

Abstract
Background
Chronic alcohol use disorders (AUD) are associated with white matter (WM) degeneration with altered myelin integrity. Matrix assisted laser desorption ionization-imaging mass spectrometry (MALDI-IMS) enables high through-put analysis of myelin lipid biochemical histopathology to help characterize disease mechanisms.
Purpose
This study utilized MALDI-IMS to interrogate frontal lobe WM myelin lipid abnormalities in AUD.
Methods
Standardized cores of formalin fixed WM from Brodmann Area 4 (BA4) and BA8/9 of 20 postmortem AUD and 19 control adult human brains were embedded in carboxymethyl-cellulose, cryo-sectioned (8 μm), thaw-mounted onto indium tin oxide (ITO)-coated glass slides, and sublimed with 2,5-dihydroxybenzxoic acid (DHB) matrix. Lipids were imaged by MALDI-time of flight in the negative ionization mode. Data were visualized with FlexImaging software v4.0 and analyzed with ClinProTools v3.0.
Results
Principal component analysis (PCA) and data bar plots of MALDI-IMS data differentiated AUD from control WM. The dominant effect of AUD was to broadly reduce expression of sphingolipids (sulfatides and ceramides) and phospholipids. Data bar plots demonstrated overall similar responses to AUD in BA4 and BA8/9. However, differential regional effects of AUD on WM lipid profiles were manifested by non-overlapping expression or discordant responses to AUD for a subset of lipid ions.
Conclusions
Human AUD is associated with substantial inhibition of frontal lobe WM lipid expression with regional variability in these effects. MALDI-IMS can be used to characterize the nature of AUD-associated lipid biochemical abnormalities for correlation with lifetime exposures and WM degeneration, altered gene expression, and responses to abstinence or treatment.
Introduction
In adults, chronic alcohol abuse causes brain atrophy (C. Harper, 1982) with selective loss of white matter (WM) (S. M. de la Monte, 1988) and impairments in executive function (Chanraud et al., 2007). Degrees of WM atrophy correlate with maximum daily and lifetime alcohol exposures (S. M. de la Monte & Kril, 2014; C. Harper, Dixon, Sheedy, & Garrick, 2003; Sutherland et al., 2013). Neuroimaging studies showed that the corpus callosum is a vulnerable target of atrophy in people with alcohol use disorders (AUD) (Estruch et al., 1997; Pfefferbaum, Rosenbloom, Adalsteinsson, & Sullivan, 2007). Other notable targets of neurodegeneration in AUD include frontal, temporal, and cerebellar WM (S. M. de la Monte & Kril, 2014; Kril & Halliday, 1999; Phillips, Harper, & Kril, 1987). Diffusion tensor imaging studies predict that the underlying basis of atrophy is disruption of WM micro-structural integrity (Pfefferbaum, Adalsteinsson, & Sullivan, 2006; Schulte, Sullivan, Muller-Oehring, Adalsteinsson, & Pfefferbaum, 2005).
Cerebral WM is largely composed of myelin, a lipid-rich membrane synthesized and maintained by oligodendrocytes. Wrapping of oligodendrocyte myelin sheaths around central nervous system (CNS) axons enables rapid and efficient neuroconductivity. Correspondingly, loss of myelin or impaired myelin homeostasis leads to deficits in CNS functions, including cognition. Major CNS WM lipids include cholesterol, glycosphingolipids, i.e. cerebrosides (galactosylceramide, galactocerebroside), sulfatides (sulfated galactocerebroside, sulfogalactosylceramide), and gangliosides, and phospholipids, consisting of glycerophospholipids (phosphatidic acid (PA), phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylglycerol (PG), phosphatidylinositol (PI), phosphatidylserine (PS) and plasmalogens) and sphingomyelin (Quarles, Macklin, & Morell, 2006). Sphingomyelin is composed of ceramide plus a phosphocholine or phosphoethanolamine polar head group (Quarles et al., 2006).
Abnormal metabolism and expression of phospholipids and sulfatides occur in a broad range of CNS diseases (Takahashi & Suzuki, 2012) including experimental alcohol mediated WM degeneration (Roux et al., 2015; E. B. Yalcin, Nunez, Tong, & de la Monte, 2015). The mechanisms and consequences of aberrant myelin lipid expression are not well understood. However, some effects can be predicted based on specific functions of major lipid subtypes. Since membrane phospholipids regulate lipid rafts and receptor functions, their deficiencies could lead to impairments in intracellular signaling. Sulfatides, localized on the extracellular leaflet of myelin plasma membranes and synthesized by oligodendrocytes (Vos, Lopes-Cardozo, & Gadella, 1994) through sulfonation of galactocerobroside, regulate neuronal plasticity, memory, myelin maintenance, protein trafficking, adhesion, glial-axonal signaling, insulin secretion, and oligodendrocyte survival (Takahashi & Suzuki, 2012). Correspondingly, reductions in membrane sulfatide disrupt myelin sheath structure and function, and compromise neuronal conductivity (Kolesnick & Kronke, 1998). Sulfatide degradation via increased galactosylceramidase, sulfatidase or aryl sulfatase activities yields ceramides (Eckhardt, 2008; Sundaram, Fan, & Lev, Ziemann; Vos et al., 1994) that promote neuroinflammation, reactive oxygen species formation, apoptosis, and dysregulated signaling through cell survival and metabolic pathways (Kolesnick & Kronke, 1998).
Despite abundant information about ethanol’s adverse effects on WM, details about the biochemical nature of degeneration have not been well characterized due to the lack of suitable tools to efficiently study pathologic alterations in lipid-rich myelin. Fortunately, over the past several years, major advances in technology and computational science have facilitated extension of Matrix Assisted Laser Desorption Ionization Imaging Mass Spectrometry (MALDI-IMS) to human research. MALDI-IMS is used for in situ imaging of lipids, proteins, and adducts for correlation with histopathology and molecular pathology (in situ hybridization and immunohistochemistry) (Caprioli, Farmer, & Gile, 1997). Instruments equipped with an Nd:YAG Smartbeam laser enable time of flight (TOF; m/z) analysis for specific identification of molecules (E. B. Yalcin & de la Monte, 2015). For this study, we utilized MALDI-IMS to characterize AUD-associated alterations in frontal lobe WM lipid ion profiles in human postmortem brains.
Methods
Human Subjects
The use of human subject tissue was approved by the Institutional Review Boards at the Rhode Island Hospital and University of Sydney. Postmortem formalin-fixed human adult brain tissue samples from 20 patients with AUD and 19 without CNS disease (controls) were obtained from the New Southwales Brain Tissue Resource Center in Sydney, Australia. The mean ages, proportions of men and women, durations of alcohol exposure, high rates of regular tobacco use, and mean postmortem brain pH were similar in the control and alcoholic groups (Table 1). In contrast, the mean lifetime quantity of alcohol consumed was significantly greater, postmortem delay was significantly longer, and mean brain weight was significantly lower in the alcoholic group.
Table 1.
Frontal lobe white matter was obtained from postmortem human adult brains banked in the New Southwales Brain Tissue Resource Center in Sydney, Australia. Intergroup statistical comparisons of mean values were made by T-test analysis and proportions were compared using Chi-Square tests.
| Characteristic | Alcoholics | Controls | P-Value |
|---|---|---|---|
| Number of Cases | 20 | 19 | N.S. |
| Age (mean ± SD; Range) | 55.2 ± 7.2 (40-67) | 54.4 ± 6.9 (40-67) | N.S. |
| Male/Female | 11/9 | 11/8 | N.S. |
| Duration of drinking (yrs) | 32.6 ± 5.2 | 29.8 ± 9.9 | N.S. |
| Lifetime alcohol consumption (kg) | 7833 ± 6064 | 447 ± 521 | <0.0001 |
| Tobacco abuse (y/n) | 10/10 | 9/10 | N.S. |
| Postmortem interval | 39.5 ± 15.4 | 26.6 ± 10.2 | 0.004 |
| Brain pH | 6.6 ± 0.2 | 6.5 ± 0.3 | N.S. |
| Brain Weight (g) | 1322 ± 161 | 1432 ± 131 | 0.02 |
Sample Preparation
At the initial brain dissection, one hemisphere was fixed for 3 weeks in 15% formalin and then embedded in agar to generate 3-mm interval sequential coronal slices that were stored in 10% formalin (C. Harper et al., 2003; Sutherland, Sheedy, & Kril, 2014). Two standardized blocks from the superior frontal gyrus (Brodmann Area 8/9; BA8/9) and the middle frontal gyrus (Brodmann Area 4; BA4) were dissected from each case and used in this study. Although BA8/9 is generally regarded as the frontal eye field, its functions are quite broad and diverse, including memory (short-term, spatial, semantic and perceptual, and recognizing emotions of others), language skills (verbal fluency), sustained attention, and executive functions (deductive and inductive reasoning, planning) {Watanabe, 2017 #4906; Technologies, 2012 #4905;Abernathy, 2010 #4907}. In contrast, BA4, located just anterior to the central fissure, is the primary motor cortex and responsible for relaying voluntary motor functions along pyramidal pathways through the brainstem and spinal cord {Technologies, 2012 #4905}. BA8/9 was of interest because of its role in drug and alcohol addiction and well-documented adverse effects of alcohol abuse on the prefrontal cortex, including BA8/9 {Abernathy, 2010 #4907}. Alcohol also affects the primary motor cortex by enhancing paired associative transcranial magnetic stimulation of long term depression-like plasticity, enhancing intracortical inhibition and suppressing intracortical facilitation {Ziemann, 1995 #4909;Srivanitchapoom, 2016 #4910}. Although the data suggest that the functional consequences of alcohol differ for the prefrontal and primary motor cortex, microarray studies showed that chronic alcohol misuse significantly alters myelin gene expression in both BA8/9 and BA4 {Mayfield, 2002 #4908}.
Matrix-assisted laser desorption/ionization imaging mass spectrometry (MALDI-IMS)
After thorough rinsing in phosphate buffered saline (PBS) for 48 hours at 4°C, the formalin-fixed tissue blocks were blotted dry and a 6-mm diameter disc of cortex-free WM was excised using a disposable punch biopsy coring tool. Color-coding and orientation of each specimen were achieved by micro-dot (1 μl) patterned labeling of the edges with surgical biopsy ink (MarginMarker; Vector Surgical, Waukesha, WI). The tissue disks were cryo-embedded in 2% sodium carboxymethylcellulose (CMC). Cryosections (8 μm) were thaw-mounted onto indium tin oxide (ITO)-coated slides (Delta Technologies, Loveland, CO) and coated by sublimation with 2,5-dehydroxybenzoic acid (DHB; Sigma-Aldrich Co, St. Louis, MO) as matrix (Angel, Spraggins, Baldwin, & Caprioli, 2012; Emine B. Yalcin, Nunez, Cornett, & de la Monte, 2015; E.B. Yalcin, Nunez, Tong, Cornett, & de la Monte, 2015). The sections were imaged in the negative ion mode using a reflectron geometry MALDI-time-of-flight (TOF)/TOF mass spectrometer (Ultraflextreme, Bruker Daltonics, Bremen, Germany), and analyzed by focusing a Smartbeam II Nd:YAG laser onto ~100 μm2 areas of white matter (Jackson, Wang, & Woods, 2007; Emine B. Yalcin et al., 2015; E.B. Yalcin et al., 2015). Negative ion mode imaging is optimum for detecting sulfatides and most phospholipids. In contrast, ceramides, phosphotidylcholine, sphingomyelin, and cholesterol were not studied because they are optimally imaged in positive rather than negative ionization mode mass spectrometry (Berry et al., 2011; Lohmann, Schachmann, Dandekar, Villmann, & Becker, 2010).
