

SUMMARY
Non-enzymatic protein modification driven by thioester reactivity is thought to play a major role in the establishment of cellular lysine acylation. However, the specific protein targets of this process are largely unknown. Here we report an experimental strategy to investigate non-enzymatic acylation in cells. Specifically, we develop a chemoproteomic method that separates thioester reactivity from enzymatic utilization, allowing selective enrichment of non-enzymatic acylation targets. Applying this method to cancer cell lines identifies numerous candidate targets of non-enzymatic acylation, including several enzymes in lower glycolysis. Functional studies highlight malonyl-CoA as a reactive thioester metabolite that can modify and inhibit glycolytic enzyme activity. Finally, we show that synthetic thioesters can be used as novel reagents to probe non-enzymatic acylation in living cells. Our studies provide new insights into the targets and drivers of non-enzymatic acylation, and demonstrate the utility of reactivity-based methods to experimentally investigate this phenomenon in biology and disease.
eTOC Blurb
Kulkarni et al. describe a reactivity-based method to track targets of non-enzymatic acylation. This approach leads to the discovery that malonyl-CoA is a hyperreactive thioester metabolite that can covalently antagonize glycolytic enzyme activity.

Introduction
Lysine acylation encompasses a diverse family of posttranslational modifications (PTMs) derived from acyl-CoA thioesters (Huang et al., 2014a; Lin et al., 2012). The prototypical PTM of this family is lysine acetylation. Lysine acetylation has been mapped to >8000 sites in human cells, and plays a critical role in many biological processes, including the regulation of epigenetic signaling and adaptive metabolic responses (Scholz et al., 2015; Verdin and Ott, 2015). Besides acetylation, several additional acyl-CoA derived lysine modifications have also been described, including butyrylation, succinylation, crotonylation, malonylation, and palmitoylation. Numerous studies have characterized the biological function of these alternative acylations, as well as regulation of their removal by NAD+-dependent histone deacetylase enzymes (Jiang et al., 2013; Lin et al., 2012). In contrast, the specific lysine acyltransferases (KATs) responsible for catalyzing many of these PTMs remain unknown.
A unique feature of acyl-CoA metabolites is their intrinsic electrophilicity. For example, nucleophilic addition of water to acetyl-CoA’s electrophilic thioester bond results in hydrolysis, and the net release of 35.8 kJ/mol of energy (Guynn et al., 1973). These favorable thermodynamics have led to the hypothesis that a subset of cellular acylation may be established in an enzyme-independent fashion, via direct reaction of nucleophilic lysines with acyl-CoAs (Wagner and Hirschey, 2014). This view is supported by the observation that acyl-CoAs and proteins display spontaneous reactivity in vitro (Olia et al., 2015; Wagner and Payne, 2013), and that many protein acylations occur at low stoichiometry, as expected for a non-enzymatic process (Weinert et al., 2014). Identifying the protein targets of intrinsic thioester reactivity could provide critical insights into how cellular acylation is established, as well as how these modifications are functionally linked to physiological or pathophysiological metabolism. However, a fundamental challenge lies in a lack of methods to specifically define and manipulate non-enzymatic acylation in living cells.
Here we describe a chemoproteomic approach to identify targets of intrinsic thioester reactivity. Our strategy employs a bioorthogonal thioester, which reports on the reactivity of acyl-CoAs, but is not utilized as an enzyme cofactor. Exploration of glycolysis as a model pathway susceptible to non-enzymatic acylation facilitates the discovery of a functional interaction between glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and malonyl-CoA, a hyperreactive thioester metabolite. Finally, we apply this knowledge to design cell-permeable thioesters that can be used to probe the effect of non-enzymatic acylation on glycolytic enzyme function in cells. These studies represent a first step towards the development of experimental methods to directly study non-enzymatic acylation in cells, and highlight lysine malonylation as a prototypical reactivity-driven PTM whose manipulation may prove therapeutically useful in cancer.
Results
A chemical reporter of intrinsic thioester reactivity
To develop an experimental approach to characterize cellular targets of non-enzymatic acylation, we envisioned a chemoproteomic probe comprising three general features: 1) an electrophilic thioester which mimics the reactivity of endogenous acyl-CoA cofactors, 2) a bioorthogonal reporter, capable of labeling reactive protein targets via S- to N- transacylation chemistry, and 3) a chemical structure incompatible with utilization as a KAT cofactor (Figure 1). This last feature is of utmost importance, as it is necessary in order to differentiate enzymatic and non-enzymatic acylation. Inspection of a number of KAT structures in complex with CoA-derived ligands revealed in almost all cases a high proportion of hydrogen bonding and van der Waals interactions originated from the 3′,5′-ADP moiety of CoA (Figure 1a) (Trievel et al., 1999; Wang et al., 2008). This suggested that synthetic thioesters lacking the phosphoadenine portion of endogenous acyl-CoA metabolites would not strongly interact, nor be used as cofactors, by KATs.
Figure 1.
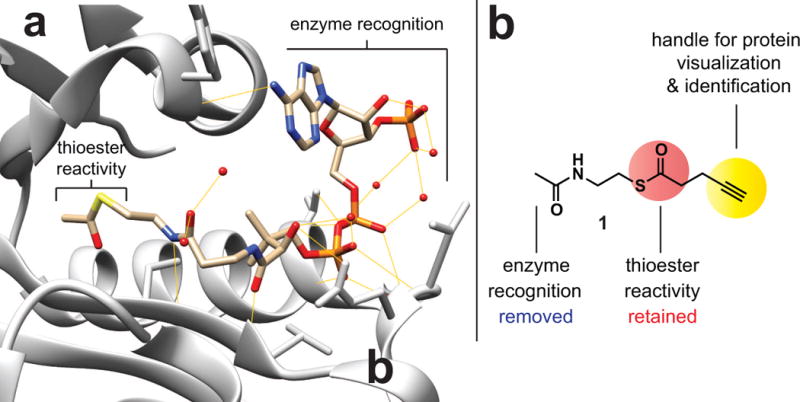
Design of a reactivity-based approach for profiling non-enzymatic acetylation. (a) Structure of acetyl-CoA bound to the prototypical lysine acetyltransferase GCN5L2 (PDB 1Z4R). Yellow lines indicate hydrogen bonds made between Gcn5 and the acetyl-CoA cofactor. (b) Reactivity-based profiling reagent 1.
To test this hypothesis, we synthesized a small panel of acetyl- and 4-pentynoyl thioesters containing varying degrees of steric bulk at the N-acyl group (Figure S1). These molecules were assayed against two phylogenetically distinct KAT enzymes, p300 and Gcn5, using a recently reported microfluidic mobility shift assay (Sorum et al., 2016). The results indicate that none of the molecules display significant cofactor utilization in this assay, even when utilized at concentrations >1000-fold higher than acetyl-CoA (Figure S1). In the case of the prototypical thioester reporter 4-pentynoyl-N-acetylcystamine (1), this lack of cofactor utilization was further confirmed using an orthogonal assay that monitors CoA release (Figure S1) (Trievel et al., 2000). The lack of utilization by p300 is notable, as p300 has been found to utilize a wide-range of acyl-CoA cofactors (Chen et al., 2007; Sabari et al., 2015). These findings indicate truncated bioorthogonal thioesters may be capable of decoupling enzymatic and non-enzymatic acylation.
Next, we performed gel-based labeling experiments to define the protein reactivity of thioester reporter 1. Following incubation with cancer cell lysates, proteins labeled by 1 were ligated to a fluorescent reporter using copper-catalyzed azide-alkyne cycloaddition, and detected via SDS-PAGE (Figure 2) (Speers and Cravatt, 2004). Probe labeling for almost all proteins was concentration-dependent and not saturatable, as expected for a non-enzymatic reaction (Figure 2b) (Olia et al., 2015). Of note, robust labeling was observed even at 100 μM which lies in the range of cellular acetyl-CoA levels (estimated 20–200 μM) (Henry et al., 2015). Using competition studies, we found that labeling of proteins by thioester 1 was strongly impeded by pre-incubating lysates with excess acetyl-CoA (Figure 2c). Furthermore, protein labeling by 1 was enhanced in lysates prepared from the p300-deficient cancer cell line SU-DHL-6 (Pasqualucci et al., 2011), indicating that 1-labeling is correlated with unoccupied protein lysine residues, rather than KAT activity (Figure S2a). These data suggest bioorthogonal thioesters are capable of detecting cellular proteins susceptible to reaction with endogenous acyl-CoA metabolites.
