

Abstract
BACKGROUND
Left ventricular (LV) hypertrophy (LVH) plays an important role in hypertensive heart disease, and may be accompanied by myocardial autophagy. However, the pattern of autophagy during evolution of LVH is unclear. We hypothesized that autophagy activation indicates advancing cardiac LVH with tissue remodeling.
METHODS
Ten domestic pigs with a 10-week unilateral renovascular hypertension (HTN) were classified as mild or moderate HTN ( n = 5 each group) based on the degree of renal artery stenosis (above or below 75%). Seven normal pigs served as controls. Left ventricular remodeling, function, and microvascular density were assessed using multi-detector- and micro-computed tomography and histology. Markers of myocardial autophagic and endoplasmic reticulum (ER) stress-related unfolded protein response (UPR), apoptosis, and fibrosis were examined ex vivo .
RESULTS
Both HTN groups had increased myocyte cross-sectional area, but it was greater in moderate HTN, accompanied by elevated LV muscle-mass. Moderate, but not mild HTN, also showed impaired microvascular density and impaired myocardial perfusion. Autophagy mediators were unaltered in mild HTN but UPR markers were increased, while in moderate HTN they were all upregulated, whereas UPR markers were suppressed. Myocardial apoptosis and fibrosis were also greater in moderate HTN. Autophagic proteins were correlated with LVH and fibrosis.
CONCLUSIONS
Autophagic activity is stimulated during the exacerbation of LVH, following a transient early increase in ER stress, and may be involved in the progression of cardiac remodeling in renovascular hypertensive heart disease.
Keywords: autophagy, blood pressure, hypertension, left ventricular hypertrophy, renal artery stenosis, renin-angiotensin-aldosterone system, unfolded protein response.
Almost 30% adults within the United States are affected by hypertension (HTN), 1 , 2 a leading risk factor for mortality and closely associated with chronic cardiac dysfunction. 3 Development of left ventricular (LV) hypertrophy (LVH), an increase in LV muscle mass in response to elevated afterload mostly caused by HTN, contributes to heart failure. The extent of LVH is directly linked to the magnitude and duration of HTN, 4 and its persistence associates with cardiac fibrosis and diastolic dysfunction that may advance to systolic heart failure.
The mechanisms that account for myocardial pathophysiological events in LVH are yet undetermined. Microvascular remodeling and oxidative stress are known contributors to tissue injury in HTN, 5–7 and recent studies have also linked it to autophagy. 8 During adaptive left ventricular remodeling toward LVH, a compensatory increase in protein synthesis causes accumulation of toxic misfolded molecules and protein aggregates. To maintain cardiac integrity, 9 , 10 autophagy serves as a major cellular mechanism for clearing misfolded proteins and/or dysfunctional organelles in response to stress induced by pressure overload and resultant tissue hypoxia. 11 Interestingly, however, autophagy has been observed to be triggered in both LV dysfunction 12 , 13 and LVH regression, 14 suggesting that the activity of myocardial autophagy follows a dynamic pattern that could vary depending on the phases of LVH, serving as a protective or deleterious mechanism. In addition, in response to accumulation of unfolded or misfolded proteins in the endoplasmic reticulum (ER), a coordinated adaptive program of unfolded protein response (UPR) is also activated. The UPR aims to alleviate ER stress by suppression of protein synthesis, facilitation of protein folding via induction of ER chaperones including glucose-related protein (GRP)78 and GRP94, and degradation of unfolded proteins. Indeed, ER stress has been implicated in ischemic heart disease and hypertrophy. 15 , 16
Renovascular HTN, the leading cause of secondary HTN due to renal artery stenosis, may lead to hypertensive heart diseases, 17–19 given the marked activation of the renin-angiotensin-aldosterone system that contributes to LVH. 20 We have previously shown that the renovascular hypertensive heart exhibits enhanced autophagy accompanying LVH. 18 However, the pattern and relationship of autophagy and ER stress activation at different stages of LVH progression are unclear. We hypothesized that autophagic activity is increased during the evolution of LVH induced by renovascular HTN in a swine model, and associated with escalated tissue remodeling.
METHODS
Seventeen domestic pigs were randomized into normal ( n = 7) and renovascular HTN ( n = 10), achieved by unilateral renal artery stenosis, and the HTN pigs were further arbitrarily classified at 10 week as mild (stenosis <75%, n = 5) or moderate (stenosis ≥75%, n = 5) HTN. Left ventricular remodeling and microvascular density were studied by using multi-detector computer tomography in vivo and micro-computer tomography ex vivo , respectively. Plasma renin activity, aldosterone, and serum creatinine were assessed. Myocardial autophagy and apoptosis were measured ex vivo in the myocardium, and ER stress markers in isolated ER. (Detailed descriptions of all experimental methods are included in the Supplementary Methods .)
