches. Trends Biotechnol. 2014;32:271–280. doi: 10.1016/j.tibtech.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 12.Bell TH, El-Din Hassan S, Lauron-Moreau A, Al-Otaibi F, Hijri M, Yergeau E, St-Arnaud M. Linkage between bacterial and fungal rhizosphere communities in hydrocarbon-contaminated soils is related to plant phylogeny. ISME J. 2014;8:331–343. doi: 10.1038/ismej.2013.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bissonnette L, St-Arnaud M, Labrecque M. Phytoextraction of heavy metals by two Salicaceae clones in symbiosis with arbuscular mycorrhizal fungi during the second year of a field trial. Plant Soil. 2010;332:55–67. doi: 10.1007/s11104-009-0273-x. [DOI] [Google Scholar]
- 14.FCM . (The Federation of Canadian Municipalities) brownfields, sustainability snapshot 2009. 2009. [Google Scholar]
- 15.Ray M, Brereton N, Shield I, Karp A, Murphy R. Variation in cell wall composition and accessibility in relation to biofuel potential of short rotation coppice willows. Bioenergy Res. 2012;5:1–14. doi: 10.1007/s12155-011-9177-8. [DOI] [Google Scholar]
- 16.Heller MC, Keoleian GA, Mann MK, Volk TA. Life cycle energy and environmental benefits of generating electricity from willow biomass. Renew Energy. 2004;29:1023–1042. doi: 10.1016/j.renene.2003.11.018. [DOI] [Google Scholar]
- 17.Gnansounou E, Dauriat A. Techno-economic analysis of lignocellulosic ethanol: a review. Bioresour Technol. 2010;101:4980–4991. doi: 10.1016/j.biortech.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 18.Galbe M, Sassner P, Wingren A, Zacchi G. Process engineering economics of bioethanol production. Biofuels. 2007;108:303–327. doi: 10.1007/10_2007_063. [DOI] [PubMed] [Google Scholar]
- 19.Hamelinck CN, van Hooijdonk G, Faaij APC. Ethanol from lignocellulosic biomass: techno-economic performance in short-, middle- and long-term. Biomass Bioenergy. 2005;28:384–410. doi: 10.1016/j.biombioe.2004.09.002. [DOI] [Google Scholar]
- 20.Bonfante P, Anca I-A. Plants, mycorrhizal fungi, and bacteria: a network of interactions. Annu Rev Microbiol. 2009;63:363–383. doi: 10.1146/annurev.micro.091208.073504. [DOI] [PubMed] [Google Scholar]
- 21.Hassan SED, Bell TH, Stefani FOP, Denis D, Hijri M, St-Arnaud M. Contrasting the community structure of arbuscular mycorrhizal fungi from hydrocarbon-contaminated and uncontaminated soils following willow (Salix spp. L.) planting. PLoS One. 2014;9:e102838. doi: 10.1371/journal.pone.0102838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Almeida-Rodríguez AM, Gómes MP, Loubert-Hudon A, Joly S, Labrecque M. Symbiotic association between Salix purpurea L. and Rhizophagus irregularis: modulation of plant responses under copper stress. Tree Physiol. 2015;36(4):407–420. doi: 10.1093/treephys/tpv119. [DOI] [PubMed] [Google Scholar]
- 23.Wang B, Qiu Y-L. Phylogenetic distribution and evolution of mycorrhizas in land plants. Mycorrhiza. 2006;16:299–363. doi: 10.1007/s00572-005-0033-6. [DOI] [PubMed] [Google Scholar]
- 24.Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42(Database issue):D699–704. [DOI] [PMC free article] [PubMed]
- 25.Abbasian F, Lockington R, Palanisami T, Ramadass K, Megharaj M, Naidu R. Microbial diversity and hydrocarbon degrading gene capacity of a crude oil field soil as determined by metagenomics analysis. Biotechnol Prog. 2016;32(3):638–648. doi: 10.1002/btpr.2249. [DOI] [PubMed] [Google Scholar]
- 26.Bamforth SM, Singleton I. Bioremediation of polycyclic aromatic hydrocarbons: current knowledge and future directions. J Chem Technol Biotechnol. 2005;80:723–736. doi: 10.1002/jctb.1276. [DOI] [Google Scholar]
- 27.Yergeau E, Sanschagrin S, Maynard C, St-Arnaud M, Greer CW. Microbial expression profiles in the rhizosphere of willows depend on soil contamination. ISME J. 2014;8:344–358. doi: 10.1038/ismej.2013.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taktek S, St-Arnaud M, Piché Y, Fortin JA, Antoun H. Igneous phosphate rock solubilization by biofilm-forming mycorrhizobacteria and hyphobacteria associated with Rhizoglomus irregulare DAOM 197198. Mycorrhiza. 2017;27(1):13–22. doi: 10.1007/s00572-016-0726-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frey-Klett P, Garbaye J, Tarkka M. The mycorrhiza helper bacteria revisited. New Phytol. 2007;176:22–36. doi: 10.1111/j.1469-8137.2007.02191.x. [DOI] [PubMed] [Google Scholar]
- 30.Garbaye J. Helper bacteria—a new dimension to the mycorrhizal symbiosis. New Phytol. 1994;128:197–210. doi: 10.1111/j.1469-8137.1994.tb04003.x. [DOI] [PubMed] [Google Scholar]
- 31.Taghavi S, Van Der Lelie D, Hoffman A, Zhang Y-B, Walla MD, Vangronsveld J, Newman L, Monchy S. Genome sequence of the plant growth promoting endophytic bacterium Enterobacter sp. 638. PLoS Genet. 2010;6:e1000943. doi: 10.1371/journal.pgen.1000943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang S, Puryear J, Cairney J. A simple and efficient method for isolating RNA from pine trees. Plant Mol Biol Report. 1993;11:113–116. doi: 10.1007/BF02670468. [DOI] [Google Scholar]
- 33.Gambino G, Perrone I, Gribaudo I. A rapid and effective method for RNA extraction from different tissues of grapevine and other woody plants. Phytochem Anal. 2008;19:520–525. doi: 10.1002/pca.1078. [DOI] [PubMed] [Google Scholar]
- 34.Stewart FJ, Ottesen EA, DeLong EF. Development and quantitative analyses of a universal rRNA-subtraction protocol for microbial metatranscriptomics. ISME j. 2010;4:896–907. doi: 10.1038/ismej.2010.18. [DOI] [PubMed] [Google Scholar]
- 35.Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 2010;2010:t5448. doi: 10.1101/pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- 36.Lohse M, Bolger AM, Nagel A, Fernie AR, Lunn JE, Stitt M, Usadel B. RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res. 2012;40:W622–W627. doi: 10.1093/nar/gks540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–U354. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langmead B. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics. 2010;CHAPTER 11:Unit 11.17. doi: 10.1002/0471250953.bi1107s32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villacorta-Martin C, Núñez de Cáceres González FF, de Haan J, Huijben K, Passarinho P, Lugassi-Ben Hamo M, Zaccai M. Whole transcriptome profiling of the vernalization process in Lilium longiflorum (cultivar White Heaven) bulbs. BMC Genomics. 2015;16:550. doi: 10.1186/s12864-015-1675-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods. 2013;10:71–U99. doi: 10.1038/nmeth.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BMG, Haag JD, Gould MN, Stewart RM, Kendziorski C. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29:1035–1043. doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soneson C, Delorenzi M. A comparison of methods for differential expression analysis of RNA-seq data. Bmc Bioinformatics. 2013;14:91. doi: 10.1186/1471-2105-14-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gonzalez E, Brereton NJB, Marleau J, Guidi Nissim W, Labrecque M, Pitre FE, Joly S. Meta-transcriptomics indicates biotic cross-tolerance in willow trees cultivated on petroleum hydrocarbon contaminated soil. BMC Plant Biol. 2015;15:246. doi: 10.1186/s12870-015-0636-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ondov BD, Bergman NH, Phillippy AM. Interactive metagenomic visualization in a Web browser. Bmc Bioinformatics. 2011;12:385. doi: 10.1186/1471-2105-12-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mao F, Dam P, Chou J, Olman V, Xu Y. DOOR: a database for prokaryotic operons. Nucleic Acids Res. 2009;37:D459–D463. doi: 10.1093/nar/gkn757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hultman J, Waldrop MP, Mackelprang R, David MM, McFarland J, Blazewicz SJ, Harden J, Turetsky MR, McGuire AD, Shah MB. Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature. 2015;521:208–212. doi: 10.1038/nature14238. [DOI] [PubMed] [Google Scholar]
