

Abstract
Background
Phosphatase and tensin homolog (PTEN) is one of the most frequently inactivated tumor suppressors in breast cancer. While PTEN itself is not considered a druggable target, PTEN synthetic-sick or synthetic-lethal (PTEN-SSL) genes are potential drug targets in PTEN-deficient breast cancers. Therefore, with the aim of identifying potential targets for precision breast cancer therapy, we sought to discover PTEN-SSL genes present in a broad spectrum of breast cancers.
Methods
To discover broad-spectrum PTEN-SSL genes in breast cancer, we used a multi-step approach that started with (1) a genome-wide short interfering RNA (siRNA) screen of ~ 21,000 genes in a pair of isogenic human mammary epithelial cell lines, followed by (2) a short hairpin RNA (shRNA) screen of ~ 1200 genes focused on hits from the first screen in a panel of 11 breast cancer cell lines; we then determined reproducibility of hits by (3) identification of overlaps between our results and reanalyzed data from 3 independent gene-essentiality screens, and finally, for selected candidate PTEN-SSL genes we (4) confirmed PTEN-SSL activity using either drug sensitivity experiments in a panel of 19 cell lines or mutual exclusivity analysis of publicly available pan-cancer somatic mutation data.
Results
The screens (steps 1 and 2) and the reproducibility analysis (step 3) identified six candidate broad-spectrum PTEN-SSL genes (PIK3CB, ADAMTS20, AP1M2, HMMR, STK11, and NUAK1). PIK3CB was previously identified as PTEN-SSL, while the other five genes represent novel PTEN-SSL candidates. Confirmation studies (step 4) provided additional evidence that NUAK1 and STK11 have PTEN-SSL patterns of activity. Consistent with PTEN-SSL status, inhibition of the NUAK1 protein kinase by the small molecule drug HTH-01-015 selectively impaired viability in multiple PTEN-deficient breast cancer cell lines, while mutations affecting STK11 and PTEN were largely mutually exclusive across large pan-cancer data sets.
Conclusions
Six genes showed PTEN-SSL patterns of activity in a large proportion of PTEN-deficient breast cancer cell lines and are potential specific vulnerabilities in PTEN-deficient breast cancer. Furthermore, the NUAK1 PTEN-SSL vulnerability identified by RNA interference techniques can be recapitulated and exploited using the small molecule kinase inhibitor HTH-01-015. Thus, NUAK1 inhibition may be an effective strategy for precision treatment of PTEN-deficient breast tumors.
Electronic supplementary material
The online version of this article (10.1186/s13058-018-0949-3) contains supplementary material, which is available to authorized users.
Keywords: Synthetic lethality, Synthetic sickness, Precision medicine, PTEN, Breast cancer, Targeted cancer therapy, NUAK1, STK11, LKB1
Background
The discovery of specific vulnerabilities in phosphatase and tensin homolog (PTEN)-deficient cancers is clinically important because PTEN is one of the most frequently inactivated tumor suppressors in human cancer [1, 2]. Inactivation of PTEN can occur through loss-of-function genetic mutations, epigenetic silencing and transcriptional regulation, post-transcriptional regulation by non-coding RNAs, and through post-translational modifications and protein-protein interactions [3]. Germline PTEN mutations that result in loss of PTEN function confer an increased risk of developing benign and malignant tumors of the breast, thyroid, and endometrium [4]. Significantly, 67 to 85% of women with germline PTEN mutations develop breast cancer [5]. Although somatic PTEN mutations occur in only 5% of sporadic breast cancers, PTEN protein expression is significantly reduced in 25 to 37% of all breast tumors [6, 7]. PTEN loss in breast cancer is also associated with more aggressive disease and worse outcomes [8]. In particular, PTEN deficiency occurs more frequently in triple-negative breast cancers, which are not responsive to targeted cancer therapies [6, 8–11]. Therefore, the identification of specific vulnerabilities in PTEN-deficient breast cancer may suggest potential drug targets for an aggressive subset of breast cancers for which there is no effective therapy.
It has been challenging to clinically target PTEN-deficiency in cancer despite the well-established rationale for doing so. This is because PTEN function cannot directly be restored using small molecule drugs. The best-characterized function of PTEN is in antagonizing the phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway, which is essential for cell survival. PI3K activity is responsible for the formation of phosphatidylinositol (3,4,5)-trisphosphate (PIP3), a key second messenger that promotes phosphorylation and activation of the AKT kinase. AKT in turn phosphorylates and regulates multiple downstream processes. PTEN acts as a brake on this pathway by dephosphorylating PIP3 and suppressing AKT activation [1]. Consequently, loss of PTEN function removes a molecular brake on this pathway and allows PI3K to unabatedly activate downstream AKT signaling, thereby promoting cell survival and tumor formation [12]. As PTEN is a well-characterized PI3K pathway regulator, most drugs currently in clinical development for the treatment of PTEN-deficient cancers are kinase inhibitors that attempt to compensate for the loss of PTEN by suppressing PI3K/AKT signaling [13]. However, PTEN regulates multiple cell processes, including growth, proliferation, survival, chromosome stability, and DNA damage repair through mechanisms that are both dependent and independent of the PI3K pathway [14]. Thus, it is our hypothesis that there are unidentified therapeutic opportunities among the largely unexplored, PI3K-independent vulnerabilities of PTEN-deficient breast cancers.
Some of these therapeutic opportunities may exist in the form of PTEN synthetic-sick or synthetic-lethal (PTEN-SSL) genes. Synthetic lethality and synthetic sickness are terms used to describe gene-gene interactions that result in reduced cell viability or fitness [15]. A synthetic lethal relationship exists between two genes if their simultaneous perturbation results in death. Analogously, there is a synthetic sick relationship if simultaneous perturbation leads to reduced growth. In the context of cancer therapy, synthetic sick/lethal (SSL) interactions are of potential importance if SSL partners of oncogenes or tumor suppressor genes can be identified. In such cases, targeting the SSL partner by small molecule inhibitors or RNA interference (RNAi) might inhibit tumor growth or survival. This is well illustrated in the treatment of BRCA-deficient cancers with poly(ADP-ribose) polymerase (PARP) inhibitors, which exploit synthetic lethality between the PARP and BRCA genes [16]. A PTEN-SSL gene is, by definition, essential for cell survival or proliferation in PTEN-deficient (PTEN-) cells but not in PTEN-expressing (PTEN+) cells. PTEN-SSL genes would thus constitute potential vulnerabilities that could be exploited for the treatment of PTEN-deficient breast cancer.
Here, we present the results of a multi-step strategy to identify PTEN-SSL genes in breast cancer (Fig. 1). We began with a two stage RNAi screening strategy of a large number of genes and followed up with reproducibility and confirmation studies of the most promising hits. First, to comprehensively detect a large set of possible PTEN-SSL genes, we carried out a PTEN-SSL screen of essentially all human genes (~ 21,000 genes) in an isogenic, non-malignant, breast epithelial cell line model. Second, to select genes with evidence of PTEN-SSL activity across multiple cell lines, we screened hits from the first screen combined with other previously reported PTEN-SSL candidate genes (~ 1200 genes total) in 11 genetically diverse breast cancer cell lines, of which 8 were PTEN-deficient. To identify candidate PTEN-SSL genes that have reproducible and general PTEN-SSL activity in breast cancer, we identified hit overlaps between our results and reanalyzed data from three publicly available gene-essentiality screens carried out in large panels of breast cancer cell lines [17–19]. Finally, where small molecule inhibitors of gene function or somatic gene mutation data in large tumor cohorts were available, we further investigated the PTEN-SSL activity of selected candidate genes. Namely, we assessed drug sensitivity in an expanded panel of 19 breast cancer cell lines and also looked for independent evidence of PTEN-SSL activity in the form of mutual exclusivity of mutations in large tumor cohorts.
Fig. 1.
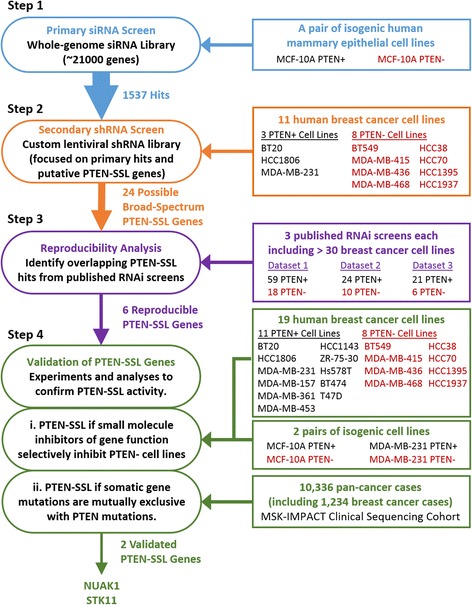
Overview of the phosphatase and tensin homolog-synthetic sick/lethal (PTEN-SSL) screening approach: the multi-step strategy used in this study to identify and validate broad-spectrum PTEN-SSL genes in breast cancer. siRNA, small interfering RNA; shRNA, short hairpin RNA; RNAi, RNA interference; NUAK1, NUAK family kinase 1; STK11, serine/threonine kinase 11
Methods
Cell lines
We utilized a pair of isogenic PTEN+ and PTEN- MCF-10A cell lines (obtained from Horizon Discovery, HD 101–006) for our primary short interfering RNA (siRNA) screen. MCF-10A PTEN+ cells express the wild-type PTEN protein, while MCF-10A PTEN- cells were engineered to be PTEN-deficient [20]. The parental MCF-10A cell line was derived from benign proliferative breast tissue and shows normal expression of wild-type PTEN [21–24]. Both MCF-10A cell lines were cultured in DMEM/F-12 including 2.5 mM L-glutamine and 15 mM HEPES (Gibco), supplemented with 10% fetal bovine serum (Hyclone), 10 μg/ml insulin (Sigma), 20 ng/ml epidermal growth factor (Sigma), and 500 ng/ml hydrocortisone (Calbiochem) [25].
