

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
This is the first trial to evaluate the clinical utility of Glu-plasminogen in children and adults with congenital plasminogen deficiency.
Glu-plasminogen achieves physiological levels of plasminogen activity coinciding with improved clinical efficacy and disease management.
Abstract
Congenital plasminogen deficiency is caused by mutations in PLG, the gene coding for production of the zymogen plasminogen, and is an ultrarare disorder associated with abnormal accumulation or growth of fibrin-rich pseudomembranous lesions on mucous membranes. Left untreated, these lesions may impair organ function and impact quality of life. Plasminogen replacement therapy should provide an effective treatment of the manifestations of congenital plasminogen deficiency. An open-label phase 2/3 study of human Glu-plasminogen administered IV at 6.6 mg/kg every 2 to 4 days in 15 patients with congenital plasminogen deficiency is ongoing. Reported here are data on 14 patients who completed at least 12 weeks of treatment. The primary end point was an increase in trough plasminogen activity levels by at least an absolute 10% above baseline. The secondary end point was clinical success, defined as ≥50% improvement in lesion number/size or functionality impact from baseline. All patients achieved at least an absolute 10% increase in trough plasminogen activity above baseline. Clinical success was observed in all patients with clinically visible (conjunctiva and gingiva), nonvisible (nasopharynx, bronchus, colon, kidney, cervix, and vagina), and wound-healing manifestations of the disease. Therapeutic effects were rapid, as all but 2 lesions resolved or improved after 4 weeks of treatment. Human Glu-plasminogen was well tolerated in both children and adults. This study provides critical first evidence of the clinical utility of ongoing replacement therapy with human Glu-plasminogen for the treatment of children and adults with congenital plasminogen deficiency. This trial was registered at www.clinicaltrials.gov as #NCT02690714.
Visual Abstract
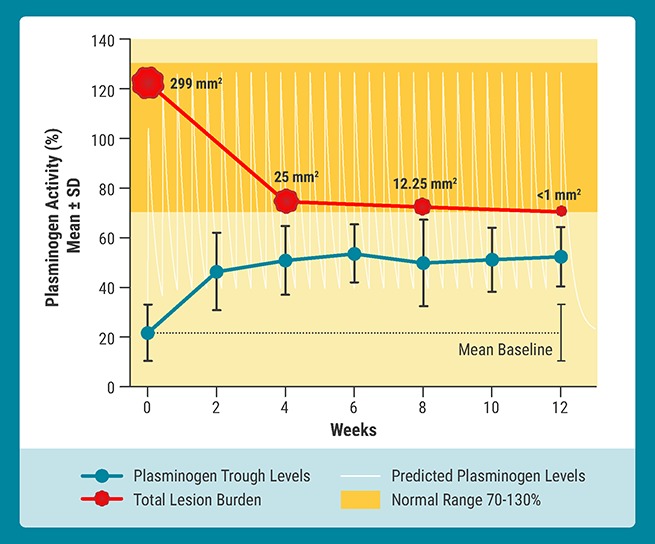
Introduction
Congenital plasminogen deficiency (also referred to as type I plasminogen deficiency, plasminogen deficiency, or hypoplasminogenemia) is an ultrarare autosomal-recessive disorder of the fibrinolytic system whose primary manifestation is the development of abnormal extravascular accumulation or growth of fibrin-rich, woody (ligneous) pseudomembranous lesions on mucous membranes throughout the body.1-3 Ligneous conjunctivitis (LC) appears to be the most common clinical manifestation and is characterized by inflamed, woody growths on the conjunctival membranes that, if left untreated, can result in visual impairment or blindness.4,5 Other disease manifestations may be observed in the central nervous system (eg, congenital occlusive hydrocephalus), ears, nasopharynx, oral cavity (eg, ligneous gingivitis or tonsillitis), and respiratory, gastrointestinal, and genitourinary tracts.1-3,5 Abnormal wound healing and infertility have also been reported.1,6,7 Although most affected patients survive into adulthood, the disease appears to be most severe in infants and children, where manifestations can lead to death, blindness, respiratory failure due to bronchial obstruction, or complications from hydrocephalus.3,5,8,9 Absent from these manifestations is the observance of thrombosis.2-7 Congenital plasminogen deficiency can impact self-image, quality of life, and ability to achieve full potential in school and/or work.