Lipid assignments
Phospholipids and sphingolipids were identified by generating product ion spectra with tandem mass spectrometry (LIFT-TOF/TOF) and fragment ion searches in LIPID MAPS (http://www.lipidmaps.org/tools/index.html). Lipid ion assignments/identifications were made by comparing the precursor and product ion m/z values with catalogued data in LIPID MAPS and confirmed by TOF as previously described (Emine B. Yalcin et al., 2015; E.B. Yalcin et al., 2015). Alternatively, lipid ion assignments were based on published reports (Dreisewerd et al., 2007; Eckhardt et al., 2007; Fernandez-Lima et al., 2011; Gode & Volmer, 2013; Hsu & Turk, 2000; Shanta et al., 2011).
Data Analysis
MALDI data were processed using FlexAnalysis v3.4 (Bruker Daltonics, Billerica, MA) and visualized with FlexImaging software v4.0 (Bruker Daltonics, Billerica, MA). Results were normalized to total ion count and analyzed using ClinProTools v3.0 (Bruker Daltonics, Billerica, MA). Principal component analysis (PCA) was used to generate 2-D and 3-D plots and compare the AUD and control groups with respect to their overall patterns of lipid ion expression. Inter-group comparisons of the mean ion expression levels were made using T-tests and applying a 5% false discovery rate. Effects of AUD were displayed with R-generated data bar plots (Version 3.2; ggplot2 module) that were scaled by calculating the mean percentage changes in lipid ion levels associated with AUD, focusing on m/z’s between 600 and 1200 Daltons. Overall effects of AUD on different subclasses of lipids were evaluated by Chi-square analysis. P-values less than 0.05 were considered statistically significant.
Results
Frontal Lobe WM Lipid Ion Profiles
The Peak Statistic report identified 200 lipid ions that had mass/charge (m/z) ratios between 600 and 1200 Daltons. The lipids were categorized as: 1) sphingolipids (n=44; 22%), including 28 (14%) sulfatides, 6 (3%) ceramides, and 10 (5%) glycosphingolipids; 2) phospholipids (n=136; 68%), including 16 (8%) phosphatidic acids, 22 (11%) phosphatidylethanolamines, 23 (11.5%) phosphatidylglycerols, 15 (7.5%) phosphatidylserines, and 54 (27%) phosphatidylinositides; 3) glycerophospho-inositolglycans (n=5; 2.5%); 4) glycerophospholipid (n=1; 0.5%); 5) other (Cytidine 5’-diphosphate glycerol or fatty esters) (n=3; 1.5%); or 6) unidentified (n=17; 8.5%) based on the LIPID MAPS database and published literature (Table 2). The catalog included C13-isotopes of sulfatides, lactosylceramides, and phospholipids. In contrast, most ceramides, phosphotidylcholine, sphingomyelin, and cholesterol were not detected because they are only accessible via positive rather than negative ionization mode mass spectrometry (Berry et al., 2011; Lohmann et al., 2010).
Table 2.
Postmortem formalin-fixed frontal lobe samples from Brodmann Areas (BA) 4 and 8/9 were used for MALDI-IMS to characterize white matter (WM) lipid composition. Lipids were identified based on mass/charge (m/z) and time of flight. Overall and regional WM lipid compositions are listed. Note the regional differences in WM lipid ion profiles.
| Lipid Type | Total- No (%) | BA4- No (%) | BA8/9- No (%) |
|---|---|---|---|
| All Lipids | 200 | 146 | 157 |
| Sphingolipids/Glyceroceramides | 44 (22.0) | 35 (24.0) | 36 (22.9%) |
| Sulfatides | 28 (14.0%) | 24 (16.4%) | 25 (15.9%) |
| Sulfatide | 19 (9.5%) | 15 (10.3%) | 17 (10.8%) |
| C13-Isotope of Sulfatide | 9 (4.5%) | 9 (6.2%) | 8 (5.1%) |
| Ceramides (CER) | 6 (3%) | 6 (4.1%) | 5 (3.2%) |
| Ceramide | 2 (1.0%) | 2 (1.4%) | 3 (1.3%) |
| CerP | 1 (0.5%) | 1 (0.7%) | 1 (0.6%) |
| MIPC-Ceramide phosphoinositol | 3 (1.5%) | 3 (2.0%) | 2 (1.3%) |
| Glycosphingolipids (GSL) | 10 (5.0%) | 5 (3.4%) | 6 (3.8%) |
| Glycosphingolipid | 4 (2.0%) | 2 (1.4%) | 3 (1.9%) |
| LacCer (LACCER) | 3 (1.5%) | 2 (1.4%) | 1 (1.3%) |
| C13-LacCer | 1 (0.5%) | 0 | 1 (1.3%) |
| Galactosyl Ceramide (GLCCER) | 2 (1.0%) | 1 (0.7%) | 1 (1.3%) |
| Phospholipids | 136 (68.0%) | 101 (69.2%) | 104 (66.2%) |
| Phosphatidic acid (PA) | 16 (8.0%) | 12 (8.2%) | 13 (8.3%) |
| C13-Isotope of Phosphatidic acid | 0 | 0 | 0 |
| Phosphatidylethanolamine (PE) | 20 (10.0%) | 13 (8.9%) | 15 (9.6%) |
| C13-Isotope of Phosphatidylethanolamine | 2 (1.0%) | 1 (0.7%) | 1 (1.3%) |
| Phosphatidylglycerol (PG) | 22 (11.0%) | 18 (12.3%) | 17 (10.8%) |
| C13-Isotope of Phosphatidylglycerol | 1 (0.5%) | 0 | 0 |
| Phosphatidylserine (PS) | 14 (7.0%) | 11 (7.5%) | 12 (7.6%) |
| C13-Isotope of Phosphadidylserine | 1 (0.5%) | 0 | 1 (1.3%) |
| Phosphatidylinositide (PI) | 47 (23.5%) | 34 (23.3%) | 32 (20.4%) |
| C13-Isotope of Phosphatidylinositide | 7 (3.5%) | 7 (4.8%) | 7(4.5%) |
| Glycerophsophoinositolglycans | 5 (2.5%) | 4 (2.7%) | 5 (3.2%) |
| Glycerophospholipid | 1 (0.5%) | 1 (0.7%) | 1 (1.3%) |
| Other | 3 (1.5%) | 3 (2.05%) | 3 (1.9%) |
| Unidentified | 17 (8.5%) | 7 (4.8%) | 14 (8.9%) |
Regional Differences in WM Lipid Expression
To optimally control the experimental conditions in comparing lipid ion expression within different regions and between AUD and control samples, 8 standardized paired samples of BA4 and BA8/9 WM from control and AUD patients were mounted onto individual ITO-coated slides for simultaneous imaging and data acquisition. The 10 slides used to accommodate all samples were imaged sequentially under identical conditions. Graphic display of the Peak Statistics report revealed consistent within-region lipid profiles and AUD effects across the data set. The main source of variability was that several of the 200 lipid ions identified were not detected in every sample due to very low abundance. To simplify the data presentation, only the lipids detected in both control and AUD samples from the same frontal lobe region were included in the evaluations of AUD’s effects.
Although the profiles were quite similar in BA4 and BA8/9, fewer lipid ions were detected in both control and AUD samples from BA4 (n=146) compared with BA8/9 (n=157) (Table 3). BA8/9 expressed 23 ions that were not detected in BA4, and BA4 expressed 11 lipid ions that were not detected in BA8/9 (Table 3). Corresponding with the greater number of detected lipid ions, BA4 WM differentially expressed 5 sphingolipids, 10 phospholipids, CDP-DG(40:4), and 7 unidentified lipids that were not observed in BA8/9. BA8/9 differentially expressed just 1 sphingolipid and 10 phospholipids that were not expressed in BA4 (Table 3). Therefore, the WM lipid profiles were not identical in BA4 and BA8/9. These findings were unrelated to AUD.
Table 3.
Results from Table 2 were culled to demonstrate discordant regional expression (presence or absence) of specific lipid ions.
| Mass | Identification | BA4+ Only | BA8/9+ Only |
|---|---|---|---|
| Sphingolipids | |||
| 877.67 | ST(40:2)(OH)- C13 Isotope | YES | |
| 860.20 | ST(40:2) | YES | |
| 874.16 | ST(41:2) | YES | |
| 886.69 | ST(42:3) | YES | |
| 915.66 | LacCer(d38:2)- C13 Isotope | YES | |
| 1063.63 | Glycosphingolipid (GalNAcβ1-4Galβ1-4Glcβ-Cer(d18:1/16:0)) | YES | |
| Phospholipids | |||
| 633.85 | PA(31:0) | YES | |
| 706.42 | PE(34:6) | YES | |
| 689.78 | PG(30:2) | YES | |
| 731.96 | PG(33:2) | YES | |
| 817.25 | PI(33:3) | YES | |
| 863.01 | PI(36:1) | YES | |
| 883.74 | PI(38:5) | YES | |
| 875.09 | PI(38:9) | YES | |
| 843.21 | PI(O-36:4) | YES | |
| 937.90 | PS(48:12)- C13 Isotope | YES | |
| 699.38 | PA(36:2) | YES | |
| 731.57 | PA(39:7) | YES | |
| 696.36 | PE(33:4) | YES | |
| 718.13 | PE(34:0) | YES | |
| 736.98 | PE(36:5) | YES | |
| 879.04 | PG(44:5) | YES | |
| 931.95 | PI(41:2) | YES | |
| 941.87 | PI(42:4) | YES | |
| 951.94 | PI: PIP(37:4) | YES | |
| 900.66 | PS(44:1) | YES | |
| Other | |||
| 1056.16 | CDP-DG(40:4) | YES | |
| 950.91 | Not Identified | YES | |
| 952.95 | Not Identified | YES | |
| 1068.11 | Not Identified | YES | |
| 1079.12 | Not Identified | YES | |
| 1092.68 | Not Identified | YES | |
| 1100.61 | Not Identified | YES | |
| 1107.65 | Not Identified | YES |
ST = sulfatide; PA = phosphatidic acid; PE = phosphatidylethanolamine; PG = phosphatidylglycerol; PI = phosphatidylinositol; PS = phosphatidylserine; not identified = not yet listed in lipid databases or literature.