In addition to the MCF-10A cell lines, we used 19 human breast cancer cell lines in this study. Of these cell lines, 11 were screened with a short hairpin shRNA (shRNA) library and all 19 were tested in drug sensitivity experiments. The human breast cancer cell lines HCC38, HCC70, HCC1395, HCC1806, HCC1937, BT20, BT549, MDA-MB-231, MDA-MB-436, MDA-MB-468, and Hs578T were obtained directly from American Type Culture Collection (ATCC) while BT474, HCC1143, MDA-MB-157, MDA-MB-361, MDA-MB-415, MDA-MB-453, T47D, and ZR-75-30 were obtained through the Duke University Cell Culture Facility. Breast cancer cell lines were cultured using growth medium as previously reported (Additional file 1: Table S1) [26]. All cells were cultured in a humidified atmosphere at 37 °C in 5% CO2 and all cell culture medium was supplemented with 50 units/ml penicillin and 50 mg/ml streptomycin.
We generated the MDA-MD-231 PTEN+ and PTEN- isogenic cell lines used in drug sensitivity experiments by lentivirally transducing MDA-MB-231 cells with a non-silencing shRNA (Open Biosystems - GE Dharmacon, RHS4351) and a shRNA pool targeting human PTEN (Open Biosystems - GE Dharmacon, RHS4533-NM_000314), respectively. Lentivirus particles were produced in HEK293T cells using the Trans-Lentiviral pGIPZ Packaging System (Open Biosystems - GE Dharmacon, TLP4615) according to the manufacturer’s protocol. We selected for cells with viral integration using puromycin. We confirmed stable knockdown of PTEN protein in the MDA-MB-231 PTEN- cell line through western blotting: the MDA-MB-231 PTEN+ cell line expressed PTEN at wild-type levels while the MDA-MB-231 PTEN- cell line expressed lower PTEN protein levels.
PTEN protein detection and quantification
PTEN protein abundance in 19 breast cancer cell lines was assayed using western blots. Cells were lysed with lysis buffer containing 2% SDS (sodium dodecyl sulfate) when they reached 80% confluence. Lysate protein concentration was determined using micro BCA assay (Pierce). The same amount of protein (15 μg) from each cell line was prepared in Laemmli sample buffer and loaded into each well of hand-cast 10% SDS-polyacrylamide gels. SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting of protein to nitrocellulose membranes was carried out using the Mini-PROTEAN Tetra system (Bio-Rad) according to the manufacturer’s instructions. The resulting membranes were probed with antibodies targeting PTEN (Cell Signaling Technology, #9552) and Actin (Chemicon, MAB1501R) according to the manufacturers’ instructions. DyLight 800 conjugated goat anti-rabbit and anti-mouse secondary antibodies were used in visualization of protein bands using the LI-COR Odyssey infrared imaging system (LI-COR Biosciences). There were 8 cell lines (HCC38, HCC70, HCC1395, HCC1937, BT549, MCA-MB-415, MDA-MB-436, and MDA-MB-468) with no visible PTEN protein bands that were categorized as PTEN- while the other 11 cell lines with visible PTEN bands were considered PTEN+.
For quantification of protein abundance, we analyzed the western blot images using the Image Studio software (LI-COR) and protein bands were quantified using densitometric analysis. The densities of PTEN protein bands for each cell line were normalized to the actin protein bands (loading controls) from the same cell line. Two reference cell lines, one PTEN+ (HCC1806) and one PTEN- (BT549), were included on all gels/membranes and data from these cell lines were used to normalize protein quantities across membranes (Additional file 1: Table S2). We also obtained publicly available quantitative PTEN protein abundance data that were measured by reverse-phase protein arrays (RPPA) for 75 breast cancer cell lines [17], which included all 19 cell lines that we analyzed by western blot. We identified good correlation between RPPA-measured PTEN abundance values and our quantitative western blot measurements across cell lines, which were assayed using both methods (Spearman’s rho 0.87, p < 0.0001). Based on this correlation, we established a quantitative cutoff for identifying PTEN+ and PTEN- cell lines. We thus categorized 18 breast cancer cell lines as PTEN- and the remaining 57 cell lines as PTEN+ (Additional file 1: Table S3).
Primary siRNA screen protocol
We carried out the primary siRNA screen in PTEN+ and PTEN- MCF-10A cell lines. Cell lines were screened using the whole-genome siGENOME siRNA library from Dharmacon (now Thermo-Fisher). The entire library, comprising 21,121 siRNA SMARTpools (pools containing 4 different siRNAs targeting each gene) was arrayed (one SMARTpool per well) in 68 black-walled 384-well plates (Greiner, #781091). Three types of control were included on all screening plates: non-silencing negative controls (48 wells containing Qiagen AllStars Negative Control siRNA; SI03650318), essential gene-positive controls (8 wells containing a siRNA pool targeting PLK1), and PTEN-SSL gene-positive controls (8 wells containing a siRNA pool targeting CDC25A). The screen was done by reverse transfecting 250 cells with 25 nM siRNA and 0.25 μl HiPerFect transfection reagent (Qiagen) in each well and then culturing the cells for 5 days. After 5 days, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% triton-x, and stained with Hoechst 33,342 (Molecular Probes). Stained nuclei were imaged and automatically counted using an ImageXpress Micro high-content imaging system (Molecular Devices) to generate raw cell counts after siRNA perturbation.
Primary siRNA screen analysis
Percentage cell viability after siRNA perturbation was calculated as:
where x is the cell count for a given siRNA pool, μneg is the mean cell count for non-silencing negative controls (interpreted as indicating highest possible viability), and μpos is the mean cell count for essential-gene, positive controls (interpreted as indicating highest possible lethality) arrayed on each individual plate. Two replicate PTEN-SSL siRNA screens, each comprising parallel screens in PTEN+ and PTEN- cell lines, were carried out. We looked for PTEN-SSL hits by assessing the PTEN-dependent activity attributable to each siRNA SMARTpool. We first calculated the change in cell viability (∆viability) attributable to PTEN loss by subtracting PTEN- cell line viability from PTEN+ cell line viability. We then scored PTEN-SSL activity of a specific siRNA pool by calculating a z score based on the ∆viability of that siRNA relative to the ∆viability distribution (mean and standard deviation) of all 3264 non-silencing negative controls screened. Primary screen results (∆viability and z score data) can be found in Additional file 1: Table S4.
Primary siRNA screen quality assessment
For screen quality assessment, we used “Z-prime factors”, as described previously [27], to determine how well separated the on-plate negative and positive controls were, thus estimating the dynamic range of each 384-well screening plate. Z-prime factors were calculated as:
where μ and σ are the mean and standard deviation of the positive (pos) and negative (neg) control cell counts for each plate, respectively. A Z-prime factor between 0.5 and 1 would indicate an excellent assay while a Z-prime factor between 0 and 0.5 is marginally acceptable. A Z-prime factor <0 would indicate that the overlap between the positive and negative controls is too great for the assay to be useful. We used a threshold of Z-prime factor >0 to define acceptable screening plates, since RNAi assays with Z-prime factors >0 have been successful in identifying validated hits in duplicate or triplicate lethality screens [28]. When Z-prime factors were calculated using the essential-gene positive control that drastically inhibits cell viability in both cell lines, all plates screened passed quality assessment with a mean Z-prime factor of 0.70 across both cell lines and replicates. Similarly, all PTEN- cell line screening plates passed quality assessment when Z-prime factors were calculated using the PTEN-SSL positive control. Their mean Z-prime factor was 0.59. Thus, our primary screen had sufficient dynamic range to identify genes that had an impact on cell viability when they were knocked down in individual cell lines.
Secondary shRNA screen protocol
To identify genes with broad-spectrum PTEN-SSL activity, we carried out a pooled lentiviral shRNA dropout viability screen in a panel of 11 breast cancer cell lines, of which 8 were PTEN- (HCC38, HCC70, HCC1395, HCC1937, BT549, MCA-MB-415, MDA-MB-436, and MDA-MB-468) and 3 were PTEN+ (BT20, HCC1806, MDA-MB-231). The custom shRNA library (Cellecta) comprised a total of 6500 individually barcoded lentiviral shRNA constructs targeting ~ 1200 genes (Additional file 1: Table S5). Genes targeted included ~ 1000 PTEN-SSL hits identified in the primary screen, together with 9 putative PTEN-SSL genes, which were reported in other studies but failed to score as hits in our primary screen. The remaining ~ 200 genes represent non-PTEN-SSL hits, which were included as controls. The custom-designed shRNA library included multiple shRNA constructs (5–10 individual shRNA sequences) to target each gene. The shRNA sequences were designed using Cellecta’s proprietary shRNA design algorithm and database of validated shRNAs, which have been optimized for RNAi genetic screens in pooled format (sequences provided in Additional file 1: Table S5). The library was constructed in the pRSI16-U6-(sh)-HTS6-UbiC-TagRFP-2A-Puro vector, and provided as pre-packaged ready-to-transfect VSV-g pseudotyped lentiviral particles by the manufacturer.
