

Abstract
Peripheral neurons regenerate their axons after injury. Transcriptional regulation by microRNAs (miRNAs) is one possible mechanism controlling regeneration. We profiled miRNA expression in mouse dorsal root ganglion (DRG) neurons after a sciatic nerve crush, and identified 49 differentially expressed miRNAs. We evaluated the functional role of each miRNA using a phenotypic analysis approach. To predict the targets of the miRNAs we employed RNA-Sequencing and examined transcription at the isoform level. We identify thousands of differentially expressed isoforms and bioinformatically associate the miRNAs that modulate neurite growth with their putative target isoforms to outline a network of regulatory events underlying peripheral nerve regeneration. MiR-298, let-7a and let-7f enhance neurite growth and target the majority of isoforms in the differentially expressed network.
Keywords: dorsal root ganglion, high content analysis, regeneration, miRNA, RNA-Seq, PNS, axon growth, mRNA isoforms, C57BL/6
Introduction
Neurons of the central nervous system (CNS) generally fail to regenerate their axons after an injury [1]. In contrast, neurons of the peripheral nervous system (PNS) display a robust re-growth capacity after axotomy [1]. Peripheral sensory neurons located in dorsal root ganglia (DRG) extend bifurcating axons with one branch extending via dorsal roots into the spinal cord and the other branch extending into the periphery. Injury of the peripheral branch induces activation of transcriptional programs associated with a regenerative response [2]. It is of fundamental importance to understand the transcriptional changes and their regulatory events associated with PNS regeneration, because one approach to promote CNS regeneration might be to activate similar transcriptional programs [3–5].
Our laboratory has studied PNS gene expression with different approaches to identify transcriptional programs that might promote axonal regeneration [4,6,7]. Despite the identification of factors like STAT3, SRF and KLFs, which showed functional roles in regeneration [5,8–10], transcriptional strategies to induce regeneration in CNS neurons have had limited success, both in the number of responding cells and in the extent of growth past an injury site. Therefore identifying the transcriptional mechanisms associated with nerve regeneration continues to be important.
MicroRNAs (miRNAs) are short RNA molecules, 18-22 nucleotides in length [11–13], that inhibit mRNA translation [14,15] by mediating the interaction of the RNA-induced Silencing Complex (RISC) with a target sequence on the 3′ untranslated region (UTR) of mRNAs [16]. MiRNA target recognition happens through an imperfect match [17,18], allowing a single miRNA to target hundreds of transcripts simultaneously [19–22]. At the same time the 3′ UTR of an mRNA isoform can contain targets for several miRNAs, which might synergistically affect the levels of that mRNA. Several studies used genomic approaches to identify miRNAs whose expression change after peripheral nerve injury [23–33]. However only a handful have tried to functionally characterize those miRNAs with an active role in the process of peripheral neuron axon regeneration [24,31–33]. In most cases functional studies in DRG neurons attributed miRNA growth-regulating activities to downregulation of a single gene; e.g., SPRY2 [31] or PTEN [33]. Moreover, targeting has been inferred using only the conventional annotated 3′UTRs of mRNAs.
Next Generation Sequencing applied to the transcriptome (RNA-Seq) allows a deeper level of observation of gene expression as it identifies differential expression of known and unknown isoforms [34]. Traditionally gene expression and functional studies have focused on conventional isoforms without considering cell type-specific isoforms. When we applied RNA-Seq to DRG neurons in culture and compared their isoform expression patterns to cerebellar granule neurons (CGNs) we found enormous transcriptional diversity [4]. This diversity happens at the coding sequence level but more often at the level of regulatory sequences, indicating the use of cell specific promoters, transcription start sites or 3′UTRs. This observation suggests that functional gene targets of miRNAs can be more reliably predicted by analyzing gene expression at the isoform level. We have used RNA-Seq and real time quantitative PCR to identify expression changes in mRNA isoforms and miRNAs in adult mouse DRG neurons 7d after a sciatic nerve crush injury. This information was used to screen 47 differentially expressed miRNAs in PNS neurons to identify those that regulate neurite growth. These combined data sets were then used to design a putative network of transcriptional events underlying peripheral nerve regeneration. Overall, our functional genomic approach reveals the global gene and miRNA expression patterns in vivo after peripheral nerve crush.
Materials and Methods
Additional experimental procedures are described in SI
Sciatic nerve crush and animal handling
All procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of Miami. Male C57BL/6 mice between 6 and 8 weeks of age were used. Twenty-two animals underwent surgery for this study. In 11 animals the sciatic nerve was exposed in the left thigh and was crushed with Dumont forceps for 10 sec before closing the incision. Eleven control animals underwent a sham surgery where the nerve was exposed, and the incision closed after 10 sec. Metadata for the experiment has been documented in Attachment S1 in compliance with the MIASCI reporting standard [35] and deposited in the RegenBase Knowledgebase [36] as recommended by the FAIR guiding principles [37].
Laser Capture Microdissection of DRG neurons, RNA isolation and preparation for miRNA expression analysis
Animals were euthanized with CO2 inhalation 7 days after surgery. Lumbar level 3, 4 and 5 DRGs, comprising the sciatic nerve [38], were dissected, embedded in freezing media and flash-frozen on a layer of isopentane floating on liquid nitrogen. 16 μm cryosections were taken and stained with Toluidine Blue (see SI). All the recognizable DRG neuronal bodies (Fig. S1) in all sections of each ganglion were microdissected (Leica LMD6000, USA) [39]. Total RNA isolation was performed with the miRNeasy Micro Kit (QIAGEN, USA). 20 ng of RNA per panel per sample was reverse transcribed. For qPCR panels cDNA was combined with SYBR Green Master mix and added to the microRNA Ready-to-Use PCR, Mouse&Rat panels I + II (Exiqon, USA). Reactions were run on a Roche LightCycler® 480 Instrument. Poorly expressed miRNAs with a Cycle Threshold (Ct) higher than 38 were not included in the analysis.
Plasmid preparation
Cloning of the individual miRNAs into the expression vector was performed as described [40]. Briefly the minimum possible complete loop including the mature form of the miRNA (see Table S1 for oligos sequences) was synthesized as a single-stranded DNA oligomer with flanking Bbs1 restriction sites (Integrated DNA Technologies, USA), and cloned into the Bbs1 site of the UI4-GFP-SIBR [41] kindly provided by Dr. David Turner (University of Michigan, USA). As a negative control we created an anti-Luciferase siRNA expressing vector with the same procedure. Clones were verified by test digestion and sequencing.
Isolation and transfection of DRG neurons
Cervical, thoracic and lumbar level DRGs were isolated from adult C57BL/6 male mice. All procedures were performed in accordance with the protocols approved by the Institutional Animal Care and Use Committee at the Ohio State University. Ganglia were trimmed of axons and cells were isolated as described in SI. Cells were then resuspended in Nucleofector Buffer from the Amaxa P3 Primary Cell kit (Lonza, USA), transfected with plasmid DNA and plated.
DRG neuron neurite outgrowth detection and analysis
After 24 hours in culture the cells were trypsinized, resuspended and replated. After 15 additional hours in culture cells were fixed and immunostained with rabbit polyclonal anti beta-III (BIII) tubulin (Sigma, TT2200), washed and incubated with anti-rabbit Alex Fluor®555 (Abcam: ab150086) and Hoechst 33258 to identify nuclei (Sigma, 861405). Images were obtained using an ArrayScan XTI Automated Microscope (ThermoFisher, USA). Neurite outgrowth was assessed using the Neuronal Profiling 4.1 Algorithm (ThermoFisher, USA). Cells transfected with each miRNA were identified by GFP intensity as previously described [42]. In each experiment the robust Z-scores were determined using each well of cells transfected with the control vector independently and pooling together wells transfected with each miRNA clone. Robust Z-score is: (median total neurite length for miRNA – median total neurite length for control) / (median absolute deviation across control wells).