RESULTS
Animal characteristics
Ten weeks after induction of renal artery stenosis, mean arterial pressure was elevated in both HTN groups, but was higher in moderate HTN ( Table 1 ), while plasma renin activity and plasma aldosterone were elevated only in moderate HTN. Serum creatinine was similarly elevated in both HTN groups ( Table 1 ).
Table 1.
Systemic characteristics and cardiac function and structure (mean ± SEM) of normal ( n = 7), mild ( n = 5), and moderate ( n = 5) renovascular hypertension (HTN) pigs
| Normal | Mild HTN | Moderate HTN | |
|---|---|---|---|
| Body weight (kg) | 47.7±1.3 | 46.2±2.7 | 49.8±3.0 |
| Degree of stenosis (%) | — | 73±6 | 98±2 † |
| MAP (mm Hg) | 87.1±2.1 | 93.1±2.0* | 131.4±6.7* ,† |
| Plasma creatinine (mg/dl) | 1.2±0.1 | 1.7±0.1* | 1.6±0.1* |
| PRA (ng/ml/h) | 0.2±0.1 | 0.2±0.1 | 10.6±4.8* |
| Plasma aldosterone (pg/ml) | 555.6±33.7 | 519.2±42.3 | 1,044.5±44.5* |
| E/A ratio | 1.4±0.1 | 0.8±0.1* | 0.9±0.1* |
| LVEDV (ml) | 1.9±0.1 | 1.8±0.1 | 1.7±0.1 |
| Stroke volume (ml) | 49.9±1.2 | 54.7±3.5 | 54.5±5.0 |
| Cardiac output (l/min) | 5.3±0.6 | 5.2±0.5 | 4.5±0.5 |
| Ejection fraction (%) | 55±3.0 | 56±4.0 | 63±8.0 |
Abbreviations: LVEDV, left ventricular end-diastolic volume; MAP, mean arterial pressure; PRA, plasma renin activity. * P < 0.05 vs. normal; †P < 0.05 vs. mild HTN.
LV remodeling, function, and microvasculature
HTN increased myocyte cross-sectional area in both groups, suggesting myocyte hypertrophy, but to a greater extent in moderate HTN; LV muscle mass increased only in moderate HTN ( Figure 1A , B ). The E/A ratios were similarly lower in both mild and moderate HTN compared to normal ( Table 1 ), indicating diastolic dysfunction, while LV end-diastolic volume, stroke volume, and ejection fraction were unaltered ( Table 1 ). However, moderate but not mild HTN decreased the density of microvessels (diameter <500 μm, Figure 1C ), and upregulated hypoxia-inducible factor-1α ( Figure 1D ). Moderate HTN also blunted the response of myocardial perfusion to adenosine ( Figure 2A ).
Figure 1.
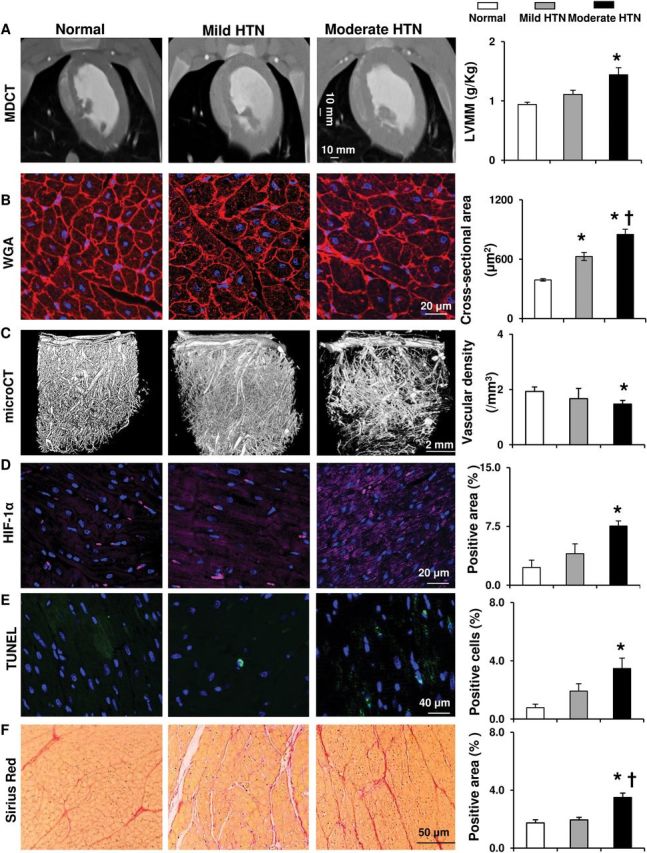
Alterations in left ventricular structure in hypertension. ( A , B ) Representative multi-detector computed tomography (CT) and wheat germ agglutinin (WGA) staining images establishing left ventricular and myocyte hypertrophy in pigs without or with mild or moderate hypertension (HTN), and their quantification. ( C ) Representative micro-CT images of the myocardial microvasculature and quantification of vascular density (<500 μm in diameter). ( D–F ) Representative staining and quantification of hypoxia-inducible factor (HIF)-1α, dUTP nick-end labeling (TUNEL), and Sirius Red. * P < 0.05 vs. normal, †P < 0.05 vs. mild HTN.