- 50.Tveit A, Schwacke R, Svenning MM, Urich T. Organic carbon transformations in high-Arctic peat soils: key functions and microorganisms. ISME j. 2013;7:299–311. doi: 10.1038/ismej.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bent SJ, Pierson JD, Forney LJ. Measuring species richness based on microbial community fingerprints: the emperor has no clothes. Appl Environ Microbiol. 2007;73:2399–2401. doi: 10.1128/AEM.02383-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bellemain E, Carlsen T, Brochmann C, Coissac E, Taberlet P, Kauserud H. ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiol. 2010;10:189. doi: 10.1186/1471-2180-10-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lindner DL, Banik MT. Intragenomic variation in the ITS rDNA region obscures phylogenetic relationships and inflates estimates of operational taxonomic units in genus Laetiporus. Mycologia. 2011;103:731–740. doi: 10.3852/10-331. [DOI] [PubMed] [Google Scholar]
- 54.Vetrovsky T, Baldrian P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One. 2013;8(2):e57923. doi: 10.1371/journal.pone.0057923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo LL, Sui ZH, Zhang S, Ren YY, Liu Y. Comparison of potential diatom ‘barcode’ genes (the 18S rRNA gene and ITS, COI, rbcL) and their effectiveness in discriminating and determining species taxonomy in the Bacillariophyta. Int J Syst Evol Microbiol. 2015;65:1369–1380. doi: 10.1099/ijs.0.000076. [DOI] [PubMed] [Google Scholar]
- 56.Maslunka C, Gifford B, Tucci J, Gurtler V, Seviour RJ. Insertions or deletions (Indels) in the rrn 16S-23S rRNA gene internal transcribed spacer region (ITS) compromise the typing and identification of strains within the Acinetobacter calcoaceticus-baumannii (Acb) complex and closely related members. PLoS One. 2014;9(8):e105390. 10.1371/journal.pone.0105390. [DOI] [PMC free article] [PubMed]
- 57.Porras-Alfaro A, Liu KL, Kuske CR, Xie G. From genus to phylum: large-subunit and internal transcribed spacer rRNA operon regions show similar classification accuracies influenced by database composition. Appl Environ Microbiol. 2014;80:829–840. doi: 10.1128/AEM.02894-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iffis B, St-Arnaud M, Hijri M. Petroleum hydrocarbon contamination, plant identity and arbuscular mycorrhizal fungal (AMF) community determine assemblages of the AMF spore-associated microbes. Environ Microbiol. 2016;18:2689–2704. doi: 10.1111/1462-2920.13438. [DOI] [PubMed] [Google Scholar]
- 59.Yergeau E, Bell TH, Champagne J, Maynard C, Tardif S, Tremblay J, Greer CW. Transplanting soil microbiomes leads to lasting effects on willow growth, but not on the rhizosphere microbiome. Front Microbiol. 2015;6:1436. doi: 10.3389/fmicb.2015.01436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brereton NJB, Ahmed F, Sykes D, Ray MJ, Shield I, Karp A, Murphy RJ. X-ray micro-computed tomography in willow reveals tissue patterning of reaction wood and delay in programmed cell death. BMC Plant Biol. 2015;15:83. doi: 10.1186/s12870-015-0438-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sandermann H. Plant metabolism of xenobiotics. Trends Biochem Sci. 1992;17:82–84. doi: 10.1016/0968-0004(92)90507-6. [DOI] [PubMed] [Google Scholar]
- 62.Noguchi A, Saito A, Homma Y, Nakao M, Sasaki N, Nishino T, Takahashi S, Nakayama T. A UDP-glucose: isoflavone 7-O-glucosyltransferase from the roots of soybean (glycine max) seedlings purification, gene cloning, phylogenetics, and an implication for an alternative strategy of enzyme catalysis. J Biol Chem. 2007;282:23581–23590. doi: 10.1074/jbc.M702651200. [DOI] [PubMed] [Google Scholar]
- 63.Dixon R, Wright G, Behrns G, Teskey R, Hinckley T. Water deficits and root growth of ectomycorrhizal white oak seedlings. Can J For Res. 1980;10:545–548. doi: 10.1139/x80-089. [DOI] [Google Scholar]
- 64.Dixon R, Pallardy S, Garrett H, Cox G, Sander I. Comparative water relations of container-grown and bare-root ectomycorrhizal and nonmycorrhizal Quercus velutina seedlings. Can J Bot. 1983;61:1559–1565. doi: 10.1139/b83-168. [DOI] [Google Scholar]
- 65.Lehto T, Zwiazek JJ. Ectomycorrhizas and water relations of trees: a review. Mycorrhiza. 2011;21:71–90. doi: 10.1007/s00572-010-0348-9. [DOI] [PubMed] [Google Scholar]
- 66.Marjanović Ž, Uehlein N, Kaldenhoff R, Zwiazek JJ, Weiß M, Hampp R, Nehls U. Aquaporins in poplar: what a difference a symbiont makes! Planta. 2005;222:258–268. doi: 10.1007/s00425-005-1539-z. [DOI] [PubMed] [Google Scholar]
- 67.Fischer WN, Loo DD, Ludewig U, Boorer KJ, Tegeder M, Rentsch D, Wright EM, Frommer WB. Low and high affinity amino acid H+-cotransporters for cellular import of neutral and charged amino acids. Plant J. 2002;29:717–731. doi: 10.1046/j.1365-313X.2002.01248.x. [DOI] [PubMed] [Google Scholar]
- 68.Garcia K, Doidy J, Zimmermann SD, Wipf D, Courty P-E. Take a trip through the plant and fungal transportome of mycorrhiza. Trends Plant Sci. 2016;21(11):937–950. doi: 10.1016/j.tplants.2016.07.010. [DOI] [PubMed] [Google Scholar]
- 69.Grennan AK. The role of trehalose biosynthesis in plants. Plant Physiol. 2007;144:3–5. doi: 10.1104/pp.104.900223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bago B, Pfeffer PE, Douds DD, Brouillette J, Bécard G, Shachar-Hill Y. Carbon metabolism in spores of the arbuscular mycorrhizal fungusGlomus intraradices as revealed by nuclear magnetic resonance spectroscopy. Plant Physiol. 1999;121:263–272. doi: 10.1104/pp.121.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ferreira AS, Tótola MR, Borges AC. Physiological implications of trehalose in the ectomycorrhizal fungus Pisolithus sp. under thermal stress. J Therm Biol. 2007;32:34–41. doi: 10.1016/j.jtherbio.2006.08.009. [DOI] [Google Scholar]