AUD Effects on Frontal WM Lipid Profiles Demonstrated by Principal Component Analysis (PCA)
PCA was used to compare control and AUD groups with respect to the patterns of lipid ion expression. The 3-D (Figure 1A) and 2-D (Figures 1B-1D) plots of BA4 profiles demonstrated excellent separation of the groups in the PC1xPC2 (Figure 1B) and PC1xPC3 (Figure 1C) planes, but moderate overlap in the PC2xPC3 (Z) plane (Figure 1D). The clearly separated points depict differential effects of AUD on white matter lipids, whereas the overlapping regions reflect shared lipid ion profiles. Therefore, AUD was associated with substantial alterations in lipid ion profiles in BA4 WM. For BA8/9, PCA revealed well-delineated regions of extensive overlap or clear separation between the AUD and control groups (Figures 1E-1H). The regionally disparate PCA results (BA4 versus BA8/9) suggest that the effects of chronic alcohol misuse on WM lipid profiles are not uniform throughout the brain.
Figure 1.
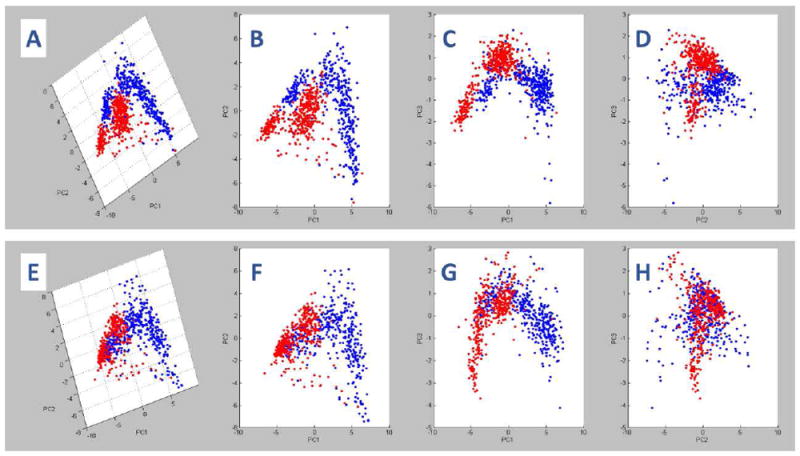
Principal component analysis (PCA) of imaging mass spectrometry (IMS) data acquired in the negative ionization mode of MALDI mass spectrometry: ClinProTools was used for PCA of the total IMS spectra generated from (A-D) BA4 and (E-H) BA8/9 control (blue) and AUD (red) frontal lobe white matter (WM) regions of interest (ROIs). Panels A and E represent the 3-D PCA images. B-D and F-H correspond to 2-D rotated images of A and E, respectively. In each region, two distinct but overlapping clusters were identified corresponding to control and AUD samples. Non-overlapping components of each spectra reflect differential effects of AUD on frontal WM lipid expression in BA4 and BA8/9.
AUD Effects on Frontal Lobe WM Lipid Ion Expression-Sorted by m/z
Comparative analysis of results obtained from 5 independent probings of 20 AUD and 19 control paired BA4 and BA8/9 samples revealed consistent within-region lipid profiles and relative effects of AUD for m/z’s ranging from 611.62 to 1107 Da. To simplify the data presentation, representative results from 8 paired and simultaneously imaged samples from two regions each are shown and discussed. To assess the effects of AUD on lipid expression, the mean peak intensities (reflecting lipid ion abundance) were compared by T-test analysis with a 5% false discovery rate correction. The calculated percentage differences in mean lipid ion abundance were graphed in data bar plots to illustrate and compare relative effects of AUD on BA4 and BA8/9 WM lipid profiles. Results were sorted by ascending m/z values (Figure 2). Bars to the left of the median axis convey AUD-associated reductions in lipid expression, whereas those to the right indicate AUD-associated increases in lipid expression. Differences below 5% were regarded as unchanged. Significant differences (P<0.05) obtained by T-test analysis are color-coded in the columns to the right of the corresponding data bars. The overall effects of AUD on the expression levels of different lipid classes are summarized in Tables 4-5.
Figure 2.
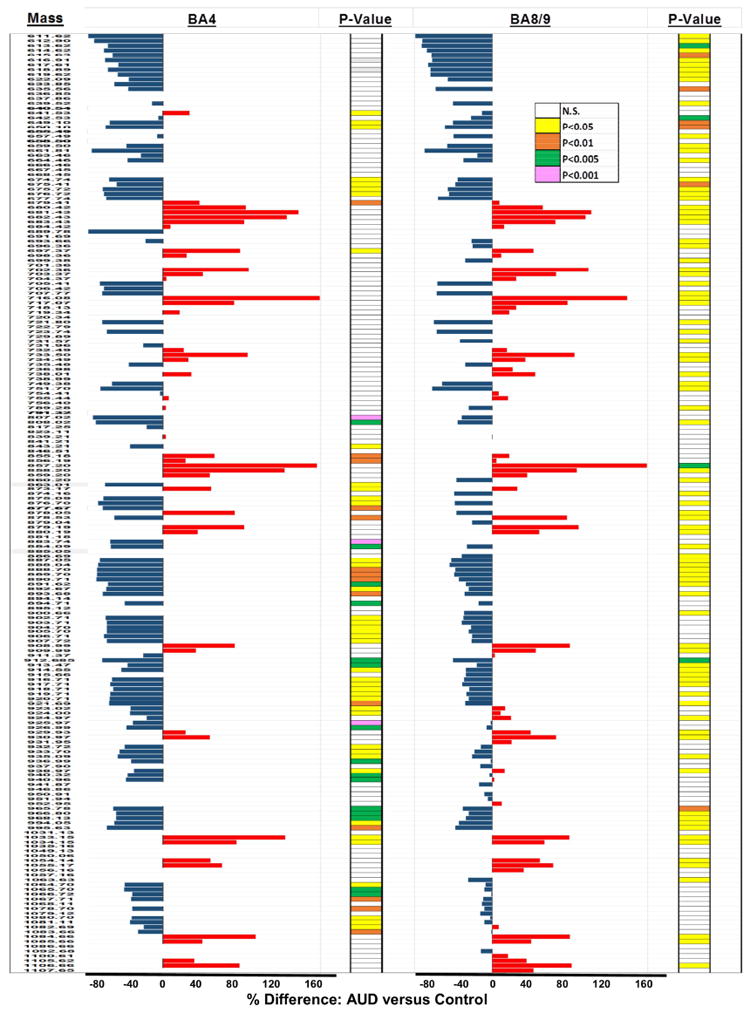
Data Bar Plots illustrating effects of AUD on lipid ion expression in BA4 and BA8/9 human frontal lobe WM as detected by MALDI-IMS in the negative ionization mode: Plots depict the calculated percentage differences in the mean levels of each lipid ion expressed. The scale bars depict the range of responses from -80% to +160% relative to control. T-test results (P-Values) from comparing the mean levels of each lipid ion in control and AUD groups are color-coded to the right of each data bar plot. Results are organized with respect to increasing m/z values. See Supplementary Table 1 for lipid identifications. Blue bars to the left of the vertical axis reflect AUD-associated reductions in lipid expression, and red bars to the right correspond to mean percentage increases in lipid expression.
Table 4.
Effects of AUD on subtypes of lipid ions expressed in the BA4 region of frontal lobe white matter.
| Lipid Type | Number (%) | AUD Effects on Lipid Ion Expression | ||
|---|---|---|---|---|
| Increased | No Change | Decreased | ||
| All Lipids | 146 | 38 (26.0%) | 8 (5.5%) | 100 (68.5%) |
| Sphingolipids/Glyceroceramides | 35 (24.0) | 5 (14.3%) | 2 (5.7%) | 28 (80.0%) |
| Sulfatides | 24 (16.4%) | 3 (12.5%) | 0 | 21 (87.5%) |
| Sulfatide | 15 (10.3%) | 3 (20.0%) | 0 | 12 (80.0%) |
| C13-Isotope of Sulfatide | 9 (6.2%) | 0 | 0 | 9 (100%) |
| Ceramides (CER) | 6 (4.1%) | 1 (16.7%) | 1 (16.7%) | 4 (67.0%) |
| Ceramide | 2 (1.4%) | 0 | 0 | 2 (100%) |
| CerP | 1 (0.7%) | 0 | 0 | 1 (100%) |
| MIPC-Ceramide phosphoinositol | 3 (2.0%) | 1 (33.3%) | 1 (33.3%) | 1 (33.3%) |
| Glycosphingolipids (GSL) | 5 (3.4%) | 1 (10.0%) | 1 (50.0%) | 3 (40.0%) |
| Glycosphingolipid | 2 (1.4%) | 1 (50.0%) | 1 (50.0%) | |
| LacCer (LACCER) | 2 (1.4%) | 0 | 0 | 2 (100%) |
| C13-LacCer | 0 | 0 | 0 | 0 |
| Galactosyl Ceramide (GLCCER) | 1 (0.7%) | 0 | 1 (100%) | 0 |
| Phospholipids | 101 (69.2%) | 30 (29.7%) | 6 (5.9%) | 65 (64.4%) |
| Phosphatidic acid (PA) | 12 (8.2%) | 3 (25.0%) | 1 (8.3%) | 8 (66.7%) |
| C13-Isotope of Phosphatidic acid | 0 | 0 | 0 | 0 |
| Phosphatidylethanolamine (PE) | 13 (8.9%) | 7 (53.8%) | 1 (7.7%) | 5 (38.5%) |
| C13-Isotope of Phosphatidylethanolamine | 1 (0.7%) | 1 (50%) | 0 | 0 |
| Phosphatidylglycerol (PG) | 18 (12.3%) | 4 (22.2%) | 1 (5.6%) | 13 (72.2%) |
| C13-Isotope of Phosphatidylglycerol | 0 | 0 | 0 | 0 |
| Phosphatidylserine (PS) | 11 (7.5%) | 2 (18.1%) | 2 (18.1%) | 7 (63.6%) |
| C13-Isotope of Phosphadidylserine | 0 | 0 | 0 | 0 |
| Phosphatidylinositide (PI) | 34 (23.3%) | 9 (26.5%) | 1 (2.9%) | 24 (70.6%) |
| C13-Isotope of Phosphatidylinositide | 7 (4.8%) | 1 (14.3%) | 0 | 6 (85.7%) |
| Glycerophsophoinositolglycans | 4 (2.7%) | 2 (50%) | 0 | 2 (50%) |
| Glycerophospholipid | 1 (0.7%) | 1 (100%) | 0 | 0 |
| Other | 3 (2.05%) | 1 (33%) | 0 | 2 (67%) |
| Unidentified | 7 (4.8%) | 2 (28.6%) | 0 | 5 (71.4%) |
These data summarize results depicted in the databar plot shown in Figure 2 (left side).
No change: Percentage difference in lipid ion abundances was less than 5% for AUD versus control.