RNA-isolation and preparation for RNA-Seq
DRG dissection, tissue processing and cell collection was performed in the same way as described for miRNA expression analysis. RNA isolation was performed as described in Danzi et al [39]. Briefly, neurons were collected in 50 μl of extraction buffer from the Arcturus® PicoPure® RNA Isolation Kit (Life Technologies, USA). RNA extraction was performed following the manufacturer’s instructions. The whole procedure was performed in RNAse free conditions and all solutions were made with diethyl pyrocarbonate (DEPC; Sigma) treated water. RNA quality was checked via Bioanalyzer 2100 (Agilent, USA), and only samples with an RNA Index Number (RIN) above 7 were used. RNA-Seq was performed at the Hussman Institute for Human Genomics Sequencing Core Facility (University of Miami, Miami, FL).
RNA-Seq data analysis
The Tuxedo collection of software [43,44] was used as described [4]. The following versions were used: Tophat 1.3.2, Cufflinks 1.1.0, Cuffcompare 1.1.0 and Cuffdiff 1.2.1. Logistic regression analysis was performed to define reliable quantification of isoform expression [4]. Reliability cutoffs for the individual samples were averaged to define a unique threshold (Table S4). All raw data files, output table, gtf file and sample information can be found in the GEO repository (accession number: GSE59547).
Isoform validation and re-sequencing
Isoform structural details were recovered from the “.gtf” output of Cufflinks (GEO accession number GSE59547). For validation, RNA was isolated from intact DRGs, reverse transcribed and amplified with primers designed to uniquely capture the selected newly identified splice sites (see SI). The amplified material was then run on agarose gels. The band corresponding to the expected amplicon size was cut from the gel and DNA was extracted. The fragments were then sent for sequencing by GENEWIZ (www.genewiz.com, USA). Sequences were then BLASTed against the isoform sequences to confirm the fragments actually corresponded to the newly identified splice site and were not the product of non-specific amplification.
Quantitative Real Time PCR
DRG dissection, tissue processing and cell collection was performed in the same way as described for miRNA expression analysis. Total RNA was isolated as described for RNA-Seq analysis. From each total RNA sample, 200 ng was reverse transcribed. All primers used for qPCR were designed to be intron spanning and isoform specific and are listed in Table S5. Reactions were performed using a 7300 Real-Time PCR System (Applied Biosystems, USA). Relative expression was calculated using the –ΔΔCt method [45].
MiRNA target analysis
We obtained 3′UTRs by identifying stop codons for each coding sequence and extracting the rest of the sequence to the end of the transcript. The 3′UTRs were submitted to TargetScan for identification of the miRNA response elements (MREs) [46] and calculations of the context scores [18,47]. To increase specificity a similar approach was used with a different predictive tool: Miranda [48]. Interactions predicted by both tools were used for follow-up analysis. Cytoscape software was used to create graphical representations of the network [49].
Results
Axotomy of DRG neurons alters miRNA expression
DRG neurons respond to a peripheral axon lesion with robust regeneration [1,50]. Changes in expression of numerous genes have been described during this process [2,51]. These changes are orchestrated by different regulators of gene expression [1,6]. One important class of gene expression regulators is represented by miRNAs. In mice, the peripheral neuron injury response is dependent on the expression of Dicer, the enzyme responsible for the synthesis of miRNAs [52,53], indicating the importance of miRNA expression in peripheral regeneration. A complete analysis of differentially expressed miRNAs in mouse PNS neurons after injury has not been performed. To define the expression of miRNAs in regenerating DRG neurons, we used a sciatic nerve crush to elicit transcriptional changes associated with axon regeneration. We selected 7 days post-crush for analysis because transcriptional programs regulating regeneration are still active at this time [2], and we hypothesize that expression changes induced by post-injury processes such as inflammation in the neuronal bodies are dampened, since the peak of acute inflammation in the distal nerve ends by this time point [54]. DRG neurons were isolated by laser capture microdissection and total RNA was extracted from 4 crushed mice and 4 controls. We used a commercially available miRNA PCR panel to profile the expression of 752 rodent miRNAs. To determine the consistency of miRNA expression among animals of the same group we clustered the samples by Euclidean distance. We found that miRNA expression patterns correlated perfectly with the treatment group (Fig. S2). This strongly suggests the presence of injury-regulated miRNA expression changes. A 2-tailed t-test revealed the presence of 49 miRNAs significantly differentially expressed in DRG neurons 7DPI (Fig. 1 and Table S3). Forty-four miRNAs were upregulated post-axotomy and 5 were downregulated (Fig. 1 and Table S3). The majority of them were mouse miRNAs, but in a few cases we could detect differential expression for miRNAs annotated in rat. We hypothesize that miRNAs differentially expressed after injury, are functionally associated with different ongoing processes in the axotomized neurons, such as injury response, inflammation and axonal regeneration.
Fig. 1. Numerous miRNAs are differentially expressed in DRG neurons 7 days post axotomy.
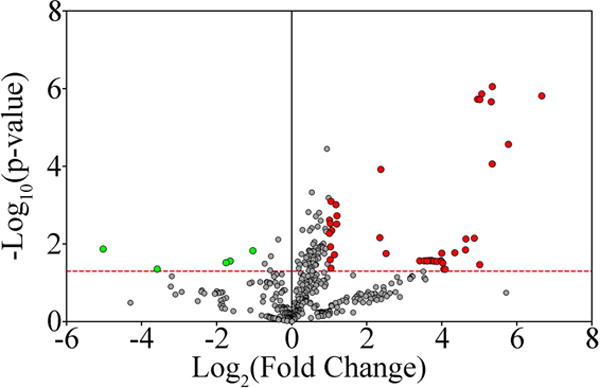
MiRNA expression in DRG neurons during axon regeneration was examined. All the miRNAs tested are reported as dots in the volcano plot, plotted as the –Log10 of the p-value for differential expression (applying a t-test), as a function of the Log2 of the fold change. The red dotted line represents a p-value of 0.05. All red dots are miRNAs that are considered differentially expressed (p-value < 0.05, Log2 (Fold Change) <−1 or >1). Green dots are miRNAs considered non-differentially expressed (p-value > 0.05, Log2 (Fold Change) >−1 and <1). A total of 49 miRNAs were determined to be differentially expressed: 44 upregulated and 5 downregulated.
A phenotypic screen identifies miRNAs that regulate neuronal morphology
We used a gain-of-function screen of the differentially expressed miRNAs to examine their ability to alter neurite outgrowth from DRG neurons in vitro. For miRNAs potentially involved in axon regeneration, we hypothesized that those upregulated in DRG neurons after injury would increase Total Neurite Length (TNL) when overexpressed in vitro, and those downregulated after injury would decrease TNL when overexpressed. We used the UI4-GFP-SIBR vector, originally designed for siRNA expression [41], to overexpress miRNAs, using our previous strategy [40]. The genomic fragments corresponding to the pre-miRNA hairpins [55] for all the miRNAs that were differentially expressed were individually cloned into the vector (Table S1). Adult mouse DRG neurons were dissociated, electroporated with the miRNA vectors and cultured for 24 hours to allow miRNA expression before being trypsinized and replated for 15 hours to test neurite growth ability. Since DRG neurons in culture activate transcriptional programs of axon elongation similar to those induced by peripheral axotomy in as little as 12 hours, and extend processes within 24 hours [56], 24 hours should be sufficient for miRNAs involved in axon extension programs to down-regulate their relevant targets. We used an automated microscope to identify transfected neurons and trace their neurites (see Fig. 2). We measured TNL for 25-150 neurons per miRNA and calculated the robust Z-score [57] compared to neurons in the same plate transfected with a control anti-Luciferase sequence [42]. By using a cutoff of Robust Z-score ± 2 we identified 13 miRNAs that increase TNL and 9 miRNAs that decreased TNL in DRG neurons (Fig. 3). MiRNAs that had the same effect in vitro as they were predicted to have in vivo were called “consistent” in our analysis. Out of 22 miRNAs with an effect on DRG morphology, 12 were called consistent; 11 were upregulated and only 1 was downregulated (mmu-miR-466d). MiR-1188 consistently impaired neuronal survival in several experiments (data not shown) and was therefore eliminated from the study. To confirm the observed effects were dependent specifically on the overexpression of the miRNAs we generated new clones containing the 12 miRNAs with a mutated seed core region (nucleotides 2 to 8 of the original miRNA sequence, see Supplementary Information and Table S2 for details on clone generation), the miRNA portion most responsible for target recognition [18]. The 12 mutant miRNAs were overexpressed in DRG neurons in culture in the same process described for the wild type miRNAs. When overexpressing the mutant forms, 11 out of 12 of the miRNAs did not show a significant effect on neurite length (see Fig. S3). Only in one case (miR-544) the mutant was able to induce an increase in Total Neurite Length, but to a lesser degree than the wild type. We hypothesized that this effect could be due to residual activity of the scrambled region that was still able to recognize some of the miRNA targets but with reduced strength or specificity. Overall this experiment confirmed the specificity of the miRNA effect. We propose that changes in expression of these 12 miRNAs play a role in regulating transcriptional events leading to axon regeneration in DRG neurons after axotomy.