Figure 2.
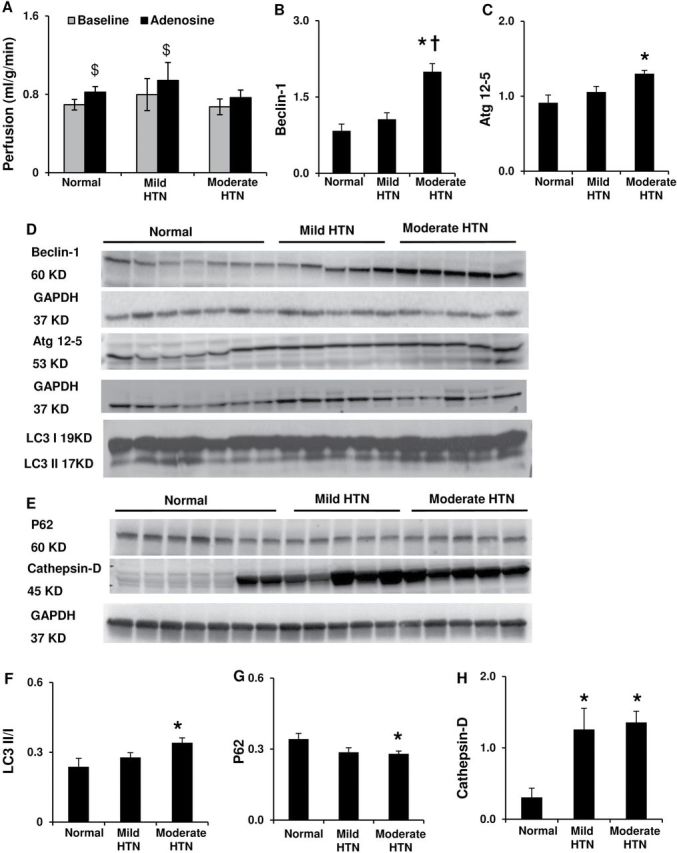
Left ventricular myocardial perfusion and autophagic activity. ( A ) Myocardial perfusion at baseline and in response to adenosine. ( B – D , F ) Myocardial protein expression and quantification for Beclin-1, autophagy-related (Atg) 12–5, and microtubule-associated light-chain (LC) 3-II and LC3-I ratio. ( E , G , H ) Myocardial protein expression and quantification of P62 and cathepsin-D. * P < 0.05 vs. normal, †P <0.05 vs. mild HTN. $P < 0.05 vs. baseline.
LV autophagy, UPR, apoptosis, and fibrosis
Mild HTN did not alter autophagic mediators like Beclin-1, autophagosome formation hallmarks autophagy-related (Atg) 12–5, or the microtubule-associated protein-1 light-chain (LC)3-II/LC3-I ratio ( Figure 2B–D ,F), whereas moderate HTN increased them all. The lysosomal protease cathepsin-D was elevated in both HTN groups, but P62 decreased only in moderate HTN, suggesting enhanced lysosomal and proteasome degradation 21 , 22 ( Figure 2E , G , H ). Mild HTN also increased the autophagy inhibitors Akt, mammalian target of rapamycin, and its effector pS6 ( Figure 3A ,C–E), which were lower in moderate compared to mild HTN. These results are consistent with magnified autophagic activity in more severe LVH. Interestingly, expression of the UPR marker GRP78 was elevated, and GRP94 also tended ( P = 0.07) to increase only in mild HTN, while in moderate HTN both were not different from normal and lower than in mild HTN ( Figure 3B , F , G ), suggesting a transient ER stress response in the early stage of LVH.