We infected cell lines with a single pool of lentiviral particles at a multiplicity of infection (MOI) between 0.1 and 0.5 and at 500–1000-fold representation of the library. Under these conditions, most cells were infected with either one or no shRNA constructs. Medium was replenished 24 h post infection. Infection efficiency was monitored using the percentage of virus-integrated red fluorescent protein positive (RFP+) cells tracked by fluorescence-activated cell sorting (FACS). We confirmed that MOI did not exceed 0.5 for all cell lines screened. At 2 days after infection, when viral integration was presumed complete, we exposed the cells to puromycin for 3 days to select for cells with viral integration. A start sample was collected from the population when puromycin selection was completed. Cells were subcultured when they reached 80% confluence to ensure continued logarithmic growth. Sufficient cells (determined by cell counting) were maintained in each passage so that on average there would be at least 500 cells per shRNA. We did this by seeding > 500 × 6500 = 3.25 million cells in each passage, with 6500 being the size of the library. When cells reached 4–6 population doublings, an end sample was taken. Genomic DNA was extracted from the start and end samples and sent to Cellecta for barcode amplification, next-generation sequencing, and enumeration of shRNA barcode counts from raw sequenced data (Additional file 1: Table S6).
Secondary shRNA screen analysis
Screening data for all cell lines was provided by Cellecta as normalized shRNA-barcode counts adjusted to 20 M reads. To track changes in shRNA abundance over the course of the screen, we calculated the fold change in normalized barcode counts between the start and end of each screen (Additional file 1: Table S7). Since each gene was targeted by multiple shRNAs, we aggregated shRNA-level fold-change data into gene-level viability scores using the ATARiS (analytic technique for assessment of RNAi by similarity) algorithm [29]. ATARiS analyzes data from multiple shRNAs targeting each gene across all cell lines to select shRNAs with consistent activity profiles across all cell lines (Additional file 1: Table S8). ATARiS then uses the shRNAs with consistent profiles to calculate the gene-level viability score for each gene in each cell line (Additional file 1: Table S9). We ran the ATARiS version 2 algorithm using the GenePattern public server [30]. ATARiS sometimes generates > 1 solution per gene depending on the number of shRNA clusters showing the same activity profile across cell lines. We used all gene solutions generated by ATARiS. To detect broad-spectrum PTEN-SSL genes, we analyzed differences in cell line sensitivity attributable to PTEN loss by comparing the gene-level ATARiS scores of PTEN- cell lines against those of PTEN+ cell lines.
Reanalysis of data from previous, independent RNAi screens
We assessed the reproducibility of our PTEN-SSL hits through a reanalysis of three independent, previously published RNAi screens. We accessed sensitivity scores reported for each study. Per-gene zGARP scores were downloaded from the Breast Functional Genomics study website (http://neellab.github.io/bfg/) [17] and these are available in Additional file 1: Table S11. DEMETER z scores for the Cancer Dependency Map dataset (Achilles v2.20.2 gene solutions) were downloaded from the Project Achilles website (https://portals.broadinstitute.org/achilles/datasets/all) [19] and are available in Additional file 1: Table S12. The z scores for the Kinase Dependency Profiles dataset were downloaded from Table S1 of the published study [18] and are available in Additional file 1: Table S13. We identified overlapping hit genes and tested the statistical significance of observing a given overlap between two sets of genes using the hypergeometric distribution (R function dhyper). The hypergeometric p value reflects the probability that an overlap of the observed cardinality or greater will occur as a result of randomly picking two sets of genes of the given hit-list sizes from the pool of all genes present in both datasets [31]. Overlaps with a p value <0.05 were considered statistically significant.
Drug sensitivity experiments
We treated breast cancer cell lines with NUAK inhibitors WZ4003 (S7317) and HTH-01-015 (S7318), which were purchased from Selleck Chemicals and diluted in dimethyl sulfoxide (DMSO). For determination of cell line drug sensitivity, 1000–3000 cells were seeded in each well of 96-well plates and treated with 1–30 μM of drug. Cell viability was measured using a MTS cell proliferation assay (Promega CellTiter AQueous One) when the cells reached 80–90% confluence 3–5 days after treatment. The surviving fraction of cells in each well was determined relative to DMSO-treated controls. Drug sensitivity experiments were carried out in quadruplicate. Drug sensitivity was determined by calculating the drug concentration required to reduce cell viability by 50% relative to DMSO-treated controls (IC50). IC50 values were determined from dose-response data using GraphPad Prism version 7.
Mutual exclusivity analysis of genetic alterations in patient cohorts
To look for patterns of mutual exclusivity between PTEN and candidate PTEN-SSL genes, we analyzed a breast cancer dataset (The Cancer Genome Atlas (TCGA) provisional breast invasive carcinoma cohort [6, 32]) and a pan-cancer patient dataset (the MSK-IMPACT study [33]). For each pair of genes, cBioPortal [34, 35] computes an “odds-ratio” for co-occurrence versus mutual exclusivity and a Fisher’s exact test p value of statistical significance. The “odds ratio” for two genes, G1 and G2, is computed as “(A * D) / (B * C), [w]here A = number of cases altered in both genes; B = number of cases altered in G1 but not G2; C = number of cases altered in G2 but not G1; and D = number of cases altered in neither genes (sic)” (quoted verbatim from [34]). We used the criteria of odds ratio <1 and p < 0.05 to identify mutually exclusive mutation patterns.
Statistical analysis
Data with assumed equal variance were analyzed by the t test and this was computed using GraphPad Prism version 7. For the secondary screen and the reanalysis in step 3, we used the one-sided Wilcoxon rank-sum test (R function wilcox.test) for comparing ATARiS scores between two groups of cell lines (PTEN+ and PTEN-). We used this non-parametric test because the ATARiS scores in our secondary screen and the sensitivity measures in step 3 had non-normal distributions (all p < 0.0001 using the D’Agostino-Pearson omnibus normality test in GraphPad Prism) and were compared in the same way. The alternative hypothesis was that the median of the PTEN- cell line ATARiS scores is lower than that of PTEN+ cell line scores.
Results
Primary siRNA screen in isogenic cell lines
To identify PTEN-SSL genes in breast cancer, we first carried out a primary small interfering RNA (siRNA) screen in a pair of isogenic cell lines (Step 1 in Fig. 1). These were derived from the MCF-10A human mammary epithelial cell line and differed only in PTEN status [20]. MCF-10A is an immortalized, but non-tumorigenic, breast epithelial cell line with a basal-like gene expression profile [21, 22]. Wild-type MCF-10A cells (MCF-10A PTEN+) express PTEN protein, while the PTEN knockout cells (MCF-10A PTEN-) do not. We chose a non-tumorigenic cell line for screening based on the hypothesis that PTEN-SSL interactions that are independent of oncogenic alterations would more likely generalize to a broad range of PTEN- tumors. We further hypothesized that, although some of the PTEN-SSL activities detected in the primary screen might be restricted to the MCF-10A cell line, our secondary screen in multiple cell lines would filter out such restricted vulnerabilities.
In the primary screen, a genome-scale siRNA library targeting ~ 21,000 genes was assayed in the MCF-10A PTEN+ and PTEN- cells. We expressed the difference in cell viability between PTEN- and PTEN+ for each siRNA pool as a z score based on the distribution of differences in cell viability in the non-targeting (that is, negative) controls (see “Methods”). The z scores <0 indicate selective reduction of cell viability or proliferation in the PTEN- cells compared to PTEN+ cells and constitute evidence of PTEN-SSL activity. We carried out two independent PTEN-SSL screens and found screening results to be generally reproducible between replicate screens for both cell lines (Spearman’s rho 0.8, p < 0.0001) (Fig. 2a). Importantly, the positive PTEN-SSL control, CDC25A, had mean z scores < − 3 in both replicate screens (Fig. 2b). Therefore, we took a z score < − 1 in both screens as a non-stringent criterion for identifying candidate PTEN-SSL genes (Fig. 2c). A total of 1537 genes were scored as primary PTEN-SSL hits using this criterion and were prioritized for validation based on the z scores (Additional file 1: Table S4).
Fig. 2.
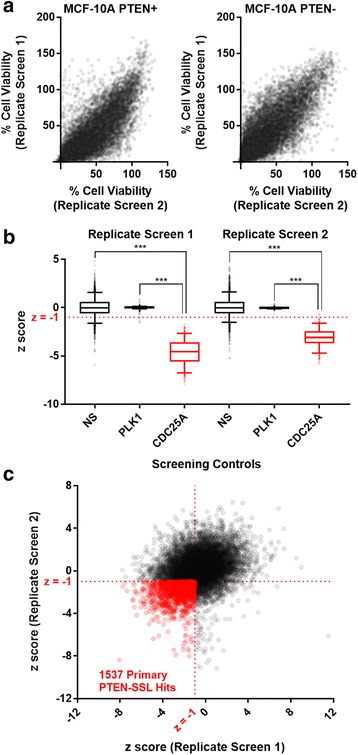
Identification of phosphatase and tensin homolog-synthetic sick/lethal (PTEN-SSL) genes in the MCF-10A cell line. a Cell line viability responses to ~ 21,000 short interfering RNA (siRNA) pools in two replicate screens. Each dot represents siRNA pool. b The z scores for three types of screening control: non-silencing (NS), essential gene (PLK1), and PTEN-SSL gene (CDC25A). Whiskers indicate 95th and 5th percentiles; ***p < 0.0001 (Wilcoxon rank-sum test). Each dot represents one control siRNA pool. c siRNA z scores from two replicate screens. Each dot represents one siRNA pool
We used a non-stringent z score cutoff for primary hit selection to capture “true positive” PTEN-SSL genes, which would otherwise be excluded by a more stringent cutoff. This is because PTEN synthetic sick genes, by definition, will result in subtle but meaningful reductions in cell viability when silenced by RNAi reagents. However, since off-target gene silencing is well-known to be a considerable source of error in RNAi screens, we expected the 1537 primary hits to include a large number of “false positives” attributable to off-target siRNA effects. Furthermore, because most synthetic sickness and synthetic lethal relationships exist only in certain genetic backgrounds or under specific cellular conditions [36], another subset of primary hits would be PTEN-SSL only in the MCF-10A genetic background and would not be reproduced in other cell lines. We thus followed the primary siRNA screen with a secondary screen designed to filter out the majority of false positive hits. The secondary screen differed from the primary screen in two key ways: (1) the secondary screen was performed using orthogonal RNAi technology, because siRNA off-target effects are unlikely to be reproduced by multiple shRNAs targeting the same gene and (2) the secondary screen was carried out in multiple cell lines to identify genes with possible PTEN-SSL activity in a large proportion of cell lines.