Figure 2.

Assessing the reactivity of a thioester reporter in complex proteomes. (a) Strategy for ex situ (lysate) and in situ (cellular) labeling using thioester 1. (b) Dose-dependent ex situ labeling of cancer cell lysates by thioester 1 (A549, 15 h, 0, 100, 200, 400, 800 μM). (c) Labeling by 1 is competed by acetyl-CoA (1 h pre-incubation with 0, 100, 200, 400, or 800 μM acetyl-CoA, then 400 μM 1 for 15 h). (d) Dose-dependent in situ labeling of cancer cell by thioester 1 (A549, 15 h, 0, 100, 200, 400, 800 μM).
Given the small size and neutral charge of 1, we hypothesized it might also be capable of reporting on thioester reactivity in intact cells. Many groups have found that the protein targets of reactive small molecules can differ substantially when identified in situ (Bachovchin et al., 2014; Martinez Molina et al., 2013; Speers et al., 2003). Accordingly, we treated A549 cells with thioester 1, isolated proteomes, and analyzed protein labeling via SDS-PAGE (Figure 2). Cellular administration of 1 resulted in dose-dependent protein labeling (Figure 2d). The overall patterns of labeling were very similar to those observed in lysates, with a few notable exceptions. To assess whether differences in labeling may results from the metabolism of 1, we also assessed protein labeling by 4-pentynoic acid, a hydrolysis product of 1 that can form a known KAT substrate (4-pentynoyl-CoA) in cells (Yang et al., 2010b). Dosed at equivalent concentrations (400 μM), 4-pentynoic acid resulted in negligible labeling of cellular proteins compared to thioester 1 (Figure S2b). This suggests the major mechanism by which 1 labels proteins in cells is via direct transacylation, rather than metabolism. Overall, these studies highlight the ability of thioester reporters to differentiate enzymatic and non-enzymatic mechanisms of protein acylation in complex proteomes and living cells.
Reactivity-based profiling identifies cellular pathways susceptible to non-enzymatic acylation
We next set out to develop a mass spectrometry (MS) method capable of identifying cellular proteins susceptible to non-enzymatic acylation. To accomplish this, soluble proteomes from the human lung cancer cell line A549 were first labeled with reactive thioester 1, either directly or via preparation of lysates from in situ treated cells. Labeled proteomes were then subjected to chemoselective ligation with biotin azide, enriched over streptavidin-agarose, tryptically digested, and analyzed by LC-MS/MS (Figure 3) (Jessani et al., 2005). Quantification by spectral counting led to the identification of 100 candidate non-enzymatic acylation targets from lysates treated with 1 (> 10 spectral counts, >2-fold enrichment compared to an untreated control). Applying similar criteria to in situ treated samples led to the identification of 53 candidate non-enzymatic acylation targets (Tables S1a–b). Extension of this approach to identify reactive thioester targets in a second cancer cell line (Raji cells) increased our proteome coverage, resulting in the identification of 78 and 82 proteins for ex situ and in situ treatments, respectively (Tables S1c–d). Enrichments were validated by reinterrogating a subset of these candidate targets via affinity purification and immunoblot (Figure 3b). Validated LC-MS/MS identified targets of 1 displayed either acetyl-CoA competitive (ex situ) or concentration-dependent (in situ) enrichment when analyzed (Figure 3b). To ask whether targets of 1 constitute physiologically acetylated proteins, we analyzed their Uniprot annotations. This analysis revealed proteins containing acetylated lysines were heavily enriched in our probe dataset (~80%) compared to the overall proteome (~18%). Gene ontology analyses of enriched datasets also revealed acetylation to be the most significantly associated term (Tables S1a–d). These results demonstrate that thioester reactivity probes can be used to discover reactive, physiological acetylation targets.
Figure 3.
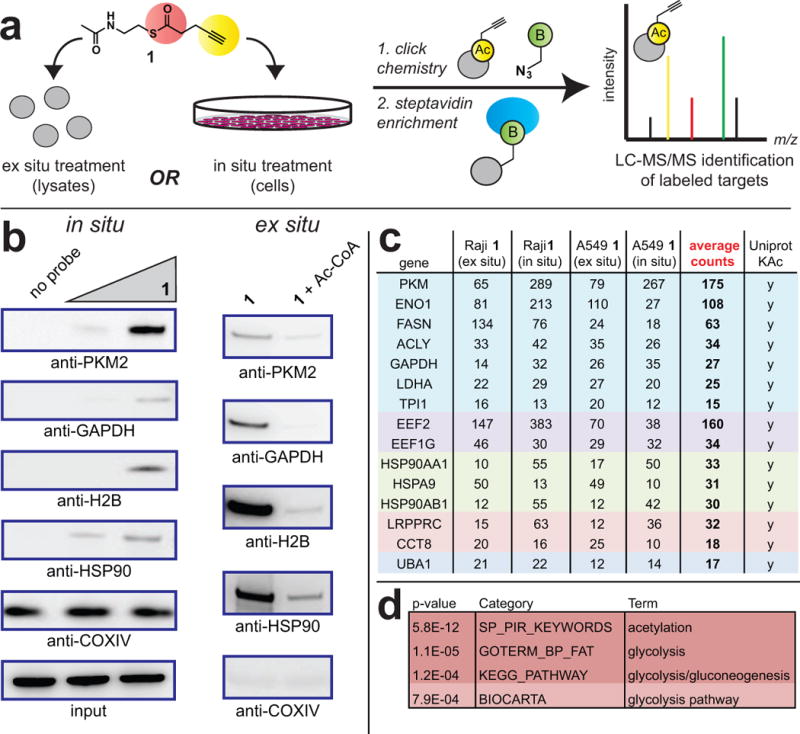
Assessing the targets of a thioester reporter in complex proteomes. (a) Strategy for ex situ (lysate) and in situ (cellular) labeling of thioester 1 targets. Following click chemistry to biotin-azide, tagged proteins are subjected to on-bead tryptic digest and identified by LC-MS/MS. (b) Validation of targets by affinity capture/immunoblot. Left: In situ enrichment of Raji proteins by thioester 1 (15 h; 0, 100, 400 μM). Right: Ex situ enrichment of Raji proteins by thioester 1 is competed by acetyl-CoA (30 min, 1 mM acetyl-CoA; then 15 h, 100 μM 1). (c) Spectral count data for universally identified thioester-reactive proteins enriched by thioester probe 1 (15 h, 400 μM). Data represents the output of individual LC-MS/MS experiments. Listed proteins were enriched ≥2-fold in each cancer cell line specified, using both in situ and ex situ labeling protocols, respectively. Functionally related proteins are grouped according to color. Complete lists of enriched targets are given in Supplementary Table S1. (d) Terms strongly enriched during gene ontology analysis of universally identified thioester-reactive proteins.
Next, we performed comparative analyses of our datasets. The majority of proteins enriched by 1 using in situ treatment overlapped with ex situ identified targets (72% in A549, 62% in Raji). One noteworthy difference between in situ and ex situ targets was the identification of the histone variants HIS1H2AH and HIST2H2AC exclusively from in situ labeled Raji cell proteomes (Table S1d). This may indicate that histone H2A variants possess increased reactivity in cellular contexts, or signify unanticipated changes in protein abundance and reactivity triggered by exposure of cells to 1. Focusing on overlapping targets, sixteen proteins were identified in all four datasets (in situ and ex situ, A549 and Raji; Figure 3c and Table S1e). The majority of these targets are cytosolic, and the lack of mitochondrial proteins may indicate that mitochondrial non-enzymatic acylation is driven by high acyl-CoA concentrations, rather than intrinsic protein side chain reactivity (Olia et al., 2015). Prominent among these candidate targets were abundant ribosomal proteins (EEF2, EEF1G), molecular chaperones (HSP90AA1, HSP90AB1, HSPA9), and metabolic enzymes (GAPDH, PKM2, ENO1, LDHA, FASN, ACLY). This bias towards abundant species is expected for a reactivity-driven phenomenon (Weinert et al., 2013), and suggests these proteins may function as major “sinks” for electrophilic acyl-CoAs in cells (Figure 3).