Congenital plasminogen deficiency is caused by mutations in PLG, the gene coding for the production of the zymogen plasminogen, with resultant decrease in antigenic and functional levels.3,5 A large epidemiologic study performed in blood donors in the United Kingdom demonstrated an observed rate of heterozygous (asymptomatic) plasminogen deficiency of 25 out of 9611 patients (0.26%).10 Based on these data, the theoretical prevalence of homozygous or compound heterozygotes is 1.6 affected individuals per million population.1,5
The zymogen plasminogen is activated to plasmin by either tissue-type plasminogen activator or urokinase-type plasminogen activator and serves as the pivotal enzyme in the fibrinolytic pathway involved in both intravascular and extravascular lysis of fibrin in addition to its role in wound healing, cell migration, tissue remodeling, angiogenesis, and embryogenesis.11,12 Plasminogen is primarily synthesized in the liver, although other areas include adrenal glands, kidney, brain, testis, heart, lung, uterus, spleen, thymus, gastrointestinal tract, and cornea.13,14 There are 2 main forms of native plasminogen, one with glutamic acid at its N terminus (Glu-plasminogen) and one with lysine at its N terminus (Lys-plasminogen).15 Glu-plasminogen has a longer half-life than Lys-plasminogen (2 to 2.5 days vs 0.8 days, respectively, in healthy subjects) and is the predominant (>95%) form of circulating native plasminogen.12 Upon binding to a fibrin clot, Glu-plasminogen is cleaved and converted into the more readily activated Lys-plasminogen.11,12 Plasminogen (human) is a highly purified Glu-plasminogen derived from human plasma. It is being developed as an IV replacement therapy for children and adults with congenital plasminogen deficiency.
Currently, there is no specific replacement therapy approved for the treatment of congenital plasminogen deficiency. As such, treatment options have been limited to nonspecific therapies and interventions (including surgery), all of which have proved to be less than optimal.1,5,16,17 There are several reports of the systemic use of Lys-plasminogen in patients with LC in which clinical success was described in patients who achieved plasminogen activity trough levels between 7% and 20% above baseline.7,18-20 Lys-plasminogen is not available and represents a limited treatment option because of its relatively short half-life as compared with Glu-plasminogen.
A multinational, multicenter, open-label, phase 2/3 study (2002C011G) evaluating the pharmacokinetics (PK), effectiveness, and safety of human Glu-plasminogen was initiated in patients with congenital plasminogen deficiency and is ongoing. Data on 14 patients who completed at least 12 weeks of treatment are reported to evaluate whether replacement therapy with human Glu-plasminogen (1) increases trough plasminogen activity levels by at least an absolute 10% above baseline (primary outcome) and (2) improves the clinical manifestations of the disease (secondary outcome).
Patients and methods
Participants
Children (2 to <18 years of age) and adults (18 to 80 years of age) with a documented history of lesions and symptoms consistent with a diagnosis of congenital plasminogen deficiency and a plasminogen activity level ≤45% were eligible for study inclusion. Patients who received exogenous plasminogen (ocular or IV), such as laboratory grade plasminogen, fresh frozen plasma, or plasminogen (human) within 2 weeks of the screening visit were excluded.
Study design and treatment
This is an ongoing open-label study with a 21-day screening period (for eligibility) and 3 treatment segments (Figure 1). Patients were given human Glu-plasminogen at 6.6 mg/kg administered as a 10- to 30-minute IV infusion every second, third, or fourth day based upon individual PK. Patients will be treated for 48 weeks in Norway or until product licensing or study termination by the sponsor (whichever is later) in the United States. The dosing intervals were investigator determined based upon individual patient plasminogen activity PK curves, obtained from patients who received study drug in the 6-mg/kg cohort of the previous phase 1 study (2002C005G) or from new-to-study patients who received study drug on day −4 of the current study (segment 1). The study drug underwent manufacturing process characterization after the start of the phase 2/3 study, with subsequent target dose change from 6 mg/kg to 6.6 mg/kg without change to study product. To determine the dosing interval, investigators selected the approximate longest time point that the patient’s plasminogen activity trough level remained an absolute 10% above baseline, taking into account known variability in plasminogen activity assay measurements of ±3%. For example, if a patient’s plasminogen activity was 25% above baseline at 48 hours, 12% above baseline at 72 hours, and <10% above baseline at 96 hours, then the patient was to be dosed every second day due to the assay variability at 72 hours. After the initial 12 weeks of treatment (segment 2), patients had the option to continue treatment of another 36 weeks (segment 3) or longer or withdraw from the study. In segment 3, patients started with the same dose and frequency as in segment 2, with the investigator having the option to modify the dosing schedule based on clinical response and plasminogen activity trough levels.