- 72.Chen L-Q, Hou B-H, Lalonde S, Takanaga H, Hartung ML, Qu X-Q, Guo W-J, Kim J-G, Underwood W, Chaudhuri B. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature. 2010;468:527–532. doi: 10.1038/nature09606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Doidy J, Grace E, Kühn C, Simon-Plas F, Casieri L, Wipf D. Sugar transporters in plants and in their interactions with fungi. Trends Plant Sci. 2012;17:413–422. doi: 10.1016/j.tplants.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 74.Manck-Götzenberger J, Requena N. Arbuscular mycorrhiza symbiosis induces a major transcriptional reprogramming of the potato SWEET sugar transporter family. Front Plant Sci. 2016;7:487. doi: 10.3389/fpls.2016.00487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen HY, Huh JH, Yu YC, Ho LH, Chen LQ, Tholl D, Frommer WB, Guo WJ. The Arabidopsis vacuolar sugar transporter SWEET2 limits carbon sequestration from roots and restricts Pythium infection. Plant J. 2015;83:1046–1058. doi: 10.1111/tpj.12948. [DOI] [PubMed] [Google Scholar]
- 76.Floß DS, Hause B, Lange PR, Kuester H, Strack D, Walter MH. Knock-down of the MEP pathway isogene 1-deoxy-D-xylulose 5-phosphate synthase 2 inhibits formation of arbuscular mycorrhiza-induced apocarotenoids, and abolishes normal expression of mycorrhiza-specific plant marker genes. Plant J. 2008;56:86–100. doi: 10.1111/j.1365-313X.2008.03575.x. [DOI] [PubMed] [Google Scholar]
- 77.Salzer P, Hubner B, Sirrenberg A, Hager A. Differential effect of purified spruce chitinases and [beta]-1, 3-glucanases on the activity of elicitors from ectomycorrhizal fungi. Plant Physiol. 1997;114:957–968. doi: 10.1104/pp.114.3.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Albrecht C, Burgess T, Dell B, Lapeyrie F. Chitinase and peroxidase activities are induced in eucalyptus roots according to aggressiveness of Australian ectomycorrhizal strains of Pisolithus sp. New Phytol. 1994;127:217–222. doi: 10.1111/j.1469-8137.1994.tb04273.x. [DOI] [PubMed] [Google Scholar]
- 79.Stefani FO, Tanguay P, Pelletier G, Piché Y, Hamelin RC. Impact of endochitinase-transformed white spruce on soil fungal biomass and ectendomycorrhizal symbiosis. Appl Environ Microbiol. 2010;76:2607–2614. doi: 10.1128/AEM.02807-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Le Quéré A, Wright DP, Söderström B, Tunlid A, Johansson T. Global patterns of gene regulation associated with the development of ectomycorrhiza between birch (Betula pendula Roth.) and Paxillus involutus (Batsch) Fr. Mol Plant-Microbe Interact. 2005;18:659–673. doi: 10.1094/MPMI-18-0659. [DOI] [PubMed] [Google Scholar]
- 81.Chisholm ST, Coaker G, Day B, Staskawicz BJ. Host-microbe interactions: shaping the evolution of the plant immune response. Cell. 2006;124:803–814. doi: 10.1016/j.cell.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 82.Plett JM, Daguerre Y, Wittulsky S, Vayssières A, Deveau A, Melton SJ, Kohler A, Morrell-Falvey JL, Brun A, Veneault-Fourrey C. Effector MiSSP7 of the mutualistic fungus Laccaria bicolor stabilizes the Populus JAZ6 protein and represses jasmonic acid (JA) responsive genes. Proc Natl Acad Sci. 2014;111:8299–8304. doi: 10.1073/pnas.1322671111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bonito GM, Gryganskyi AP, Trappe JM, Vilgalys R. A global meta-analysis of Tuber ITS rDNA sequences: species diversity, host associations and long-distance dispersal. Mol Ecol. 2010;19:4994–5008. doi: 10.1111/j.1365-294X.2010.04855.x. [DOI] [PubMed] [Google Scholar]
- 84.Leonardi M, Iotti M, Oddis M, Lalli G, Pacioni G, Leonardi P, Maccherini S, Perini C, Salerni E, Zambonelli A. Assessment of ectomycorrhizal fungal communities in the natural habitats of Tuber magnatum (Ascomycota, Pezizales) Mycorrhiza. 2013;23:349–358. doi: 10.1007/s00572-012-0474-7. [DOI] [PubMed] [Google Scholar]
- 85.Sundaram S, Kim S, Suzuki H, Mcquattie C, Hiremath S, Podila G. Isolation and characterization of a symbiosis-regulated ras from the ectomycorrhizal fungus Laccaria bicolor. Mol Plant-Microbe Interact. 2001;14:618–628. doi: 10.1094/MPMI.2001.14.5.618. [DOI] [PubMed] [Google Scholar]
- 86.Matheny PB, Curtis JM, Hofstetter V, Aime MC, Moncalvo J-M, Ge Z-W, Yang Z-L, Slot JC, Ammirati JF, Baroni TJ. Major clades of Agaricales: a multilocus phylogenetic overview. Mycologia. 2006;98:982–995. doi: 10.1080/15572536.2006.11832627. [DOI] [PubMed] [Google Scholar]
- 87.Hou W, Lian B, Dong H, Jiang H, Wu X. Distinguishing ectomycorrhizal and saprophytic fungi using carbon and nitrogen isotopic compositions. Geosci Front. 2012;3:351–356. doi: 10.1016/j.gsf.2011.12.005. [DOI] [Google Scholar]
- 88.Pertsemlidis A, Fondon JW. Having a BLAST with bioinformatics (and avoiding BLASTphemy) Genome Biol. 2001;2:reviews2002.2001–reviews2002.2010. doi: 10.1186/gb-2001-2-10-reviews2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Koide RT, Sharda JN, Herr JR, Malcolm GM. Ectomycorrhizal fungi and the biotrophy–saprotrophy continuum. New Phytol. 2008;178:230–233. doi: 10.1111/j.1469-8137.2008.02401.x. [DOI] [PubMed] [Google Scholar]
- 90.Pellegrin C, Morin E, Martin FM, Veneault-Fourrey C. Comparative analysis of secretomes from ectomycorrhizal fungi with an emphasis on small-secreted proteins. Front Microbiol. 2015;6:1278. doi: 10.3389/fmicb.2015.01278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kohler A, Kuo A, Nagy LG, Morin E, Barry KW, Buscot F, Canbäck B, Choi C, Cichocki N, Clum A. Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat Genet. 2015;47:410–415. doi: 10.1038/ng.3223. [DOI] [PubMed] [Google Scholar]
- 92.Shah F, Nicolás C, Bentzer J, Ellström M, Smits M, Rineau F, Canbäck B, Floudas D, Carleer R, Lackner G. Ectomycorrhizal fungi decompose soil organic matter using oxidative mechanisms adapted from saprotrophic ancestors. New Phytol. 2016;209:1705–1719. doi: 10.1111/nph.13722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Navarre C, Goffeau A. Membrane hyperpolarization and salt sensitivity induced by deletion of PMP3, a highly conserved small protein of yeast plasma membrane. EMBO J. 2000;19:2515–2524. doi: 10.1093/emboj/19.11.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.De Block J, Szopinska A, Guerriat B, Dodzian J, Villers J, Hochstenbach J-F, Morsomme P. Yeast Pmp3p has an important role in plasma membrane organization. J Cell Sci. 2015;128:3646–3659. doi: 10.1242/jcs.173211. [DOI] [PubMed] [Google Scholar]
- 95.Peng R-H, Xiong A-S, Xue Y, Fu X-Y, Gao F, Zhao W, Tian Y-S, Yao Q-H. Microbial biodegradation of polyaromatic hydrocarbons. FEMS Microbiol Rev. 2008;32:927–955. doi: 10.1111/j.1574-6976.2008.00127.x. [DOI] [PubMed] [Google Scholar]
- 96.Chikere CB, Okpokwasili GC, Chikere BO. Monitoring of microbial hydrocarbon remediation in the soil. Biotech. 2011;1:117–138. doi: 10.1007/s13205-011-0014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sipilä TP, Keskinen A-K, Åkerman M-L, Fortelius C, Haahtela K, Yrjälä K. High aromatic ring-cleavage diversity in birch rhizosphere: PAH treatment-specific changes of IE 3 group extradiol dioxygenases and 16S rRNA bacterial communities in soil. ISME j. 2008;2:968–981. doi: 10.1038/ismej.2008.50. [DOI] [PubMed] [Google Scholar]