Fig. 2. Overexpressing microRNAs in DRG neurons in culture influences neurite length.
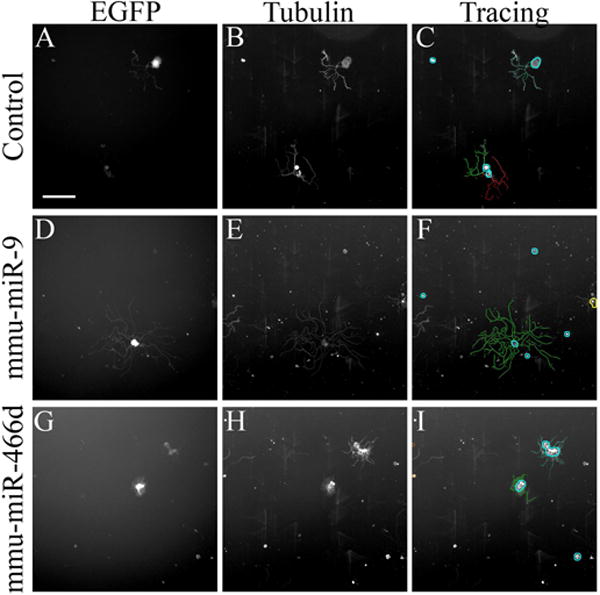
DRG neurons were transfected with a control EGFP-anti-luciferase-siRNA expressing vector (A, B, C) or EGFP-microRNA expressing vectors (D-I) and let grow in vitro to analyze their effect on neurite growth. The neurons were immunostained for βIII tubulin (B, E, H) and automatically traced (C, F, I). The figure reports an example of one miRNA increasing the neurite length, miR-9 (D, E, F), and one decreasing length miR-466d (G, H, I). Scale bar is 200 μm.
Fig. 3. A phenotypic screen revealed the effects of miRNA overexpression in neurite growth of DRG neurons in culture.
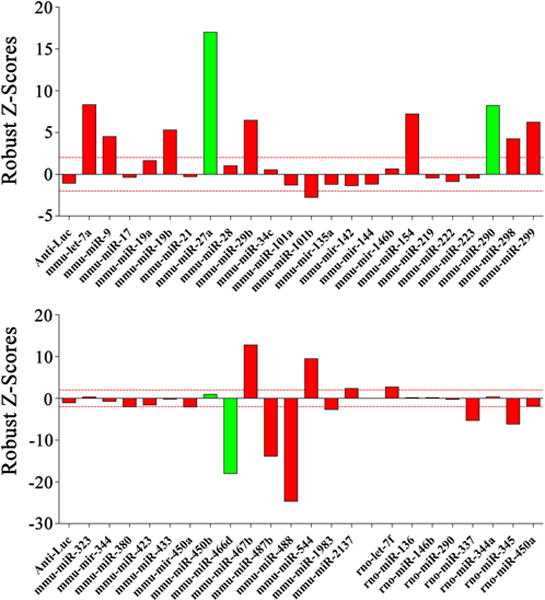
All differentially expressed miRNAs were cloned into expression vectors and individually transfected in adult mouse DRG neurons. Neurite length was measured for each transfected cell 39 hours post transfection (15 hours after re-plating) and robust Z-scores were calculated comparing to cells transfected with a control vector expressing an anti-Luciferase siRNA. The barplot reports robust Z-scores for each miRNA. Green bars are miRNAs that were downregulated in vivo in DRG neurons post-axotomy; red bars are miRNAs that were upregulated. For each miRNA between 25 and 100 cells were measured. Red dotted lines represent arbitrary threshold for significance (> 2, < -2). Overexpression of 9 miRNAs resulted in a decrease in neurite length (robust Z-score < -2), while overexpression of 13 different miRNAs resulted in an increase in neurite length (robust Z-score > 2).
RNA-Seq reveals differentially expressed isoforms in regenerating PNS neurons
One useful step towards identifying the gene targets by which miRNAs regulate axon regeneration is to analyze gene expression in the same cells. Past studies were limited to the most commonly known isoform for each gene, overlooking the concept of isoform diversity. We hypothesized that numerous transcriptional events important for axon regeneration remain unidentified. In the last few years the application of RNA-Seq has revealed a vast landscape of isoform diversity in different organisms [58–61] and cell types [4,62–65]. The identification of previously undetected isoforms and splicing events [4,44,66] supports the idea that the full spectrum of transcriptional events underlying biological processes is considerably more complex than had been appreciated. The use of cell specific isoforms is fundamental to cellular processes [67–69] including axon growth [70,71]. To define the full repertoire of isoform expression in regenerating DRG neurons, we used a sciatic nerve crush injury to elicit transcriptional changes associated with axon regeneration and used RNA-Seq to define these changes. DRG neuron RNA from crushed and control animals, was isolated by laser capture microdissection and almost 80 million reads per sample were sequenced (see Methods and SI). On average ~70% of the reads aligned correctly to the mouse genome (see Table S6); mRNA isoforms were assembled by Cufflinks [72]. Expression levels of individual isoforms were quantified in Fragments per Kilobase of transcript per Million mapped reads (FPKM). We used a statistical approach to define a cutoff for reliability of expression measured as described previously [4], and filtered all isoforms with FPKM below the cutoff (Table S4). We identified about 60,000 expressed isoforms in DRG neurons transcribed from 17,000 individual loci (Table S7). When compared to a combined reference annotation assembled from three different databases (UCSC Genome Browser, RefSeq and ENSEMBL) almost 43,000 of the isoforms presented at least one element (splicing event, transcription start site (TSS) or alternative 3′untranslated region (UTR)) that has not been described before (labeled as “_j” in Table S7). Euclidean distance between patterns of isoform expression was used to cluster the samples. The crushed samples clustered together as did 2 of the control samples (see Fig. S4). One of the control samples, ‘Control_2’, clustered separately from all other samples. For this and other reasons, including a significant difference in the expression distribution compared to the other samples (see Fig. S5), we excluded Control_2 sample in the following analysis. To evaluate differential expression between Crushed and Control samples we used Cuffdiff [66]. We considered only isoforms with an FDR corrected p-value below 0.05 and a fold change <-1 or >1 on a base 2 logarithmic scale. More than 2500 isoforms met our criteria for differential expression with ~1400 isoforms being upregulated and ~1200 downregulated in DRG neurons 7 days post axotomy (Fig. 4). To test our hypothesis that the initial inflammatory response is lost from the neuronal cell bodies at 7dpi, we examined the expression of inflammatory markers Il6, Nos1 (aka iNos), Tlr3 and 5, and Hspb1 in our RNAseq data. Expression levels in crushed DRG neurons are unchanged compared to non-injured sham mice (Table S7). This observation confirms that the changes we observe are not associated with inflammatory mechanisms.
Fig. 4. Thousands of isoforms are differentially expressed in DRG neurons 7 days after axotomy.