Figure 3.
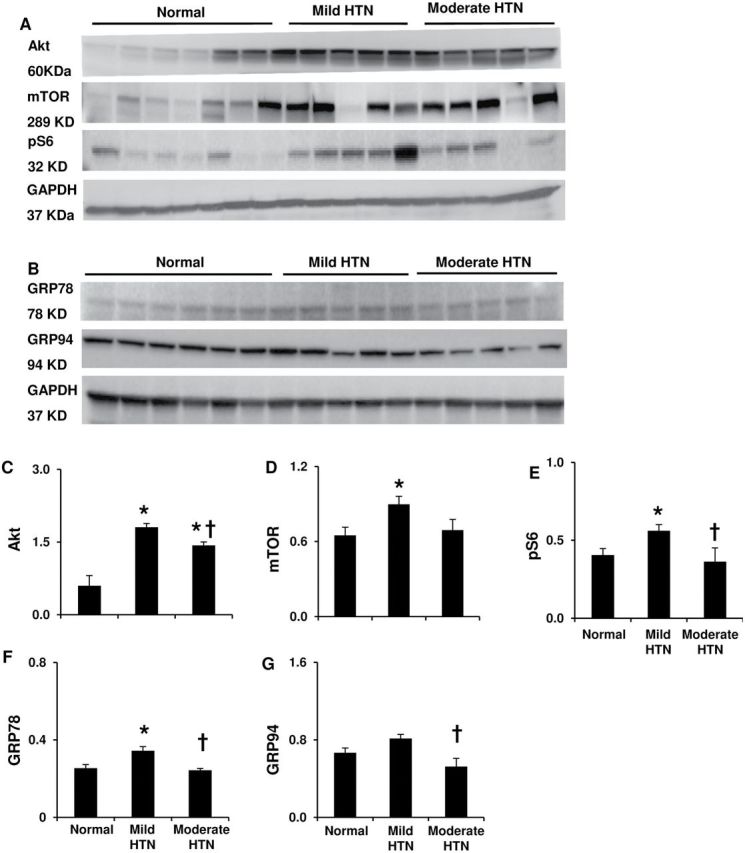
Left ventricular autophagic activity, and endoplasmic reticulum stress. ( A , C – E ) Myocardial protein expression and quantification for phos-/total Akt, mammalian target of rapamycin (mTOR) and phos-s6. ( B , F – G ) Protein expression and quantification for unfolded protein response markers glucose-regulated protein (GRP)-78 and GRP94 in isolated endoplasmic reticulum. * P < 0.05 vs. normal, †P < 0.05 vs. mild HTN.
HTN increased the number of apoptotic cells only in moderate HTN ( Figure 1E ). Both HTN groups decreased expression of anti-apoptotic B-cell lymphoma extra-large (Bcl-xl), but moderate HTN additionally decreased Bcl-2, while Bax remained unchanged ( Figure 4A ,C–E), suggesting that the enhanced apoptosis in moderate HTN was possibly due to attenuated anti-apoptotic activities.
Figure 4.
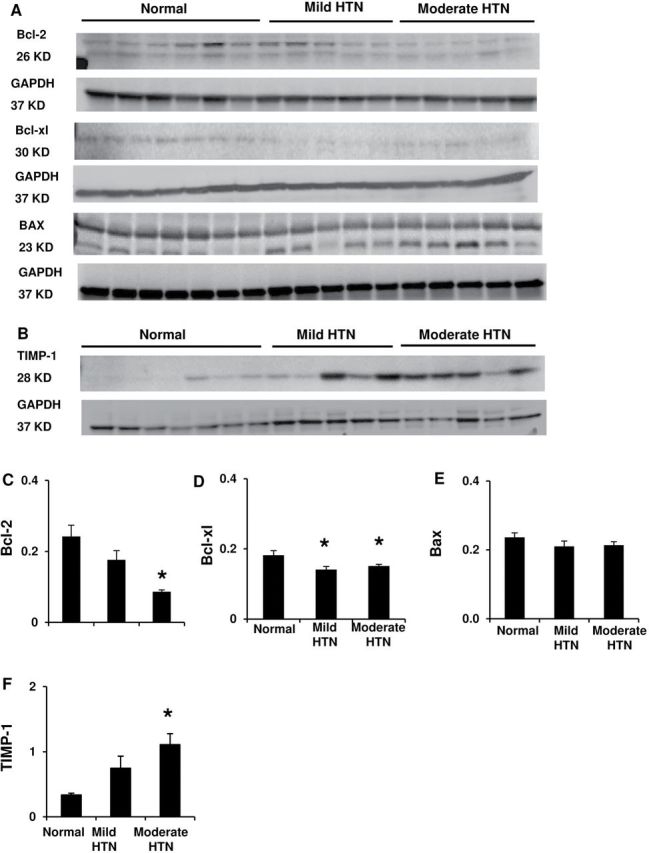
Left ventricular apoptotic and pro-fibrotic activities. ( A , C–E ) Protein expression and quantification of the anti-apoptotic B-cell lymphoma (Bcl)-2, Bcl-xl, and pro-apoptotic Bax. ( B , F ) Tissue inhibitor of metalloproteinase (TIMP)-1 expression. * P < 0.05 vs. normal.