- 98.Carmel-Harel O, Storz G. Roles of the glutathione-and thioredoxin-dependent reduction systems in the Escherichia coli and Saccharomyces cerevisiae responses to oxidative stress. Annu Rev Microbiol. 2000;54:439–461. doi: 10.1146/annurev.micro.54.1.439. [DOI] [PubMed] [Google Scholar]
- 99.Nehls U, Grunze N, Willmann M, Reich M, Kuester H. Sugar for my honey: carbohydrate partitioning in ectomycorrhizal symbiosis. Phytochemistry. 2007;68:82–91. doi: 10.1016/j.phytochem.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 100.Nehls U, Wiese J, Guttenberger M, Hampp R. Carbon allocation in ectomycorrhizas: identification and expression analysis of an Amanita muscaria monosaccharide transporter. Mol Plant-Microbe Interact. 1998;11:167–176. doi: 10.1094/MPMI.1998.11.3.167. [DOI] [PubMed] [Google Scholar]
- 101.Fajardo López M, Dietz S, Grunze N, Bloschies J, Weiß M, Nehls U. The sugar porter gene family of Laccaria bicolor: function in ectomycorrhizal symbiosis and soil-growing hyphae. New Phytol. 2008;180:365–378. doi: 10.1111/j.1469-8137.2008.02539.x. [DOI] [PubMed] [Google Scholar]
- 102.Hynson NA, Weiss M, Preiss K, Gebauer G, Treseder KK. Fungal host specificity is not a bottleneck for the germination of Pyroleae species (Ericaceae) in a Bavarian forest. Mol Ecol. 2013;22:1473–1481. doi: 10.1111/mec.12180. [DOI] [PubMed] [Google Scholar]
- 103.Deshmukh S, Hückelhoven R, Schäfer P, Imani J, Sharma M, Weiss M, Waller F, Kogel K-H. The root endophytic fungus Piriformospora indica requires host cell death for proliferation during mutualistic symbiosis with barley. Proc Natl Acad Sci. 2006;103:18450–18457. doi: 10.1073/pnas.0605697103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zuccaro A, Lahrmann U, Güldener U, Langen G, Pfiffi S, Biedenkopf D, Wong P, Samans B, Grimm C, Basiewicz M. Endophytic life strategies decoded by genome and transcriptome analyses of the mutualistic root symbiont Piriformospora indica. PLoS Pathog. 2011;7:e1002290. doi: 10.1371/journal.ppat.1002290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Roy A, Hashmi S, Li Z, Dement AD, Cho KH, Kim J-H. The glucose metabolite methylglyoxal inhibits expression of the glucose transporter genes by inactivating the cell surface glucose sensors Rgt2 and Snf3 in yeast. Mol Biol Cell. 2016;27:862–871. doi: 10.1091/mbc.E15-11-0789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Martin F, Aerts A, Ahrén D, Brun A, Danchin E, Duchaussoy F, Gibon J, Kohler A, Lindquist E, Pereda V. The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature. 2008;452:88–92. doi: 10.1038/nature06556. [DOI] [PubMed] [Google Scholar]
- 107.Nikolaidis N, Doran N, Cosgrove DJ. Plant expansins in bacteria and fungi: evolution by horizontal gene transfer and independent domain fusion. Mol Biol Evol. 2014;31:376–386. doi: 10.1093/molbev/mst206. [DOI] [PubMed] [Google Scholar]
- 108.Rytioja J, Hildén K, Yuzon J, Hatakka A, de Vries RP, Mäkelä MR. Plant-polysaccharide-degrading enzymes from basidiomycetes. Microbiol Mol Biol Rev. 2014;78:614–649. doi: 10.1128/MMBR.00035-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sillo F, Fangel JU, Henrissat B, Faccio A, Bonfante P, Martin F, Willats WG, Balestrini R. Understanding plant cell-wall remodelling during the symbiotic interaction between Tuber melanosporum and Corylus avellana using a carbohydrate microarray. Planta. 2016;244(2):347–59. doi: 10.1007/s00425-016-2507-5. [DOI] [PubMed] [Google Scholar]
- 110.Veneault-Fourrey C, Kohler A, Morin E, Balestrini R, Plett J, Danchin E, Coutinho P, Wiebenga A, De Vries RP, Henrissat B. Genomic and transcriptomic analysis of Laccaria bicolor CAZome reveals insights into polysaccharides remodelling during symbiosis establishment. Fungal Genet Biol. 2014;72:168–181. doi: 10.1016/j.fgb.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 111.Peter M, Kohler A, Ohm RA, Kuo A, Krützmann J, Morin E, Arend M, Barry KW, Binder M, Choi C, et al. Ectomycorrhizal ecology is imprinted in the genome of the dominant symbiotic fungus Cenococcum geophilum. Nat Commun. 2016;7:12662. doi: 10.1038/ncomms12662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Avigad G, Amaral D, Asensio C, Horecker B. The D-galactose oxidase of Polyporus circinatus. J Biol Chem. 1962;237:2736–2743. [PubMed] [Google Scholar]
- 113.Donaldson LA, Knox JP. Localization of cell wall polysaccharides in normal and compression wood of radiata pine: relationships with lignification and microfibril orientation. Plant Physiol. 2012;158:642–653. doi: 10.1104/pp.111.184036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Massicotte H, Ackerley C, Peterson R. Localization of three sugar residues in the interface of ectomycorrhizae synthesized between Alnus crispa and Alpova diplophloeus as demonstrated by lectin binding. Can J Bot. 1987;65:1127–1132. doi: 10.1139/b87-157. [DOI] [Google Scholar]
- 115.Giollant M, Guillot J, Damez M, Dusser M, Didier P, Didier E. Characterization of a lectin from Lactarius deterrimus (research on the possible involvement of the fungal lectin in recognition between mushroom and spruce during the early stages of mycorrhizae formation) Plant Physiol. 1993;101:513–522. doi: 10.1104/pp.101.2.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.FUJIMOTO Z. Structure and function of carbohydrate-binding module families 13 and 42 of glycoside hydrolases, comprising a β-trefoil fold. Biosci Biotechnol Biochem. 2013;77:1363–1371. doi: 10.1271/bbb.130183. [DOI] [PubMed] [Google Scholar]
- 117.Martin F, Ramstedt M, Söderhäll K. Carbon and nitrogen metabolism in ectomycorrhizal fungi and ectomycorrhizas. Biochimie. 1987;69:569–581. doi: 10.1016/0300-9084(87)90176-3. [DOI] [PubMed] [Google Scholar]
- 118.Deveau A, Kohler A, Frey-Klett P, Martin F. The major pathways of carbohydrate metabolism in the ectomycorrhizal basidiomycete Laccaria bicolor S238N. New Phytol. 2008;180:379–390. doi: 10.1111/j.1469-8137.2008.02581.x. [DOI] [PubMed] [Google Scholar]
- 119.Ceccaroli P, Buffalini M, Saltarelli R, Barbieri E, Polidori E, Ottonello S, Kohler A, Tisserant E, Martin F, Stocchi V. Genomic profiling of carbohydrate metabolism in the ectomycorrhizal fungus Tuber melanosporum. New Phytol. 2011;189:751–764. doi: 10.1111/j.1469-8137.2010.03520.x. [DOI] [PubMed] [Google Scholar]
- 120.Shah F, Rineau F, Canbäck B, Johansson T, Tunlid A. The molecular components of the extracellular protein-degradation pathways of the ectomycorrhizal fungus Paxillus involutus. New Phytol. 2013;200:875–887. doi: 10.1111/nph.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lucic E, Fourrey C, Kohler A, Martin F, Chalot M, Brun-Jacob A. A gene repertoire for nitrogen transporters in Laccaria bicolor. New Phytol. 2008;180:343–364. doi: 10.1111/j.1469-8137.2008.02580.x. [DOI] [PubMed] [Google Scholar]
- 122.Sundaram S, Brand JH, Hymes MJ, Hiremath S, Podila GK. Isolation and analysis of a symbiosis-regulated and Ras-interacting vesicular assembly protein gene from the ectomycorrhizal fungus Laccaria bicolor. New Phytol. 2004;161:529–538. doi: 10.1046/j.1469-8137.2003.00935.x. [DOI] [PubMed] [Google Scholar]