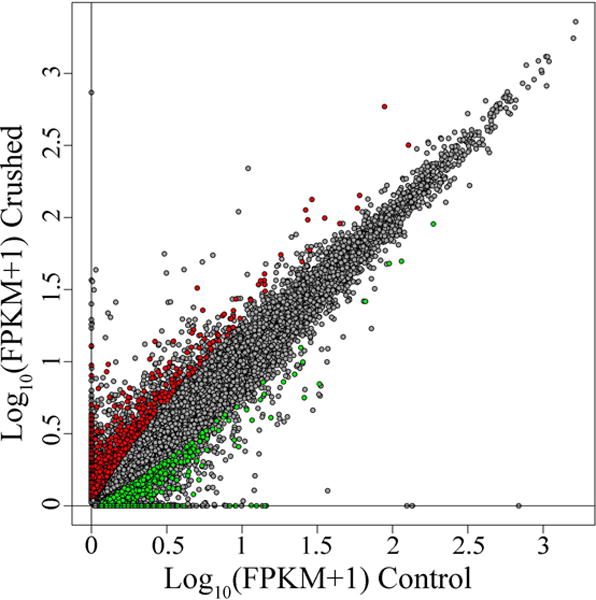
Adult mice underwent sciatic nerve crush (Crushed, y-axis) or sham surgery (Controls, x-axis). 7 days post injury the ganglia at the L3, L4 and L5 levels were dissected and neurons were collected by laser microdissection. RNA-Seq technology was used to analyze changes in RNA expression at the isoform level. Grey dots in the scatterplot represent isoforms. The x axis represents the log10 of the expression value in the control samples (reported in FPKM + 1). The y-axis represents the log10 of the expression value in the crushed samples. In red are the isoforms that passed a Jensen-Shannon divergence test for differential expression (FDR corrected p-value < 0.05) and meet our arbitrary threshold for meaningful change (Log2 of the Fold Change <-1 or >1). In green are the isoforms that did not pass the statistical test (FDR corrected p-value > 0.05) or did not meet the Fold Change threshold (Log2 of the Fold Change >-1 and <1). A total of 2682 isoforms were called differentially expressed.
Newly identified splicing events are confirmed with direct amplification
The majority of the differentially expressed isoforms are classified as new isoforms because of a new splice site, alternative TSS or 3′UTR shorter or longer than the conventional annotated one. To confirm the presence of some of these isoform-specific features, we designed primers flanking specific splice sites in a randomly chosen subset of differentially expressed isoforms [4]. We selected isoforms with splice sites not shared with any other isoform of the same gene. We did this for more than 20 newly identified isoform specific splice sites (Table S8). These primers were used in PCR reactions with cDNA made from RNA extracted from intact DRGs (see Methods and SI). Sequencing of the amplification products confirmed the presence of the newly identified splice sites for each of the primers selected (data not shown). This result supports the validity of our analysis.
Real Time qPCR confirms differential expression of selected isoforms
Next we validated our differential expression analysis by examining changes of some isoforms by Real Time qPCR. We selected (see Methods and SI) 8 isoforms: 4 up- and 4 down-regulated (Table S9). Half of these isoforms were already used to confirm splicing events. We tested isoform expression levels in RNA extracts from laser captured neurons collected from 6 additional biological replicates: 3 crushed and 3 control animals. Two isoforms with remarkably stable expression across all samples (Canx and Psmb2) were used as housekeeping genes. We calculated fold changes in crushed vs. controls normalizing to one or the other of these housekeeping genes (Table S9). There was a strong correlation between the RNA-Seq results and the qPCR results (Fig. 5: Pearson correlation R2 = 0.89 for Canx and R2 = 0.9 for Psmb2, p = 0.0004 and p=0.0003, respectively). To measure the correspondence of the results we used Kendall’s correlation analysis, which makes minimal distributional assumptions [4]. This resulted in a tau coefficient of 0.71 with a statistically significant p-value of 0.019 (Fig. 5). These data support the consistency and reproducibility of our RNA-Seq analysis for isoform differential expression.
Fig. 5. qPCR confirms isoform differential expression.
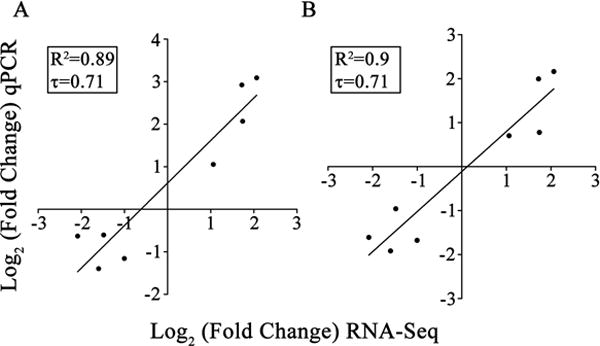
We used quantitative Real Time PCR to confirm differential expression for 8 selected isoforms. (A) Fold Changes of differential expression obtained using the Canx gene isoform for normalization strongly correlates (Pearson correlation R2 = 0.89, Kendall correlation tau coefficient = 0.71) with results reported by RNA-Seq analysis. (B) Similar results were obtained when using Psmb2 for normalization (Pearson correlation R2 = 0.9, Kendall tau coefficient = 0.71).
MiRNA-mRNA interaction analysis predicts a large network of transcriptional events controlling axonal regeneration
MiRNAs regulate transcript expression by targeting MREs mostly located in the 3′UTRs of mRNAs [16]. The targeting recognition happens through an imperfect match [18,46]. In addition, several physical properties in the targets have been identified [18,47] making it possible to predict the presence of target sites and to calculate an “interaction score”, which is directly proportional to the predicted downregulation effect of the miRNA on the messenger RNA (the more negative the score, the stronger the downregulation) [18,47]. We hypothesize that the miRNAs affecting axonal growth regulate differential isoform expression. To test this idea we used a computational approach to predict a network of transcriptional regulation among differentially expressed isoforms and miRNAs. To build the network we examined the 3′UTRs of the differentially expressed isoforms for MREs recognizing the 12 miRNAs that significantly altered neurite growth in DRGs. We isolated the 3′UTRs for the 2500 differentially expressed isoforms and analyzed the sequences with two tools that predict the presence of MREs: TargetScan [47,73] and MiRanda [48]. Only interactions predicted by both tools (see Methods) were taken into consideration. We were able to predict thousands of miRNA-mRNA interactions with diverse scores (Table S10). Interestingly, we found that miR-466d-5p, the miRNA that decreased neurite growth, had a very distinct set of isoform targets compared to the other miRNAs (Fig. S6). To generate the network diagram we filtered to retain only the interactions connecting divergent nodes, defined as: upregulated miRNA to downregulated mRNA and vice versa, with the assumption that these interactions are active in the regeneration process because they are consistent with the predicted effect of the miRNA on the specific isoform. We obtained a very dense network (Fig. S7). miRNAs likely exert their effect on gene expression by targeting numerous mRNAs at the same time; thus perturbing targets individually would likely not result in a robust phenotypic effect [74,75].
MiRNAs that increase axonal growth would likely share critical targets that represent the core mechanism important for axon regeneration. To identify this core network we set up filtering criteria: for upregulated miRNAs we identified isoforms that were targeted by more than one miRNA and then limited this network to isoforms that had 6 or more interactions with these miRNAs. We restricted this to 6 or more interactions because with this criterion each of the upregulated miRNAs had at least one target (see Fig. 6, right side). MiR-466d and its upregulated targets represent a separate network (Fig. S7); since miR-466d is the only down regulated miRNA that was identified we could not apply these same criteria. We noticed that several genes expressed both up- and downregulated isoforms after injury (Table S11). For some of these genes one or more of their upregulated isoforms were predicted targets of miR-466d and one or more of their downregulated isoforms were predicted targets of the other, upregulated miRNAs (upregulated and downregulated isoforms of the same gene are connected by blue edges in Fig. 6 and Fig. S7). This observation suggests that co-regulatory mechanisms between up and downregulated miRNAs provide the switch between different isoforms of these genes. We decided to include those miR-466d targets connected by blue edges to downregulated isoforms that passed the ‘6 or more interactions’ criteria. By applying these filters we predicted a core network of interactions (see Fig. 6) that includes 12 miRNAs (544, 299,154, 29b, 9, let-7a, let-7f, 298, 19b, 467b, 2137 and 466d), which target 71 isoforms from 47 genes. MiR-298, let-7a and let-7f are the most involved miRNAs, each targeting more than 30 isoforms at the same time. Gene Ontology and pathway analysis of the 47 genes with isoforms in the network reveals (Table S12) that the network is significantly associated with morphogenesis, polarity, GTPase regulatory activity, actin cytoskeleton reorganization and neural tube formation, all biological process that are strongly associated with axon growth. There are 21 genes that have at least one upregulated isoform targeted by miR-466d and one downregulated isoform targeted by one or more of the other miRNAs (Fig. 6). Interestingly, when we compared the open reading frames of the isoforms of the 21 genes, all isoforms in the network from the same gene showed diverse protein coding sequences, suggesting that miRNA mediated regulation of mRNAs results in different protein isoform expression. The large number of co-regulatory effects predicted in the network suggests that a robust strategy to enhance nerve regeneration would potentially require simultaneously modulating the expression of all these miRNAs.