Protein acylation driven directly by thioester reactivity has the potential to either impact biological function, or constitute non-functional background. Reasoning that pathways enriched in thioester reactive proteins may have evolved to be functionally regulated by non-enzymatic acylation, we sought to identify such pathways using the Database for Annotation, Visualization and Integrated Discovery (DAVID) gene ontology tool. Analyzing proteins identified in overlapping datasets in DAVID revealed the recurrent enrichment of terms related to glycolysis (Figure 3d, Table S1f). Glycolytic enzymes labeled by 1 cluster in the lower reaction chain of glycolysis, which is responsible for the production and processing of the 3-carbon metabolite glyceraldehyde-3-phosphate to pyruvate. The thioester reactivity of glycolytic enzymes is intriguing, as glucose metabolism is the major source of acyl-CoA thioester metabolites found in the nucleus and cytosol, which is also where these enzymes localize (Wellen et al., 2009). However, while many glycolytic enzymes are characterized targets of acetylation, the effect of non-enzymatic acylation on their function has not been specifically explored (Lv et al., 2013). These factors prompted us to focus on glycolysis as a model to understand the functional effects of protein acylation driven by intrinsic thioester reactivity.
Malonyl-CoA is a hyperreactive cytosolic thioester that inhibits glycolytic enzymes
To narrow the scope of our functional studies, we focused on glycolytic enzymes that were: 1) strongly enriched by 1 (>2-fold) in additional cell lines (Tables S1g–i), 2) known to be susceptible to non-enzymatic modification by reactive metabolites, and 3) known to be regulated by lysine acylation. These criteria led us to explore GAPDH and PKM2 as prototypical targets of thioester reactivity. (Anastasiou et al., 2011; Moellering and Cravatt, 2013; Mustafa et al., 2009). To study this phenomenon, we first compared the ability of acyl-CoA metabolites to inhibit purified GAPDH. We analyzed a wide panel of different acyl-CoA metabolites for GAPDH inhibition, reasoning that their unique physiochemical properties (charge, steric bulk, hydrophobicity) may alter their reactivity and/or effects on protein function (Lin et al., 2012; Montgomery et al., 2015b). While the majority of acyl-CoAs had at most modest effects on GAPDH catalysis, we found that pre-incubation of GAPDH with malonyl-CoA (200 μM for 30 minutes) caused potent inhibition of enzyme activity (Figure 4a). Similarly, we found that malonyl-CoA, but not acetyl-CoA, also strongly inhibited pyruvate kinase activity in A549 cell lysates (Figure S3a). Notably, this inhibitory effect was not universal, as under identical conditions malonyl-CoA did not strongly inhibit recombinant fumarate hydratase (FH) or the lysine acetyltransferase p300 (Figure S3b–c). These findings prompted us to further explore the scope and mechanistic basis for malonyl-CoA’s effects on glycolytic enzyme function.
Figure 4.
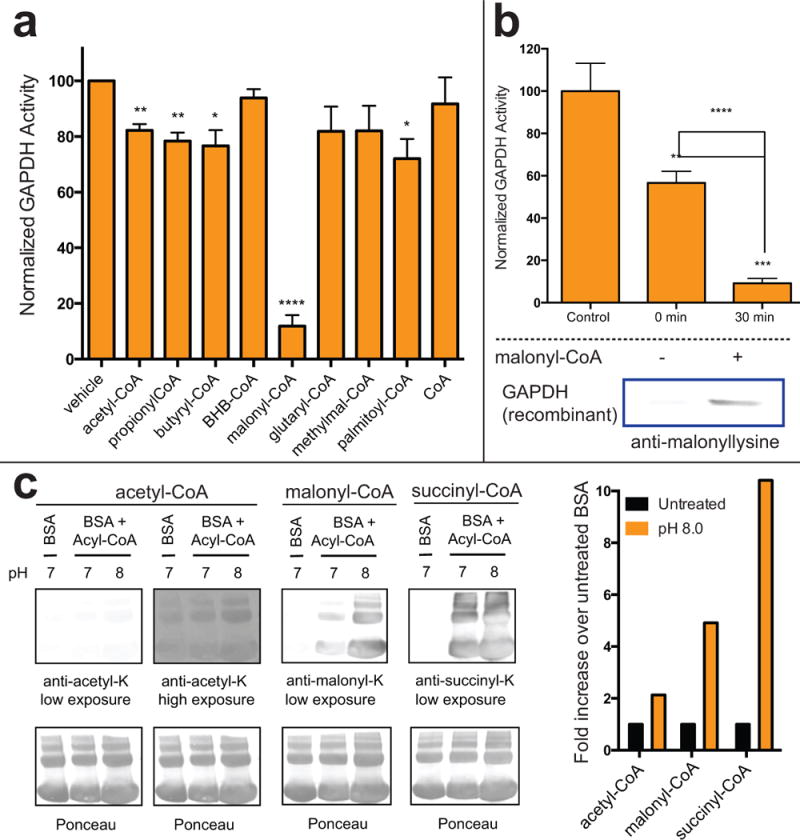
Thioester reactivity-mediated inhibition of glycolytic enzyme activity. (a) Effect of cytosolic acyl-CoA thioesters (200 μM) on recombinant GAPDH activity after 30 min pre-incubation. (b) Inhibition of recombinant GAPDH by malonyl-CoA (200 μM) is time-dependent and correlates with increased lysine malonylation. (c) Gel-based comparison of acetyl-CoA, malonyl-CoA and succinyl-CoA reactivity. Acyl-CoAs (200 μM) were incubated with BSA at 37 °C for 6 h prior to western blotting. Enzyme activity represents the average of ≥3 replicates, with significance analyzed by unpaired Student’s t test (ns = not significant, * = P < 0.05, ** = P < 0.01, and *** = P < 0.001)
Malonyl-CoA serves as the essential cofactor for lysine malonylation, an alternative acylation that has been recently proposed to play a role in regulation of glycolysis (Nishida et al., 2015). To understand whether malonyl-CoA’s effects on glycolytic enzymes may stem from covalent non-enzymatic malonylation, we performed parallel assessments of GAPDH malonylation and activity as a function of time. Time-dependent enzyme activity loss was minimized by performing these analyses at 200 μM, which likely represents the upper range of physiologically relevant malonyl-CoA concentrations (Henry et al., 2015; Tokutake et al., 2012; Tokutake et al., 2010). Consistent with a covalent inhibition mechanism, malonyl-CoA reduces GAPDH activity in a time-dependent manner (Figure 4b). This loss of activity correlates with the time required for lysine malonylation, as assessed by immunoblotting (Figure 4b, bottom). GAPDH activity was not recovered by desalting or hydroxylamine treatment, also consistent with a covalent, lysine-directed modification (Figures S3d–g). Analysis of recombinant human and yeast GAPDH revealed several lysines that displayed increased malonylation and reduced proteolytic cleavage by trypsin upon exposure to malonyl-CoA (Table S2a–b), including K215 which is highly conserved and known to exhibit increased malonylation in physiological models of malonyl-CoA decarboxylase deficiency (Tables S2c–d) (Colak et al., 2015; Moellering and Cravatt, 2013; Peng et al., 2011). These results indicate that covalent malonylation driven by intrinsic thioester activity can have functional effects on enzyme activity.