- 123.Rajashekar B, Kohler A, Johansson T, Martin F, Tunlid A, Ahrén D. Expansion of signal pathways in the ectomycorrhizal fungus Laccaria bicolor–evolution of nucleotide sequences and expression patterns in families of protein kinases and RAS small GTPases. New Phytol. 2009;183:365–379. doi: 10.1111/j.1469-8137.2009.02860.x. [DOI] [PubMed] [Google Scholar]
- 124.De Camilli P, Emr SD, McPherson PS, Novick P. Phosphoinositides as regulators in membrane traffic. Science. 1996;271:1533. doi: 10.1126/science.271.5255.1533. [DOI] [PubMed] [Google Scholar]
- 125.Kim S-J, Hiremath ST, Podila GK. Cloning and identification of symbiosis-regulated genes from the ectomycorrhizal Laccaria bicolor. Mycol Res. 1999;103:168–172. doi: 10.1017/S0953756298006881. [DOI] [Google Scholar]
- 126.de Freitas PM, Betancourth BML, Teixeira JA, Zubieta MP, de Queiroz MV, Kasuya MCM, Costa MD, de Araújo EF. In vitro Scleroderma laeve and Eucalyptus grandis mycorrhization and analysis of atp6, 17S rDNA, and ras gene expression during ectomycorrhizal formation. J Basic Microbiol. 2014;54:1358–1366. doi: 10.1002/jobm.201400253. [DOI] [PubMed] [Google Scholar]
- 127.Inada N, Ueda T. Membrane trafficking pathways and their roles in plant–microbe interactions. Plant Cell Physiol. 2014;55:672–686. doi: 10.1093/pcp/pcu046. [DOI] [PubMed] [Google Scholar]
- 128.Sarkar N. Polyadenylation of mRNA in bacteria. Microbiol-Uk. 1996;142:3125–3133. doi: 10.1099/13500872-142-11-3125. [DOI] [PubMed] [Google Scholar]
- 129.Nakazato H, Venkatesan S, Edmonds M. Polyadenylic acid sequences in E. coli messenger RNA. Nature. 1975;256:144–146. doi: 10.1038/256144a0. [DOI] [PubMed] [Google Scholar]
- 130.Hajnsdorf E, Braun F, Haugel-Nielsen J, Regnier P. Polyadenylylation destabilizes the rpsO mRNA of Escherichia coli. Proc Natl Acad Sci U S A. 1995;92:3973–3977. doi: 10.1073/pnas.92.9.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.O'Hara EB, Chekanova JA, Ingle CA, Kushner ZR, Peters E, Kushner SR. Polyadenylylation helps regulate mRNA decay in Escherichia coli. Proc Natl Acad Sci U S A. 1995;92:1807–1811. doi: 10.1073/pnas.92.6.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Li Z, Reimers S, Pandit S, Deutscher MP. RNA quality control: degradation of defective transfer RNA. EMBO J. 2002;21:1132–1138. doi: 10.1093/emboj/21.5.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mohanty BK, Kushner SR. Bacterial/archaeal/organellar polyadenylation. Wiley Interdiscip Rev-Rna. 2011;2:256–276. doi: 10.1002/wrna.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kushner SR. Polyadenylation in E. coli: a 20 year odyssey. RNA. 2015;21:673–674. doi: 10.1261/rna.049700.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Srinivasan PR, Ramanarayanan M, Rabbani E. Presence of polyriboadenylate sequences in pulse-labeled RNA of Escherichia coli. Proc Natl Acad Sci U S A. 1975;72:2910–2914. doi: 10.1073/pnas.72.8.2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sarkar N, Langley D, Paulus H. Isolation and characterization of polyadenylate-containing RNA from Bacillus brevis. Biochemistry. 1978;17:3468–3474. doi: 10.1021/bi00610a007. [DOI] [PubMed] [Google Scholar]
- 137.Mohanty BK, Kushner SR. The majority of Escherichia coli mRNAs undergo post-transcriptional modification in exponentially growing cells. Nucleic Acids Res. 2006;34:5695–5704. doi: 10.1093/nar/gkl684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jasiecki J, Wegrzyn G. Growth-rate dependent RNA polyadenylation in Escherichia coli. EMBO Rep. 2003;4:172–177. doi: 10.1038/sj.embor.embor733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Maes A, Gracia C, Brechemier D, Hamman P, Chatre E, Lemelle L, Bertin PN, Hajnsdorf E. Role of polyadenylation in regulation of the flagella cascade and motility in Escherichia coli. Biochimie. 2013;95:410–418. doi: 10.1016/j.biochi.2012.10.017. [DOI] [PubMed] [Google Scholar]
- 140.Mohanty BK, Kushner SR. Analysis of the function of Escherichia coli poly(A) polymerase I in RNA metabolism. Mol Microbiol. 1999;34:1094–1108. doi: 10.1046/j.1365-2958.1999.01673.x. [DOI] [PubMed] [Google Scholar]
- 141.Brouwer RW, Kuipers OP, van Hijum SA. The relative value of operon predictions. Brief Bioinform. 2008;9:367–375. doi: 10.1093/bib/bbn019. [DOI] [PubMed] [Google Scholar]
- 142.Mao XZ, Ma Q, Liu BQ, Chen X, Zhang HY, Xu Y. Revisiting operons: an analysis of the landscape of transcriptional units in E. coli. Bmc Bioinformatics. 2015;16:356. doi: 10.1186/s12859-015-0805-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Heitkamp MA, Freeman JP, Miller DW, Cerniglia CE. Pyrene degradation by a Mycobacterium sp.—identification of ring oxidation and ring fission-products. Appl Environ Microbiol. 1988;54:2556–2565. doi: 10.1128/aem.54.10.2556-2565.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yung PY, Lo Grasso L, Mohidin AF, Acerbi E, Hinks J, Seviour T, Marsili E, Lauro FM. Global transcriptomic responses of Escherichia coli K-12 to volatile organic compounds. Sci Rep. 2016;6:19899. doi: 10.1038/srep19899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Malinverni JC, Silhavy TJ. An ABC transport system that maintains lipid asymmetry in the Gram-negative outer membrane. Proc Natl Acad Sci U S A. 2009;106:8009–8014. doi: 10.1073/pnas.0903229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Duan J, Jiang W, Cheng Z, Heikkila JJ, Glick BR. The complete genome sequence of the plant growth-promoting bacterium Pseudomonas sp. UW4. PLoS One. 2013;8:e58640. doi: 10.1371/journal.pone.0058640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.van der Ploeg JR, Cummings NJ, Leisinger T, Connerton IF. Bacillus subtilis genes for the utilization of sulfur from aliphatic sulfonates. Microbiol-Uk. 1998;144:2555–2561. doi: 10.1099/00221287-144-9-2555. [DOI] [PubMed] [Google Scholar]
- 148.Kane SR, Chakicherla AY, Chain PS, Schmidt R, Shin MW, Legler TC, Scow KM, Larimer FW, Lucas SM, Richardson PM. Whole-genome analysis of the methyl tert-butyl ether-degrading beta-proteobacterium Methylibium petroleiphilum PM1. J Bacteriol. 2007;189:1931–1945. doi: 10.1128/JB.01259-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pohnlein M, Hausmann R, Lang S, Syldatk C. Enzymatic synthesis and modification of surface-active glycolipids. Eur J Lipid Sci Technol. 2015;117:145–155. doi: 10.1002/ejlt.201400418. [DOI] [Google Scholar]
- 150.Müller MM, Kügler JH, Henkel M, Gerlitzki M, Hörmann B, Pöhnlein M, Syldatk C, Hausmann R. Rhamnolipids—next generation surfactants? J Biotechnol. 2012;162:366–380. doi: 10.1016/j.jbiotec.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 151.Gutknecht R, Beutler R, Garcia-Alles LF, Baumann U, Erni B. The dihydroxyacetone kinase of Escherichia coli utilizes a phosphoprotein instead of ATP as phosphoryl donor. EMBO J. 2001;20:2480–2486. doi: 10.1093/emboj/20.10.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li H, Su H, Kim SB, Chang YK, Hong S-K, Seo Y-G, Kim C-J. Enhanced production of trehalose in Escherichia coli by homologous expression of otsBA in the presence of the trehalase inhibitor, validamycin A, at high osmolarity. J Biosci Bioeng. 2012;113:224–232. doi: 10.1016/j.jbiosc.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 153.Cabrera-Valladares N, Richardson A-P, Olvera C, Treviño LG, Déziel E, Lépine F, Soberón-Chávez G. Monorhamnolipids and 3-(3-hydroxyalkanoyloxy) alkanoic acids (HAAs) production using Escherichia coli as a heterologous host. Appl Microbiol Biotechnol. 2006;73:187–194. doi: 10.1007/s00253-006-0468-5. [DOI] [PubMed] [Google Scholar]