Fig. 6. MRE analysis identifies core network of interactions behind DRG regeneration.
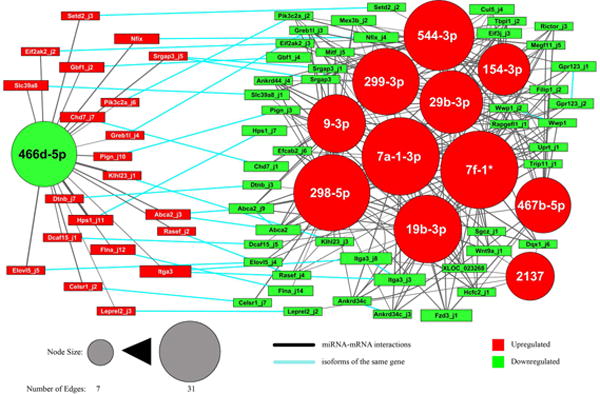
miRNA binding site analysis was used to predict interactions between miRNAs and mRNA isoforms. The isoforms were filtered to identify the core of the network of interactions (see Methods and Results). The resulting network is reported, where nodes are miRNAs (circles) and isoforms (boxes). Red nodes were upregulated in vivo after axotomy, green nodes were downregulated. Node size is proportional to the number of edges connecting it. Edge thickness is inversely correlated with the interaction score (see Methods). Blue edges connect isoforms of the same gene.
Discussion
Our long-term goal, similar to previous studies [2,51], was to examine gene expression changes in regenerating peripheral neurons as a way of elucidating the mechanisms through which these adaptive changes are controlled. We focused on the roles of miRNAs, an important class of transcriptional regulator, in regulating genes involved in regeneration of PNS axons. Previous studies have concentrated almost exclusively on single miRNAs [31,33] which, while informative, likely oversimplifies the complexity associated with miRNA-mRNA interactions. We therefore utilized a high-throughput gene expression analysis followed by a phenotypic screen in primary neurons, to systemically study expression changes and functional roles of all known mouse miRNAs relating to regeneration of DRG neuron axons. We found that of the 49 miRNAs differentially expressed in DRG neurons after injury, almost 90% were upregulated. While each of these miRNAs is a candidate regulator of the differentially expressed mRNA isoforms, not all are directly associated with the same biological processes. To determine the roles of these miRNAs in neurite extension, we used a gain-of-function approach and individually overexpressed them in DRG neurons, predicting that upregulated miRNAs would lead to increased neurite growth, and downregulated miRNAs to less growth. Of the 12 miRNAs whose activities were consistent with this functional hypothesis, only 3 have previously been linked to axon growth–miR-29b [76], miR-9 [77–79] and miR-154 [80]. Interestingly, for some miRNAs the effect observed was opposite to our hypothesis. Explanations could include changes in gene expression in cultured DRG neurons (leading to changes in miRNA target acquisition), and/or increased activities on secondary targets [47] due to overexpression of miRNAs. It is also worth noting that vector-based overexpression of miRNAs dissociates their expression from their normal complex feedback and feedforward regulatory loops, which are likely to exist in DRG neurons as in other cells [81–83].
The commercial qPCR panels we used to screen all known rodent miRNAs, include, in some cases, both the mouse and rat versions, even when they share the same sequences. An event that frequently happens since miRNAs are highly conserved across organisms [46,84]. It is not surprising that when miRNA sequences were the same between mouse and rat, we detected similar differential expression for both the rat and the mouse forms. In a few cases only the rat form of the miRNA was differentially expressed. In these cases the sequences were not similar, suggesting the presence of previously unrecognized mouse miRNAs with homologs in rats [85,86]. The number of differentially expressed miRNAs we found depends on cutoffs for significance and fold change that were arbitrarily decided. Less stringent criteria would have resulted in a larger number of differentially expressed miRNAs; in particular a less stringent fold change cutoff would have allowed small changes in miRNA expression to be called significant. Given the multi-targeting nature of miRNAs, it has previously been suggested that minor variations in expression could have a real impact on cellular functions [87,88]. However, lowering the fold change cutoff also increases false positives, mostly due to inherent variability and biases, especially with a technology such as qPCR[89,90]. Our more stringent approach, which has been also applied by others [24,34], increased our confidence in the biological significance of the 49 miRNAs we analyzed.
These explanations highlight some of the challenges associated with the gain of function approach we adopted, in particular the expression of the miRNAs at non-physiological levels, and the possibility of non-specific targeting. Thus more tests with alternative strategies (e.g. miRNA silencing) will be useful to confirm the involvement of the suggested miRNAs in axon regeneration. To understand the mechanisms by which miRNAs modulate neurite growth, we need to identify their relevant mRNA targets. Former studies have focused on a single target for each axon growth associated miRNA [31,33], even though miRNAs have the potential to simultaneously target hundreds of mRNAs [19,21,91]. Additionally, these single targets have been assumed to be represented by their best known (“conventional”) isoforms. However, recognition of the importance of cell specific isoforms in biological processes [68,69] including axon growth [70,71] has been growing. Isoform diversity often occurs at the level of the non-coding portions of mRNAs, such as the TSS or the 3′UTR. These elements are responsible for regulation of mRNA expression, suggesting the existence of isoform specific regulation of expression by miRNAs. We hypothesize that defining these regulatory processes will be critical to identify which targets comprise nodes mediating axon regeneration.
By using RNA-Seq to analyze gene expression at the isoform level in laser captured DRG neurons, we observed the expression of several thousand cell specific isoforms. Most of these were not found in major reference databases (e.g. UCSC Genome Browser, RefSeq or Ensembl) and included novel protein splice forms, 3′UTRs, and/or TSSs. More importantly from our perspective, we observed more than 2000 differentially expressed isoforms comparing DRG neurons of crushed and control nerves. In the majority of cases, several isoforms were expressed from the same gene but only some were differentially expressed (see Table S11). These observations strongly suggest that examining only conventional isoforms will not allow a detailed understanding of miRNA regulation of regeneration.
We analyzed expression changes of miRNAs and mRNAs at a single time point–7 days post injury. This approach does not take into account dynamics of gene expression over long periods of time (some of the expression levels we observe might be in a rising or declining phase). Sampling of other time points, however, would increase the identification of changes associated with biological processes outside the scope of this study, such as inflammation or re-myelination [54]. We believe that our analysis provides enough information to identify expression changes responsible for or associated with axon regeneration, which is ongoing at 7 days post injury [2]. A more comprehensive study of the totality of events in peripheral ganglia after injury would require an experimental design including multiple time points.
To predict which miRNA targets (mRNA isoforms) influence DRG axon growth, we used a bioinformatics approach to identify, in the differentially expressed mRNA isoforms, MREs for the miRNAs that altered neurite growth. We used this information to design a network of interactions that we propose influences regeneration of DRG axons. Further experiments will be required to confirm that the proposed model is valid in vivo. In the dense network we observed, we focused on a core group of isoforms, each of which is targeted by several miRNAs. The genes in the network are strongly associated with morphogenesis, cell polarity and regulation of the cytoskeleton. These biological processes are likely to be important in axon regeneration. Strategies that use a number of miRNAs to target this network offer an exciting possibility to promote regeneration of DRG neurons.