The finding that malonyl-CoA, but not acetyl-CoA, inhibits GAPDH was unexpected. We considered that this effect could, in principle, stem from two non-mutually exclusive possibilities: 1) malonyl-CoA is more reactive than acetyl-CoA, and thus causes greater GAPDH modification and inhibition, or 2) lysine malonylation inhibits GAPDH to a greater degree than acetylation. To examine malonyl-CoA’s reactivity, we used a straightforward SDS-PAGE assay to analyze its ability to modify the model protein bovine serum albumin (BSA) (Wagner and Payne, 2013). For context malonyl-CoA’s reactivity was compared to acetyl-CoA as well as succinyl-CoA, a mitochondrial thioester whose enhanced reactivity has been previously observed (Simic et al., 2015; Wagner and Payne, 2013). Applying this assay, we found malonyl-CoA and succinyl-CoA both cause enhanced acylation of BSA relative to acetyl-CoA (Figure 4c). The increased reactivity of succinyl-CoA correlated with more potent inhibition of GAPDH, consistent with a reactivity-based phenomenon (Figures 4c, S3h). Malonyl-CoA’s reactivity is notable, and represents one of the first examples of a cytosolic acyl-CoA with enhanced transacylation potential. To validate this result, we compared acetyl-CoA and malonyl-CoA’s hydrolysis using a ratiometric HPLC assay, confirming the increased reactivity of malonyl-CoA (Figure S3i). Conceptually the increased reactivity of malonyl-CoA may be rationalized on the basis of its carboxy-functionality being inductively electron withdrawing, thereby increasing the susceptibility of adjacent thioester to nucleophilic attack. Consistent with this rationale, UV spectroscopy analysis reveals a simplified malonyl thioester cleaves much more rapidly than its acetyl analogue (Figure S3j). Overall, these findings demonstrate the susceptibility of GAPDH to inhibition by non-enzymatic acylation, and highlight malonyl-CoA as a novel reactive cytosolic metabolite.
Reactive thioesters antagonize glycolytic enzymes and Warburg metabolism in cells
Having demonstrated the potential of cytosolic malonyl-CoA to inhibit glycolytic enzymes via covalent malonylation in vitro, we next set out to determine whether manipulating this PTM could similarly affect enzyme activity in cells. Since the major consumer of malonyl-CoA is fatty acid biosynthesis, we first tested whether the FASN inhibitor orlistat altered lysine malonylation and/or cellular GAPDH activity in the A549 lung cancer cell line (Figure 5) (Pemble et al., 2007). Treatment of cells with 25 μM orlistat for 24 hours resulted in an increase in proteome-wide lysine malonylation, as assessed by immunoblotting (Figure 5b). Under these same conditions, orlistat also caused a significant reduction in cellular GAPDH activity (Figure 5c). Cellular inhibition of GAPDH was partial, suggesting that FASN inhibition does not increase intracellular malonyl-CoA to levels required for complete disruption of GAPDH activity. Alternatively, covalent malonylation may be antagonized by competing acylations that do not alter GAPDH function. Malonylation and GAPDH inhibition caused by orlistat was time-dependent and not significantly observed at early time points, as expected for a reactivity-driven phenomenon (Figure S4a–b).
Figure 5.

Exploring the effects of cytosolic thioester reactivity on glycolytic enzymes. (a) Strategies for direct (left) and indirect (right) manipulation of enzyme malonylation using malonyl-NAC and orlistat, respectively. (b) Effects of malonyl-NAC (1 mM) and orlistat (25 μM) on cellular malonylation in A549 cells. (c) Effects of malonyl-NAC (1 mM) and orlistat (25 μM) on GAPDH activity in A549 cells. (d) Effects of malonyl-NAC (1 mM) on pyruvate kinase activity in A549 cells. Enzyme activity represents the average of ≥3 replicates, with significance analyzed by unpaired Student’s t test (ns = not significant, * = P < 0.05, ** = P < 0.01)
An ambiguity in our results lies in the fact that inhibition of fatty acid biosynthesis may manifest pleiotropic effects unrelated to lysine malonylation. For example, FASN plays a key role in production of signaling lipids, and also contributes to the maintenance of cellular redox balance (Benjamin et al., 2015; Nomura et al., 2010). This inspired us to develop an orthogonal chemical method to more directly probe the effects of non-enzymatic malonylation on glycolytic enzyme function. Recalling our chemical proteomic studies, we envisioned that non-enzymatic malonylation could be directly manipulated by treating cells with an exogenous reactive thioester. Accordingly, we synthesized a small molecule, ethyl malonyl-N-acetylcystamine (malonyl-NAC) designed to cross cell membranes and stimulate lysine malonylation via direct S to N acyl transfer (Figure 5a, left). Treatment of A549 cells with 1 mM malonyl-NAC for 24 hours resulted in a significant time-dependent increase in overall lysine malonylation, indicating this reagent is able to cross cell membranes and induce this physiological PTM in cells (Figure 5b, Figure S4c). One caveat to these results was the high concentration of malonyl-NAC required to induce acylation, suggesting the efficiency of this reagent may be limited by uptake, hydrolysis, or inefficient ester cleavage. While overall labeling patterns were similar, a subset of proteins demonstrated greater malonylation when treated with malonyl-NAC as opposed to orlistat (Figure 5b). These distinctions may reflect differential molecular recognition of malonyl-CoA and malonyl-NAC. Similar to the orlistat results, we found that increases in cellular malonylation caused by malonyl-NAC led to a reduction in endogenous GAPDH activity (Figure 5c). Overexpression and purification of FLAG-GAPDH followed by anti-malonyllysine immunoblot confirmed the ability of malonyl-NAC to increase GAPDH malonylation in cells (Figure S4d). Moreover, in agreement with the observed biochemical inhibition of PKM by malonyl-CoA, we found malonyl-NAC inhibited pyruvate kinase activity in treated cells (Figure 5d). In contrast to the impact on activity, orlistat and malonyl-NAC did not affect overall levels of GAPDH and PKM2 expression (Figure S4e). Finally, we performed metabolomic analyses to characterize the overall effects of reactive thioesters on glucose metabolism (Figure 6). We found that treatment of lung cancer cells with malonyl-NAC results in an accumulation of the upstream glycolytic intermediates glucose-6-phosphate and fructose-1,6,-bisphosphate (Figure 6b). Similarly, malonyl-NAC treatment resulted in a modest, but significant, reduction in lactate and acetyl-CoA metabolism (Figure 6c–d, Figure S5). Comparison of malonyl and acetyl thioesters revealed a greater disruption of glucose metabolism by the former, consistent with their increased reactivity (Figure 5c). In addition, malonyl-NAC was found to limit metabolism and proliferation of a highly glycolytic kidney cancer cell line harboring a TCA cycle mutation (Figure S6). These data are consistent with a model in which malonyl-CoA antagonizes cellular glycolytic enzyme function via reactivity-driven modifications, and suggest that synthetic thioesters may be useful cellular probes of acylation-dependent phenotypes.
Figure 6.