Secondary shRNA screen in panel of cell lines
For the secondary screen, we carried out a pooled lentiviral shRNA dropout viability screen in eight PTEN- and three PTEN+ cell lines (step 2, Fig. 1). All cell lines were screened with a library of 6500 shRNAs targeting ~ 1200 genes, of which ~ 1000 were hits from our primary screen, 9 were putative PTEN-SSL genes reported in other studies but which did not score as hits in the primary screen, and the remaining genes were non-PTEN-SSL controls. Each gene was represented by a pool of 5 to 10 shRNAs. We measured the sensitivity of each cell line to shRNA perturbation by tracking shRNA dropout over four to six population doublings (depending on the doubling time of each cell line). We used the ATARiS algorithm [29] to calculate a sensitivity score for each cell line to each shRNA pool. ATARiS scores were obtained for 727 genes that were deemed to have consistent shRNA effects across the cell lines (ATARiS scores were not obtained for the remainder of the genes, indicating no evidence of PTEN-SSL activity). We then used the one-sided Wilcoxon rank-sum test to identify genes that had lower sensitivity scores in the eight PTEN- cell lines than in the three PTEN+ cell lines (Fig. 3a). A p value <0.05 (uncorrected for multiple testing) in the Wilcoxon rank-sum test was used as a non-stringent hit-selection criterion and we identified 24 secondary hits showing evidence of broad-spectrum PTEN-SSL activity across cell lines (Fig. 3b, Additional file 1: Table S9).
Fig. 3.
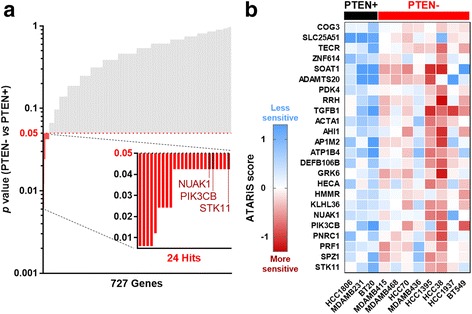
Identification of broad-spectrum phosphatase and tensin homolog-synthetic sick/lethal (PTEN-SSL) genes. a The p values from the one-sided Wilcoxon rank-sum test for the hypothesis that PTEN- cell lines are more sensitive (lower ATARiS scores) to knockdown of each gene compared to PTEN+ cell lines. Each bar represents one of 727 genes in the secondary screen with computed ATARiS gene solutions. Possible broad-spectrum PTEN-SSL genes were selected using p < 0.05 (not corrected for multiple testing) and are shown as red bars. b Heat map visualization of the sensitivity (ATARiS scores) of individual cell lines in response to on-target mRNA knockdown of 24 possible broad-spectrum PTEN-SSL genes selected in the secondary screen (red region in a)
As in the primary screen, we used a non-stringent hit criterion with the aim of identifying a reasonably small set of possible broad-spectrum PTEN-SSL genes that would contain a subset of true PTEN-SSL genes. These few true broad-spectrum PTEN-SSL genes would have to be confirmed by subsequent experiments and analyses. Non-stringent statistical criteria have been similarly used for hit selection in synthetic lethal screens in part because of the relatively low validation rate of such screens [37–39]. We used a p value-based criterion for hit selection, but, as is typical for high throughput screens, our intent was not to control the family-wise error rate or even to attain a low false discovery rate. Instead, we accepted a very high false discovery rate as a necessary condition for capturing a small number of true discoveries to be identified by subsequent experiments and analyses. We note that this criterion identified phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta (PIK3CB), a known PTEN-SSL gene [40], as one of the 24 possible broad-spectrum PTEN-SSL hits from the secondary screen. This showed that the chosen criterion was able to identify a true PTEN-SSL gene. Importantly, PIK3CB would not have scored as a hit if a more stringent statistical cutoff taking into account multiple testing were used. Thus, other novel, true positive PTEN-SSL genes could potentially be missed if a stringent criterion had been used at this step. As presented next, we subsequently assessed reproducibility of the 24 hits in independent screening datasets and followed this by drug sensitivity experiments using small molecule inhibitors of gene function and by analyses of mutual exclusivity of mutations in tumors.
PTEN-SSL genes validated in independent RNAi screening datasets
To date, several high-throughput RNAi screens have been carried out to systematically identify genetic dependencies in large collections of cancer cell lines [17–19, 41–45]. We assessed the reproducibility and generality of our PTEN-SSL hits in the data from three of these previous studies, each of which included > 20 breast cancer cell lines (step 3, Figs. 1 and 4a). The studies we reanalyzed were (1) the Breast Functional Genomics study, comprising genome-scale shRNA screens of 77 breast cancer cell lines [17, 43]; (2) the Cancer Dependency Map study, comprising genome-scale shRNA screens of 501 cancer cell lines, including 34 from breast cancer [19, 42, 44]; and (3) the Kinase Dependency Profiles study, comprising kinome-scale siRNA screens of 117 cancer cell lines, including 27 from breast cancer [18, 41].
Fig. 4.
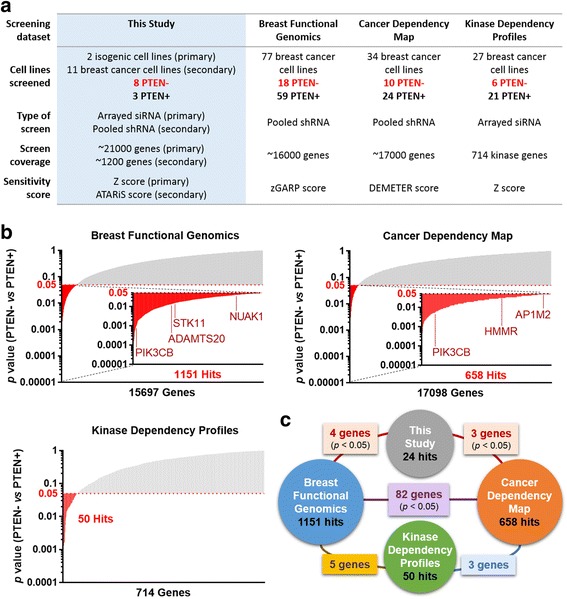
Identification of reproducible broad-spectrum phosphatase and tensin homolog-synthetic sick/lethal (PTEN-SSL) genes. a Cmparison of the secondary short hairpin RNA (shRNA) screen described in this study and three independent RNA interference (RNAi) screening datasets selected for reanalysis: Breast Functional Genomics [17], Cancer Dependency Map [19], and Kinase Dependency Profiles [18]. b The p values from the one-sided Wilcoxon rank sum test for whether PTEN- cell lines were more sensitive (lower ATARiS scores) to knockdown of each gene compared to PTEN+ cell lines. Each bar represents one of 727 genes in the secondary screen that had ATARiS gene solutions. Possible broad-spectrum PTEN-SSL hits were selected using the same criterion (p < 0.05, uncorrected for multiple testing) for each screen and are shown as red bars. PIK3CB, NUAK1, STK11, ADAMTS20, AP1M2, and HMMR were found to be hits in our reanalysis of ≥ 1 of the previous screens. c Number of overlaps in possible broad-spectrum PTEN-SSL genes that were identified by this study and in reanalysis of three previous screens; p values based on one-sided hypergeometric distribution
We reanalyzed the results of each screen to identify broad-spectrum PTEN-SSL genes by extracting gene-level sensitivity scores reported for each dataset (Fig. 4a). Although each screen was performed using different scoring approaches (zGARP, DEMETER, and z scores), increasingly negative scores indicate greater inhibition of cell growth in all cases. We determined the PTEN status of 75 breast cancer cell lines based on previously published reverse-phase protein array data [17], and confirmed that these data correlated strongly with western blot quantification in a subset of 20 cell lines (Spearman’s rho = 0.86, p < 0.0001; / Additional file 2: Figure S1). For each gene in each dataset, we selected genes with PTEN-SSL activity using the same method as in our secondary screen, that is by performing the one sided Wilcoxon rank-sum test to identify genes that had lower sensitivity scores in PTEN- cell lines than in PTEN+ cell lines. As in the secondary screen, we used p < 0.05 (without correcting for multiple testing) as a non-stringent criterion for selecting hits. This identified 1151, 658, and 50 possible broad-spectrum PTEN-SSL genes from the three studies, respectively (Fig. 4b).