Figure 1.

Study schematic of the open-label, phase 2/3 trial of human Glu-plasminogen in children and adults with congenital plasminogen deficiency. This is an ongoing open-label study with a 21-day screening period (for eligibility) and 3 treatment segments. Patients were given human Glu-plasminogen at 6.6 mg/kg administered as a 10- to 30-minute IV infusion every second, third, or fourth day based upon individual PK in segments 1 and 2. In segment 3, patients started with the same dose and frequency as in segment 2, with the investigator having the option to modify the dosing schedule based on clinical response and plasminogen activity trough levels. Patients will be treated for 48 weeks in Norway or until product licensing or study termination by the sponsor (whichever is later) in the United States.
The planned sample size was at least 10 patients who received 12 weeks of treatment, with no formal sample size calculation due to disease rarity. Data analysis is conducted in stages. An interim analysis was performed after 14 patients completed 12 weeks of treatment (segments 1 and 2); the data cutoff date of this analysis was 1 April 2017. The second analysis will be performed when all patients have either completed the week 48 visit or have withdrawn consent and completed the final safety visit (segment 3), with a planned data cutoff date in April 2018. The final analysis will be performed when all patients have completed the final safety visit.
Study outcomes
The primary outcome of the interim analysis was the number and percentage of patients who achieved target trough plasminogen activity levels, defined as an increase of individual plasminogen activity trough level by at least an absolute 10% above baseline for at least 3 measurements in 12 weeks. Primary end point success was defined as at least 80% of evaluable patients achieving target trough plasminogen activity levels. Plasminogen activity was presented by individual patients and summarized descriptively. Standard PK parameters, including area under the concentration–time curve from time 0 to the last measured time point (AUC0-t) and terminal half-life (t1/2) were calculated using noncompartmental analysis and baseline-adjusted plasminogen levels derived from segment 1 data and data collected at the end of segment 2. PK data were summarized descriptively using mean and standard deviation (SD). The secondary outcome of the interim analysis was the overall clinical success in the number and size of lesions or change in organ function at week 12 as compared with baseline assessments. Secondary end point success was defined as 50% of patients with clinically visible or other measurable lesions achieving ≥50% reduction in lesion number and/or size or improved organ function (eg, improvement in forced expiratory volume in 1 second [FEV1]) from baseline. Other secondary outcomes included the Clinical Global Impression-Global Improvement (CGI-I) Scale, which captures the clinician’s impression of the patient’s disease improvement over time21,22; a quality-of-life assessment using the American Chronic Pain Association Quality of Life Scale,23 with the investigator (or designee) asking questions to either the patient (≥12 years of age) or the parents of the patient (<12 years of age); and safety. Safety included adverse events, clinical laboratory tests, antiplasminogen antibodies, vital signs, and virology. The investigators collected adverse events in an unsolicited manner, which were graded by severity and assessed for causality.
No formal statistical analyses were performed on study outcomes due to the small sample size and known high disease variability; therefore, all data were summarized descriptively.