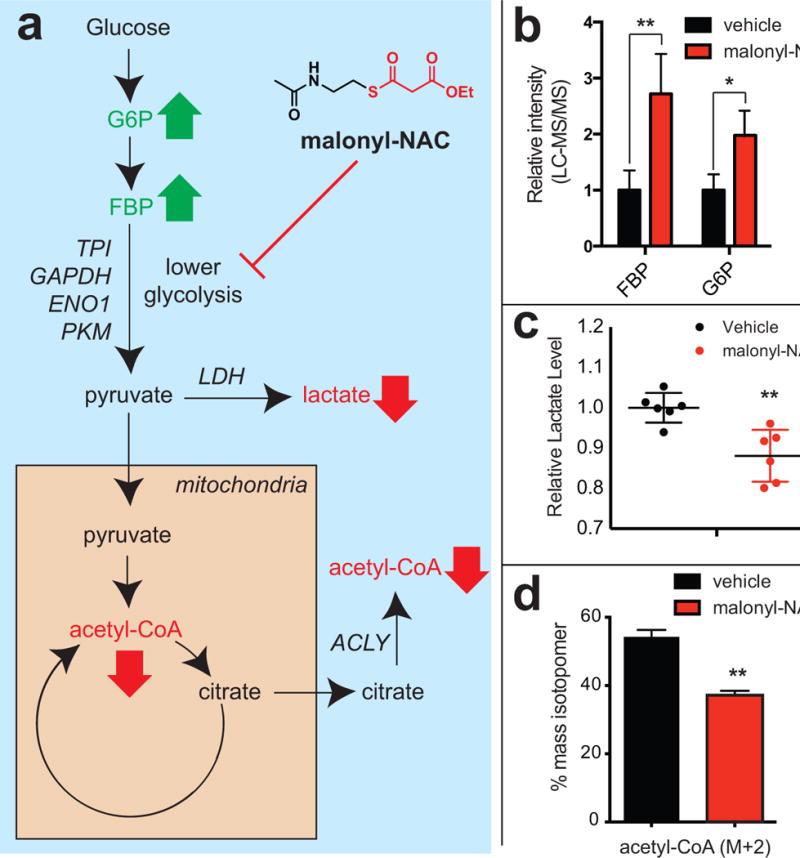
Exploring the effects of cytosolic thioester reactivity on cellular glucose metabolism. (a) Schematic of glycolysis. Bold green and red arrows reflect expected changes in abundance of metabolites that lie upstream and downstream of glycolytic enzymes targeted by non-enzymatic acylation. Gene names in italic refer to high confidence targets of non-enzymatic acylation where malonyl-NAC may intervene. (b) Effects of malonyl-NAC (1 mM, 24 h) on glucose-6-phosphate (G6P) and fructose-1,6-bisphosphate (FBP) levels in A549 cells. (c) Effects of malonyl-NAC (1 mM, 24 h) on lactate levels in A549 cells. (d) Effects of malonyl-NAC (1 mM, 24 h) on glucose-derived acetyl-CoA (M+2) in A549 cells. Metabolomic measurements represent the average of ≥3 replicates, with significance analyzed by unpaired Student’s t test (ns = not significant, * = P < 0.05, ** = P < 0.01)
Discussion
Understanding the functional contributions of non-enzymatic processes to lysine acylation remains a major challenge. Here we have reported an experimental approach that harnesses thioester reactivity to directly investigate non-enzymatic acylation. Our work builds on previous efforts by providing a synthetic strategy to separate thioester reactivity from enzymatic utilization, allowing non-enzymatic acylation to be selectively studied the first time in cells and complex proteomes. Applying this strategy to immortalized cancer cell lines led to the discovery that lower glycolysis in general, and GAPDH specifically, can be spontaneously modified and inhibited by hyperreactive thioester metabolites such as cytosolic malonyl-CoA. It is important to note that 1 is not a specific probe of malonylation or succinylation, but rather exhibits overlap with the targets of the reactive acyl-CoAs from which they derive. Small molecule manipulation of lysine malonylation disrupts GAPDH and pyruvate kinase activity, reduces cellular glucose metabolism, and inhibits the growth of cancer cells highly dependent on glycolytic Warburg metabolism.
While these findings demonstrate the ability of reactivity-based methods to provide new insights into non-enzymatic acylation, we must also note some limitations of our approach. First, in our functional studies we have narrowly focused on the role non-enzymatic acylation may play in regulation of glycolysis. However, the effects of non-enzymatic acylation on cellular function are fundamentally pleiotropic, and our studies indicate reactive thioesters target many other important cellular processes including fatty acid biosynthesis (FASN, (ACLY) and protein translation (EEF1G), folding (HSPAA901) and homeostasis (UBAE1). Consistent with the idea that non-enzymatic acylation may manifest functional effects beyond glycolysis, analysis of the NCI-60 cell line panel found that mutations to common regulators of glycolysis such as VHL or LKB1 are not by themselves predictive of cell-killing by the non-enzymatic acylation agent malonyl-NAC, and biochemical assays suggest malonyl-CoA can disrupt UBAE1 function vitro (Figure S6e–f). Second, the structural truncation of our probe may limit the detection of proteins whose non-enzymatic acylation is driven by interaction with the adenine nucleotide moiety of acyl-CoA metabolites. This class of non-enzymatic acylation targets may be more suitably explored using competitive reactivity-based profiling methods targeting lysine (Louie et al., 2016), or via the development of biorthogonal acyl-CoAs that are not substrates for KAT enzymes (Khim et al., 2010). A third limitation of the current method is its bias towards highly abundant proteins, which may obfuscate the detection of comparatively rare signaling proteins that are subject to regulation by non-enzymatic acylation. In the future this may be addressed by integrating thioester probes with affinity enrichment strategies that use isotopically-tagged cleavable linkers (Qian et al., 2013; Weerapana et al., 2007), which would enable detection and quantification of non-enzymatic acylation at the peptide level, or integration with recombinant protein array technologies, which can detect the ex situ thioester reactivity of many proteins in parallel independent of protein abundance (Olia et al., 2015). The development of such methods have the potential to further broaden and enrich our understanding of non-enzymatic acylation in biology.
It is also worth emphasizing some biological implications of the present study. Most notably, our results are the first to experimentally define malonyl-CoA as a hyperreactive cytosolic thioester metabolite. This substantiates earlier computational studies of malonyl thioesters, which had suggested their significantly increased reactivity (Kang and Han, 1997). While our results do not preclude the possibility of enzyme-catalyzed malonylation, the heightened reactivity of malonyl-CoA provides a mechanistic rationale for the recent observation in the literature that lysine malonylation predominantly affects nuclear and cytosolic proteins, and in particular dynamically modifies many glycolytic enzymes (Nishida et al., 2015). Importantly, proteome-wide analyses of lysine malonylation (Colak et al., 2015) provide residue-specific information that usefully complements our reactivity-based method, which is biased toward the analysis of abundant proteins. Integrating these two approaches with mutational analyses will be an important step to identifying specific lysines whose reactivity functionally contributes to the regulation of GAPDH and other cytosolic enzyme activities. Although the cellular effects of malonylation on glycolytic enzyme activity were relatively modest, several groups have found that inhibition of FASN can reduce glucose metabolism (Huang et al., 2014b; Sankaranarayanapillai et al., 2013; Zaytseva et al., 2015; Zhou et al., 2016). This suggests that in certain contexts, reactivity-driven malonylation may collaborate with other mechanisms to decrease flux through lower glycolysis in response to stimuli that inhibit FASN. Interestingly, the acetyl-CoA precursor citrate (PFK1 inhibitor) intersects glycolysis at a distinct point relative to acetyl-CoA (PKM inhibitor) and malonyl-CoA (GAPDH/PKM inhibitor). Understanding the temporal interplay of these metabolic inhibitors, as well as acyl-CoA sensitive KAT activities (Lee et al., 2014), will be necessary to elucidate the molecular logic and phenotypic significance of links between acyl-CoA metabolism and glycolysis. Finally, genomic analyses indicate that SIRT5 is upregulated in a wide-range of cancers (Gao et al., 2013). Of note, SIRT5 removes two hyperreactive thioester-derived PTMs, lysine succinylation and malonylation. Our studies suggest that in addition to its role in mitochondrial function as a desuccinylase (Du et al., 2011; Lee et al., 2014; Rardin et al., 2013), high SIRT5 activity may also facilitate cancer growth via demalonylation of enzymes essential to rapid proliferation, including those involved in glycolytic Warburg metabolism. Increases in malonyl-CoA levels have been previously invoked to explain the therapeutic effects of FASN inhibitors (Pizer et al., 2000; Zhou et al., 2007). The studies of intrinsic thioester reactivity reported here represent a critical first step in defining non-enzymatic acylation as a biomarker, and potential therapeutic target, in human malignancy.