Although no single gene scored as a PTEN-SSL hit across all screens, PIK3CB was reproducibly identified as a hit in our screens and in two out of the three reanalyzed screens. In addition, another five genes were identified in reanalysis of one or more of the previous screening studies (Fig. 4b). These genes are (ADAM metallopeptidase with thrombospondin type 1 motif 20 (ADAMTS20), (adaptor related protein complex 1 mu 2 subunit (AP1M2), hyaluronan mediated motility receptor (HMMR), NUAK family kinase 1 (NUAK1), and serine/threonine kinase 11 (STK11). These overlaps in possible broad-spectrum PTEN-SSL genes were statistically significant (p < 0.05 as determined using the hypergeometric distribution) (Fig. 4c). The identification of PIK3CB is again an indication that there may be other true positive PTEN-SSL genes among these six genes. We consider these genes candidates for further validation of PTEN-SSL activity in experimental studies and clinical datasets.
NUAK1 inhibition is PTEN-SSL in breast cancer cell lines
Two of the broad-spectrum PTEN-SSL genes identified in our screen and shown to be reproducible in reanalysis of data from other studies, NUAK1, also known as AMPK-related protein kinase 5 (ARK5), and STK11, also known as liver kinase B1 (LKB1), are functionally related, suggesting that their PTEN-SSL activity may stem from a common mechanism. NUAK1 can be inhibited by commercially available small molecules, and we assessed if two of them, HTH-01-015 and WZ4003 [46], could reduce cell viability selectively in PTEN- cells (step 4, Fig. 1). We compared the sensitivity of PTEN+ and PTEN- cell lines to inhibition by these compounds in 19 breast cancer cell lines (Fig. 5a, Additional file 1: Table S1). PTEN- cell lines were indeed more sensitive to HTH-01-015 treatment than PTEN+ cell lines, with significantly lower IC50 values (p < 0.05, two-sided Wilcoxon rank-sum test). By contrast, the PTEN- cell lines were not significantly more sensitive to WZ4003. This difference may be due to differing specificity of the two inhibitors: WZ4003 inhibits both NUAK1 and NUAK2 while the activity of HTH-01-015 is more specific to NUAK1 [46].
Fig. 5.
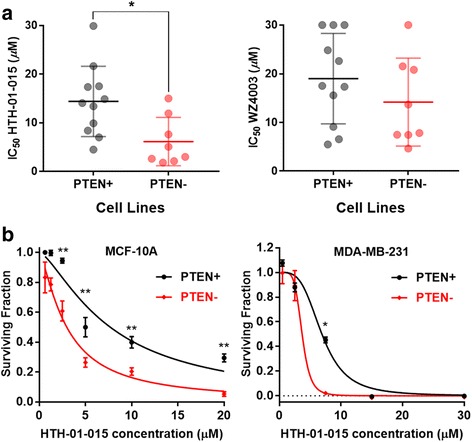
NUAK family kinase 1 (NUAK1) inhibition is phosphatase and tensin homolog-synthetic sick/lethal (PTEN-SSL) in breast cancer cell lines. a Sensitivity of 11 PTEN+ and 8 PTEN- breast cancer cell lines to HTH-01-015 and WZ4003, small molecule inhibitors of NUAK1 kinase; *p < 0.05, two-sided Wilcoxon rank-sum test. b HTH-01-015 dose-response curves in isogenic PTEN+ and PTEN- models derived from MCF-10A (left) and MDA-MB-231 (right) cell lines;**p < 0.01, *p < 0.05, two-sided t test on four biological replicates. IC50, concentration required to reduce cell viability by 50%
As a second test of whether PTEN expression determines sensitivity to NUAK1 inhibition by HTH-01-015, we assessed its effects in the MCF-10A and MDA-MB-231 PTEN+/PTEN- isogenic cell lines. In both cases, the PTEN- cell lines were more sensitive to HTH-01-015 than their PTEN+ counterparts (Fig. 5b). Therefore, we concluded that HTH-01-015 selectively targeted PTEN- cell lines and inhibition of NUAK1 has broad PTEN-SSL activity.
PTEN and STK11 mutations are mutually exclusive in tumors
Genes that are SSL with each other would have mutually exclusive loss-of-function mutations in tumors [47]. We therefore used cBioPortal [34, 35] to assess mutual exclusivity between PTEN and each of the six broad-spectrum PTEN-SSL genes (NUAK1, STK11, PIK3CB, HMMR, AP1M2, and ADAMTS20). We first examined mutation data in the TCGA breast cancer cohort [32]. None of the associations between mutations in PTEN and candidate PTEN-SSL genes was statistically significant (Additional file 3: Figure S2), possibly because of the small sample size. We then examined mutation data in the pan-cancer MSK-IMPACT study, which included > 10,000 patients [33]. Only two of our broad-spectrum PTEN-SSL genes (STK11 and PIK3CB) were assayed in this targeted study. We detected significant PTEN SSL effects for STK11 as indicated by mutual exclusivity of PTEN and STK11 (Fig. 6a). However, PTEN and PIK3CB mutations tended to co-occur. The analogous analysis of the breast cancer subset of the MSK-IMPACT study found that mutations in both STK11 and PIK3CB tended to be mutually exclusive with PTEN mutations, but was not statistically significant (Fig. 6b). Thus, there is evidence of PTEN-SSL activity of STK11 in a large pan-cancer patient cohort, and breast cancers within the cohort showed a similar pattern consistent with PTEN-STK11 SSL interaction.
Fig. 6.

Phosphatase and tensin homolog (PTEN) and serine/threonine kinase 11 (STK11) mutations are mutually exclusive in tumors. OncoPrints show deep (homozygous) deletions, fusions, small insertions and deletions, and non-silent single-base-substitution mutations detected by MSK-IMPACT in all patients studied (a) and in the subset of patients with breast cancer within the cohort (b). Mutual exclusivity of mutations was determined using odds ratios and the Fisher exact test. Only tumors with mutations are shown
Discussion
PTEN-SSL genes can be detected through loss-of-function RNAi screens that compare PTEN+ and PTEN- cell lines and several potential PTEN-SSL genes have been reported from such screens [41, 48, 49]. Nevertheless, the lack of overlap in results from these screens is obvious. This is not unique to the search for PTEN-SSL genes, as results from RAS-SSL screens have been similarly divergent [50]. The challenges to reproducibly identifying broad-spectrum SSL vulnerabilities through RNAi screens are well-documented and stem from the genetic complexity of tumor-derived cell lines and from the technical limitations of RNAi screening technologies [36, 50, 51].
We therefore used a screening strategy that started with a comprehensive primary screen in isogenic cell lines and followed up with a hits-focused secondary screen in a cell line panel. We detected 1537 possible PTEN-SSL genes through the primary siRNA screen of isogenic MCF-10A cell lines. In the follow up secondary shRNA screen, 24 genes showed evidence of broad-spectrum PTEN-SSL activity across a panel of 11 genetically diverse breast cancer cell lines. The high PTEN-SSL hit attrition rate was likely driven by genetic variability across the cell lines; in many cases the PTEN-SSL activity was restricted to only one or a small number of cell lines.
We then reanalyzed data from three published RNAi screens carried out in multiple breast cancer cell lines. Through this reanalysis, we identified six genes with reproducible and broad-spectrum PTEN-SSL activities. Among these, PIK3CB has previously been reported to be PTEN-SSL while the other five are novel PTEN-SSL genes [40]. ADAMTS20 encodes a secreted metalloproteinase with increased protein expression in invasive breast carcinoma [52]. HMMR, a breast cancer susceptibility gene in BRCA1 mutation carriers [53, 54], encodes a non-integral hyaluronan receptor that promotes breast cancer migration and invasion in concert with the integral hyaluronan receptor CD44 [55, 56]. AP1M2 encodes the beta subunit of clathrin-associated adaptor protein complex 1 (AP-1B), which regulates epithelial cell proliferation through its role in protein sorting [57]. Finally, NUAK1 and STK11 encode protein kinases, which are functionally related in the AMP-activated protein kinase (AMPK) pathway. STK11 (also known as LKB1) is a well-characterized tumor suppressor that phosphorylates and activates NUAK1 and 12 other members of the AMPK family [58]. We further validated the PTEN-SSL activities of the AMPK pathway members STK11 and NUAK1. We confirmed by small-molecule inhibition by HTH-01-015 that NUAK1 constitutes a novel PTEN-SSL vulnerability across a broad-spectrum of breast cancer cell lines. We also showed that, consistent with PTEN-SSL activity, PTEN and STK11 had mutually exclusive patterns of mutations in the MSK-IMPACT pan-cancer study. This study focused on the mutual exclusivity of somatic loss-of-function mutations. However, the functions of candidate PTEN-SSL genes could also potentially be dysregulated in cancer through epigenetic, transcriptional, translational, or post-translational mechanisms. Thus, mutual exclusivity analyses could be performed on such mechanisms if the relevant data became available.
PTEN, STK11, and NUAK1 have interconnected functions in regulating cell cycle progression and DNA damage repair. Inactivation of either STK11 or PTEN individually accelerates progression through the G1/S checkpoint [59, 60]. Conversely, reconstitution of STK11 or PTEN expression in cells with STK11 or PTEN loss, respectively, induces G1/S arrest [61–63]. Interestingly, NUAK1 functions at the intersection of cell cycle control by STK11 and PTEN. NUAK1 is directly activated by STK11 and indirectly inactivated by PTEN through the AKT pathway [64–66]. NUAK1 activity can either promote cell proliferation or induce cell cycle arrest, depending on whether it is activated by AKT or STK11. In the context of PTEN-deficiency, AKT phosphorylation of NUAK1 at Ser-600 promotes proliferation and invasion in cancer cell lines [64, 67, 68]. Conversely, NUAK1 promotes G1/S arrest through p53 regulation when it is phosphorylated at Thr-211 by STK11 in cells with functional PTEN [58]. NUAK1 overexpression also causes aneuploidy in normal human fibroblast cells, leading to senescence or cell death [69]. In addition to their roles in cell cycle regulation, PTEN, STK11, and NUAK1 are important for DNA repair. All three genes are required for the repair of ultraviolet B-induced DNA damage [70, 71]. In addition, STK11-deficient cells accumulate DNA damage [72], and PTEN-deleted cells have defective DNA double-strand break repair mechanisms [73].