- 154.Henkel M, Müller MM, Kügler JH, Lovaglio RB, Contiero J, Syldatk C, Hausmann R. Rhamnolipids as biosurfactants from renewable resources: concepts for next-generation rhamnolipid production. Process Biochem. 2012;47:1207–1219. doi: 10.1016/j.procbio.2012.04.018. [DOI] [Google Scholar]
- 155.Zhu K, Rock CO. RhlA converts beta-hydroxyacyl-acyl carrier protein intermediates in fatty acid synthesis to the beta-hydroxydecanoyl-beta-hydroxydecanoate component of rhamnolipids in Pseudomonas aeruginosa. J Bacteriol. 2008;190:3147–3154. doi: 10.1128/JB.00080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Abdel-Mawgoud AM, Lepine F, Deziel E. A stereospecific pathway diverts beta-oxidation intermediates to the biosynthesis of rhamnolipid biosurfactants. Chem Biol. 2014;21:156–164. doi: 10.1016/j.chembiol.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 157.Campos-Garcia J, Caro AD, Najera R, Miller-Maier RM, Al-Tahhan RA, Soberon-Chavez G. The Pseudomonas aeruginosa rhlG gene encodes an NADPH-dependent beta-ketoacyl reductase which is specifically involved in rhamnolipid synthesis. J Bacteriol. 1998;180:4442–4451. doi: 10.1128/jb.180.17.4442-4451.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Reis RS, Pereira AG, Neves BC, Freire DMG. Gene regulation of rhamnolipid production in Pseudomonas aeruginosa—a review. Bioresour Technol. 2011;102:6377–6384. doi: 10.1016/j.biortech.2011.03.074. [DOI] [PubMed] [Google Scholar]
- 159.Campbell JW, Morgan-Kiss RM, Cronan JE. A new Escherichia coli metabolic competency: growth on fatty acids by a novel anaerobic beta-oxidation pathway. Mol Microbiol. 2003;47:793–805. doi: 10.1046/j.1365-2958.2003.03341.x. [DOI] [PubMed] [Google Scholar]
- 160.Korea CG, Badouraly R, Prevost MC, Ghigo JM, Beloin C. Escherichia coli K-12 possesses multiple cryptic but functional chaperone-usher fimbriae with distinct surface specificities. Environ Microbiol. 2010;12:1957–1977. doi: 10.1111/j.1462-2920.2010.02202.x. [DOI] [PubMed] [Google Scholar]
- 161.Warmink J, Nazir R, Van Elsas J. Universal and species-specific bacterial ‘fungiphiles’ in the mycospheres of different basidiomycetous fungi. Environ Microbiol. 2009;11:300–312. doi: 10.1111/j.1462-2920.2008.01767.x. [DOI] [PubMed] [Google Scholar]
- 162.Dutton MV, Evans CS. Oxalate production by fungi: its role in pathogenicity and ecology in the soil environment. Can J Microbiol. 1996;42:881–895. doi: 10.1139/m96-114. [DOI] [Google Scholar]
- 163.Nishino K, Inazumi Y, Yamaguchi A. Global analysis of genes regulated by EvgA of the two-component regulatory system in Escherichia coli. J Bacteriol. 2003;185:2667–2672. doi: 10.1128/JB.185.8.2667-2672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Fontenot EM, Ezelle KE, Gabreski LN, Giglio ER, McAfee JM, Mills AC, Qureshi MN, Salmon KM, Toyota CG. YfdW and YfdU are required for oxalate-induced acid tolerance in Escherichia coli K-12. J Bacteriol. 2013;195:1446–1455. doi: 10.1128/JB.01936-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Takanao S, Honma S, Miura T, Ogawa C, Sugimoto H, Suzuki K, Watanabe T. Construction and basic characterization of deletion mutants of the genes involved in chitin utilization by Serratia marcescens 2170. Biosci Biotechnol Biochem. 2014;78:524–532. doi: 10.1080/09168451.2014.882755. [DOI] [PubMed] [Google Scholar]
- 166.Figueroa-Bossi N, Valentini M, Malleret L, Bossi L. Caught at its own game: regulatory small RNA inactivated by an inducible transcript mimicking its target. Genes Dev. 2009;23:2004–2015. doi: 10.1101/gad.541609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Toratani T, Shoji T, Ikehara T, Suzuki K, Watanabe T. The importance of chitobiase and N-acetylglucosamine (GlcNAc) uptake in N, N′-diacetylchitobiose [(GlcNAc) 2] utilization by Serratia marcescens 2170. Microbiology. 2008;154:1326–1332. doi: 10.1099/mic.0.2007/016246-0. [DOI] [PubMed] [Google Scholar]
- 168.Plumbridge J, Pellegrini O. Expression of the chitobiose operon of Escherichia coli is regulated by three transcription factors: NagC, ChbR and CAP. Mol Microbiol. 2004;52:437–449. doi: 10.1111/j.1365-2958.2004.03986.x. [DOI] [PubMed] [Google Scholar]
- 169.Francetic O, Belin D, Badaut C, Pugsley AP. Expression of the endogenous type II secretion pathway in Escherichia coli leads to chitinase secretion. EMBO J. 2000;19:6697–6703. doi: 10.1093/emboj/19.24.6697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Citterio B, Malatesta M, Battistelli S, Marcheggiani F, Baffone W, Saltarelli R, Stocchi V, Gazzanelli G. Possible involvement of Pseudomonas fluorescens and Bacillaceae in structural modifications of Tuber borchii fruit bodies. Can J Microbiol. 2001;47:264–268. doi: 10.1139/w01-005. [DOI] [PubMed] [Google Scholar]
- 171.De Boer W, Gunnewiek PJK, Kowalchuk GA, Van Veen JA. Growth of chitinolytic dune soil β-subclass Proteobacteria in response to invading fungal hyphae. Appl Environ Microbiol. 2001;67:3358–3362. doi: 10.1128/AEM.67.8.3358-3362.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bending GD, Poole EJ, Whipps JM, Read DJ. Characterisation of bacteria from Pinus sylvestris-Suillus luteus mycorrhizas and their effects on root-fungus interactions and plant growth. FEMS Microbiol Ecol. 2002;39:219–227. doi: 10.1111/j.1574-6941.2002.tb00924.x. [DOI] [PubMed] [Google Scholar]
- 173.Uroz S, Courty PE, Pierrat JC, Peter M, Buee M, Turpault MP, Garbaye J, Frey-Klett P. Functional profiling and distribution of the forest soil bacterial communities along the soil mycorrhizosphere continuum. Microb Ecol. 2013;66:404–415. doi: 10.1007/s00248-013-0199-y. [DOI] [PubMed] [Google Scholar]
- 174.Van Houdt R, Michiels CW. Role of bacterial cell surface structures in Escherichia coli biofilm formation. Res Microbiol. 2005;156:626–633. doi: 10.1016/j.resmic.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 175.Kohlmeier S, Smits TH, Ford RM, Keel C, Harms H, Wick LY. Taking the fungal highway: mobilization of pollutant-degrading bacteria by fungi. Environ sci technol. 2005;39:4640–4646. doi: 10.1021/es047979z. [DOI] [PubMed] [Google Scholar]
- 176.Sarand I, Timonen S, Nurmiaho-Lassila E-L, Koivula T, Haahtela K, Romantschuk M, Sen R. Microbial biofilms and catabolic plasmid harbouring degradative fluorescent pseudomonads in Scots pine mycorrhizospheres developed on petroleum contaminated soil. FEMS Microbiol Ecol. 1998;27:115–126. doi: 10.1111/j.1574-6941.1998.tb00529.x. [DOI] [Google Scholar]