Supplementary Material
Acknowledgments
We are grateful to members of the Lemmon-Bixby laboratory, past and present. We would like to thank Y. Martinez for her advice on technical aspects. We would also like to thank R. Morris for advice on statistical analysis. VPL holds the Walter G. Ross Distinguished Chair in Developmental Neuroscience. This work was supported by The Miami Project to Cure Paralysis, The Walter G. Ross Foundation, and grants R01 HD057632 and DOD W81XWH-05-1-0061 (VPL and JLB). The funding sources had no role in the conduct of the research or the preparation of the article.
Footnotes
Authors’ contributions
DM conceived the study, analyzed the data, designed and performed all the experiments except for the in vitro axon growth experiment that was designed and performed by JKL. MCD and FK helped with the data analysis. JHG helped with the microdissection of the DRG neurons. TIS helped with the generation of the clones. VPL and JLB supervised the study. DM, JKL, JLB and VPL wrote the manuscript.
DR. DARIO MOTTI (Orcid ID : 0000-0002-7714-4040)
References
- 1.Huebner EA, Strittmatter SM. Axon regeneration in the peripheral and central nervous systems. Results Probl Cell Differ. 2009;48:339–51. doi: 10.1007/400_2009_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zou H, Ho C, Wong K, Tessier-Lavigne M. Axotomy-induced Smad1 activation promotes axonal growth in adult sensory neurons. J Neurosci. 2009;29:7116–23. doi: 10.1523/JNEUROSCI.5397-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lerch JK, Martínez-Ondaro YR, Bixby JL, Lemmon VP. cJun promotes CNS axon growth. Mol Cell Neurosci. 2014;59:97–105. doi: 10.1016/j.mcn.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lerch JK, Kuo F, Motti D, Morris R, Bixby JL, Lemmon VP. Isoform diversity and regulation in peripheral and central neurons revealed through RNA-Seq. PLoS One. 2012;7:e30417. doi: 10.1371/journal.pone.0030417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin S, Zou Y, Zhang C-L. Cross-talk between KLF4 and STAT3 regulates axon regeneration. Nat Commun. 2013;4:2633. doi: 10.1038/ncomms3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith RP, Lerch-Haner JK, Pardinas JR, Buchser WJ, Bixby JL, Lemmon VP. Transcriptional profiling of intrinsic PNS factors in the postnatal mouse. Mol Cell Neurosci. 2011;46:32–44. doi: 10.1016/j.mcn.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buchser WJ, Smith RP, Pardinas JR, Haddox CL, Hutson T, Moon L, Hoffman SR, Bixby JL, Lemmon VP. Peripheral nervous system genes expressed in central neurons induce growth on inhibitory substrates. PLoS One. 2012;7:e38101. doi: 10.1371/journal.pone.0038101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stern S, Haverkamp S, Sinske D, Tedeschi A, Naumann U, Di Giovanni S, Kochanek S, Nordheim A, Knöll B. The transcription factor serum response factor stimulates axon regeneration through cytoplasmic localization and cofilin interaction. J Neurosci. 2013;33:18836–48. doi: 10.1523/JNEUROSCI.3029-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, Goldberg JL. KLF family members regulate intrinsic axon regeneration ability. Science. 2009;326:298–301. doi: 10.1126/science.1175737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blackmore MG, Wang Z, Lerch JK, Motti D, Zhang YP, Shields CB, Lee JK, Goldberg JL, Lemmon VP, Bixby JL. Krüppel-like Factor 7 engineered for transcriptional activation promotes axon regeneration in the adult corticospinal tract. Proc Natl Acad Sci U S A. 2012;109:7517–22. doi: 10.1073/pnas.1120684109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–9. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 12.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–8. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 13.Lim LP, Glasner ME, Yekta S, Burge CB, Bartel DP. Vertebrate microRNA genes. Science. 2003;299:1540. doi: 10.1126/science.1080372. [DOI] [PubMed] [Google Scholar]
- 14.Djuranovic S, Nahvi A, Green R. miRNA-Mediated Gene Silencing by Translational Repression Followed by mRNA Deadenylation and Decay. Science (80-) 2012;336:237–240. doi: 10.1126/science.1215691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 18.Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376–81. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- 20.Djuranovic S, Nahvi A, Green R. A Parsimonious Model for Gene Regulation by miRNAs. Science (80-) 2011;331:550–553. doi: 10.1126/science.1191138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M. Assessing the ceRNA Hypothesis with Quantitative Measurements of miRNA and Target Abundance. Mol Cell. 2014 doi: 10.1016/j.molcel.2014.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, Guenther MG, Johnston WK, Wernig M, Newman J, Calabrese JM, Dennis LM, Volkert TL, Gupta S, Love J, Hannett N, Sharp PA, Bartel DP, Jaenisch R, Young RA. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–33. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang H-L, Wang H-C, Chunag Y-T, Chou C-W, Lin I-L, Lai C-S, Chang L-L, Cheng K-I. miRNA Expression Change in Dorsal Root Ganglia After Peripheral Nerve Injury. J Mol Neurosci. 2017;61:169–177. doi: 10.1007/s12031-016-0876-7. [DOI] [PubMed] [Google Scholar]
- 24.Lu A, Huang Z, Zhang C, Zhang X, Zhao J, Zhang H, Zhang Q, Wu S, Yi X. Differential expression of microRNAs in dorsal root ganglia after sciatic nerve injury. Neural Regen Res. 2014;9:1031–40. doi: 10.4103/1673-5374.133164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li S, Yu B, Wang S, Gu Y, Yao D, Wang Y, Qian T, Ding F, Gu X. Identification and functional analysis of novel micro-rnas in rat dorsal root ganglia after sciatic nerve resection. J Neurosci Res. 2012;90:791–801. doi: 10.1002/jnr.22814. [DOI] [PubMed] [Google Scholar]
- 26.Phay M, Kim HH, Yoo S. Dynamic Change and Target Prediction of Axon-Specific MicroRNAs in Regenerating Sciatic Nerve. PLoS One. 2015;10:e0137461. doi: 10.1371/journal.pone.0137461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adilakshmi T, Sudol I, Tapinos N, Wang J, Coselli J. Combinatorial Action of miRNAs Regulates Transcriptional and Post-Transcriptional Gene Silencing following in vivo PNS Injury. PLoS One. 2012;7:e39674. doi: 10.1371/journal.pone.0039674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Viader A, Chang L-W, Fahrner T, Nagarajan R, Milbrandt J. MicroRNAs Modulate Schwann Cell Response to Nerve Injury by Reinforcing Transcriptional Silencing of Dedifferentiation-Related Genes. J Neurosci. 2011;31:17358–17369. doi: 10.1523/JNEUROSCI.3931-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu B, Zhou S, Qian T, Wang Y, Ding F, Gu X. Altered microRNA expression following sciatic nerve resection in dorsal root ganglia of rats. Acta Biochim Biophys Sin (Shanghai) 2011;43:909–915. doi: 10.1093/abbs/gmr083. [DOI] [PubMed] [Google Scholar]
- 30.Zhou S, Yu B, Qian T, Yao D, Wang Y, Ding F, Gu X. Early changes of microRNAs expression in the dorsal root ganglia following rat sciatic nerve transection. Neurosci Lett. 2011;494:89–93. doi: 10.1016/j.neulet.2011.02.064. [DOI] [PubMed] [Google Scholar]