Sirius Red staining revealed an increase of collagen deposition only in the moderate HTN myocardium ( Figure 1F ), as did tissue inhibitor of metalloproteinase-1 expression ( Figure 4B , F ).
Spearman rank correlation analysis showed a significant association of the expression of autophagy proteins with the extent of LVH as well as tissue fibrosis, whereas UPR markers was not associated with either ( Figure 5A–E ), revealing a close link of autophagic activity to cardiac remodeling in hypertensive heart. dUTP nick-end labeling (TUNEL) was also correlated to fibrosis. In addition, while an association between TUNEL and Beclin-1 did not reach a statistical significance level ( P = 0.07), the anti-apoptotic Bcl-2 was significantly related to autophagic proteins, suggesting a possible link between autophagic and apoptotic activities ( Figure 5G , H ).
Figure 5.
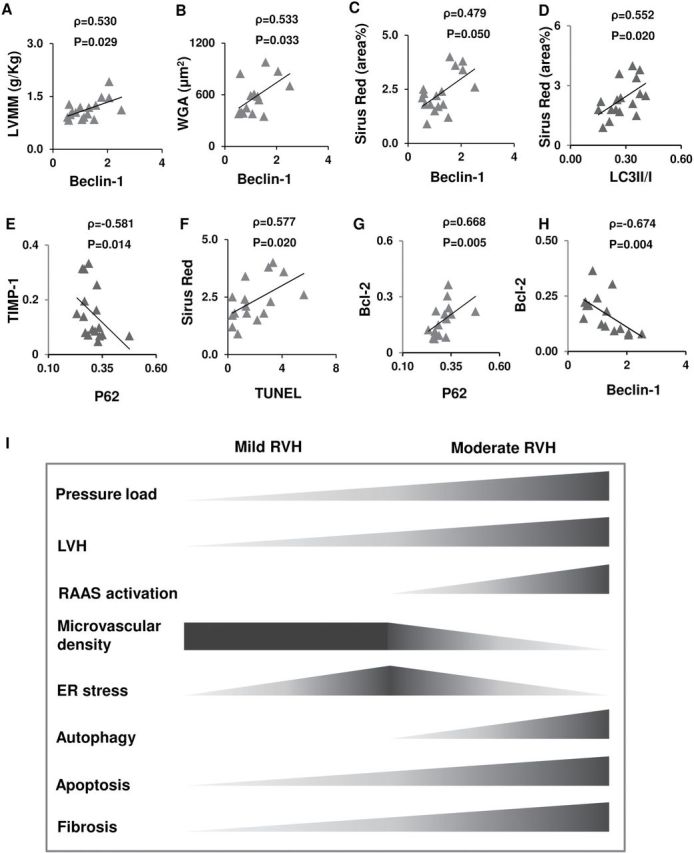
Proposed links between left ventricular remodeling and the associated pathological events. ( A – F ) Spearman rank correlation revealed associations of autophagy and apoptosis with LV myocardial remodeling. ( G , H ) Anti-apoptotic B-cell lymphoma (Bcl)-2 was correlated with autophagy proteins. ( I ) Schematic illustration of the proposed temporal pattern of pathophysiologic events during the evolution of LVH. Abbreviations: RVH, renovascular hypertension; RAAS, renin-angiotensin-aldosterone system; ER, endoplasmic reticulum.
DISCUSSION
The present study shows that progression of LVH in HTN involves a dynamic pattern of cellular turnover modulation. As LVH evolved, ER stress was upregulated as an early response, while autophagic mediators rose at a later stage (Beclin-1, Atg 12–5, LC3) as did lysosomal and proteasome degradation (increased cathepsin-D and decreased P62, respectively). The change in autophagy-related proteins paralleled magnification of apoptosis and fibrosis. Therefore, autophagic and apoptotic activities may be implicated in the pathways promoting LVH, and possibly reflect the severity of hypertensive heart disease.