Therefore, we hypothesize that STK11 and NUAK1 are selectively essential for cell cycle progression and DNA damage repair in PTEN-deficient cancers (Fig. 7). PTEN-deficient cancer cells have increased cell proliferation through accelerated cell cycle progression and also have impaired DNA damage repair. Loss of STK11 function in these cells would reduce its phosphorylation of NUAK1 at Thr-211 and thus deactivate the p53-regulated brake on cell cycle progression (Fig. 7c, top panel, faint orange activation and inhibition arcs compared to Fig. 7b). However, Ser-600 phosphorylation by the PTEN/AKT pathway would continue to promote cell cycle progression through STK11-independent mechanisms (blue arrows in Fig. 7c, top panel). Thus, the net effect of STK11 loss in these cells would be further acceleration of cell cycle. At the same time, STK11 loss would exacerbate the DNA damage repair defect (Fig. 7c, bottom panel). We hypothesize that this combination of effects (accelerated cell cycle progression and increased DNA damage) would lead to death or senescence.
Fig. 7.
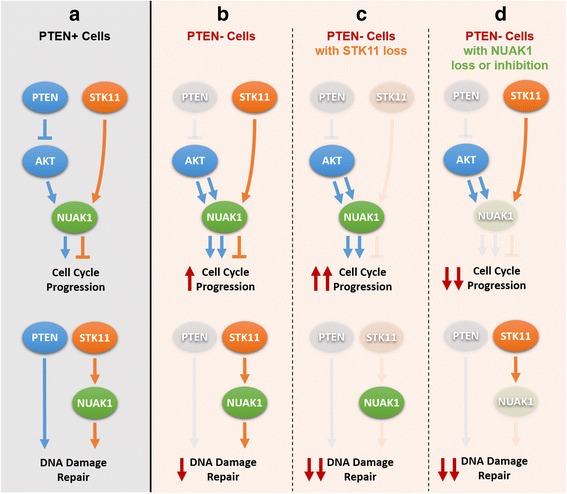
Proposed mechanisms of phosphatase and tensin homolog-serine/threonine kinase 11 (PTEN-STK11) and PTEN-NUAK family kinase 1 (NUAK1) synthetic sickness/lethality. Shown are hypothesized effects of STK11 and NUAK1 loss of function on cell cycle progression (upper panels) and DNA damage repair (lower panels) in PTEN- cells. a Roles of selected genes in PTEN+ cells for comparison. b In PTEN- cells, AKT signaling is not inhibited by PTEN, leading to increased activation of NUAK1 (double blue arrows), and increased cell-cycle progression (double blue arrows and upward-pointing red arrow in top panel). The PTEN contribution to DNA damage repair is lost (faded gray oval and arrow in bottom panel). c In PTEN- cells without STK11, cell cycle progression is even more accelerated, due to loss of STK11-NUAK1 inhibition (faint orange activation and inhibition arcs in top panel). DNA damage repair is further suppressed relative to PTEN- cells with STK11 activity because of lack of activation of NUAK1 by STK11 (faint orange arrows in bottom panel). d In PTEN cells without NUAK1, cell cycle progression is arrested (top panel). DNA damage repair is suppressed relative to PTEN- cells with NUAK1 due to loss of NUAK activity (bottom panel)
The hypothesized mechanism for PTEN-NUAK1 synthetic sickness/lethality is more direct. Since NUAK1 activation by AKT is involved in driving cell cycle progression caused by loss of PTEN, loss of NUAK1 function would directly reduce cell cycle progression. Consistent with this hypothesis, HTH-01-015 inhibition of NUAK1 in a PTEN-deficient osteosarcoma cell line has been shown to block the G1/S transition [74]. In addition, as with STK11, loss of NUAK1 would further impair DNA damage repair, thus causing further loss of cell viability. In contrast, PTEN-expressing cells would not depend on NUAK1 for cell cycle progression and would retain STK11- and NUAK1-independent DNA damage repair mechanisms. In the future, these hypothesized mechanisms for PTEN-NUAK1 and PTEN-STK11 SSL could be tested by long-term loss-of-function and gain-of-function studies in isogenic PTEN+/− cell lines and in genetically diverse breast cancer cell lines. These would include knocking out NUAK1 and STK11 individually in both PTEN+ and PTEN- cell lines using technologies such as CRISPR-Cas9. This would facilitate the study, in detail, of how these PTEN-SSL genes interact with PTEN in regulating cell cycle progression and DNA damage repair. Further chemical screens could also be carried out to identify drugs that would synergize with HTH-01-015 to selectively kill PTEN-deficient cancer cells.
Conclusions
An RNAi-based functional genomics strategy followed by validation through reanalysis of independent RNAi screens identified six genes with PTEN-SSL activity in a wide range of cell lines: PIK3CB, ADAMTS20, AP1M2, HMMR, STK11, and NUAK1. Two of these, STK11 and NUAK1, function in the AMPK pathway, and we hypothesize that their SSL relationship with PTEN stems partly from shared elements of this pathway. In addition, inhibition of NUAK1 with the small-molecule HTH-01-015 was PTEN-SSL in a wide range of cell lines, confirming the broad-spectrum PTEN-SSL activity of NUAK1. Our findings support the introduction of therapies targeting NUAK1, including inhibitors such as HTH-01-015, for treatment of PTEN-deficient breast cancers.
Additional files
Table S1. Growth media for breast cancer cell lines used in study. Table S2. PTEN protein abundance in cell lines quantified by western blot. Table S3. Classification of breast cancer cell lines as PTEN+ or PTEN- based on PTEN protein abundance. Table S4. Primary screen results and hits. Table S5. Details of shRNA library used in secondary screen. Table S6. shRNA barcode counts from secondary screen in 11 cell lines. Table S7. Fold-change in shRNA barcode count for 11 cell lines. Table S8. shRNAs used in ATARiS gene solutions, with ATARiS consistency scores per shRNA. Table S9. ATARiS gene level scores for 11 cell lines. Table S10. Secondary screen results and hits. Table S11. Screen results (zGARP scores) from Breast Functional Genomics Dataset. Table S12. Screen results (DEMETER scores) from Cancer Dependency Map Dataset. Table S13. Screen results (z scores) from Kinase Dependency Profiles Dataset. (XLSX 22688 kb)
{kind=link}
Figure S1. PTEN protein abundance of breast cancer cell lines. (A) Western blots showing PTEN and actin (loading control) abundance in 19 breast cancer cell lines. (B) Scatter plot of RPPA-measured PTEN abundance reported by Marcotte et al. [17] versus PTEN abundance that we quantified through densitometric analysis of western blot bands in (A). Cell lines were categorized as PTEN-expressing (in black) or PTEN-deficient (in red) based on PTEN protein abundance. (PNG 201 kb)
{kind=link}
Figure S2. Mutual exclusivity analysis in TCGA breast invasive carcinoma cohort. OncoPrints showing deep (homozygous) deletions, fusions, small insertions and deletions, and non-silent single-base-substitution mutations detected by TCGA. Mutual exclusivity of mutations was determined using odds ratios and the Fisher exact test. Only tumors with mutations are shown. (PNG 125 kb)
Acknowledgements
We thank past and present members of the Steve Rozen and Frederic Bard laboratories for their feedback and input through scientific discussions.
Funding
Funding was provided by the Singapore National Medical Research Council (NMRC/CIRG/1324/2012) and the Singapore Ministry of Health via the Duke-NUS Signature Research Programmes.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its Additional files.
Abbreviations
- ADAMTS20
ADAM metallopeptidase with thrombospondin type 1 motif 20
- AMPK
AMP-activated protein kinase
- AP1M2
Adaptor-related protein complex 1 mu 2 subunit
- ARK5
AMPK-related protein 5
- DMEM
Dulbecco’s modified Eagle’s medium
- DMSO
Dimethyl sulfoxide
- FACS
Fluorescence-activated cell sorting
- HEPES
2-(4-(2-Hydroxyethyl)piperazin-1-yl)ethanesulfonic acid
- HMMR
Hyaluronan-mediated motility receptor
- LKB1
Liver kinase b1
- MOI
Multiplicity of infection
- MTS
3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- NUAK1
NUAK family kinase 1
- PI3K
Phosphatidylinositol 3-kinase (PI3K)
- PIK3CB
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta
- PIP3
Phosphatidylinositol (3,4,5)-trisphosphate
- PTEN
Phosphatase and tensin homolog
- RFP
Red fluorescence protein
- RNAi
RNA interference
- RPMI
Roswell Park Memorial Institute medium
- RPPA
Reverse-phase protein array
- SDS
Sodium dodecyl sulfate
- SDS-PAGE
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- shRNA
Short hairpin RNA
- siRNA
Short interfering RNA
- SSL
Synthetic sick/lethal
- STK11
Serine/threonine kinase 11
- TGCA
The Cancer Genome Atlas
Authors’ contributions
YCT, BTT, FB, and SRG conceived and designed the project. YCT, SCH, ET, and GYG optimized and performed RNAi screens, and carried out validation experiments. YCT, AWN, and JRM analyzed and interpreted data from RNAi screens and cancer genomics studies. YCT and SRG wrote the manuscript with input from FB. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13058-018-0949-3) contains supplementary material, which is available to authorized users.