- 177.Tenorio E, Saeki T, Fujita K, Kitakawa M, Baba T, Mori H, Isono K. Systematic characterization of Escherichia coli genes/ORFs affecting biofilm formation. FEMS Microbiol Lett. 2003;225:107–114. doi: 10.1016/S0378-1097(03)00507-X. [DOI] [PubMed] [Google Scholar]
- 178.Pion M, Bshary R, Bindschedler S, Filippidou S, Wick LY, Job D, Junier P. Gains of bacterial flagellar motility in a fungal world. Appl Environ Microbiol. 2013;79:6862–6867. doi: 10.1128/AEM.01393-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Samadder P, Xicohtencatl-Cortes J, Saldana Z, Jordan D, Tarr PI, Kaper JB, Giron JA. The Escherichia coli ycbQRST operon encodes fimbriae with laminin-binding and epithelial cell adherence properties in Shiga-toxigenic E. coli O157:H7. Environ Microbiol. 2009;11:1815–1826. doi: 10.1111/j.1462-2920.2009.01906.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 180.Overhage J, Lewenza S, Marr AK, Hancock RE. Identification of genes involved in swarming motility using a Pseudomonas aeruginosa PAO1 mini-Tn5-lux mutant library. J Bacteriol. 2007;189:2164–2169. doi: 10.1128/JB.01623-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Costa TR, Felisberto-Rodrigues C, Meir A, Prevost MS, Redzej A, Trokter M, Waksman G. Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nat Rev Microbiol. 2015;13:343–359. doi: 10.1038/nrmicro3456. [DOI] [PubMed] [Google Scholar]
- 182.Gerlach RG, Hensel M. Protein secretion systems and adhesins: the molecular armory of Gram-negative pathogens. Int J Med Microbiol. 2007;297:401–415. doi: 10.1016/j.ijmm.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 183.Garnett JA, Martínez-Santos VI, Saldaña Z, Pape T, Hawthorne W, Chan J, Simpson PJ, Cota E, Puente JL, Girón JA. Structural insights into the biogenesis and biofilm formation by the Escherichia coli common pilus. Proc Natl Acad Sci. 2012;109:3950–3955. doi: 10.1073/pnas.1106733109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lehti TA, Bauchart P, Heikkinen J, Hacker J, Korhonen TK, Dobrindt U, Westerlund-Wikström B. Mat fimbriae promote biofilm formation by meningitis-associated Escherichia coli. Microbiology. 2010;156:2408–2417. doi: 10.1099/mic.0.039610-0. [DOI] [PubMed] [Google Scholar]
- 185.Otrelo-Cardoso AR, da Silva Correia MA, Schwuchow V, Svergun DI, Romão MJ, Leimkühler S, Santos-Silva T. Structural data on the periplasmic aldehyde oxidoreductase PaoABC from Escherichia coli: SAXS and preliminary X-ray crystallography analysis. Int J Mol Sci. 2014;15:2223–2236. doi: 10.3390/ijms15022223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lee PA, Tullman-Ercek D, Georgiou G. The bacterial twin-arginine translocation pathway. Annu Rev Microbiol. 2006;60:373–395. doi: 10.1146/annurev.micro.60.080805.142212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hatzixanthis K, Palmer T, Sargent F. A subset of bacterial inner membrane proteins integrated by the twin-arginine translocase. Mol Microbiol. 2003;49:1377–1390. doi: 10.1046/j.1365-2958.2003.03642.x. [DOI] [PubMed] [Google Scholar]
- 188.Barnhart MM, Chapman MR. Curli biogenesis and function. Annu Rev Microbiol. 2006;60:131–147. doi: 10.1146/annurev.micro.60.080805.142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ogasawara H, Yamamoto K, Ishihama A. Role of the biofilm master regulator CsgD in cross-regulation between biofilm formation and flagellar synthesis. J Bacteriol. 2011;193:2587–2597. doi: 10.1128/JB.01468-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kjærgaard K, Schembri MA, Ramos C, Molin S, Klemm P. Antigen 43 facilitates formation of multispecies biofilms. Environ Microbiol. 2000;2:695–702. doi: 10.1046/j.1462-2920.2000.00152.x. [DOI] [PubMed] [Google Scholar]
- 191.Edgren T, Nordlund S. Two pathways of electron transport to nitrogenase in Rhodospirillum rubrum: the major pathway is dependent on the fix gene products. FEMS Microbiol Lett. 2006;260:30–35. doi: 10.1111/j.1574-6968.2006.00297.x. [DOI] [PubMed] [Google Scholar]
- 192.Fischer H-M. Genetic regulation of nitrogen fixation in rhizobia. Microbiol Rev. 1994;58:352–386. doi: 10.1128/mr.58.3.352-386.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Eichler K, Buchet A, Bourgis F, Kleber HP, Mandrand-Berthelot MA. The fix Escherichia coli region contains four genes related to carnitine metabolism. J Basic Microbiol. 1995;35:217–227. doi: 10.1002/jobm.3620350404. [DOI] [PubMed] [Google Scholar]
- 194.Walt A, Kahn ML. The fixA and fixB genes are necessary for anaerobic carnitine reduction in Escherichia coli. J Bacteriol. 2002;184:4044–4047. doi: 10.1128/JB.184.14.4044-4047.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Jia W, Cole J. Nitrate and nitrite transport in Escherichia coli. Biochem Soc Trans. 2005;33:159–161. doi: 10.1042/BST0330159. [DOI] [PubMed] [Google Scholar]
- 196.Sparacino-Watkins C, Stolz JF, Basu P. Nitrate and periplasmic nitrate reductases. Chem Soc Rev. 2014;43:676–706. doi: 10.1039/C3CS60249D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Gardner AM, Helmick RA, Gardner PR. Flavorubredoxin, an inducible catalyst for nitric oxide reduction and detoxification in Escherichia coli. J Biol Chem. 2002;277:8172–8177. doi: 10.1074/jbc.M110471200. [DOI] [PubMed] [Google Scholar]
- 198.Harborne NR, Griffiths L, Busby SJ, Cole JA. Transcriptional control, translation and function of the products of the five open reading frames of the Escherichia coli nir operon. Mol Microbiol. 1992;6:2805–2813. doi: 10.1111/j.1365-2958.1992.tb01460.x. [DOI] [PubMed] [Google Scholar]
- 199.Boogerd FC, Ma H, Bruggeman FJ, van Heeswijk WC, García-Contreras R, Molenaar D, Krab K, Westerhoff HV. AmtB-mediated NH3 transport in prokaryotes must be active and as a consequence regulation of transport by GlnK is mandatory to limit futile cycling of NH 4+/NH 3. FEBS Lett. 2011;585:23–28. doi: 10.1016/j.febslet.2010.11.055. [DOI] [PubMed] [Google Scholar]
- 200.Kobayashi M, Ishimoto M. Aerobic inhibition of nitrate assimilation in Escherichia coli. Zeitschrift für allgemeine Mikrobiologie. 1973;13:405–413. doi: 10.1002/jobm.3630130505. [DOI] [PubMed] [Google Scholar]
- 201.Degelmann DM, Kolb S, Dumont M, Murrell JC, Drake HL. Enterobacteriaceae facilitate the anaerobic degradation of glucose by a forest soil. FEMS Microbiol Ecol. 2009;68:312–319. doi: 10.1111/j.1574-6941.2009.00681.x. [DOI] [PubMed] [Google Scholar]
- 202.Schrey SD, Schellhammer M, Ecke M, Hampp R, Tarkka MT. Mycorrhiza helper bacterium Streptomyces AcH 505 induces differential gene expression in the ectomycorrhizal fungus Amanita muscaria. New Phytol. 2005;168:205–216. doi: 10.1111/j.1469-8137.2005.01518.x. [DOI] [PubMed] [Google Scholar]
- 203.Sen R, Nurmiaho-Lassila E, Haahtela K, Korhonen K. Specificity and mode of primary attachment of Pseudomonas fluorescens strains to the cell walls of ectomycorrhizal fungi. Mycorrhizas in integrated systems: from genes to plant development ECSC-EC-EAEC. Brussels: ECSC-EC-EAEC Press; 1996. p. 661–4.