Myocardial autophagy has been largely considered a defensive response to pressure overload. 18 , 23 Due to a compensatory increase in protein synthesis during adaptive cardiac remodeling toward LVH, autophagy is triggered to clear accumulating toxic or misfolded/dysfunctional organelles and maintain cardiac integrity. 9 In addition, HTN-induced LVH impairs coronary flow reserve 24 possibly due to the regression of microvessels that is particularly discernible in moderate HTN. Although basal myocardial perfusion was relatively preserved by compensatory mechanisms, amplified oxidative stress by persistent HTN and activated renin-angiotensin system 18 might have interfered with microvascular function to diminish the response to adenosine. Insufficient microcirculation subsequently limits oxygen (reflected in increased hypoxia-inducible factor-1α) and nutrient supply, 5 both of which may stimulate autophagic signals for recycling energy substrates. 25 Activation of the renin-angiotensin system (elevated plasma renin activity and aldosterone) in moderate HTN might not only exacerbate autophagy via aggravation of hypertrophy, but also directly modulate its activity. 26 , 27 Interestingly, autophagy inhibitors were also upregulated in mild HTN. Given their contributions to mediating hypertrophy in various tissues, activated Akt/mammalian target of rapamycin/s6 may reflect elevated protein synthesis that initiates LVH. 28–30 However, production of toxic proteins requires activation of autophagy to limit their accumulation. As LVH continues to progress in moderate LVH even though the mammalian target of rapamycin pathway is lower than mild HTN, other mechanisms leading to progression of LVH in RVH warrant further investigation.
The levels of misfolded proteins and damaged organelles during cardiac hypertrophy are also sensed by the ER, which subsequently elicits the UPR to reduce the stress. 31 , 32 In the present study, ER stress appears to be involved transiently in the relatively early stage of HTN, while in the later stage autophagy became predominant. This phenomenon supports the notion that ER stress mediates autophagy, 33 , 34 and suggests its potential role as the first-step reaction to the adaptive process in maintenance of intracellular homeostasis. Besides, as a survival mechanism, the UPR may extensively upregulate the autophagy machinery using the ER itself to provide the membrane needed for autophagosome formation. 34 , 35 Particularly, GRP78 has been shown to be obligatory for autophagy in mammalian cells, 36 possibly in an Atg protein-dependent manner. 34 Indeed, the present study found no association between ER stress and LVH or fibrosis, whereas autophagic and apoptotic proteins were correlated to LV myocardial remodeling. Previous studies have shown that autophagic and apoptotic proteins may interact. Bcl-2 inhibits autophagy by binding to Beclin-1, 37 and P62 modulates the level of autophagy by interfering with the Bcl-2-Beclin-1interaction. 38 Therefore, these results implicate a sequential activation of ER stress followed by autophagy and apoptosis, which may play a role in the progression of LVH. While ER stress represents an early response to facilitate control of toxic or misfolded cellular components in LVH, autophagy might be stimulated later to dispose damaged whole cells. As angiotensin receptor blockers attenuate ER stress in cardiac injury, 39 the interaction of the renin-angiotensin-aldosterone system with ER stress and autophagy in RVH also warrants further investigation.
Limitations
This study includes relatively small group sizes without comorbidities, which might affect the severity and time-course of the evolution of LVH and myocardial tissue injury in HTN. Nevertheless, cardiovascular physiology and pathophysiology in our model resemble those in humans and thus our study has translational power. Longitudinal studies with extended and successive observation and the use of specific inhibitors of ER stress and autophagy may assist in identification of the dynamic interaction and their impact in LVH.
CONCLUSIONS
As summarized schematically in Figure 5I , our findings suggest that autophagic activity may participate in the evolution of LVH along with enhanced apoptosis, and is associated with progression of tissue remodeling including microvascular rarefaction and fibrosis. Contrarily, ER stress seems to be a transient early process during LVH. Autophagy activation may be implicated in hypertrophic cardiac injury under substantial pressure overload.
SUPLEMENTARY MATERIAL
Supplementary materials are available at American Journal of Hypertension ( Supplementary Data ).
DISCLOSURE
The authors declared no conflict of interest.
Supplementary Material
ACKNOWLEDGMENTS
This work was partly supported by the NIH grants DK73608, HL77131, DK104273, HL121561, DK102325, HL123160, and C06-RR018898; and the American Heart Association.