Contributor Information
Yew Chung Tang, Email: yewchung.tang@duke-nus.sg.
Szu-Chi Ho, Email: husky@duke-nus.edu.sg.
Elisabeth Tan, Email: Elisabeth.tan@duke-nus.edu.sg.
Alvin Wei Tian Ng, Email: alvinwtng@u.nus.edu.
John R. McPherson, Email: john.mcpherson.nz@gmail.com
Germaine Yen Lin Goh, Email: ylgoh@imcb.a-star.edu.sg.
Bin Tean Teh, Email: teh.bin.tean@singhealth.com.sg.
Frederic Bard, Email: fbard@imcb.a-star.edu.sg.
Steven G. Rozen, Email: steve.rozen@duke-nus.edu.sg
References
- 1.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133(3):403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 2.Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11(4):289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13(5):283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 4.Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16(1):64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 5.Ngeow J, Sesock K, Eng C. Breast cancer risk and clinical implications for germline PTEN mutation carriers. Breast Cancer Res Treat. 2017;165(1):1–8. doi: 10.1007/s10549-015-3665-z. [DOI] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas N Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perez-Tenorio G, Alkhori L, Olsson B, Waltersson MA, Nordenskjold B, Rutqvist LE, Skoog L, Stal O. PIK3CA mutations and PTEN loss correlate with similar prognostic factors and are not mutually exclusive in breast cancer. Clin Cancer Res. 2007;13(12):3577–3584. doi: 10.1158/1078-0432.CCR-06-1609. [DOI] [PubMed] [Google Scholar]
- 8.Li S, Shen Y, Wang M, Yang J, Lv M, Li P, Chen Z, Yang J. Loss of PTEN expression in breast cancer: association with clinicopathological characteristics and prognosis. Oncotarget. 2017;8(19):32043–32054. doi: 10.18632/oncotarget.16761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saal LH, Gruvberger-Saal SK, Persson C, Lovgren K, Jumppanen M, Staaf J, Jonsson G, Pires MM, Maurer M, Holm K, et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat Genet. 2008;40(1):102–107. doi: 10.1038/ng.2007.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Millis SZ, Gatalica Z, Winkler J, Vranic S, Kimbrough J, Reddy S, O’Shaughnessy JA. Predictive biomarker profiling of > 6000 breast cancer patients shows heterogeneity in TNBC, with treatment implications. Clin Breast Cancer. 2015;15(6):473–481. doi: 10.1016/j.clbc.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 11.Kawazu M, Kojima S, Ueno T, Totoki Y, Nakamura H, Kunita A, Qu W, Yoshimura J, Soda M, Yasuda T, et al. Integrative analysis of genomic alterations in triple-negative breast cancer in association with homologous recombination deficiency. PLoS Genet. 2017;13(6):e1006853. doi: 10.1371/journal.pgen.1006853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang S, Yu D. PI(3)king apart PTEN’s role in cancer. Clin Cancer Res. 2010;16(17):4325–4330. doi: 10.1158/1078-0432.CCR-09-2990. [DOI] [PubMed] [Google Scholar]
- 13.Dillon LM, Miller TW. Therapeutic targeting of cancers with loss of PTEN function. Curr Drug Targets. 2014;15(1):65–79. doi: 10.2174/1389450114666140106100909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Worby CA, Dixon JE. PTEN. Annu Rev Biochem. 2014;83:641–669. doi: 10.1146/annurev-biochem-082411-113907. [DOI] [PubMed] [Google Scholar]
- 15.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5(9):689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 16.Lord CJ, Tutt AN, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66:455–470. doi: 10.1146/annurev-med-050913-022545. [DOI] [PubMed] [Google Scholar]
- 17.Marcotte R, Sayad A, Brown KR, Sanchez-Garcia F, Reimand J, Haider M, Virtanen C, Bradner JE, Bader GD, Mills GB, et al. Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell. 2016;164(1–2):293–309. doi: 10.1016/j.cell.2015.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campbell J, Ryan CJ, Brough R, Bajrami I, Pemberton HN, Chong IY, Costa-Cabral S, Frankum J, Gulati A, Holme H, et al. Large-scale profiling of kinase dependencies in cancer cell lines. Cell Rep. 2016;14(10):2490–2501. doi: 10.1016/j.celrep.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. Defining a cancer dependency map. Cell. 2017;170(3):564–576. doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vitolo MI, Weiss MB, Szmacinski M, Tahir K, Waldman T, Park BH, Martin SS, Weber DJ, Bachman KE. Deletion of PTEN promotes tumorigenic signaling, resistance to anoikis, and altered response to chemotherapeutic agents in human mammary epithelial cells. Cancer Res. 2009;69(21):8275–8283. doi: 10.1158/0008-5472.CAN-09-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10(6):515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Charafe-Jauffret E, Ginestier C, Monville F, Finetti P, Adelaide J, Cervera N, Fekairi S, Xerri L, Jacquemier J, Birnbaum D, et al. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene. 2006;25(15):2273–2284. doi: 10.1038/sj.onc.1209254. [DOI] [PubMed] [Google Scholar]
- 23.Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50(18):6075–6086. [PubMed] [Google Scholar]
- 24.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30(3):256–268. doi: 10.1016/S1046-2023(03)00032-X. [DOI] [PubMed] [Google Scholar]
- 25.Di Nicolantonio F, Arena S, Gallicchio M, Zecchin D, Martini M, Flonta SE, Stella GM, Lamba S, Cancelliere C, Russo M, et al. Replacement of normal with mutant alleles in the genome of normal human cells unveils mutation-specific drug responses. Proc Natl Acad Sci U S A. 2008;105(52):20864–20869. doi: 10.1073/pnas.0808757105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weigelt B, Warne PH, Downward J. PIK3CA mutation, but not PTEN loss of function, determines the sensitivity of breast cancer cells to mTOR inhibitory drugs. Oncogene. 2011;30(29):3222–3233. doi: 10.1038/onc.2011.42. [DOI] [PubMed] [Google Scholar]
- 27.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 28.Birmingham A, Selfors LM, Forster T, Wrobel D, Kennedy CJ, Shanks E, Santoyo-Lopez J, Dunican DJ, Long A, Kelleher D, et al. Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods. 2009;6(8):569–575. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shao DD, Tsherniak A, Gopal S, Weir BA, Tamayo P, Stransky N, Schumacher SE, Zack TI, Beroukhim R, Garraway LA, et al. ATARiS: computational quantification of gene suppression phenotypes from multisample RNAi screens. Genome Res. 2013;23(4):665–678. doi: 10.1101/gr.143586.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38(5):500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 31.Fury W, Batliwalla F, Gregersen PK, Li W. Overlapping probabilities of top ranking gene lists, hypergeometric distribution, and stringency of gene selection criterion. Conf Proc IEEE Eng Med Biol Soc. 2006;1:5531–5534. doi: 10.1109/IEMBS.2006.260828. [DOI] [PubMed] [Google Scholar]
- 32.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163(2):506–519. doi: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nat Rev Genet. 2017;18:613–23. doi: 10.1038/nrg.2017.47. [DOI] [PubMed] [Google Scholar]
- 37.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, Silver SJ, Tamayo P, Wadlow RC, Ramaswamy S, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137(5):821–834. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 39.Jones SE, Fleuren EDG, Frankum J, Konde A, Williamson CT, Krastev DB, Pemberton HN, Campbell J, Gulati A, Elliott R, et al. ATR Is a therapeutic target in synovial sarcoma. Cancer Res. 2017;77(24):7014–7026. doi: 10.1158/0008-5472.CAN-17-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A. 2008;105(35):13057–13062. doi: 10.1073/pnas.0802655105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brough R, Frankum JR, Sims D, Mackay A, Mendes-Pereira AM, Bajrami I, Costa-Cabral S, Rafiq R, Ahmad AS, Cerone MA, et al. Functional viability profiles of breast cancer. Cancer Discov. 2011;1(3):260–273. doi: 10.1158/2159-8290.CD-11-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, East A, Ali LD, Lizotte PH, Wong TC, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A. 2011;108(30):12372–12377. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marcotte R, Brown KR, Suarez F, Sayad A, Karamboulas K, Krzyzanowski PM, Sircoulomb F, Medrano M, Fedyshyn Y, Koh JL, et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012;2(2):172–189. doi: 10.1158/2159-8290.CD-11-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cowley GS, Weir BA, Vazquez F, Tamayo P, Scott JA, Rusin S, East-Seletsky A, Ali LD, Gerath WF, Pantel SE, et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci Data. 2014;1:140035. doi: 10.1038/sdata.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McDonald ER, 3rd, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E, Gampa K, et al. Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell. 2017;170(3):577–592. doi: 10.1016/j.cell.2017.07.005. [DOI] [PubMed] [Google Scholar]