Table 5.
Effects of AUD on subtypes of lipid ions expressed in the BA8/9 region of frontal lobe white matter.
| Lipid Type | Number (%) | AUD Effects on Lipid Ion Expression | ||
|---|---|---|---|---|
| Increased | No Change | Decreased | ||
| All Lipids | 157 | 52 (33.1%) | 12 (7.6%) | 93 (59.2%) |
| Sphingolipids/Glyceroceramides | 36 (22.9%) | 6 (16.2%) | 1 (2.7%) | 29 (78.4%) |
| Sulfatides | 25 (15.9%) | 3 (12.0%) | 1 (4.0%) | 21 (84.0%) |
| Sulfatide | 17 (10.8%) | 3 (17.6%) | 0 | 14 (82.4%) |
| C13-Isotope of Sulfatide | 8 (5.1%) | 0 | 1 (12.5%) | 7 (87.5%) |
| Ceramides (CER) | 5 (3.2%) | 2 (40.0%) | 0 | 3 (60.0%) |
| Ceramide | 3 (1.3%) | 0 | 0 | 2 (100%) |
| CerP | 1 (0.6%) | 0 | 0 | 1 (100%) |
| MIPC-Ceramide phosphoinositol | 2 (1.3%) | 2 (100%) | 0 | 0 |
| Glycosphingolipids (GSL) | 6 (3.8%) | 1 (16.7%) | 0 | 5 (83.3%) |
| Glycosphingolipid | 3 (1.9%) | 1 (33.3%) | 0 | 2 (66.7%) |
| LacCer (LACCER) | 1 (1.3%) | 0 | 0 | 1 (100%) |
| C13-LacCer | 1 (1.3%) | 0 | 0 | 1 (100%) |
| Galactosyl Ceramide (GLCCER) | 1 (1.3%) | 0 | 0 | 1 (100%) |
| Phospholipids | 104 (66.2%) | 39 (37.5%) | 10 (9.6%) | 55 (52.9%) |
| Phosphatidic acid (PA) | 13 (8.3%) | 4 (30.8%) | 0 | 9 (69.2%) |
| C13-Isotope of Phosphatidic acid | 0 | 0 | 0 | 0 |
| Phosphatidylethanolamine (PE) | 15 (9.6%) | 10 (66.7%) | 0 | 5 (33.3%) |
| C13-Isotope of Phosphatidylethanolamine | 1 (1.3%) | 1 (100%) | 0 | 0 |
| Phosphatidylglycerol (PG) | 17 (10.8%) | 4 (23.5%) | 1 (5.9%) | 12 (70.6%) |
| C13-Isotope of Phosphatidylglycerol | 0 | 0 | 0 | 0 |
| Phosphatidylserine (PS) | 12 (7.6%) | 4 (33.3%) | 3 (25.0%) | 5 (41.7%) |
| C13-Isotope of Phosphadidylserine | 1 (1.3%) | 0 | 0 | 1 (100%) |
| Phosphatidylinositide (PI) | 32 (20.4%) | 11 (34.4%) | 3 (9.4%) | 18 (56.3%) |
| C13-Isotope of Phosphatidylinositide | 7(4.5%) | 2 (28.6%) | 1 (14.3%) | 4 (57.1%) |
| Glycerophosphoinositolglycans | 5 (3.2%) | 2 (40%) | 2 (40%) | 1 (20%) |
| Glycerophospholipid | 1 (1.3%) | 1 (100%) | 0 | 0 |
| Other | 3 (1.9%) | 2 (67%) | 0 | 1 (33%) |
| Unidentified | 14 (8.9%) | 5 (35.7%) | 1 (7.1%) | 8 (57.1%) |
These data summarize results depicted in the databar plot shown in Figure 2 (right side).
No change: Percentage difference in lipid ion abundances was less than 5% for AUD versus control.
The data bar plot for BA4 revealed that AUD reduced expression of 100 (68.5%) of the 146 ions detected, increased expression 38 (26%), and had no effect on 8 (5.5%) relative to control (Figure 2). These inter-group differences were statistically significant (P<0.05 or better) for 74 (50.7%). Furthermore, the data bar plot revealed clustering of AUD’s effects on lipid expression such that groups with narrowly different m/z values were found to have similar responses relative to control, i.e. increased, decreased, or minimally altered (less than 5%).
The data bar plot for BA8/9 closely resembled the one generated for BA4, including the clustered patterns of AUD-associated increases, reductions, and minimal effects on lipid expression (Figure 2). Among the 157 lipids detected, 93 (59.2%) were expressed at lower levels, 55 (33.1%) were expressed at higher levels, and 12 (7.6%) were minimally modulated (<5%) by AUD. These inter-group differences in lipid expression were statistically significant for 95 (60.5%) lipid ions. Therefore, the WM lipid profiles were substantially altered by AUD in both BA4 and BA8/9 such that the dominant effect was inhibitory across the full m/z range evaluated.
Differential Effects of AUD on Lipid Ion Expression in BA4 and BA8/9
Side-by-side comparison of the data bar plots for BA4 and BA8/9 revealed that although the directional effects of AUD on relative lipid ion abundance were quite similar, there were clear differences in the magnitudes and directions of responses. First we assessed regional AUD responses by comparing the percentages of each lipid subtype whose relative abundances were significantly altered (P<0.05 or smaller) versus those that were not significantly modulated relative to control (Figure 3). The results were analyzed with Chi-square tests. AUD’s differential effects on sphingolipid versus phospholipid expression were significant in both BA4 and BA8/9 (Figures 3A, 3B). However, AUD disproportionately modulated expression of sphingolipids more that phospholipids in BA4 (Chi-square = 20.74, 1 df; P < 0.0001) but not BA8/9 (Chi-square = 0.15, 1 df; NS).
Figure 3.
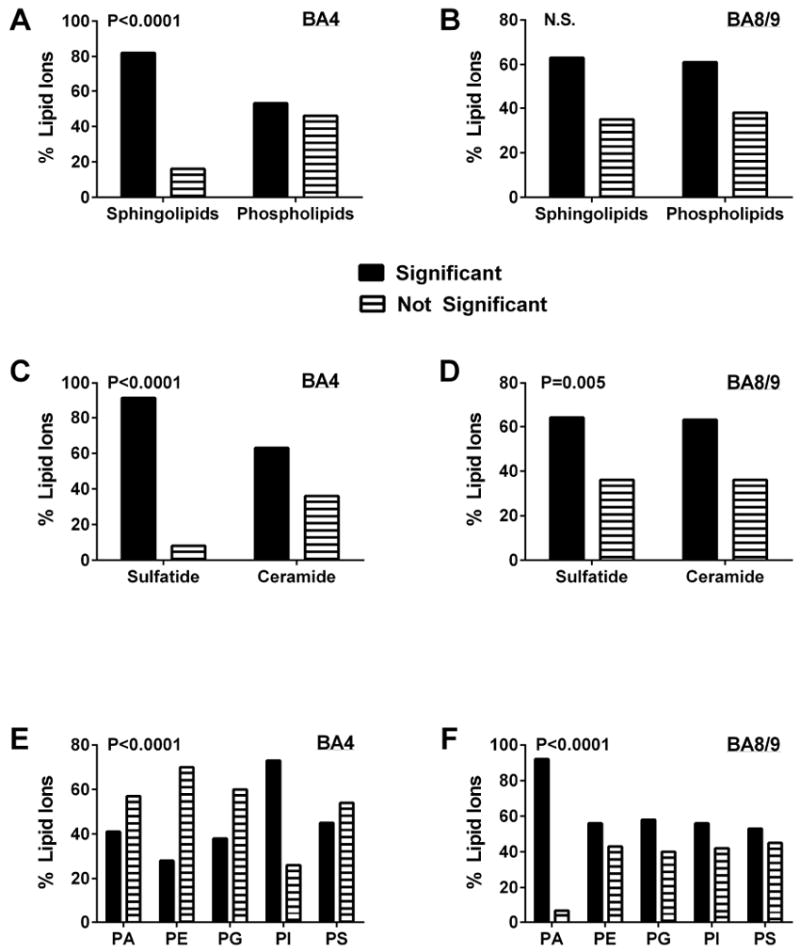
AUD-associated changes (increases or decreases) in the expression of different lipid subtypes: Databar results shown in Figure 2 were sorted and culled to graphically summarize effects of AUD on different lipid subtypes in BA4 (A, C, E) and BA8/9 (B, D, F) of human frontal lobe WM. The graphs depict the percentages of each lipid subtype that were significantly altered in expression level relative to control (increases and decreases were combined). Results were analyzed using Chi-square tests to determine if AUD differentially altered expression of (A, B) sphingolipids versus phospholipids, (C, D) sulfatides versus ceramides, and (E, F) different phospholipid subtypes (PA = phosphatidic acid; PE = phosphatidylethanolamine; PG = phosphatidylglycerol; PI = phosphatidylinositol; PS = phosphatidylserine); P-values are shown within each panel. Phosphatidylcholine (PC) was not detected in the negative ionization mode of mass spectrometry.
AUD was associated with higher percentages of significantly altered versus unaffected levels of sulfatide and ceramide expression in both BA4 (Chi-square = 22.91; 1 df; P<0.0001) and BA8/9 (Chi-square = 12.83, 1 df; P = 0.005). Comparing regional effects of AUD on sulfatides versus ceramides revealed that in BA4, the dominant effect was to significantly alter expression of sulfatides, whereas its effect on ceramides was less robust (Figure 3C). In BA8/9 AUD resulted in proportionally similar significant modulation of sulfatide and ceramide expression (Figure 3D). The most striking differences in regional responses occurred with respect to phospholipids (Figure 3E, 3F). In BA4, the proportions of PA, PE, PG, and PS that were significantly modulated by AUD were similar or lower than the percentages that had no significant change, whereas for PI, nearly twice as many lipids were significantly altered as unaffected (Chi-square = 45.43, 4 df; P<0.0001) (Figure 2E). In contrast, in BA8/9, the main effect of AUD was to significantly modulate expression of PA, PE, PG and PI, but not PS (Chi-square = 45.52, 4 df; P<0.0001) (Figure 3F).
Further characterization of the differential effects of AUD on lipid expression/abundance was performed by tabulating the responses within each lipid category as increased or decreased by greater than 5%, or unchanged (less than 5% difference) relative to control. In BA4, the dominant effect of AUD was to inhibit expression of sphingolipids including sulfatides, C13 isotopes of sulfatides, and ceramides, most phospholipids, other lipids, and unidentified lipids (Table 4, Figures 4A, 4C, 4E). Chi-square analysis of the differential inhibitory effects of AUD on sphingolipids compared with phospholipids failed to reach statistical significance (Chi-square = 3.33, 1 df; P=0.07; Figure 4A). In contrast, among sphingolipids, AUD had disproportionate inhibitory effects on sulfatide compared with ceramide (Chi-square = 24.14, 1 df; P <0.0001) (Figure 4C. Regarding phospholipids, AUD mainly inhibited PA, PG, PI and PS, but disproportionately stimulated PE (Figure 4E). However, these discordant directional effects of AUD on the different subtypes of phospholipids did not reach statistical significance by Chi-square analysis (Chi-square = 9.79, 4 df; N.S.).