- 31.Strickland IT, Richards L, Holmes FE, Wynick D, Uney JB, Wong L-F. Axotomy-induced miR-21 promotes axon growth in adult dorsal root ganglion neurons. PLoS One. 2011;6:e23423. doi: 10.1371/journal.pone.0023423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hancock ML, Preitner N, Quan J, Flanagan JG. MicroRNA-132 is enriched in developing axons, locally regulates Rasa1 mRNA, and promotes axon extension. J Neurosci. 2014;34:66–78. doi: 10.1523/JNEUROSCI.3371-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou S, Shen D, Wang Y, Gong L, Tang X, Yu B, Gu X, Ding F. microRNA-222 targeting PTEN promotes neurite outgrowth from adult dorsal root ganglion neurons following sciatic nerve transection. PLoS One. 2012;7:e44768. doi: 10.1371/journal.pone.0044768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lemmon VP, Ferguson AR, Popovich PG, Xu X-M, Snow DM, Igarashi M, Beattie CE, Bixby JL, MIASCI Consortium Minimum information about a spinal cord injury experiment: a proposed reporting standard for spinal cord injury experiments. J Neurotrauma. 2014;31:1354–61. doi: 10.1089/neu.2014.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Callahan A, Abeyruwan SW, Al-Ali H, Sakurai K, Ferguson AR, Popovich PG, Shah NH, Visser U, Bixby JL, Lemmon VP. RegenBase: a knowledge base of spinal cord injury biology for translational research. Database (Oxford) 2016;2016 doi: 10.1093/database/baw040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilkinson MD, Dumontier M, Aalbersberg IJJ, Appleton G, Axton M, Baak A, Blomberg N, Boiten J-W, da Silva Santos LB, Bourne PE, Bouwman J, Brookes AJ, Clark T, Crosas M, Dillo I, Dumon O, Edmunds S, Evelo CT, Finkers R, Gonzalez-Beltran A, Gray AJG, Groth P, Goble C, Grethe JS, Heringa J, ’t Hoen PAC, Hooft R, Kuhn T, Kok R, Kok J, Lusher SJ, Martone ME, Mons A, Packer AL, Persson B, Rocca-Serra P, Roos M, van Schaik R, Sansone S-A, Schultes E, Sengstag T, Slater T, Strawn G, Swertz MA, Thompson M, van der Lei J, van Mulligen E, Velterop J, Waagmeester A, Wittenburg P, Wolstencroft K, Zhao J, Mons B. The FAIR Guiding Principles for scientific data management and stewardship. Sci data. 2016;3:160018. doi: 10.1038/sdata.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rigaud M, Gemes G, Barabas M-E, Chernoff DI, Abram SE, Stucky CL, Hogan QH. Species and strain differences in rodent sciatic nerve anatomy: implications for studies of neuropathic pain. Pain. 2008;136:188–201. doi: 10.1016/j.pain.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Danzi MC, Motti D, Avison DL, Bixby JL, Lemmon VP. Treatment with analgesics after mouse sciatic nerve injury does not alter expression of wound healing-associated genes. Neural Regen Res. 2016;11:144–9. doi: 10.4103/1673-5374.169637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Truettner JS, Motti D, Dietrich WD. MicroRNA overexpression increases cortical neuronal vulnerability to injury. Brain Res. 2013;1533:122–30. doi: 10.1016/j.brainres.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chung K-H, Hart CC, Al-Bassam S, Avery A, Taylor J, Patel PD, Vojtek AB, Turner DL. Polycistronic RNA polymerase II expression vectors for RNA interference based on BIC/miR-155. Nucleic Acids Res. 2006;34:e53. doi: 10.1093/nar/gkl143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blackmore MG, Moore DL, Smith RP, Goldberg JL, Bixby JL, Lemmon VP. High content screening of cortical neurons identifies novel regulators of axon growth. Mol Cell Neurosci. 2010;44:43–54. doi: 10.1016/j.mcn.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010 doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 46.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 47.Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol. 2011;18:1139–46. doi: 10.1038/nsmb.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Z-L, Yu W-M, Strickland S. Peripheral regeneration. Annu Rev Neurosci. 2007;30:209–33. doi: 10.1146/annurev.neuro.30.051606.094337. [DOI] [PubMed] [Google Scholar]
- 51.Stam FJ, MacGillavry HD, Armstrong NJ, de Gunst MC, Zhang Y, van Kesteren RE, Smit AB, Verhaagen J. Identification of candidate transcriptional modulators involved in successful regeneration after nerve injury. Eur J Neurosci. 2007;25:3629–3637. doi: 10.1111/j.1460-9568.2007.05597.x. [DOI] [PubMed] [Google Scholar]
- 52.Wu D, Raafat A, Pak E, Clemens S, Murashov AK. Dicer-microRNA pathway is critical for peripheral nerve regeneration and functional recovery in vivo and regenerative axonogenesis in vitro. Exp Neurol. 2012;233:555–65. doi: 10.1016/j.expneurol.2011.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu D, Raafat M, Pak E, Hammond S, Murashov AK. MicroRNA machinery responds to peripheral nerve lesion in an injury-regulated pattern. Neuroscience. 2011;190:386–97. doi: 10.1016/j.neuroscience.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gaudet AD, Popovich PG, Ramer MS. Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J Neuroinflammation. 2011;8:110. doi: 10.1186/1742-2094-8-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bartel DP, Lee R, Feinbaum R. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function Genomics. The miRNA Genes. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 56.Smith DS, Skene JH. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J Neurosci. 1997;17:646–658. doi: 10.1523/JNEUROSCI.17-02-00646.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Birmingham A, Selfors LM, Forster T, Wrobel D, Kennedy CJ, Shanks E, Santoyo-Lopez J, Dunican DJ, Long A, Kelleher D, Smith Q, Beijersbergen RL, Ghazal P, Shamu CE. Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods. 2009;6:569–75. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Röder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Falconnet E, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena H, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Luo OJ, Park E, Persaud K, Preall JB, Ribeca P, Risk B, Robyr D, Sammeth M, Schaffer L, See L-H, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Ruan X, Hayashizaki Y, Harrow J, Gerstein M, Hubbard T, Reymond A, Antonarakis SE, Hannon G, Giddings MC, Ruan Y, Wold B, Carninci P, Guigó R, Gingeras TR. Landscape of transcription in human cells. Nature. 2012;489:101–8. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brown JB, Boley N, Eisman R, May GE, Stoiber MH, Duff MO, Booth BW, Wen J, Park S, Suzuki AM, Wan KH, Yu C, Zhang D, Carlson JW, Cherbas L, Eads BD, Miller D, Mockaitis K, Roberts J, Davis CA, Frise E, Hammonds AS, Olson S, Shenker S, Sturgill D, Samsonova AA, Weiszmann R, Robinson G, Hernandez J, Andrews J, Bickel PJ, Carninci P, Cherbas P, Gingeras TR, Hoskins RA, Kaufman TC, Lai EC, Oliver B, Perrimon N, Graveley BR, Celniker SE. Diversity and dynamics of the Drosophila transcriptome. Nature. 2014 doi: 10.1038/nature12962. advance on. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 61.Stark KL, Xu B, Bagchi A, Lai W-S, Liu H, Hsu R, Wan X, Pavlidis P, Mills AA, Karayiorgou M, Gogos JA. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat Genet. 2008;40:751–60. doi: 10.1038/ng.138. [DOI] [PubMed] [Google Scholar]