REFERENCES
- 1. Cutler JA, Sorlie PD, Wolz M, Thom T, Fields LE, Roccella EJ . Trends in hypertension prevalence, awareness, treatment, and control rates in United States adults between 1988-1994 and 1999-2004 . Hypertension 2008. ; 52 : 818 – 827 . [DOI] [PubMed] [Google Scholar]
- 2. Yoon SS, Burt V, Louis T, Carroll MD . Hypertension among adults in the United States, 2009–2010 . NCHS Data Brief 2012. ; ( 107 ): 1 – 8 . [PubMed] [Google Scholar]
- 3. Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB . Heart disease and stroke statistics--2012 update: a report from the American Heart Association . Circulation 2012. ; 125 ( 1 ): e2 – e220 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Verdecchia P, Schillaci G, Borgioni C, Ciucci A, Gattobigio R, Zampi I, Porcellati C . Prognostic value of a new electrocardiographic method for diagnosis of left ventricular hypertrophy in essential hypertension . J Am Coll Cardiol 1998. ; 31 : 383 – 390 . [DOI] [PubMed] [Google Scholar]
- 5. Zhu XY, Daghini E, Chade AR, Napoli C, Ritman EL, Lerman A, Lerman LO . Simvastatin prevents coronary microvascular remodeling in renovascular hypertensive pigs . J Am Soc Nephrol 2007. ; 18 : 1209 – 1217 . [DOI] [PubMed] [Google Scholar]
- 6. Debbabi H, Uzan L, Mourad JJ, Safar M, Levy BI, Tibiriçà E . Increased skin capillary density in treated essential hypertensive patients . Am J Hypertens 2006. ; 19 : 477 – 483 . [DOI] [PubMed] [Google Scholar]
- 7. Polizio AH, Balestrasse KB, Yannarelli GG, Noriega GO, Gorzalczany S, Taira C, Tomaro ML . Angiotensin II regulates cardiac hypertrophy via oxidative stress but not antioxidant enzyme activities in experimental renovascular hypertension . Hypertens Res 2008. ; 31 : 325 – 334 . [DOI] [PubMed] [Google Scholar]
- 8. Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA . Cardiac autophagy is a maladaptive response to hemodynamic stress . J Clin Invest 2007. ; 117 : 1782 – 1793 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K . The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress . Nat Med 2007. ; 13 : 619 – 624 . [DOI] [PubMed] [Google Scholar]
- 10. Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J . Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy . Circ Res 2007. ; 100 : 914 – 922 . [DOI] [PubMed] [Google Scholar]
- 11. Rothermel BA, Hill JA . Autophagy in load-induced heart disease . Circ Res 2008. ; 103 : 1363 – 1369 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kostin S, Pool L, Elsässer A, Hein S, Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klövekorn WP, Schaper J . Myocytes die by multiple mechanisms in failing human hearts . Circ Res 2003. ; 92 : 715 – 724 . [DOI] [PubMed] [Google Scholar]
- 13. Hein S, Arnon E, Kostin S, Schönburg M, Elsässer A, Polyakova V, Bauer EP, Klövekorn WP, Schaper J . Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms . Circulation 2003. ; 107 : 984 – 991 . [DOI] [PubMed] [Google Scholar]
- 14. Goswami SK, Das DK . Autophagy in the myocardium: Dying for survival? Exp Clin Cardiol 2006. ; 11 : 183 – 188 . [PMC free article] [PubMed] [Google Scholar]
- 15. Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M . Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis . Circulation 2004. ; 110 : 705 – 712 . [DOI] [PubMed] [Google Scholar]
- 16. Minamino T, Kitakaze M . ER stress in cardiovascular disease . J Mol Cell Cardiol 2010. ; 48 ( 6 ): 1105 – 1110 . [DOI] [PubMed] [Google Scholar]
- 17. Losito A, Fagugli RM, Zampi I, Parente B, de Rango P, Giordano G, Cao P . Comparison of target organ damage in renovascular and essential hypertension . Am J Hypertens 1996. ; 9 : 1062 – 1067 . [DOI] [PubMed] [Google Scholar]
- 18. Zhang X, Li ZL, Crane JA, Jordan KL, Pawar AS, Textor SC, Lerman A, Lerman LO . Valsartan regulates myocardial autophagy and mitochondrial turnover in experimental hypertension . Hypertension 2014. ; 64 : 87 – 93 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barbaro NR, Fontana V, Moreno H . Relationship of left ventricular hypertrophy, age, and renal artery stenosis . J Am Soc Hypertens 2014. ; 8 : 360 . [DOI] [PubMed] [Google Scholar]
- 20. Mehta PK, Griendling KK . Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system . Am J Physiol Cell Physiol 2007. ; 292 : C82 – C97 . [DOI] [PubMed] [Google Scholar]