Study procedures
Blood samples for plasminogen activity/antigen and d-dimer levels were collected at screening, baseline (day −4 for patients treated in segment 1; day 0 for patients who directly entered segment 2); prior to dosing at weeks 2, 4, 6, 8, 10, and 12; and prior to dosing every 12 weeks thereafter. Blood samples for plasminogen activity/antigen and d-dimer were collected immediately prior to plasminogen (human) administration and postdose between 5 and 15 minutes and at 6, 24, 48, 72, and 96 hours on day −4 and at week 12. Plasminogen activity was measured using a commercially available chromogenic assay (STA-Stachrom Plasminogen, Diagnostica Stago, Inc.), with a normal reference range of 70% to 130% and a coefficient of variation of ∼3%. Plasminogen antigen was measured using a commercially available enzyme-linked immunosorbent assay (Human Plasminogen Total Antigen ELISA Kit, Cell Sciences), with a normal reference range of 6 to 20 mg/dL and a coefficient of variation of ∼8%. Plasminogen activity/antigen analyses were performed centrally by Machaon Diagnostics (Oakland, CA). The investigators evaluated the clinical manifestations of congenital plasminogen deficiency and defined these as either clinically visible or nonvisible lesions. Clinically visible lesions of the eyes and gingiva and manifestations of abnormal wound healing were imaged and analyzed via digital photography, with each photograph including a millimeter scale to measure length and width. Clinically visible lesions that were too small to measure using the millimeter scale were described as nonmeasurable. The investigators assessed clinically visible lesions at baseline; weeks 4, 8, and 12 and every 12 weeks thereafter; and at 30 days after the last dose of study drug. Nonvisible lesions of the nasopharynx, lungs, abdomen, kidney, colon, cervix, and vagina could be assessed per investigator’s discretion by medical imaging studies (eg, computed tomography, magnetic resonance imaging, ultrasound), functional assessments (eg, spirometry, audiogram, oximetry), or report of clinical symptoms. The investigators assessed nonvisible lesions at baseline and at their discretion thereafter. The CGI-I Scale and quality-of-life assessment were performed at the same time as the visible lesion assessment. The investigators assessed adverse events, clinical laboratory tests, and antiplasminogen antibodies at the same time as the visible lesion assessment. A screening enzyme-linked immunosorbent assay was used to detect antiplasminogen antibodies; this test was performed centrally by Machaon Diagnostics. Vital signs were measured at each visit. Virology was measured every 12 weeks.
Study oversight and review
The study was designed, performed, and sponsored by Prometic Biotherapeutics Inc. (Rockville, MD). The protocol was approved by the institutional review board (St Vincent’s Hospital, Indianapolis, IN) or ethics committee (REC South-East A, Oslo, Norway) at each study center. The study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki and are consistent with the International Conference on Harmonization Tripartite Guidelines for Good Clinical Practice. All patients provided written informed consent, with assents obtained from children between the ages of 7 and <18 years. The sponsor collected and analyzed the data in conjunction with the authors, who had full access to the data. The sponsor’s responsible Medical Officer (third author) vouches for the accuracy of the data and analyses, and all authors vouch for the fidelity of this report.
A Safety Monitoring Committee composed of the sponsor’s responsible Medical Officer (third author), medical monitor, and pharmacovigilance officer, as well as an independent medical monitor reviewed the safety data biweekly during the first 12 weeks and periodically thereafter for the remainder of the study.
Results
Study participants
Between May 2016 and November 2016, 14 patients (5 children and 9 adults) with congenital plasminogen deficiency were enrolled at 2 sites. An interim analysis was conducted after all 14 patients received at least 12 weeks of treatment and were eligible for the primary and secondary outcomes of the interim analysis. All 14 patients were eligible for safety. Historical disease characteristics are listed in supplemental Table 1 (available on the Blood Web site), and baseline characteristics are summarized in Table 1. All patients had plasminogen activity levels ≤45% and ≥1 mutations in PLG (supplemental Table 2). All but 3 patients (3, 5, and 11) presented with clinical manifestations of the disease, with LC as the most common.
Table 1.
Summary of baseline characteristics
| Parameter | Adults (n = 9) | Children (n = 5) | Combined (N = 14) |
|---|---|---|---|
| Age, y | |||
| Mean (SD) | 31.9 (7.56) | 10.8 (5.26) | 24.4 (12.39) |
| Median | 33.0 | 11.0 | 24.0 |
| Min, max | 20, 42 | 5, 16 | 5, 42 |
| Sex, n (%) | |||
| Male | 3 (33) | 1 (20) | 4 (29) |
| Female | 6 (67) | 4 (80) | 10 (71) |
| Baseline plasminogen activity, n (%) | |||
| 0% to 10% | 3 (33) | 0 | 3 (21) |
| 11% to 20% | 1 (11) | 2 (40) | 3 (21) |
| 21% to 30% | 3 (33) | 3 (60) | 6 (43) |
| 31% to 40% | 1 (11) | 0 | 1 (7) |
| 41% to 45% | 1 (11) | 0 | 1 (7) |
| >45% | 0 | 0 | 0 |
| Site of clinical manifestations, n (%) | |||
| Any* | 8 (89) | 3 (60) | 11 (79) |
| Conjunctiva | 6 (67) | 2 (40) | 8 (57) |
| Gingiva | 3 (33) | 0 | 3 (21) |
| Abnormal wound healing | 3 (33) | 0 | 3 (21) |
| Bronchus | 2 (22) | 0 | 2 (14) |
| Colon | 0 | 2 (40) | 2 (14) |
| Cervix | 2 (22) | 0 | 2 (14) |
| Vagina | 0 | 2 (40) | 2 (14) |
| Nasopharynx | 1 (11) | 0 | 1 (7) |
| Kidney | 1 (11) | 0 | 1 (7) |
| Multisystem | 6 (67) | 3 (60) | 9 (64) |
Max, maximum; min, minimum.