- 62.Ramsköld D, Luo S, Wang Y-C, Li R, Deng Q, Faridani OR, Daniels GA, Khrebtukova I, Loring JF, Laurent LC, Schroth GP, Sandberg R. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30:777–82. doi: 10.1038/nbt.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu JQ, Habegger L, Noisa P, Szekely A, Qiu C, Hutchison S, Raha D, Egholm M, Lin H, Weissman S, Cui W, Gerstein M, Snyder M. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proc Natl Acad Sci U S A. 2010;107:5254–9. doi: 10.1073/pnas.0914114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xue Z, Huang K, Cai C, Cai L, Jiang C, Feng Y, Liu Z, Zeng Q, Cheng L, Sun YE, Liu J, Horvath S, Fan G. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature. 2013;500:593–7. doi: 10.1038/nature12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lovatt D, Ruble BK, Lee J, Dueck H, Kim TK, Fisher S, Francis C, Spaethling JM, Wolf JA, Grady MS, Ulyanova AV, Yeldell SB, Griepenburg JC, Buckley PT, Kim J, Sul J-Y, Dmochowski IJ, Eberwine J. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat Methods. 2014;11:190–6. doi: 10.1038/nmeth.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Polo-Parada L, Bose CM, Plattner F, Landmesser LT. Distinct roles of different neural cell adhesion molecule (NCAM) isoforms in synaptic maturation revealed by analysis of NCAM 180 kDa isoform-deficient mice. J Neurosci. 2004;24:1852–64. doi: 10.1523/JNEUROSCI.4406-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.An JJ, Gharami K, Liao G-Y, Woo NH, Lau AG, Vanevski F, Torre ER, Jones KR, Feng Y, Lu B, Xu B. Distinct role of long 3′ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell. 2008;134:175–87. doi: 10.1016/j.cell.2008.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baj G, Leone E, Chao MV, Tongiorgi E. Spatial segregation of BDNF transcripts enables BDNF to differentially shape distinct dendritic compartments. Proc Natl Acad Sci U S A. 2011;108:16813–8. doi: 10.1073/pnas.1014168108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.He H, Kise Y, Izadifar A, Urwyler O, Ayaz D, Parthasarthy A, Yan B, Erfurth M-L, Dascenco D, Schmucker D. Cell-Intrinsic Requirement of Dscam1 Isoform Diversity for Axon Collateral Formation. Science (80-) 2014 doi: 10.1126/science.1251852. [DOI] [PubMed] [Google Scholar]
- 71.Barnat M, Enslen H, Propst F, Davis RJ, Soares S, Nothias F. Distinct roles of c-Jun N-terminal kinase isoforms in neurite initiation and elongation during axonal regeneration. J Neurosci. 2010;30:7804–16. doi: 10.1523/JNEUROSCI.0372-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Poos K, Smida J, Nathrath M, Maugg D, Baumhoer D, Korsching E. How microRNA and transcription factor co-regulatory networks affect osteosarcoma cell proliferation. PLoS Comput Biol. 2013;9:e1003210. doi: 10.1371/journal.pcbi.1003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nazarov PV, Reinsbach SE, Muller A, Nicot N, Philippidou D, Vallar L, Kreis S. Interplay of microRNAs, transcription factors and target genes: linking dynamic expression changes to function. Nucleic Acids Res. 2013;41:2817–31. doi: 10.1093/nar/gks1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shioya M, Obayashi S, Tabunoki H, Arima K, Saito Y, Ishida T, Satoh J. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol Appl Neurobiol. 2010;36:320–330. doi: 10.1111/j.1365-2990.2010.01076.x. [DOI] [PubMed] [Google Scholar]
- 77.Wang Y, Wang H, Li X, Li Y. Epithelial microRNA-9a regulates dendrite growth through Fmi-Gq signaling in Drosophila sensory neurons. Dev Neurobiol. 2016;76:225–237. doi: 10.1002/dneu.22309. [DOI] [PubMed] [Google Scholar]
- 78.Dajas-Bailador F, Bonev B, Garcez P, Stanley P, Guillemot F, Papalopulu N. microRNA-9 regulates axon extension and branching by targeting Map1b in mouse cortical neurons. Nat Neurosci. 2012 doi: 10.1038/nn.3082. [DOI] [PubMed] [Google Scholar]
- 79.Haramati S, Chapnik E, Sztainberg Y, Eilam R, Zwang R, Gershoni N, McGlinn E, Heiser PW, Wills A-M, Wirguin I, Rubin LL, Misawa H, Tabin CJ, Brown R, Chen A, Hornstein E. miRNA malfunction causes spinal motor neuron disease. Proc Natl Acad Sci. 2010;107:13111–13116. doi: 10.1073/pnas.1006151107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tapocik JD, Luu TV, Mayo CL, Wang B-D, Doyle E, Lee AD, Lee NH, Elmer GI. Neuroplasticity, axonal guidance and micro-RNA genes are associated with morphine self-administration behavior. Addict Biol. 2013;18:480–95. doi: 10.1111/j.1369-1600.2012.00470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9:775–89. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ramalingam S, Subramaniam D, Anant S. Manipulating miRNA Expression: A Novel Approach for Colon Cancer Prevention and Chemotherapy. Curr Pharmacol reports. 2015;1:141–153. doi: 10.1007/s40495-015-0020-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gulyaeva LF, Kushlinskiy NE. Regulatory mechanisms of microRNA expression. J Transl Med. 2016;14:143. doi: 10.1186/s12967-016-0893-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, Lin C, Socci ND, Hermida L, Fulci V, Chiaretti S, Foà R, Schliwka J, Fuchs U, Novosel A, Müller R-U, Schermer B, Bissels U, Inman J, Phan Q, Chien M, Weir DB, Choksi R, De Vita G, Frezzetti D, Trompeter H-I, Hornung V, Teng G, Hartmann G, Palkovits M, Di Lauro R, Wernet P, Macino G, Rogler CE, Nagle JW, Ju J, Papavasiliou FN, Benzing T, Lichter P, Tam W, Brownstein MJ, Bosio A, Borkhardt A, Russo JJ, Sander C, Zavolan M, Tuschl T. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–14. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.D’Ambrogio A, Gu W, Udagawa T, Mello CC, Richter JD. Specific miRNA stabilization by Gld2-catalyzed monoadenylation. Cell Rep. 2012;2:1537–45. doi: 10.1016/j.celrep.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wyman SK, Knouf EC, Parkin RK, Fritz BR, Lin DW, Dennis LM, Krouse MA, Webster PJ, Tewari M. Post-transcriptional generation of miRNA variants by multiple nucleotidyl transferases contributes to miRNA transcriptome complexity. Genome Res. 2011;21:1450–61. doi: 10.1101/gr.118059.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Calin GA, Croce CM. MicroRNA-Cancer Connection: The Beginning of a New Tale. Cancer Res. 2006;66:7390–7394. doi: 10.1158/0008-5472.CAN-06-0800. [DOI] [PubMed] [Google Scholar]
- 88.Peltier HJ, Latham GJ. Normalization of microRNA expression levels in quantitative RT-PCR assays: identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA. 2008;14:844–52. doi: 10.1261/rna.939908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sanders R, Mason DJ, Foy CA, Huggett JF. Considerations for accurate gene expression measurement by reverse transcription quantitative PCR when analysing clinical samples. Anal Bioanal Chem. 2014;406:6471–83. doi: 10.1007/s00216-014-7857-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McCarthy DJ, Smyth GK. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics. 2009;25:765–71. doi: 10.1093/bioinformatics/btp053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hashimoto Y, Akiyama Y, Yuasa Y, Calin G, Sevignani C, Dumitru C, Hyslop T, Noch E, Kim Y, Yu J, Han T, Park S, Namkoong B, Bonci D, Coppola V, Musumeci M, Addario A, Giuffrida R, Creighton C, Hernandez-Herrera A, Jacobsen A, Levine D, Mankoo P, Mavrakis K, Van Der Meulen J, Wolfe A, Liu X, Mets E, Ooi C, Oh H, Wang H, Tan A, Wu J, Jemal A, Yuasa Y, Shimada S, Mimata A, Sekine M, Mogushi K, Akiyama Y, Egger G, Liang G, Aparicio A, Jones P, Lujambio A, Ropero S, Ballestar E, Fraga M, Cerrato C, Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K, Hashimoto Y, Akiyama Y, Otsubo T, Shimada S, Yuasa Y, Cheung H, Davis A, Lee T, Pang A, Nagrani S, Grady W, Parkin R, Mitchell P, Lee J, Kim Y, Jimeno A, Messersmith W, Hirsch F, Franklin W, Eckhardt S, Merritt W, Bar-Eli M, Sood A, White N, Chow T, Mejia-Guerrero S, Diamandis M, Rofael Y, Mencia N, Selga E, Noé V, Ciudad C, Wang Y, Lee A, Ma J, Wang J, Ren J, Gokhale A, Kunder R, Goel A, Sarin R, Moiyadi A, Tahimic C, Tomimatsu N, Nishigaki R, Fukuhara A, Toda T, Cummins J, He Y, Leary R, Pagliarini RLD, Jr, Yanagihara K, Tanaka H, Takigahira M, Ino Y, Yamaguchi Y, Yanagihara K, Kamada N, Tsumuraya M, Amano F, Wen X, Miyake S, Akiyama Y, Yuasa Y. Multiple-to-Multiple Relationships between MicroRNAs and Target Genes in Gastric Cancer. PLoS One. 2013;8:e62589. doi: 10.1371/journal.pone.0062589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.