- 21. Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC . Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates . Mol Cell 2009. ; 33 : 517 – 527 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tatti M, Motta M, Di Bartolomeo S, Scarpa S, Cianfanelli V, Cecconi F, Salvioli R . Reduced cathepsins B and D cause impaired autophagic degradation that can be almost completely restored by overexpression of these two proteases in Sap C-deficient fibroblasts . Hum Mol Genet 2012. ; 21 : 5159 – 5173 . [DOI] [PubMed] [Google Scholar]
- 23. Wang ZV, Rothermel BA, Hill JA . Autophagy in hypertensive heart disease . J Biol Chem 2010. ; 285 : 8509 – 8514 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hamasaki S Al Suwaidi J Higano ST Miyauchi K Holmes DR Jr, Lerman A . Attenuated coronary flow reserve and vascular remodeling in patients with hypertension and left ventricular hypertrophy . J Am Coll Cardiol 2000. ; 35 : 1654 – 1660 . [DOI] [PubMed] [Google Scholar]
- 25. Mizushima N, Levine B, Cuervo AM, Klionsky DJ . Autophagy fights disease through cellular self-digestion . Nature 2008. ; 451 : 1069 – 1075 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Porrello ER, D'Amore A, Curl CL, Allen AM, Harrap SB, Thomas WG, Delbridge LM . Angiotensin II type 2 receptor antagonizes angiotensin II type 1 receptor-mediated cardiomyocyte autophagy . Hypertension 2009. ; 53 : 1032 – 1040 . [DOI] [PubMed] [Google Scholar]
- 27. Garcia AG, Wilson RM, Heo J, Murthy NR, Baid S, Ouchi N, Sam F . Interferon-γ ablation exacerbates myocardial hypertrophy in diastolic heart failure . Am J Physiol Heart Circ Physiol 2012. ; 303 : H587 – H596 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, Izumo S . Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload . Circulation 2004. ; 109 : 3050 – 3055 . [DOI] [PubMed] [Google Scholar]
- 29. Chen JK, Chen J, Thomas G, Kozma SC, Harris RC . S6 kinase 1 knockout inhibits uninephrectomy- or diabetes-induced renal hypertrophy . Am J Physiol Renal Physiol 2009. ; 297 : F585 – F593 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gao XM, Wong G, Wang B, Kiriazis H, Moore XL, Su YD, Dart A, Du XJ . Inhibition of mTOR reduces chronic pressure-overload cardiac hypertrophy and fibrosis . J Hypertens 2006. ; 24 : 1663 – 1670 . [DOI] [PubMed] [Google Scholar]
- 31. Dickhout JG, Carlisle RE, Austin RC . Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: endoplasmic reticulum stress as a mediator of pathogenesis . Circ Res 2011. ; 108 ( 5 ): 629 – 642 . [DOI] [PubMed] [Google Scholar]
- 32. Kassan M, Galan M, Partyka M, Saifudeen Z, Henrion D, Trebak M, Matrougui K . Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice . Arteriosclerosis, thrombosis, and vascular biology 2012. ; 32 ( 7 ): 1652 – 1661 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K . Autophagy is activated for cell survival after endoplasmic reticulum stress . Mol Cell Biol 2006. ; 26 : 9220 – 9231 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yorimitsu T, Nair U, Yang Z, Klionsky DJ . Endoplasmic reticulum stress triggers autophagy . J Biol Chem 2006. ; 281 : 30299 – 30304 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT . Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum . J Cell Biol 2008. ; 182 : 685 – 701 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS . The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells . Cell Death Differ 2008. ; 15 : 1460 – 1471 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B . Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy . Cell 2005. ; 122 : 927 – 939 . [DOI] [PubMed] [Google Scholar]
- 38. Zhou L, Wang HF, Ren HG, Chen D, Gao F, Hu QS, Fu C, Xu RJ, Ying Z, Wang GH . Bcl-2-dependent upregulation of autophagy by sequestosome 1/p62 in vitro . Acta Pharmacol Sin 2013. ; 34 : 651 – 656 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sukumaran V, Watanabe K, Veeraveedu PT, Gurusamy N, Ma M, Thandavarayan RA, Lakshmanan AP, Yamaguchi K, Suzuki K, Kodama M . Olmesartan, an AT1 antagonist, attenuates oxidative stress, endoplasmic reticulum stress and cardiac inflammatory mediators in rats with heart failure induced by experimental autoimmune myocarditis . Int J Biol Sci 2011. ; 7 : 154 – 167 . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.