Three patients (2 children and 1 adult) had no clinical manifestations at baseline.
PK
All 14 patients achieved target trough plasminogen activity levels (≥ absolute 10% above baseline) across the initial 12-week treatment period (Table 2). These target trough levels were maintained at all visits, except for week 10 (in patient 1, who missed 12 days of treatment because of a cluster of adverse events of severe intensity that occurred after the 20th infusion [week 8]) and week 2 (in patient 14, who missed 1 day of treatment). Five patients had 1-day delays in dosing, which decreased trough plasminogen activity levels at 6 visits (patient 2 at week 12, patient 10 at week 8, patient 12 at week 8, patient 13 at weeks 8 and 12, and patient 14 at week 8), although target trough levels were still maintained at these visits.
Table 2.
Trough plasminogen activity levels after treatment with plasminogen (human)
| Patient | Age (y) | Prescribed infusion interval (d) | Plasminogen activity (%)* | Plasminogen activity trough levels ≥ absolute 10% above baseline | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Screening | Baseline† | Trough levels | ||||||||||
| Wk 2 | Wk 4 | Wk 6 | Wk 8 | Wk 10 | Wk 12 | Any occurrence | ≥3 times | |||||
| 1 | 39 | 3 | 26 | 29 | 57‡ | 58 | 52 | 61 | 30§ | 48 | 5 | Yes |
| 2 | 35 | 2 | 29 | 43 | 72 | 81 | 78 | 79 | 83 | 63|| | 6 | Yes |
| 3 | 16 | 4 | 30 | 28 | 55 | 52 | 42 | 53 | 49 | 55 | 6 | Yes |
| 4 | 24 | 3 | 32 | 28 | 49 | 61 | 62 | 55 | 56 | 53 | 6 | Yes |
| 5 | 20 | 3 | 18 | 22 | 45 | 44 | 44 | 41 | 39 | 45 | 6 | Yes |
| 6 | 37 | 2 | <5 | <5 | 50 | 63 | 60 | 64 | 62 | 70 | 6 | Yes |
| 7 | 24 | 3 | 26 | 31 | 58 | 46 | 58 | 67 | 57 | 62 | 6 | Yes |
| 8 | 5 | 3 | 23 | 22 | 41 | 51 | 38 | 34 | 43 | 34 | 6 | Yes |
| 9 | 16 | 3 | 24 | 20 | 55 | 44 | 64 | 63 | 57 | 64 | 6 | Yes |
| 10 | 11 | 3 | 18 | 17 | 53 | 55 | 51 | 32|| | 50 | 50 | 6 | Yes |
| 11 | 6 | 3 | 36 | 29 | 39 | 49 | 52 | 59 | 54 | 61 | 6 | Yes |
| 12 | 33 | 3 | <5 | <5 | 19 | 33 | 46 | 26|| | 44 | 53 | 6 | Yes |
| 13 | 33 | 4 | 15 | 15 | 38 | 47 | 61 | 38|| | 45 | 30|| | 6 | Yes |
| 14 | 42 | 2 | 4 | <5 | 14|| | 24 | 38 | 22|| | 44 | 41 | 5 | Yes |
Plasminogen activity normal range is 70% to 130%.
Baseline is day −4 for patients starting treatment in segment 1 and day 0 for patients starting treatment in segment 2.
Unscheduled at week 3.
Lower trough plasminogen activity level was due to a 12-day interruption of seek 8 dosing due to a cluster of predominantly severe adverse events that occurred immediately after the patient’s 20th infusion.