Experimental Procedures
Compounds, enzymes, cell lines, materials
Recombinant p300 (1195–1662) and Gcn5 (497–662) were obtained from Enzo. Qubit Protein Assay kit was purchased from Life Technologies (Q33212). Streptavidin agarose resin was purchased from ThermoFisher Scientific (20353). SDS-PAGE was performed using Bis-Tris NuPAGE gels (4–12%, Invitrogen #NP0322), and MES running buffer (Life technologies #NP0002) in Xcell SureLock MiniCells (Invitrogen) according to the manufacturer’s instructions. SDS-PAGE fluorescence was visualized using an ImageQuant Las4010 Digitial Imaging System (GE Healthcare). Total protein content on SDS-PAGE gels was visualized by Blue-silver Coomassie stain, made according to the published procedure (Candiano et al., 2004). GAPDH (5174S), PKM2 (3198S), histone H2B (12364P), HSP90 (4877P), Cox IV (4850P), acetyllysine (9441S) and malonyllysine (14942S) antibodies were purchased from Cell Signaling Technologies. Recombinant PKM2 (6372-100) was obtained from BioVision. GAPDH from human erythrocytes (G6019) and fumarate hydratase (fumarase) from porcine heart (F1757) were obtained from Sigma. Acetyl-CoA, propionyl-CoA, and butyryl-CoA were synthesized according to the literature procedure (Padmakumar et al., 1997) and purified by HPLC. Malonyl-CoA (63410), succinyl-CoA (S1129), glutaryl-CoA (G9510), methylmalonyl-CoA (M1762), DL-β-hydroxybutyryl-CoA (BHB-CoA, H0261) palmitoyl-CoA (P9716), and iodoacetamide-biotin (B2059) were purchased from Sigma. All acyl-CoAs were analyzed for purity by LC-MS prior to utilization, and re-purified via HPLC if necessary. Succinyl-CoA stock solutions (1–10 mM in PBS) were freshly prepared for all experiments to limit hydrolysis. Orlistat (O461467-9) was purchased from Cayman chemical. Anti-FLAG pulldown was performed using immunoprecipitation kit (KBA-319-383) from Rockland Immunochemicals, Inc. U-13C6-glucose was obtained from Cambridge Isotope Laboratories (CLM-1396). A549 and Raji cells were obtained from the NCI Tumor Cell Repository. HepG2 and H1299 cells were obtained from ATCC (Manassas VA). U87 cells were a gift from Ji Ming Wang (NCI, Cancer and Inflammation Program). UOK262, UOK262EV, and UOK262WT cells were obtained from Linehan lab (Yang et al., 2010a). NAD/NADH assay kit was purchased from Promega (G9071).
Cell culture and isolation of whole-cell lysates
All cell lines were cultured at 37 °C under 5% CO 2 atmosphere in a growth medium of RPMI supplemented with 10% FBS and 2 mM glutamine, with the exception of UOK262 cell lines, which were cultured in DMEM supplemented with 10% FBS, 2 mM glutamine, 1 mM pyruvate and 0.3 mg/mL of G418. Cells were dosed with thioesters (in situ treatment) by adding stock solutions (1000× stock in DMSO) directly to growth medium at the specified concentration. Unfractionated proteomes were harvested from cell lines (80–90% confluency) by washing adherent cells 3× with ice cold PBS, scraping cells into a Falcon tube, and centrifuging (1400 rcf × 3 min, 4 °C) to form a cell pellet. After removal of PBS supernatant, cell pellets were either stored at −80 °C or immediately lysed by sonication. For lysis, cells were first resuspended in 1–2 mL ice cold PBS (10–20 × 106 cells/mL) containing protease inhibitor cocktail (1X, EDTA-free, Cell Signaling Technology # 5871S) and PMSF (1 mM, Sigma # 78830). These samples were then lysed by sonication using a 100 W QSonica XL2000 sonicator (3 × 1s pulse, amplitude 1, 60s resting on ice between pulses). Lysates were pelleted by centrifugation (14,000 rcf × 30 min, 4 °C) and quantified on a Qubit 2.0 Fluorometer using a Qubit Protein Assay Kit. Quantified proteomes were diluted to 2 mg/mL and stored in 1 mg aliquots at −80 °C for chemoproteomic or enzyme activity analyses.
Chemoproteomic analyses of thioester reactivity
In situ labeled samples were prepared by treating A549 and Raji cells with thioester reporter 1 as described above. Following treatment at the specified concentration, cells were incubated with 1 for 15 hours unless indicated otherwise. Ex situ labeled samples were prepared by treating isolated proteomes with thioester 1 for 15 hours unless indicated otherwise. For both ex situ and in situ samples, 20 μg of labeled proteome was used for SDS-PAGE target analysis, and 2000 μg of labeled proteome was used for LC-MS/MS target analysis. Proteins labeled by 1 were visualized by SDS-PAGE via Cu(I)-catalyzed [3 + 2] cycloaddition with a fluorescent azide as previously reported (Montgomery et al., 2015a; Speers and Cravatt, 2004). Gels were fixed and destained in a solution of 50% MeOH/40% H2O/10% AcOH overnight to remove excess probe fluorescence, rehydrated with water, and visualized using an ImageQuant Las4010 (GE Healthcare) with green LED excitation (λmax 520–550 nm) and a 575DF20 filter. For competition experiments, proteomes were pretreated with acetyl-CoA (0–800 μM) for 1 hour prior to incubation with thioester 1. For proteomic studies, labeled proteomes were enriched via Cu(I)-catalyzed [3 + 2] cycloaddition with a biotin azide, desalted, and analyzed by LC-MS/MS as previously reported (Montgomery et al., 2015a). Extended protocols for fluorescence analyses and enrichments are provided in the Supplemental Information.
GAPDH activity assay
GAPDH activity was assayed with GAPDH assay kit (BioVision # K680) following the manufacturer’s instruction. Briefly, GAPDH from human erythrocytes (0.03 U per well) was incubated with the indicated concentrations of each acyl-CoA in the manufacturer-provided GAPDH assay buffer (total reaction volume 20 μL). Acyl-CoAs were incubated with GAPDH for 30 min at 37°C, whereupon the GAPDH reaction was initiated by addition of 20 μL of a mixture of GAPDH developer and substrate in the assay buffer. GAPDH turnover was quantified by reading absorbance at 450 nm in kinetic mode for 15 min at 37°C. All conditions were analyzed in quadruplicate in a 384-well plate. The initial rate from treated samples was normalized as a percentage of untreated control, and corrected for background absorbance. For the analysis of covalent inhibition by desalting, GAPDH (5 μg) was incubated with malonyl-CoA (200 μM) in PBS at 37 °C for 30 min. Unreacted malonyl-CoA was then removed by buffer exchange (3×) through Amicon Ultra centrifugal filters (EMD Millipore, molecular weight cut-off 3KDa). GAPDH activity was determined from the recovered samples using 0.03 U of enzyme per well as described above. Hydroxylamine-mediated rescue experiments were performed analogously to this desalting experiment, the only change being that hydroxylamine (50 mM) at 37 °C fo r 10 min was added immediately following malonyl-CoA incubation, just prior to desalting.
Western blot analyses of acyl-CoA reactivity
Western blot analyses of acyl-CoA reactivity with the model protein bovine serum albumin (BSA) were performed as described previously (Wagner and Payne, 2013) Non-acetylated BSA (Sigma #B6917) was dissolved at a final concentration of 2 mg/mL in pH 7.0 or pH 8.0 buffer (50 mM Tris-HCl,150 mM NaCl). For each non-enzymatic reaction, 40 μg of BSA was incubated with 200 μM acetyl-CoA, malonyl-CoA, or succinyl-CoA at 37°C for 6 h (total reaction volume 20 μL). Following incubation, samples were quenched with 2× loading buffer at room temperature and analyzed by SDS-PAGE. Immunoblotting was performed according to standard procedures using anti-acetyllysine according (CST 9441S) or anti-malonyllysine (CST 14942S) antibodies.
Cellular analysis of lysine malonylation
A549 cells were plated in 6-well dishes (4 ×105 cells/well in 3 ml RPMI media/well), and allowed to adhere and grow for 24 h. At this point, cells were treated with malonyl-NAC (1 mM final concentration, 3 μL of 1M stock in DMSO was added to the extracellular media) or orlistat (25 μM final concentration, 3 μL of 25 mM stock in DMSO was added to the extracellular media) or DMSO (3 μL, vehicle control) and incubated for 24 h at cultured at 37 °C under 5% CO2 atmosphere. The experiment was performed in quadruplicate. After the treatment, cells were harvested, soluble proteome isolated and quantified as described above. For the western blot analysis of lysine malonylation and expression levels of GAPDH and PKM2, 20 μg of lysates were loaded per lane of the gel. GAPDH and PKM2 activity was determined as described above using 5 μg and 1 μg of lysate per replicate, respectively.
Lactate assay
A549 cells were plated in 6-well dishes (1 ×106 cells/well in 3 ml RPMI media/well), and allowed to adhere and grow for 24 h. At this point, media was removed and cells were treated 3 mL of serum-free media containing with malonyl-NAC (1 mM final concentration, 3 μL of 1M stock in DMSO) or DMSO (3 μL, vehicle control) and incubated for 24 h at 37 °C under 5% CO2 atmosphere. The experiment was performed in biological triplicate. Extracellular lactate levels were measured in technical duplicates using a fluorimetric L-lactate assay kit (Cayman chemical # 700510) according to the manufacturer’s protocol.