- 46.Banerjee S, Buhrlage SJ, Huang HT, Deng X, Zhou W, Wang J, Traynor R, Prescott AR, Alessi DR, Gray NS. Characterization of WZ4003 and HTH-01-015 as selective inhibitors of the LKB1-tumour-suppressor-activated NUAK kinases. Biochem J. 2014;457(1):215–225. doi: 10.1042/BJ20131152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao D, DePinho RA. Synthetic essentiality: targeting tumor suppressor deficiencies in cancer. Bioessays. 2017;39(8):1700076. doi: 10.1002/bies.201700076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mendes-Pereira AM, Lord CJ, Ashworth A. NLK is a novel therapeutic target for PTEN deficient tumour cells. PLoS One. 2012;7(10):e47249. doi: 10.1371/journal.pone.0047249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vizeacoumar FJ, Arnold R, Vizeacoumar FS, Chandrashekhar M, Buzina A, Young JT, Kwan JH, Sayad A, Mero P, Lawo S, et al. A negative genetic interaction map in isogenic cancer cell lines reveals cancer cell vulnerabilities. Mol Syst Biol. 2013;9:696. doi: 10.1038/msb.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Downward J. RAS synthetic lethal screens revisited: still seeking the elusive prize? Clin Cancer Res. 2015;21(8):1802–1809. doi: 10.1158/1078-0432.CCR-14-2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paul JM, Templeton SD, Baharani A, Freywald A, Vizeacoumar FJ. Building high-resolution synthetic lethal networks: a ‘Google map’ of the cancer cell. Trends Mol Med. 2014;20(12):704–715. doi: 10.1016/j.molmed.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 52.Porter S, Clark IM, Kevorkian L, Edwards DR. The ADAMTS metalloproteinases. Biochem J. 2005;386(Pt 1):15–27. doi: 10.1042/BJ20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maxwell CA, Benitez J, Gomez-Baldo L, Osorio A, Bonifaci N, Fernandez-Ramires R, Costes SV, Guino E, Chen H, Evans GJ, et al. Interplay between BRCA1 and RHAMM regulates epithelial apicobasal polarization and may influence risk of breast cancer. PLoS Biol. 2011;9(11):e1001199. doi: 10.1371/journal.pbio.1001199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pujana MA, Han JD, Starita LM, Stevens KN, Tewari M, Ahn JS, Rennert G, Moreno V, Kirchhoff T, Gold B, et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet. 2007;39(11):1338–1349. doi: 10.1038/ng.2007.2. [DOI] [PubMed] [Google Scholar]
- 55.Wang C, Thor AD, Moore DH, 2nd, Zhao Y, Kerschmann R, Stern R, Watson PH, Turley EA. The overexpression of RHAMM, a hyaluronan-binding protein that regulates ras signaling, correlates with overexpression of mitogen-activated protein kinase and is a significant parameter in breast cancer progression. Clin Cancer Res. 1998;4(3):567–576. [PubMed] [Google Scholar]
- 56.Hamilton SR, Fard SF, Paiwand FF, Tolg C, Veiseh M, Wang C, McCarthy JB, Bissell MJ, Koropatnick J, Turley EA. The hyaluronan receptors CD44 and Rhamm (CD168) form complexes with ERK1,2 that sustain high basal motility in breast cancer cells. J Biol Chem. 2007;282(22):16667–16680. doi: 10.1074/jbc.M702078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hase K, Nakatsu F, Ohmae M, Sugihara K, Shioda N, Takahashi D, Obata Y, Furusawa Y, Fujimura Y, Yamashita T, et al. AP-1B-mediated protein sorting regulates polarity and proliferation of intestinal epithelial cells in mice. Gastroenterology. 2013;145(3):625–635. doi: 10.1053/j.gastro.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 58.Hou X, Liu JE, Liu W, Liu CY, Liu ZY, Sun ZY. A new role of NUAK1: directly phosphorylating p53 and regulating cell proliferation. Oncogene. 2011;30(26):2933–2942. doi: 10.1038/onc.2011.19. [DOI] [PubMed] [Google Scholar]
- 59.Radu A, Neubauer V, Akagi T, Hanafusa H, Georgescu MM. PTEN induces cell cycle arrest by decreasing the level and nuclear localization of cyclin D1. Mol Cell Biol. 2003;23(17):6139–6149. doi: 10.1128/MCB.23.17.6139-6149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liang X, Wang P, Gao Q, Xiang T, Tao X. Endogenous LKB1 knockdown accelerates G(1)/S transition through p53 and p16 pathways. Cancer Biol Ther. 2010;9(2):156–160. doi: 10.4161/cbt.9.2.10452. [DOI] [PubMed] [Google Scholar]
- 61.Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, Sellers WR. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1999;96(5):2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, Gavrilova N, Mueller B, Liu X, Wu H. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1999;96(11):6199–6204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tiainen M, Ylikorkala A, Makela TP. Growth suppression by Lkb1 is mediated by a G(1) cell cycle arrest. Proc Natl Acad Sci U S A. 1999;96(16):9248–9251. doi: 10.1073/pnas.96.16.9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suzuki A, Lu J, Kusakai G, Kishimoto A, Ogura T, Esumi H. ARK5 is a tumor invasion-associated factor downstream of Akt signaling. Mol Cell Biol. 2004;24(8):3526–3535. doi: 10.1128/MCB.24.8.3526-3535.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zagorska A, Deak M, Campbell DG, Banerjee S, Hirano M, Aizawa S, Prescott AR, Alessi DR. New roles for the LKB1-NUAK pathway in controlling myosin phosphatase complexes and cell adhesion. Sci Signal. 2010;3(115):ra25. doi: 10.1126/scisignal.2000616. [DOI] [PubMed] [Google Scholar]
- 66.Suzuki A, Kusakai G, Kishimoto A, Lu J, Ogura T, Lavin MF, Esumi H. Identification of a novel protein kinase mediating Akt survival signaling to the ATM protein. J Biol Chem. 2003;278(1):48–53. doi: 10.1074/jbc.M206025200. [DOI] [PubMed] [Google Scholar]
- 67.Chen P, Li K, Liang Y, Li L, Zhu X. High NUAK1 expression correlates with poor prognosis and involved in NSCLC cells migration and invasion. Exp Lung Res. 2013;39(1):9–17. doi: 10.3109/01902148.2012.744115. [DOI] [PubMed] [Google Scholar]
- 68.Chang XZ, Yu J, Liu HY, Dong RH, Cao XC. ARK5 is associated with the invasive and metastatic potential of human breast cancer cells. J Cancer Res Clin Oncol. 2012;138(2):247–254. doi: 10.1007/s00432-011-1102-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Humbert N, Navaratnam N, Augert A, Da Costa M, Martien S, Wang J, Martinez D, Abbadie C, Carling D, de Launoit Y, et al. Regulation of ploidy and senescence by the AMPK-related kinase NUAK1. EMBO J. 2010;29(2):376–386. doi: 10.1038/emboj.2009.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Esteve-Puig R, Gil R, Gonzalez-Sanchez E, Bech-Serra JJ, Grueso J, Hernandez-Losa J, Moline T, Canals F, Ferrer B, Cortes J, et al. A mouse model uncovers LKB1 as an UVB-induced DNA damage sensor mediating CDKN1A (p21WAF1/CIP1) degradation. PLoS Genet. 2014;10(10):e1004721. doi: 10.1371/journal.pgen.1004721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ming M, Feng L, Shea CR, Soltani K, Zhao B, Han W, Smart RC, Trempus CS, He YY. PTEN positively regulates UVB-induced DNA damage repair. Cancer Res. 2011;71(15):5287–5295. doi: 10.1158/0008-5472.CAN-10-4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu Y, Marks K, Cowley GS, Carretero J, Liu Q, Nieland TJ, Xu C, Cohoon TJ, Gao P, Zhang Y, et al. Metabolic and functional genomic studies identify deoxythymidylate kinase as a target in LKB1-mutant lung cancer. Cancer Discov. 2013;3(8):870–879. doi: 10.1158/2159-8290.CD-13-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128(1):157–170. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 74.Banerjee S, Zagorska A, Deak M, Campbell DG, Prescott AR, Alessi DR. Interplay between Polo kinase, LKB1-activated NUAK1 kinase, PP1betaMYPT1 phosphatase complex and the SCFbetaTrCP E3 ubiquitin ligase. Biochem J. 2014;461(2):233–245. doi: 10.1042/BJ20140408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Growth media for breast cancer cell lines used in study. Table S2. PTEN protein abundance in cell lines quantified by western blot. Table S3. Classification of breast cancer cell lines as PTEN+ or PTEN- based on PTEN protein abundance. Table S4. Primary screen results and hits. Table S5. Details of shRNA library used in secondary screen. Table S6. shRNA barcode counts from secondary screen in 11 cell lines. Table S7. Fold-change in shRNA barcode count for 11 cell lines. Table S8. shRNAs used in ATARiS gene solutions, with ATARiS consistency scores per shRNA. Table S9. ATARiS gene level scores for 11 cell lines. Table S10. Secondary screen results and hits. Table S11. Screen results (zGARP scores) from Breast Functional Genomics Dataset. Table S12. Screen results (DEMETER scores) from Cancer Dependency Map Dataset. Table S13. Screen results (z scores) from Kinase Dependency Profiles Dataset. (XLSX 22688 kb)
Figure S1. PTEN protein abundance of breast cancer cell lines. (A) Western blots showing PTEN and actin (loading control) abundance in 19 breast cancer cell lines. (B) Scatter plot of RPPA-measured PTEN abundance reported by Marcotte et al. [17] versus PTEN abundance that we quantified through densitometric analysis of western blot bands in (A). Cell lines were categorized as PTEN-expressing (in black) or PTEN-deficient (in red) based on PTEN protein abundance. (PNG 201 kb)
Figure S2. Mutual exclusivity analysis in TCGA breast invasive carcinoma cohort. OncoPrints showing deep (homozygous) deletions, fusions, small insertions and deletions, and non-silent single-base-substitution mutations detected by TCGA. Mutual exclusivity of mutations was determined using odds ratios and the Fisher exact test. Only tumors with mutations are shown. (PNG 125 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its Additional files.