Lower trough plasminogen activity level due to a 1-day delay in the next planned dose of study drug.
Individual dosing intervals were established using individual plasminogen activity PK curves generated at study onset. Summaries of these curves are presented in Figure 2 for the first and week 12 doses of study drug. Physiological levels of plasminogen activity (70% to 130%) were observed in both children and adults immediately after the first dose and were sustained for ∼24 hours after dosing (Figure 2A). After 12 weeks, physiological levels in children and adults were sustained for ∼48 hours after dosing (Figure 2B).
Figure 2.

Mean plasminogen activity levels after the first and week 12 doses of plasminogen (human). (A) Mean plasminogen activity levels after the first dose of plasminogen (human) in 14 patients with hypoplasminogenemia; this includes 6 patients (1 child and 5 adults) who received a single 6-mg/kg dose in the phase 1 study (2002C005G) and 8 patients (4 children and 4 adults) who received a single 6.6-mg/kg dose in the phase 2/3 study. (B) Mean plasminogen activity levels after the week 12 dose of 6.6 mg/kg plasminogen (human) in the same 14 patients.
Mean ± SD t1/2 was 34.2 ± 12.1 hours after the first dose and slightly longer (40.3 ± 7.9 hours) at week 12. Mean ± SD t1/2 in adults was 32.4 ± 13.1 hours after the first dose and 38.5 ± 7.1 hours after 12 weeks. The mean ± SD t1/2 in children was 37.3 ± 10.8 hours after the first dose and 43.4 ± 9.1 hours after 12 weeks. Taking these results together, steady state was most likely reached in all patients after 2 weeks of study drug administration. Mean ± SD AUC was 3063 ± 808 hours × % after the first dose that increased to 4797 ± 933 hours × % after 12 weeks; this change corresponded to an accumulation ratio of ∼1.5. Although some interpatient variability was observed, PK parameters were generally similar between children and adults.
Plasminogen antigen levels behaved similarly to plasminogen activity levels, although there was greater interpatient variability.
Three patients with extremely low plasminogen activity levels (<5%) and large disease burden had clinically significant elevations in d-dimer, starting shortly after initial treatment and lasting up to 6 weeks in duration. These elevations were temporally associated with treatment response (reduction or resolution of lesions).
Clinical outcomes
An overall success rate of 100% was achieved in all patients with clinically visible and/or nonvisible manifestations of congenital plasminogen deficiency assessed at baseline. A total of 9 patients had 23 clinically visible lesions, which consisted of 9 measurable lesions in the conjunctiva and 14 nonmeasurable lesions (lesions that were no longer measurable using the millimeter scale) in the conjunctiva and gingiva (Table 3). Patient 6 had 7 nonmeasurable lesions, patient 12 had 4 measurable and nonmeasurable lesions, and patient 14 had 4 nonmeasurable lesions; these subjects had the lowest plasminogen activity at baseline (<5%). All 23 measurable and nonmeasurable lesions of the conjunctiva and gingiva resolved or improved by week 12, with 12 resolving (Figure 3) and 9 improving by week 4, 15 resolving and 7 improving by week 8, and 18 resolving and 5 improving by week 12. This response was sustained through the initial 12 weeks of treatment and, for 13 of these lesions, through at least 24 weeks of treatment (for those patients who had 24-week data as of the data cutoff date of the for this analysis). Two visible lesions (conjunctiva) were in a 5-year-old girl (patient 8) whose disease burden was the largest among all assessed patients (total size of 125 mm2). These lesions decreased in size by over 90% at week 4, with 1 lesion completely resolving at week 8 and the other lesion decreasing in size by over 98% at week 12 to a point where it was no longer measurable using the millimeter scale. By week 24, this latter lesion resolved completely. Three patients had 8 manifestations of abnormal wound healing (acne, wounds, scars, and palmar/plantar warts), with 6 showing improvement by week 4 and all 8 showing improvement by week 12.
Table 3.