- 204.Leveau JH, Preston GM. Bacterial mycophagy: definition and diagnosis of a unique bacterial-fungal interaction. New Phytol. 2008;177:859–876. doi: 10.1111/j.1469-8137.2007.02325.x. [DOI] [PubMed] [Google Scholar]
- 205.Lehembre F, Doillon D, David E, Perrotto S, Baude J, Foulon J, Harfouche L, Vallon L, Poulain J, Da Silva C, et al. Soil metatranscriptomics for mining eukaryotic heavy metal resistance genes. Environ Microbiol. 2013;15:2829–2840. doi: 10.1111/1462-2920.12143. [DOI] [PubMed] [Google Scholar]
- 206.Brosche M, Vinocur B, Alatalo ER, Lamminmaki A, Teichmann T, Ottow EA, Djilianov D, Afif D, Bogeat-Triboulot MB, Altman A, et al. Gene expression and metabolite profiling of Populus euphratica growing in the Negev desert. Genome Biol. 2005;6(12):R101. doi: 10.1186/gb-2005-6-12-r101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Nanjo T, Sakurai T, Totoki Y, Toyoda A, Nishiguchi M, Kado T, Igasaki T, Futamura N, Seki M, Sakaki Y, et al. Functional annotation of 19,841 Populus nigra full-length enriched cDNA clones. BMC Genomics. 2007;8:448. doi: 10.1186/1471-2164-8-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sterky F, Bhalerao RR, Unneberg P, Segerman B, Nilsson P, Brunner AM, Charbonnel-Campaa L, Lindvall JJ, Tandre K, Strauss SH, et al. A Populus EST resource for plant functional genomics. Proc Natl Acad Sci U S A. 2004;101:13951–13956. doi: 10.1073/pnas.0401641101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kohler A, Delaruelle C, Martin D, Encelot N, Martin F. The poplar root transcriptome: analysis of 7000 expressed sequence tags. FEBS Lett. 2003;542:37–41. doi: 10.1016/S0014-5793(03)00334-X. [DOI] [PubMed] [Google Scholar]
- 210.Ralph S, Oddy C, Cooper D, Yueh H, Jancsik S, Kolosova N, Philippe RN, Aeschliman D, White R, Huber D. Genomics of hybrid poplar (Populus trichocarpa× deltoides) interacting with forest tent caterpillars (Malacosoma disstria): normalized and full-length cDNA libraries, expressed sequence tags, and a cDNA microarray for the study of insect-induced defences in poplar. Mol Ecol. 2006;15:1275–1297. doi: 10.1111/j.1365-294X.2006.02824.x. [DOI] [PubMed] [Google Scholar]
- 211.Varshney RK, Hiremath PJ, Lekha P, Kashiwagi J, Balaji J, Deokar AA, Vadez V, Xiao YL, Srinivasan R, Gaur PM, et al. A comprehensive resource of drought- and salinity-responsive ESTs for gene discovery and marker development in chickpea (Cicer arietinum L.) BMC Genomics. 2009;10:523. doi: 10.1186/1471-2164-10-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Gupta V, Raghuvanshi S, Gupta A, Saini N, Gaur A, Khan MS, Gupta RS, Singh J, Duttamajumder SK, Srivastava S, et al. The water-deficit stress- and red-rot-related genes in sugarcane. Funct Integr Genomics. 2010;10:207–214. doi: 10.1007/s10142-009-0144-9. [DOI] [PubMed] [Google Scholar]
- 213.Li HY, Wang YC, Jiang J, Liu GF, Gao CQ, Yang CP. Identification of genes responsive to salt stress on Tamarix hispida roots. Gene. 2009;433:65–71. doi: 10.1016/j.gene.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 214.Lambilliotte R, Cooke R, Samson D, Fizames C, Gaymard F, Plassard C, Tatry MV, Berger C, Laudié M, Legeai F. Large-scale identification of genes in the fungus Hebeloma cylindrosporum paves the way to molecular analyses of ectomycorrhizal symbiosis. New Phytol. 2004;164:505–513. doi: 10.1111/j.1469-8137.2004.01185.x. [DOI] [Google Scholar]
- 215.Brown DW, Cheung F, Proctor RH, Butchko RA, Zheng L, Lee Y, Utterback T, Smith S, Feldblyum T, Glenn AE. Comparative analysis of 87,000 expressed sequence tags from the fumonisin-producing fungus Fusarium verticillioides. Fungal Genet Biol. 2005;42:848–861. doi: 10.1016/j.fgb.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 216.Karim N, Shibuya H, Kikuchi T. Analysis of expressed sequence tags from the wood-decaying fungus Fomitopsis palustris and identification of potential genes involved in the decay process. J Microbiol Biotechnol. 2011;21:347–358. [PubMed] [Google Scholar]
- 217.Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. 2013;14:699–712. doi: 10.1038/nrm3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ferracin M, Gautheret D, Hubé F, Mani SA, Mattick JS, Andersson Ørom U, Santulli G, Slotkin RK, Szweykowska-Kulinska Z, Taube JH. The non-coding RNA journal club: highlights on recent papers. Non-Coding RNA. 2015;1:87–93. doi: 10.3390/ncrna1010087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Ponting CP, Belgard TG. Transcribed dark matter: meaning or myth? Hum Mol Genet. 2010;19:R162–R168. doi: 10.1093/hmg/ddq362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Palazzo AF, Lee ES. Non-coding RNA: what is functional and what is junk? Front Genet. 2015;6:2. doi: 10.3389/fgene.2015.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Mora C, Tittensor DP, Adl S, Simpson AGB, Worm B. How many species are there on earth and in the ocean? PLoS Biol. 2011;9(8):e1001127. doi: 10.1371/journal.pbio.1001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Ellegren H. Genome sequencing and population genomics in non-model organisms. Trends Ecol Evol. 2014;29:51–63. doi: 10.1016/j.tree.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 223.Nesme J, Achouak W, Agathos SN, Bailey M, Baldrian P, Brunel D, Frostegård Å, Heulin T, Jansson JK, Jurkevitch E. Back to the future of soil metagenomics. Front Microbiol. 2016;7:73. doi: 10.3389/fmicb.2016.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Spribille T, Tuovinen V, Resl P, Vanderpool D, Wolinski H, Aime MC, Schneider K, Stabentheiner E, Toome-Heller M, Thor G. Basidiomycete yeasts in the cortex of ascomycete macrolichens. Science. 2016;353:488–492. doi: 10.1126/science.aaf8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Custom scripts. (DOCX 133 kb)
Unknown DE spreadsheet. (XLSX 2375 kb)
Total annotation spreadsheet. (XLSX 65570 kb)
Salix DE spreadsheet. (XLSX 2169 kb)
Fungi DE spreadsheet. (XLSX 6048 kb)
Upregulated DE Basidiomycota spreadsheet. (XLSX 1898 kb)
Upregulated DE Basidiomycota blastn spreadsheet. (XLSX 958 kb)
Total DE spreadsheet. (XLSX 7230 kb)
Bacteria DE transdecoded (annotation of polycistronic contigs) spreadsheet. (XLSX 2104 kb)
Data Availability Statement
Sequence data from this article can be found in the European Nucleotide Archive online repository (PRJEB16316).