Figure 4.
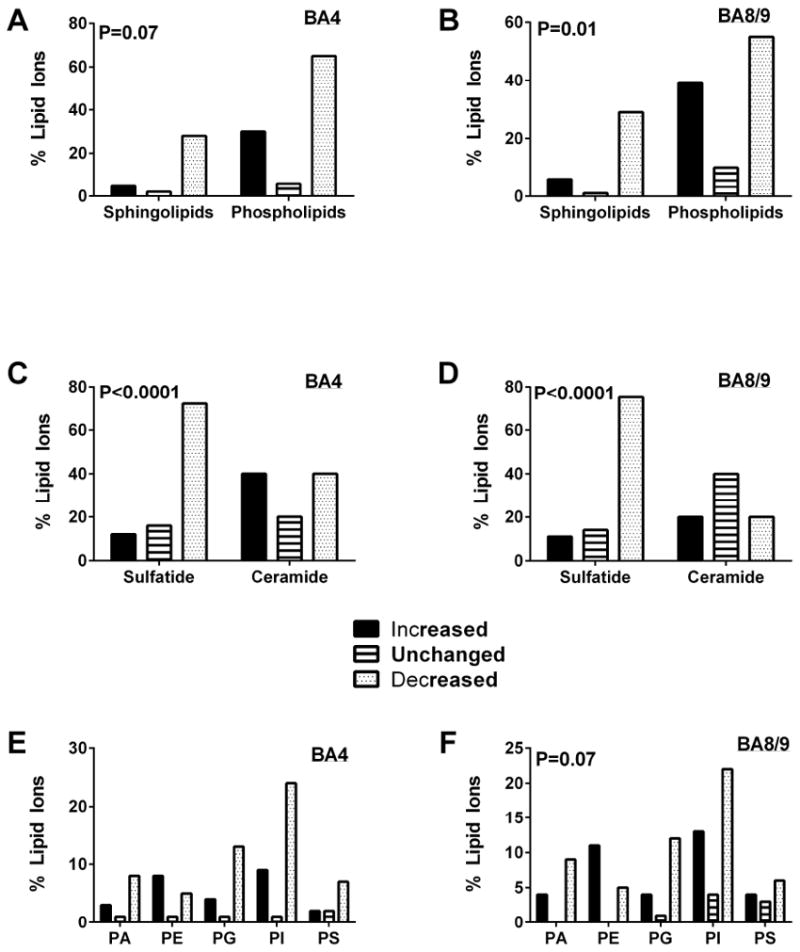
Inhibitory versus stimulatory effects of AUD on relative lipid ion expression in the (A, C, E) BA4 and (B, D, F) BA8/9 regions of human frontal lobe WM. Results in Figure 2 are summarized and graphically displayed to compare the percentages of (A, B) sphingolipids and phospholipids, (C, D) sulfatides and ceramides, and (E, F) different phospholipid subtypes that were significantly increased, unchanged, or significantly decreased in AUD relative to control samples. Data were analyzed using Chi-square tests and corresponding P-values are shown within the panels. Phosphatidylcholine was not detected in the negative ionization mode of mass spectrometry.
In BA8/9, the overwhelmingly dominant effect of AUD was to inhibit sphingolipid expression, whereas the effects on phospholipids, other lipids, and unidentified lipids were more heterogeneous than in BA4 (Table 5, Figures 4B, 4D, 4F). Chi-square analysis confirmed that the differential inhibitory effect of AUD on sphingolipid versus phospholipid expression in BA8/9 was statistically significant (Chi-square = 8.62, 2 df; P=0.01) (Figure 4B). The selective inhibitory effect of AUD on sulfatide versus ceramide was more pronounced in BA8/9 than BA4 (Chi-square = 45.31, 2 df; P < 0.0001) (Figure 4D). In BA8/9, AUD dominantly inhibited expression of PA, PG, and PI by at least two-fold, and either increased or had neutral effects on PE, PS, glycerophosphoinositolglycans, and other lipids (Figure 4F). However, overall, Chi-square analysis of these differential phospholipid responses did not reach statistical significance (Chi-square = 14.94, 8 df; P=0.06). The regional differences in AUD’s effects were related to discordant directional responses of 12 lipids, including 4 sphingolipids and 8 phospholipids (Table 6). AUD increased expression of one sulfatide and two phospholipids (phosphoinositols) in BA4 but inhibited their expression in BA8/9. In addition, AUD increased expression of 1 ceramide phosphoinositol (MIPC(44:0)), 2 sulfatides, one of which was a C13-isotope, and 6 phospholipids (phosphoinositides and phosphatidylserines), one of which was a C13-isotope in BA8/9 while inhibiting their expression in BA4.
Table 6.
Discordant, i.e. directionally opposite effects of AUD on BA4 and BA8/9 frontal lobe WM expression of 12 specific lipid ions, including 4 sphingolipids and 8 phospholipids were observed.
| Mass | Identification | BA4 | BA8/9 |
|---|---|---|---|
| Sphingolipids | |||
| 1082.69 | MIPC(44:0) | Decreased | Increased |
| 878.05 | ST(40:1)(OH) | Increased | Decreased |
| 878.53 | ST(40:2)(OH) | Decreased | Increased |
| 940.96 | ST(44:2)(OH)- C13 isotope | Decreased | Increased |
| Phospholipids | |||
| 641.53 | PI(21:0) | Increased | Decreased |
| 789.28 | PI(31:3) | Increased | Decreased |
| 911.37 | PI(40:5) | Decreased | Increased |
| 923.02 | PI(41:6) | Decreased | Increased |
| 924.00 | PI(41:6)- C13 isotope | Decreased | Increased |
| 924.97 | PI-Cer(42:0(2OH)) | Decreased | Increased |
| 754.41 | PS(34:4) | Decreased | Increased |
| 938.97 | PS(48:11) | Decreased | Increased |
Discussion
This study investigated the effects of AUD on WM lipid profiles in the prefrontal (BA8/9) and primary motor (BA4) regions of the frontal lobe in humans using formalin fixed archival tissue stored in the NIAAA-funded Brain Tissue Resource Center in Sydney, Australia. The AUD group consumed significantly greater lifetime quantities of alcohol and had significantly lower mean brain weights reflecting atrophy and neurodegeneration. Although the post-mortem interval was longer in the AUD group, the mean pH’s of the brains were similar, suggesting that the degrees to which the tissue harvesting was delayed did not significantly impact tissue integrity. There were no significant inter-group differences regarding other parameters such as age, sex, and tobacco abuse, which could potentially influence outcomes of brain studies.
MALDI-IMS detected similar proportions and spectra of sphingolipids and phospholipids as previously identified in WM with MALDI TOF IMS in the negative ion mode (Nunez et al., 2016; O’Brien & Sampson, 1965; E. B. Yalcin et al., 2015). In contrast, other lipids that are abundantly expressed in myelin including cholesterol, sphingomyelin and phosphatidylcholine were not detected because they are mainly accessible via positive ionization mode mass spectrometry (Berry et al., 2011; Lohmann et al., 2010). One of the goals of this study was to characterize AUD’s effects on sulfatide expression since previous reports indicated that alcohol disproportionately targets sphingolipids (Roux et al., 2015; E. B. Yalcin et al., 2015).
MALDI-IMS detected 200 lipid species in frontal lobe WM. The composition of 22% sphingolipids and 68% glycerophospholipids is consistent with previous analyses of WM from humans and experimental animals using mass spectrometry approaches (O’Brien & Sampson, 1965). A greater number of lipids were identified in BA8/9 than BA4. Although some of the differences were due to extremely low levels (below threshold) of lipid expression, the differentially expressed lipids were as follows: 1 sphingolipid and 10 phospholipids were detected in BA4 only, and 5 sphingolipids, 10 phospholipids, 7 unidentified lipids and CDP-DG(40:4) were detected in BA8/9 and not BA4. These findings suggest that regional heterogeneity exists with respect to brain WM lipid composition. Therefore, to effectively evaluate disease state effects on WM lipid expression, matched brain regions must be examined. One possible explanation for the regional differences in lipid ion expression is that enzymes mediating biosynthesis and degradation of sphingolipids and membrane phospholipids are differentially expressed in BA4 and BA8/9. Correspondingly, since most of the AUD effects were to either decrease or increase rather than completely inhibit or aberrantly express specific lipids, they could have been mediated by functional changes in myelin maintenance enzyme gene expression or activity. This concept is supported by previous studies linking disease-associated abnormalities in brain WM lipid composition to alterated lipid enzyme activity (Fu, Mozzi, Krakowka, Higgins, & Horrocks, 1980; Haughey, Bandaru, Bae, & Mattson, 2010; S. Kim, Steelman, Zhang, Kinney, & Li, 2012; Neu & Woelk, 1982; Ohtani et al., 2004; Ramsey, Banik, Scott, & Davison, 1976).
MALDI-IMS delineated broad and significant AUD-associated alterations in frontal lobe WM myelin lipid profiles with several sub-region distinctions. The PCA plots demonstrated clearer delineations between control and AUD lipid ion profiles in BA4 compared with BA8/9, suggesting that the alterations in WM lipid ion expression were greater in BA4. AUD inhibited expression of nearly twice as many WM lipids as it stimulated in both frontal lobe regions. AUD disproportionately altered expression of sphingolipids versus phospholipids, and sulfatides versus ceramides in BA4, whereas in BA8/9, the proportions of lipids in which expression levels were significantly changed were similar for sphingolipids versus phospholipids and sulfatides versus ceramides. Furthermore, higher percentages of lipid ions were decreased and lower percentages were increased by AUD in BA4 compared with BA8/9.
The above findings illustrate non-uniform effects of AUD on WM lipid composition in the frontal lobe, and suggest that there are regional differences in vulnerability to AUD-mediated WM degeneration. This concept is reinforced by the finding of several regionally discordant effects of AUD. For example, in AUD, the expression levels of 3 sphingolipids and 6 phospholipids were significantly reduced in BA4 but increased in BA8/9, and 1 sphingolipid and 2 phospholipids were increased in BA4 but reduced in BA8/9. Whether these regional differences reflect different stages of WM degeneration or differential responses to AUD cannot be determined by this cross-sectional study and instead requires time course analysis of WM target vulnerability and probably interrogation of experimental models.