Supplementary Material
SIGNIFICANCE.
Recent studies have identified a myriad of protein posttranslational modifications derived from metabolic acyl-CoAs. However, the mechanisms by which “non-acetyl” lysine acylations are established are largely enigmatic, leading to speculation these modifications may be driven by intrinsic thioester reactivity. Here we have developed the first chemical proteomic strategy capable of identifying proteins susceptible to acylation by reactive thioester metabolites. These studies led to the identification of several abundant proteins involved in glycolysis, fatty acid biosynthesis, and protein translation as candidate targets of non-enzymatic acylation. Further interrogation of specific enzyme/acyl-CoA interactions led to the unexpected finding that malonyl-CoA is a hyperreactive thioester that can functionally inhibit glycolytic enzymes. Finally, we demonstrate that non-enzymatic malonylation can be manipulated directly using reactive thioesters or agents targeting fatty acid metabolism. Our studies indicate a substantial proportion of cellular malonylation may be driven by chemical reactivity, rather than enzymatic catalysis, and specify new physiological processes that may be sensitive to deacylase inhibition. In addition, these studies illustrate the utility of reactivity-based profiling methods to rapidly characterize non-enzymatic acylation in living systems, and suggest stimulation of hypermalonylation may warrant exploration as a therapeutic strategy to disrupt pathways required for cancer cell proliferation.
Highlights.
-
-
Non-enzymatic reactivity is an important input into cellular protein acylation.
-
-
Minimal thioesters can mechanistically resolve enzymatic and non-enzymatic acylation.
-
-
Thioester probes can identify candidate proteomic targets of non-enzymatic acylation.
-
-
Malonyl-CoA is a hyperreactive thioester that acylates and inhibits glycolytic enzymes.
Acknowledgments
The authors thank Dr. Carissa Grose (Protein Expression Laboratory) for assisting with cloning and preparation of plasmid DNA, Dr. David Evans and Lori Bowles (Developmental Therapeutics Program) for NCI-60 cell lines, and Dr. Daniel McVicar (Cancer Inflammation Program) and Dr. Martin Schnermann (Chemical Biology Laboratory) for helpful discussions. This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (ZIA BC011488-02).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions:
R.A.K. and J.L.M. designed experiments. R.A.K. performed all chemical proteomic, biochemical assays and cell-based experiments. R.A.K. and T.T.Z synthesized compounds. J.H.S. performed enrichment/immunoblot experiments for PKM2 and GAPDH. J.M.G. and A.M.R. assisted with chemical proteomic and immunoblot analyses. D.C.M. assisted with proteomic and biochemical experiments. A.J.W., I.A.B., C.M., and N.W.S. performed metabolomic analyses. M.Z. T.A., K.C, and S.D. performed LC-MS/MS studies and data analysis. C.S., B.K.G., and W.M.L. performed experiments in UOK262 cells. Y.C.T. and A.M.W. performed ubiquitinylation analyses. R.A.K. and J.L.M. analyzed data and wrote the manuscript.
Supplementary Information:
References
- Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334:1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachovchin DA, Koblan LW, Wu W, Liu Y, Li Y, Zhao P, Woznica I, Shu Y, Lai JH, Poplawski SE, et al. A high-throughput, multiplexed assay for superfamily-wide profiling of enzyme activity. Nature chemical biology. 2014;10:656–663. doi: 10.1038/nchembio.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin DI, Li DS, Lowe W, Heuer T, Kemble G, Nomura DK. Diacylglycerol Metabolism and Signaling Is a Driving Force Underlying FASN Inhibitor Sensitivity in Cancer Cells. ACS chemical biology. 2015;10:1616–1623. doi: 10.1021/acschembio.5b00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candiano G, Bruschi M, Musante L, Santucci L, Ghiggeri GM, Carnemolla B, Orecchia P, Zardi L, Righetti PG. Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis. 2004;25:1327–1333. doi: 10.1002/elps.200305844. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, Falck JR, Peng J, Gu W, Zhao Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Molecular & cellular proteomics : MCP. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak G, Pougovkina O, Dai L, Tan M, Te Brinke H, Huang H, Cheng Z, Park J, Wan X, Liu X, et al. Proteomic and Biochemical Studies of Lysine Malonylation Suggest Its Malonic Aciduria-associated Regulatory Role in Mitochondrial Function and Fatty Acid Oxidation. Molecular & cellular proteomics : MCP. 2015;14:3056–3071. doi: 10.1074/mcp.M115.048850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guynn RW, Gelberg HJ, Veech RL. Equilibrium constants of the malate dehydrogenase, citrate synthase, citrate lyase, and acetyl coenzyme A hydrolysis reactions under physiological conditions. The Journal of biological chemistry. 1973;248:6957–6965. [PubMed] [Google Scholar]
- Henry RA, Kuo YM, Bhattacharjee V, Yen TJ, Andrews AJ. Changing the selectivity of p300 by acetyl-CoA modulation of histone acetylation. ACS chemical biology. 2015;10:146–156. doi: 10.1021/cb500726b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Sabari BR, Garcia BA, Allis CD, Zhao Y. SnapShot: histone modifications. Cell. 2014a;159:458–458 e451. doi: 10.1016/j.cell.2014.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nature immunology. 2014b;15:846–855. doi: 10.1038/ni.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessani N, Niessen S, Wei BQ, Nicolau M, Humphrey M, Ji Y, Han W, Noh DY, Yates JR, Jeffrey SS, 3rd, Cravatt BF. A streamlined platform for high-content functional proteomics of primary human specimens. Nature methods. 2005;2:691–697. doi: 10.1038/nmeth778. [DOI] [PubMed] [Google Scholar]
- Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, et al. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature. 2013;496:110–113. doi: 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YK, Han SJ. Ab Initio Molecular Orbital Calculations on N-β-Mercaptoethylacetamide and Its Derivatives as Model Compounds of Coenzyme A (CoA), Acetyl-CoA, and Malonyl-CoA. Journal of Physical Chemistry B. 1997;101:7001–7006. [Google Scholar]
- Khim L, Han J, Willetts L, Brady K, Gillece P, Rached O, Thomas NR, Stylianou E. Complementary PCAF-coenzyme A mutagenesis: chemoenzymatic synthesis of a novel enlarged coenzyme A analogue and evaluation of its biological activity. Chembiochem : a European journal of chemical biology. 2010;11:2100–2103. doi: 10.1002/cbic.201000286. [DOI] [PubMed] [Google Scholar]
- Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell metabolism. 2014;20:306–319. doi: 10.1016/j.cmet.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Su X, He B. Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS chemical biology. 2012;7:947–960. doi: 10.1021/cb3001793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie SM, Grossman EA, Crawford LA, Ding L, Camarda R, Huffman TR, Miyamoto DK, Goga A, Weerapana E, Nomura DK. GSTP1 Is a Driver of Triple-Negative Breast Cancer Cell Metabolism and Pathogenicity. Cell Chem Biol. 2016;23:567–578. doi: 10.1016/j.chembiol.2016.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv L, Xu YP, Zhao D, Li FL, Wang W, Sasaki N, Jiang Y, Zhou X, Li TT, Guan KL, et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Molecular cell. 2013;52:340–352. doi: 10.1016/j.molcel.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- Moellering RE, Cravatt BF. Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science. 2013;341:549–553. doi: 10.1126/science.1238327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery DC, Sorum AW, Guasch L, Nicklaus MC, Meier JL. Metabolic Regulation of Histone Acetyltransferases by Endogenous Acyl-CoA Cofactors. Chemistry & biology. 2015a;22:1030–1039. doi: 10.1016/j.chembiol.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery DC, Sorum AW, Meier JL. Defining the orphan functions of lysine acetyltransferases. ACS chemical biology. 2015b;10:85–94. doi: 10.1021/cb500853p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH. H2S signals through protein S-sulfhydration. Science signaling. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida Y, Rardin MJ, Carrico C, He W, Sahu AK, Gut P, Najjar R, Fitch M, Hellerstein M, Gibson BW, Verdin E. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Molecular cell. 2015;59:321–332. doi: 10.1016/j.molcel.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140:49–61. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olia AS, Barker K, McCullough CE, Tang HY, Speicher DW, Qiu J, LaBaer J, Marmorstein R. Nonenzymatic Protein Acetylation Detected by NAPPA Protein Arrays. ACS chemical biology. 2015;10:2034–2047. doi: 10.1021/acschembio.5b00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmakumar R, Padmakumar R, Banerjee R. Large-scale synthesis of coenzyme A esters. Methods in enzymology. 1997;279:220–224. doi: 10.1016/s0076-6879(97)79026-8. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–195. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemble CWt, Johnson LC, Kridel SJ, Lowther WT. Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by Orlistat. Nature structural & molecular biology. 2007;14:704–709. doi: 10.1038/nsmb1265. [DOI] [PubMed] [Google Scholar]