On the other hand, general overlapping or shared effects of AUD in BA4 and BA8/9 were observed as manifested by reduced expression of sphingolipids as well as membrane phospholipids, with disproportionately greater inhibitory effects on sphingolipids. In addition, AUD had predominantly inhibitory effects on sulfatide versus ceramide expression in BA4 and BA8/9; the proportions of inhibited or stimulated ceramides were comparable for both regions. Finally, the trends regarding AUDs effects on different phospholipid subtypes were similar in BA4 and BA8/9 such that PA, PG, PI, and PS were mainly inhibited, whereas PE was mainly stimulated. Therefore, it is important to emphasize that despite regional differences in lipid ion detection, levels of lipid expression, and stimulatory versus inhibitory effects on specific lipids, the overall responses in BA4 and BA8/9 WM were in many respects comparable, highlighting fundamental effects of AUD on WM lipid biochemistry. It is noteworthy that the inhibitory effects of AUD on WM sphingolipid and phospholipid expression correspond with previous observations in experimental models of alcohol and/or tobacco-specific nitrosamine exposures (E. B. Yalcin et al., 2015). The broad range over which lipid ion intensities were reduced by AUD could reflect myelin loss or demyelination associated with WM atrophy in alcoholics (S. M. de la Monte, 1988; S. M. de la Monte & Kril, 2014; C. G. Harper, Kril, & Holloway, 1985).
Sulfatides, synthesized from galactose, ceramide and sulfate, are important structural and functional constituents of myelin, and have critical roles in its formation (Honke, 2013; Takahashi & Suzuki, 2012) and stability (Coetzee et al., 1996). Therefore, the striking reductions in sulfatide caused by AUD may have contributed to atrophy of WM (McCorkindale, Sheedy, Kril, & Sutherland, 2016). In the context of normal myelin turnover, AUD may compromise both its de novo biosynthesis and structural stability, and result in hypomyelination during recovery and enhanced myelin degradation with continued exposures. Increased degradation of sulfatides leads to increased levels of ceramide. Ceramides modulate signaling pathways involved in differentiation, senescence, proliferation, inflammation, and apoptosis (Adam, Heinrich, Kabelitz, & Schutze, 2002; Kolesnick & Kronke, 1998; Summers, 2006; Venable). In addition, their neurotoxic effects impair insulin signaling, and thereby drive oxidative injury, metabolic dysfunction, impairments in neuronal plasticity, myelin maintenance and neuronal and glial cell survival in the brain (S. M. de la Monte et al., 2010). Correspondingly, increased ceramide levels have been reported in the context of neurodegeneration (S. M. de la Monte, 2012; S. M. de la Monte, Re, Longato, & Tong, 2012; S. M. de la Monte et al., 2010; Yu et al., 2015), including that caused by alcohol exposures (S. de la Monte, Derdak, & Wands, 2012; S. M. de la Monte, 2013; S. M. de la Monte, Longato, Tong, DeNucci, & Wands, 2009).
The data bar plots illustrated clustered patterns of AUD-associated inhibitory and stimulatory effects on lipid ion expression over the full m/z range that was interrogated. Most of inter-group differences in lipid expression were statistically significant or had statistical trends. The strikingly greater AUD effects on lipid expression compared with the findings in an experimental rat model (E. B. Yalcin et al., 2015) may have been due to the considerably longer durations of alcohol exposure in humans, i.e. years versus weeks, as well as potential cofactor exposures such as smoking (Yu et al., 2015) or nutritional deficiencies (S. M. de la Monte & Kril, 2014). Analysis of the effects of AUD on different lipid subtypes revealed that in BA4, 60% or more of the sulfatides, ceramides, phosphatidic acids, phosphatidylglycerols, phosphatidylserines, and phosphatidylinositides were inhibited while more than 50% of the phosphatidylethanolamines were stimulated. In BA8/9, 60% or more of the sulfatides, ceramides, glycosphingolipids, phosphatidic acids, and phosphatidylglycerols were inhibited while 50% or more of the phosphatidylethanolamines and phosphatidylinositides were increased by AUD. Therefore, although most of the responses overlapped, differential inhibition of phosphatidylserines and phosphatidylinositides in BA4 and stimulation of phosphatidylinositides in BA8/9 provide additional evidence for somewhat selective regional WM myelin lipid vulnerability to AUD.
Phospholipids regulate signal transduction at the plasma membrane. PSs activate Akt networks to promote cell survival, growth, proliferation, and metabolism via insulin-like growth factor, type 1 (IGF-1) signaling (Huang, Akbar, Kevala, & Kim, 2011) and enhance CNS WM myelination (Flores et al., 2008). The AUD-associated inhibition of PS expression corresponds with the previously reported impairments in insulin and IGF-1 signaling through Akt pathways in various ethanol exposure models (Andreani, Tong, Gundogan, Silbermann, & de la Monte, 2016; S. de la Monte et al., 2012; S. M. de la Monte & Wands, 2002; Deochand, Tong, Agarwal, Cadenas, & de la Monte, 2015; Ewenczyk, Ziplow, Tong, Le, & de la Monte, 2012; Xu et al., 2003) and in humans with AUD (S. M. de la Monte, 2013; S. M. de la Monte et al., 2008) and suggests that these effects may be mediated in part by reductions in myelin PS content.
PA is one of the most important intermediates in lipid metabolism and the precursor of all glycerophospholipids in biological membranes (Carman & Henry, 2007). PA is synthesized by phospholipase D hydrolysis of phosphatidylcholine, phosphorylation of diacylglycerol and acylation of lysophosphatidic acid. PA is an essential substrate for enzymes that promote synthesis of triacylglycerols and glycerophospholipids (Athenstaedt & Daum, 1999). In addition, PA is used in the synthesis of PE and PC via diacylglycerol. Enzymes needed for de novo PA biosynthesis are localized in the endoplasmic reticulum and mitochondria (Athenstaedt & Daum, 1999). PA’s diverse roles include regulation of membrane dynamics, signal transduction (Arisz, Testerink, & Munnik, 2009; Stace et al., 2008), transcriptional synthesis of glycerophospholipid (Carman & Henry, 2007), and mitogenic activation of mammalian target of rapamycin complex 1 (mTORC1) which promotes cell growth (Yoon, Sun, Arauz, Jiang, & Chen, 2011). Given its importance in generating many membrane phospholipids, AUD-associated reductions in PA expression would likely have broad adverse effects on the structural and functional integrity of myelin and lipid signaling.
PEs comprise nearly one-quarter of all phospholipids in cells and are abundantly expressed in the brain, including white matter where they comprise 45% of all phospholipids. PE is generated by decarboxylation of PS in mitochondrial membranes, but is transported throughout the cell to other membranes for use. PE is also generated from ethanolamine in the cytosol and ER. Over 70% of the PE in WM is plasmalogen (Chrast, Saher, Nave, & Verheijen, 2011). Plasmalogens play a role in membrane fusion and dynamics (fluidity), and may be involved in myelin membrane formation or maintenance via potentiation of membrane dynamics (Chrast et al., 2011). The significance of the relatively increased levels of PE in AUD WM is not apparent as it is unclear whether the effect is pathologic or positive and compensatory. It is noteworthy that PE is significantly reduced in other neurodegenerative disorders including Alzheimer’s and Parkinson’s diseases (Han, Holtzman, & McKeel, 2001; Wood, 2012; Wood et al., 2010).
PG is synthesized in mitochondria and the precursor for cardiolipin. Cardiolipin is distributed in the inner mitochondrial membrane and required for optimum function of enzymes used in oxidative phosphorylation. Deficits in PG would likely lead to reduced levels of cardiolipin and attendant mitochondrial dysfunction. Previous studies have linked apoptotic cell death in neurodegeneration to altered cardiolipin levels and cardiolipin peroxidation (Schenkel & Bakovic, 2014). Cardiolipin is expressed in mitochondria throughout the central nervous system, including in oligodendrocytes (Pointer & Klegeris, 2016). Mitochondrial dysfunction kills or impairs oligodendrocytes (Jana & Pahan, 2007; Lismont, Nordgren, Van Veldhoven, & Fransen, 2015). In the present study, PG expression was found to be reduced in AUD WM. Mechanistically, AUD-inhibition of PG with attendant reductions in mitochondrial cardiolipin could contribute to WM atrophy due to death or dysfunction of oligodendrocytes and attendant loss of myelin or insufficient myelin maintenance.
PIs constitute a diverse class of phospholipids substrates for lipid kinases which phosphorylate hydroxyl groups, and phospholipases, generating second messengers for regulating cellular signaling, lipid signaling, and membrane fluidity, permeability, and trafficking (Cockcroft & Carvou, 2007; Crews et al., 1986; Gurtovenko & Anwar, 2009; Patra et al., 2006). Therefore, the striking reductions in WM PI content could have a mechanistic role in the impairment of broad biological functions, including PI3 kinase activation of Akt.
PS is a major acidic phospholipid enriched in the inner (cytoplasmic) leaflet of the plasma membrane and synthesized from PE or PC via exchange of its head group with serine. PS forms part of the protein docking sites needed for activation of pro-survival and pro-growth Protein kinase C, Raf-1 and Akt signaling (H. Y. Kim, Huang, & Spector, 2014). PS also regulates neurotransmitter release by exocytosis and the abundance of synaptic proteins and receptors. Previous studies showed that ethanol exposure inhibits DHA-promoted PS synthesis and accumulation in neurons (H. Y. Kim et al., 2014). Finally, PS may have a role in supporting cognitive function (H. Y. Kim et al., 2014). Inhibition of PS in alcohol-related neurodegeneration and cognitive impairment (Harris, Baxter, Mitchell, & Hitzemann, 1984).
In conclusion, this study demonstrates major abnormalities in sphingolipid and phospholipid expression in WM from individuals with AUD. The breadth of impairments in lipid expression point to oligodendrocyte dysfunction with attendant pathological alterations that challenge the structural and functional integrity WM myelin. Future studies will assess whether these considerable adverse effects of chronic alcohol misuse are mediated by impaired expression and function of upstream enzymes that regulate oligodendrocyte myelin membrane lipid homeostasis.
Supplementary Material
Highlights.
Myelin lipid abnormalities associated with frontal lobe white matter atrophy in human alcoholics were evaluated using MALDI imaging mass spectrometry.
Chronic alcohol abuse was associated with broad-ranging inhibition of sphingolipid and phospholipid expression in white matter myelin.
The sub-region differences in the effects of alcohol on white matter lipid profiles indicate that the targets of alcohol-mediated neurodegeneration are non-uniform, which may correspond with clinical and pathological disease characteristics and progression.
MALDI-IMS could be used to evaluate responses to treatments designed to slow or reverse alcohol-mediated WM degeneration.
Acknowledgments
Supported by R37AA-011431, AA024018-01, and R28AA-012725 from the National Institutes of Health
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adam D, Heinrich M, Kabelitz D, Schutze S. Ceramide: does it matter for T cells? Trends Immunol. 2002;23(1):1–4. doi: 10.1016/s1471-4906(01)02091-9. [DOI] [PubMed] [Google Scholar]
- Andreani T, Tong M, Gundogan F, Silbermann E, de la Monte S. Differential Effects of 3rd Trimester-Equivalent Binge Ethanol and Tobacco-Specific Nitrosamine Ketone Exposures on Brain Insulin Signaling in Adolescence. J Diab Rel Dis. 2016;1(1):105–114. [PMC free article] [PubMed] [Google Scholar]
- Angel PM, Spraggins JM, Baldwin HS, Caprioli R. Enhanced sensitivity for high spatial resolution lipid analysis by negative ion mode matrix assisted laser desorption ionization imaging mass spectrometry. Anal Chem. 2012;84(3):1557–1564. doi: 10.1021/ac202383m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arisz SA, Testerink C, Munnik T. Plant PA signaling via diacylglycerol kinase. Biochim Biophys Acta. 2009;1791(9):869–875. doi: 10.1016/j.bbalip.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Athenstaedt K, Daum G. Phosphatidic acid, a key intermediate in lipid metabolism. Eur J Biochem. 1999;266(1):1–16. doi: 10.1046/j.1432-1327.1999.00822.x. [DOI] [PubMed] [Google Scholar]
- Berry KA, Hankin JA, Barkley RM, Spraggins JM, Caprioli RM, Murphy RC. MALDI imaging of lipid biochemistry in tissues by mass spectrometry. Chem Rev. 2011;111(10):6491–6512. doi: 10.1021/cr200280p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprioli RM, Farmer TB, Gile J. Molecular imaging of biological samples: localization of peptides and proteins using MALDI-TOF MS. Anal Chem. 1997;69(23):4751–4760. doi: 10.1021/ac970888i. [DOI] [PubMed] [Google Scholar]
- Carman GM, Henry SA. Phosphatidic acid plays a central role in the transcriptional regulation of glycerophospholipid synthesis in Saccharomyces cerevisiae. J Biol Chem. 2007;282(52):37293–37297. doi: 10.1074/jbc.R700038200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanraud S, Martelli C, Delain F, Kostogianni N, Douaud G, Aubin HJ, et al. Brain morphometry and cognitive performance in detoxified alcohol-dependents with preserved psychosocial functioning. Neuropsychopharmacology. 2007;32(2):429–438. doi: 10.1038/sj.npp.1301219. [DOI] [PubMed] [Google Scholar]
- Chrast R, Saher G, Nave KA, Verheijen MH. Lipid metabolism in myelinating glial cells: lessons from human inherited disorders and mouse models. J Lipid Res. 2011;52(3):419–434. doi: 10.1194/jlr.R009761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockcroft S, Carvou N. Biochemical and biological functions of class I phosphatidylinositol transfer proteins. Biochim Biophys Acta. 2007;1771(6):677–691. doi: 10.1016/j.bbalip.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, et al. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 1996;86(2):209–219. doi: 10.1016/s0092-8674(00)80093-8. [DOI] [PubMed] [Google Scholar]
- Crews FT, Gonzales RA, Palovcik R, Phillips MI, Theiss C, Raizada M. Changes in receptor stimulated phosphoinositide hydrolysis in brain during ethanol administration, aging, and other pathological conditions. Psychopharmacol Bull. 1986;22(3):775–780. [PubMed] [Google Scholar]
- de la Monte S, Derdak Z, Wands JR. Alcohol, insulin resistance and the liver-brain axis. [Research Support, N.I.H., Extramural Review] J Gastroenterol Hepatol. 2012;27(Suppl 2):33–41. doi: 10.1111/j.1440-1746.2011.07023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM. Disproportionate atrophy of cerebral white matter in chronic alcoholics. Arch Neurol. 1988;45(9):990–992. doi: 10.1001/archneur.1988.00520330076013. [DOI] [PubMed] [Google Scholar]
- de la Monte SM. Metabolic derangements mediate cognitive impairment and Alzheimer’s disease: role of peripheral insulin-resistance diseases. Panminerva Med. 2012;54(3):171–178. [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM. Alcohol-induced liver and brain degeneration: roles of insulin resistance, toxic ceramides, and endoplasmic reticulum stress. In: P VR, Watson Ronald Ross, Zibadi Sherma, editors. Alcohol, Nutrition, and Health Consequences. New York: Humana Press; 2013. pp. 507–520. [Google Scholar]
- de la Monte SM, Kril JJ. Human alcohol-related neuropathology. Acta Neuropathol. 2014;127(1):71–90. doi: 10.1007/s00401-013-1233-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, Longato L, Tong M, DeNucci S, Wands JR. The liver-brain axis of alcohol-mediated neurodegeneration: role of toxic lipids. [Review] Int J Environ Res Public Health. 2009;6(7):2055–2075. doi: 10.3390/ijerph6072055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, Re E, Longato L, Tong M. Dysfunctional pro-ceramide, ER stress, and insulin/IGF signaling networks with progression of Alzheimer’s disease. [Research Support, N.I.H., Extramural] J Alzheimers Dis. 2012;30(Suppl 2):S217–229. doi: 10.3233/JAD-2012-111728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, Tong M, Cohen AC, Sheedy D, Harper C, Wands JR. Insulin and insulin-like growth factor resistance in alcoholic neurodegeneration. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] Alcohol Clin Exp Res. 2008;32(9):1630–1644. doi: 10.1111/j.1530-0277.2008.00731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, Tong M, Nguyen V, Setshedi M, Longato L, Wands JR. Ceramide-mediated insulin resistance and impairment of cognitive-motor functions. [Research Support, N.I.H., Extramural] J Alzheimers Dis. 2010;21(3):967–984. doi: 10.3233/JAD-2010-091726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, Wands JR. Chronic gestational exposure to ethanol impairs insulin-stimulated survival and mitochondrial function in cerebellar neurons. Cell Mol Life Sci. 2002;59(5):882–893. doi: 10.1007/s00018-002-8475-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deochand C, Tong M, Agarwal AR, Cadenas E, de la Monte SM. Tobacco Smoke Exposure Impairs Brain Insulin/IGF Signaling: Potential Co-Factor Role in Neurodegeneration. J Alzheimers Dis. 2015;50(2):373–386. doi: 10.3233/JAD-150664. [DOI] [PubMed] [Google Scholar]
- Dreisewerd K, Lemaire R, Pohlentz G, Salzet M, Wisztorski M, Berkenkamp S, et al. Molecular profiling of native and matrix-coated tissue slices from rat brain by infrared and ultraviolet laser desorption/ionization orthogonal time-of-flight mass spectrometry. Anal Chem. 2007;79(6):2463–2471. doi: 10.1021/ac061768q. [DOI] [PubMed] [Google Scholar]
- Eckhardt M. The role and metabolism of sulfatide in the nervous system. Mol Neurobiol. 2008;37(2-3):93–103. doi: 10.1007/s12035-008-8022-3. [DOI] [PubMed] [Google Scholar]
- Eckhardt M, Hedayati KK, Pitsch J, Lullmann-Rauch R, Beck H, Fewou SN, et al. Sulfatide storage in neurons causes hyperexcitability and axonal degeneration in a mouse model of metachromatic leukodystrophy. J Neurosci. 2007;27(34):9009–9021. doi: 10.1523/JNEUROSCI.2329-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estruch R, Nicolas JM, Salamero M, Aragon C, Sacanella E, Fernandez-Sola J, et al. Atrophy of the corpus callosum in chronic alcoholism. J Neurol Sci. 1997;146(2):145–151. doi: 10.1016/s0022-510x(96)00298-5. [DOI] [PubMed] [Google Scholar]
- Ewenczyk AE, Ziplow J, Tong M, Le T, de la Monte SM. Sustained impairments in brain insulin/IGF signaling in adolescent rats subjected to binge alcohol exposure during development. J Clinic Experiment Pathol. 2012;2(2):106. doi: 10.4172/2161-0681.1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Lima FA, Post J, DeBord JD, Eller MJ, Verkhoturov SV, Della-Negra S, et al. Analysis of native biological surfaces using a 100 kV massive gold cluster source. Anal Chem. 2011;83(22):8448–8453. doi: 10.1021/ac201481r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores AI, Narayanan SP, Morse EN, Shick HE, Yin X, Kidd G, et al. Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci. 2008;28(28):7174–7183. doi: 10.1523/JNEUROSCI.0150-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu SC, Mozzi R, Krakowka S, Higgins RJ, Horrocks LA. Plasmalogenase and phospholipase A1, A2, and L1 activities in white matter in canine distemper virus-associated demyelinating encephalomyelitis. Acta Neuropathol. 1980;49(1):13–18. doi: 10.1007/BF00692214. [DOI] [PubMed] [Google Scholar]
- Gode D, Volmer DA. Lipid imaging by mass spectrometry - a review. Analyst. 2013;138(5):1289–315. doi: 10.1039/c2an36337b. [DOI] [PubMed] [Google Scholar]
- Gurtovenko AA, Anwar J. Interaction of ethanol with biological membranes: the formation of non-bilayer structures within the membrane interior and their significance. J Phys Chem B. 2009;113(7):1983–1992. doi: 10.1021/jp808041z. [DOI] [PubMed] [Google Scholar]
- Han X, Holtzman DM, McKeel DW., Jr Plasmalogen deficiency in early Alzheimer’s disease subjects and in animal models: molecular characterization using electrospray ionization mass spectrometry. J Neurochem. 2001;77(4):1168–1180. doi: 10.1046/j.1471-4159.2001.00332.x. [DOI] [PubMed] [Google Scholar]
- Harper C. Neuropathology of brain damage caused by alcohol. Med J Aust. 1982;2(6):276–282. doi: 10.5694/j.1326-5377.1982.tb124389.x. [DOI] [PubMed] [Google Scholar]
- Harper C, Dixon G, Sheedy D, Garrick T. Neuropathological alterations in alcoholic brains. Studies arising from the New South Wales Tissue Resource Centre. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(6):951–961. doi: 10.1016/S0278-5846(03)00155-6. [DOI] [PubMed] [Google Scholar]
- Harper CG, Kril JJ, Holloway RL. Brain shrinkage in chronic alcoholics: a pathological study. Br Med J (Clin Res Ed) 1985;290(6467):501–504. doi: 10.1136/bmj.290.6467.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, Baxter DM, Mitchell MA, Hitzemann RJ. Physical properties and lipid composition of brain membranes from ethanol tolerant-dependent mice. Mol Pharmacol. 1984;25(3):401–409. [PubMed] [Google Scholar]