- Peng C, Lu Z, Xie Z, Cheng Z, Chen Y, Tan M, Luo H, Zhang Y, He W, Yang K, et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Molecular & cellular proteomics : MCP. 2011;10 doi: 10.1074/mcp.M111.012658. M111 012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizer ES, Thupari J, Han WF, Pinn ML, Chrest FJ, Frehywot GL, Townsend CA, Kuhajda FP. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer research. 2000;60:213–218. [PubMed] [Google Scholar]
- Qian Y, Martell J, Pace NJ, Ballard TE, Johnson DS, Weerapana E. An isotopically tagged azobenzene-based cleavable linker for quantitative proteomics. Chembiochem : a European journal of chemical biology. 2013;14:1410–1414. doi: 10.1002/cbic.201300396. [DOI] [PubMed] [Google Scholar]
- Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, Guo A, Gut P, Sahu AK, Li B, et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell metabolism. 2013;18:920–933. doi: 10.1016/j.cmet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Tang Z, Huang H, Yong-Gonzalez V, Molina H, Kong HE, Dai L, Shimada M, Cross JR, Zhao Y, et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Molecular cell. 2015;58:203–215. doi: 10.1016/j.molcel.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanapillai M, Zhang N, Baggerly KA, Gelovani JG. Metabolic shifts induced by fatty acid synthase inhibitor orlistat in non-small cell lung carcinoma cells provide novel pharmacodynamic biomarkers for positron emission tomography and magnetic resonance spectroscopy. Molecular imaging and biology : MIB : the official publication of the Academy of Molecular Imaging. 2013;15:136–147. doi: 10.1007/s11307-012-0587-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz C, Weinert BT, Wagner SA, Beli P, Miyake Y, Qi J, Jensen LJ, Streicher W, McCarthy AR, Westwood NJ, et al. Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nature biotechnology. 2015;33:415–423. doi: 10.1038/nbt.3130. [DOI] [PubMed] [Google Scholar]
- Simic Z, Weiwad M, Schierhorn A, Steegborn C, Schutkowski M. The ε-Amino Group of Protein Lysine Residues Is Highly Susceptible to Nonenzymatic Acylation by Several Physiological Acyl-CoA Thioesters. Chembiochem. 2015;16:2337–2347. doi: 10.1002/cbic.201500364. [DOI] [PubMed] [Google Scholar]
- Sorum AW, Shrimp JH, Roberts AM, Montgomery DC, Tiwari NK, Lal-Nag M, Simeonov A, Jadhav A, Meier JL. Microfluidic Mobility Shift Profiling of Lysine Acetyltransferases Enables Screening and Mechanistic Analysis of Cellular Acetylation Inhibitors. ACS chemical biology. 2016;11:734–741. doi: 10.1021/acschembio.5b00709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speers AE, Adam GC, Cravatt BF. Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3 + 2] cycloaddition. Journal of the American Chemical Society. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- Speers AE, Cravatt BF. Profiling enzyme activities in vivo using click chemistry methods. Chemistry & biology. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Tokutake Y, Iio W, Onizawa N, Ogata Y, Kohari D, Toyoda A, Chohnan S. Effect of diet composition on coenzyme A and its thioester pools in various rat tissues. Biochem Biophys Res Commun. 2012;423:781–784. doi: 10.1016/j.bbrc.2012.06.037. [DOI] [PubMed] [Google Scholar]
- Tokutake Y, Onizawa N, Katoh H, Toyoda A, Chohnan S. Coenzyme A and its thioester pools in fasted and fed rat tissues. Biochem Biophys Res Commun. 2010;402:158–162. doi: 10.1016/j.bbrc.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Trievel RC, Li FY, Marmorstein R. Application of a fluorescent histone acetyltransferase assay to probe the substrate specificity of the human p300/CBP-associated factor. Analytical biochemistry. 2000;287:319–328. doi: 10.1006/abio.2000.4855. [DOI] [PubMed] [Google Scholar]
- Trievel RC, Rojas JR, Sterner DE, Venkataramani RN, Wang L, Zhou J, Allis CD, Berger SL, Marmorstein R. Crystal structure and mechanism of histone acetylation of the yeast GCN5 transcriptional coactivator. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:8931–8936. doi: 10.1073/pnas.96.16.8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nature reviews Molecular cell biology. 2015;16:258–264. doi: 10.1038/nrm3931. [DOI] [PubMed] [Google Scholar]
- Wagner GR, Hirschey MD. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol Cell. 2014;54:5–16. doi: 10.1016/j.molcel.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GR, Payne RM. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem. 2013;288:29036–29045. doi: 10.1074/jbc.M113.486753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Tang Y, Cole PA, Marmorstein R. Structure and chemistry of the p300/CBP and Rtt109 histone acetyltransferases: implications for histone acetyltransferase evolution and function. Current opinion in structural biology. 2008;18:741–747. doi: 10.1016/j.sbi.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E, Speers AE, Cravatt BF. Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP)–a general method for mapping sites of probe modification in proteomes. Nat Protoc. 2007;2:1414–1425. doi: 10.1038/nprot.2007.194. [DOI] [PubMed] [Google Scholar]
- Weinert BT, Iesmantavicius V, Moustafa T, Scholz C, Wagner SA, Magnes C, Zechner R, Choudhary C. Acetylation dynamics and stoichiometry in Saccharomyces cerevisiae. Molecular systems biology. 2014;10:716. doi: 10.1002/msb.134766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert BT, Scholz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, Choudhary C. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell reports. 2013;4:842–851. doi: 10.1016/j.celrep.2013.07.024. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Valera VA, Padilla-Nash HM, Sourbier C, Vocke CD, Vira MA, Abu-Asab MS, Bratslavsky G, Tsokos M, Merino MJ, et al. UOK 262 cell line, fumarate hydratase deficient (FH−/FH−) hereditary leiomyomatosis renal cell carcinoma: in vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer genetics and cytogenetics. 2010a;196:45–55. doi: 10.1016/j.cancergencyto.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YY, Ascano JM, Hang HC. Bioorthogonal chemical reporters for monitoring protein acetylation. Journal of the American Chemical Society. 2010b;132:3640–3641. doi: 10.1021/ja908871t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaytseva YY, Harris JW, Mitov MI, Kim JT, Butterfield DA, Lee EY, Weiss HL, Gao T, Evers BM. Increased expression of fatty acid synthase provides a survival advantage to colorectal cancer cells via upregulation of cellular respiration. Oncotarget. 2015;6:18891–18904. doi: 10.18632/oncotarget.3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Jiang S, Fu Q, Smith K, Tu K, Li H, Zhao Y. FASN, ErbB2-mediated glycolysis is required for breast cancer cell migration. Oncology reports. 2016;35:2715–2722. doi: 10.3892/or.2016.4627. [DOI] [PubMed] [Google Scholar]
- Zhou W, Han WF, Landree LE, Thupari JN, Pinn ML, Bililign T, Kim EK, Vadlamudi A, Medghalchi SM, El Meskini R, et al. Fatty acid synthase inhibition activates AMP-activated protein kinase in SKOV3 human ovarian cancer cells. Cancer research. 2007;67:2964–2971. doi: 10.1158/0008-5472.CAN-06-3439. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.