- Haughey NJ, Bandaru VV, Bae M, Mattson MP. Roles for dysfunctional sphingolipid metabolism in Alzheimer’s disease neuropathogenesis. [Research Support, N.I.H., Extramural Research Support, N.I.H., Intramural Review] Biochimica et biophysica acta. 2010;1801(8):878–886. doi: 10.1016/j.bbalip.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honke K. Biosynthesis and biological function of sulfoglycolipids. Proc Jpn Acad Ser B Phys Biol Sci. 2013;89(4):129–138. doi: 10.2183/pjab.89.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu FF, Turk J. Characterization of phosphatidylinositol, phosphatidylinositol-4-phosphate, and phosphatidylinositol-4,5-bisphosphate by electrospray ionization tandem mass spectrometry: a mechanistic study. J Am Soc Mass Spectrom. 2000;11(11):986–999. doi: 10.1016/S1044-0305(00)00172-0. [DOI] [PubMed] [Google Scholar]
- Huang BX, Akbar M, Kevala K, Kim HY. Phosphatidylserine is a critical modulator for Akt activation. J Cell Biol. 2011;192(6):979–992. doi: 10.1083/jcb.201005100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SN, Wang HY, Woods AS. In situ structural characterization of glycerophospholipids and sulfatides in brain tissue using MALDI-MS/MS. J Am Soc Mass Spectrom. 2007;18(1):17–26. doi: 10.1016/j.jasms.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Jana A, Pahan K. Oxidative stress kills human primary oligodendrocytes via neutral sphingomyelinase: implications for multiple sclerosis. J Neuroimmune Pharmacol. 2007;2(2):184–193. doi: 10.1007/s11481-007-9066-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, Huang BX, Spector AA. Phosphatidylserine in the brain: metabolism and function. Prog Lipid Res. 2014;56:1–18. doi: 10.1016/j.plipres.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Steelman AJ, Zhang Y, Kinney HC, Li J. Aberrant upregulation of astroglial ceramide potentiates oligodendrocyte injury. Brain Pathol. 2012;22(1):41–57. doi: 10.1111/j.1750-3639.2011.00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnick RN, Kronke M. Regulation of ceramide production and apoptosis. Annu Rev Physiol. 1998;60:643–665. doi: 10.1146/annurev.physiol.60.1.643. [DOI] [PubMed] [Google Scholar]
- Kril JJ, Halliday GM. Brain shrinkage in alcoholics: a decade on and what have we learned? Prog Neurobiol. 1999;58(4):381–387. doi: 10.1016/s0301-0082(98)00091-4. [DOI] [PubMed] [Google Scholar]
- Lismont C, Nordgren M, Van Veldhoven PP, Fransen M. Redox interplay between mitochondria and peroxisomes. Front Cell Dev Biol. 2015;3:35. doi: 10.3389/fcell.2015.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann C, Schachmann E, Dandekar T, Villmann C, Becker CM. Developmental profiling by mass spectrometry of phosphocholine containing phospholipids in the rat nervous system reveals temporo-spatial gradients. J Neurochem. 2010;114(4):1119–1134. doi: 10.1111/j.1471-4159.2010.06836.x. [DOI] [PubMed] [Google Scholar]
- McCorkindale AN, Sheedy D, Kril JJ, Sutherland GT. The effects of chronic smoking on the pathology of alcohol-related brain damage. Alcohol. 2016;53:35–44. doi: 10.1016/j.alcohol.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu I, Woelk H. Investigations of the lipid metabolism of the white matter in multiple sclerosis: changes in glycero-phosphatides and lipid-splitting enzymes. Neurochem Res. 1982;7(6):727–735. doi: 10.1007/BF00965525. [DOI] [PubMed] [Google Scholar]
- Nunez K, Kay J, Krotow A, Tong M, Agarwal AR, Cadenas E, et al. Cigarette Smoke-Induced Alterations in Frontal White Matter Lipid Profiles Demonstrated by MALDI-Imaging Mass Spectrometry: Relevance to Alzheimer’s Disease. J Alzheimers Dis. 2016;51(1):151–163. doi: 10.3233/JAD-150916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JS, Sampson EL. Lipid composition of the normal human brain: gray matter, white matter, and myelin. J Lipid Res. 1965;6(4):537–544. [PubMed] [Google Scholar]
- Ohtani R, Tomimoto H, Kondo T, Wakita H, Akiguchi I, Shibasaki H, et al. Upregulation of ceramide and its regulating mechanism in a rat model of chronic cerebral ischemia. Brain Res. 2004;1023(1):31–40. doi: 10.1016/j.brainres.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Patra M, Salonen E, Terama E, Vattulainen I, Faller R, Lee BW, et al. Under the influence of alcohol: the effect of ethanol and methanol on lipid bilayers. Biophys J. 2006;90(4):1121–1135. doi: 10.1529/biophysj.105.062364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferbaum A, Adalsteinsson E, Sullivan EV. Dysmorphology and microstructural degradation of the corpus callosum: Interaction of age and alcoholism. Neurobiol Aging. 2006;27(7):994–1009. doi: 10.1016/j.neurobiolaging.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Rosenbloom MJ, Adalsteinsson E, Sullivan EV. Diffusion tensor imaging with quantitative fibre tracking in HIV infection and alcoholism comorbidity: synergistic white matter damage. Brain. 2007;130(Pt 1):48–64. doi: 10.1093/brain/awl242. [DOI] [PubMed] [Google Scholar]
- Phillips SC, Harper CG, Kril J. A quantitative histological study of the cerebellar vermis in alcoholic patients. Brain. 1987;110(Pt 2):301–314. doi: 10.1093/brain/110.2.301. [DOI] [PubMed] [Google Scholar]
- Pointer CB, Klegeris A. Cardiolipin in Central Nervous System Physiology and Pathology. Cell Mol Neurobiol. 2016 doi: 10.1007/s10571-016-0458-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarles RH, Macklin WB, Morell P. Myelin Formation, Structure and Biochemistry. 6. Philadelphia: Elsevier; 2006. [Google Scholar]
- Ramsey RB, Banik NL, Scott T, Davison AN. Neurochemical findings in adreno-leukodystrophy. J Neurol Sci. 1976;29(2-4):277–294. doi: 10.1016/0022-510x(76)90177-5. [DOI] [PubMed] [Google Scholar]
- Roux A, Muller L, Jackson SN, Baldwin K, Womack V, Pagiazitis JG, et al. Chronic ethanol consumption profoundly alters regional brain ceramide and sphingomyelin content in rodents. ACS Chem Neurosci. 2015;6(2):247–259. doi: 10.1021/cn500174c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel LC, Bakovic M. Formation and regulation of mitochondrial membranes. Int J Cell Biol. 2014;2014 doi: 10.1155/2014/709828. 709828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte T, Sullivan EV, Muller-Oehring EM, Adalsteinsson E, Pfefferbaum A. Corpus callosal microstructural integrity influences interhemispheric processing: a diffusion tensor imaging study. Cereb Cortex. 2005;15(9):1384–1392. doi: 10.1093/cercor/bhi020. [DOI] [PubMed] [Google Scholar]
- Shanta SR, Zhou LH, Park YS, Kim YH, Kim Y, Kim KP. Binary matrix for MALDI imaging mass spectrometry of phospholipids in both ion modes. Anal Chem. 2011;83(4):1252–1259. doi: 10.1021/ac1029659. [DOI] [PubMed] [Google Scholar]
- Stace C, Manifava M, Delon C, Coadwell J, Cockcroft S, Ktistakis NT. PA binding of phosphatidylinositol 4-phosphate 5-kinase. Adv Enzyme Regul. 2008;48:55–72. doi: 10.1016/j.advenzreg.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45(1):42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Sundaram SK, Fan JH, Lev M. A neutral galactocerebroside sulfate sulfatidase from mouse brain. J Biol Chem. 1995;270(17):10187–10192. doi: 10.1074/jbc.270.17.10187. [DOI] [PubMed] [Google Scholar]
- Sutherland GT, Sheahan PJ, Matthews J, Dennis CV, Sheedy DS, McCrossin T, et al. The effects of chronic alcoholism on cell proliferation in the human brain. Exp Neurol. 2013;247:9–18. doi: 10.1016/j.expneurol.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland GT, Sheedy D, Kril JJ. Using autopsy brain tissue to study alcohol-related brain damage in the genomic age. Alcohol Clin Exp Res. 2014;38(1):1–8. doi: 10.1111/acer.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Suzuki T. Role of sulfatide in normal and pathological cells and tissues. J Lipid Res. 2012;53(8):1437–1450. doi: 10.1194/jlr.R026682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venable ME. Role of ceramide in cellular senescence. doi: 10.1074/jbc.270.51.30701. [DOI] [PubMed] [Google Scholar]
- Vos JP, Lopes-Cardozo M, Gadella BM. Metabolic and functional aspects of sulfogalactolipids. Biochim Biophys Acta. 1994;1211(2):125–149. doi: 10.1016/0005-2760(94)90262-3. [DOI] [PubMed] [Google Scholar]
- Wood PL. Lipidomics of Alzheimer’s disease: current status. Alzheimers Res Ther. 2012;4(1):5. doi: 10.1186/alzrt103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood PL, Mankidy R, Ritchie S, Heath D, Wood JA, Flax J, et al. Circulating plasmalogen levels and Alzheimer Disease Assessment Scale-Cognitive scores in Alzheimer patients. J Psychiatry Neurosci. 2010;35(1):59–62. doi: 10.1503/jpn.090059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Yeon JE, Chang H, Tison G, Chen GJ, Wands J, et al. Ethanol impairs insulin- stimulated neuronal survival in the developing brain: role of PTEN phosphatase. J Biol Chem. 2003;278(29):26929–26937. doi: 10.1074/jbc.M300401200. [DOI] [PubMed] [Google Scholar]
- Yalcin EB, de la Monte SM. Review of matrix-assisted laser desorption ionization-imaging mass spectrometry for lipid biochemical histopathology. J Histochem Cytochem. 2015;63(10):762–771. doi: 10.1369/0022155415596202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin EB, Nunez K, Cornett DS, de la Monte SM. Differential Lipid Profiles in Experimental Steatohepatitis: Role for Imaging Mass Spectrometry as a Diagnostic Aid. Journal of Drug and Alcohol Research. 2015;4(2015):1–11. [Google Scholar]
- Yalcin EB, Nunez K, Tong M, Cornett SD, de la Monte SM. MALDI-IMS Detects Differential White Matter Degeneration-Associated Lipid Profiles in Rat Models of Chronic Alcohol, Tobacco Nitrosamine, or Both Exposures. J Am Soc Mass Spectrom. 2015;26(S1):95. [Google Scholar]
- Yalcin EB, Nunez K, Tong M, de la Monte SM. Differential Sphingolipid and Phospholipid Profiles in Alcohol and Nicotine-Derived Nitrosamine Ketone-Associated White Matter Degeneration. Alcohol Clin Exp Res. 2015;39(12):2324–2333. doi: 10.1111/acer.12909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MS, Sun Y, Arauz E, Jiang Y, Chen J. Phosphatidic acid activates mammalian target of rapamycin complex 1 (mTORC1) kinase by displacing FK506 binding protein 38 (FKBP38) and exerting an allosteric effect. J Biol Chem. 2011;286(34):29568–29574. doi: 10.1074/jbc.M111.262816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Deochand C, Krotow A, Leao R, Tong M, Agarwal AR, et al. Tobacco Smoke-Induced Brain White Matter Myelin Dysfunction: Potential Co-Factor Role of Smoking in Neurodegeneration. J Alzheimers Dis. 2015;50(1):133–148. doi: 10.3233/JAD-150751. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.