Clinical response to plasminogen (human)
| Patient* | Sex | Age (y) | Lesion site | Lesion assessment | ||||
|---|---|---|---|---|---|---|---|---|
| Baseline | Wk 4 | Wk 8 | Wk 12 | Wk 24 | ||||
| 1 | F | 39 | Left eye | 10 mm × 2 mm | Resolved | Resolved | Resolved | Resolved |
| Right eye | 10 mm × 2 mm | Resolved | Resolved | Resolved | Resolved | |||
| Cervix | Present | Not assessed | Not assessed | Not assessed | Not assessed | |||
| 2 | M | 35 | Left eye | 15 mm × 5 mm | Resolved | Resolved | Resolved | Resolved |
| 4 | F | 24 | Upper gingiva | Nonmeasurable† | Resolved | Resolved | Resolved | Resolved |
| Bronchus | Present | Resolved | Resolved | Resolved | Resolved | |||
| 6 | F | 37 | Left eye | Nonmeasurable | Resolved | Resolved | Resolved | Resolved |
| Right eye | Nonmeasurable | Resolved | Resolved | Resolved | Resolved | |||
| Gingiva (5 locations) | Nonmeasurable | Resolved | Resolved | Resolved | Resolved | |||
| Right nares | Present‡ | Resolved | Resolved | Resolved | Resolved | |||
| Bronchus | Present | Not assessed | Not assessed | Improved | Improved | |||
| Renal | Present | Not assessed | Not assessed | Improved | Improved | |||
| 7 | F | 24 | Cervix | Present | Not assessed | Not assessed | Not assessed | Resolved |
| 8 | F | 5 | Right eye, lower lid | 10 mm × 5 mm | 1 mm × 1 mm | Resolved | Resolved | Resolved |
| Right eye, upper lid | 15 mm × 5 mm | 5 mm × 2 mm | 5 mm × 2 mm | Nonmeasurable§ | Resolved | |||
| Vagina | Present | Not assessed | Not assessed | Not assessed | Not assessed | |||
| 9 | F | 16 | Colon | Present | Not assessed | Not assessed | Resolved | Resolved |
| Vagina | Present | Not assessed | Not assessed | Resolved | Resolved | |||
| 10 | F | 11 | Left eye | 5 mm × 3 mm | Resolved | Resolved | Resolved | Resolved |
| Colon | Present | Not assessed | Not assessed | Not assessed | Not assessed | |||
| 12 | M | 33 | Left eye | 4 mm × 4 mm | 3 mm × 2 mm | Resolved | Resolved | NA |
| Right eye | 4 mm × 4 mm | 2 mm × 2 mm | Resolved | Resolved | NA | |||
| Lower gingiva | Nonmeasurable | Improved | Improved | Improved | NA | |||
| Upper gingiva | Nonmeasurable | Improved | Improved | Improved | NA | |||
| Acne|| | Nonmeasurable | Improved | Improved | Improved | NA | |||
| Wounds/scars, hands|| | Nonmeasurable | Improved | Improved | Improved | NA | |||
| Plantar warts, feet|| | Nonmeasurable | Improved | Improved | Improved | NA | |||
| Palmar warts, hands|| | Nonmeasurable | Improved | Improved | Improved | NA | |||
| 13 | F | 33 | Left eye | 4 mm × 3 mm | 2 mm × 2 mm | 1.5 mm × 1.5 mm | Resolved | NA |
| Scar, right shoulder|| | Nonmeasurable | Unchanged | Unchanged | Improved | NA | |||
| Scar, right underarm|| | Nonmeasurable | Unchanged | Unchanged | Improved | NA | |||
| 14 | F | 42 | Left eye | Nonmeasurable | Improved | Improved | Resolved | NA |
| Right eye | Nonmeasurable | Improved | Improved | Resolved | NA | |||
| Lower gingiva | Nonmeasurable | Unchanged | Improved | Improved | NA | |||
| Upper gingiva | Nonmeasurable | Unchanged | Improved | Improved | NA | |||
| Tumors, wrists|| | Nonmeasurable | Improved | Improved | Improved | NA | |||
| Palmar warts, hands|| | Nonmeasurable | Improved | Improved | Improved | NA | |||
F, female; M, male; NA, not available.
Patients 3, 5, and 11 did not have lesions present at baseline.
Clinically visible lesions that were too small to measure using the millimeter scale were described as nonmeasurable.
Lesion in the right nares was not accessed via photography; therefore, it was considered a nonvisible lesion.
Size of lesion decreased by 98% at week 12 to a point where it was no longer measurable using the millimeter scale.
Manifestations representing abnormal wound healing due to congenital plasminogen deficiency.
Figure 3.

LC before and after replacement therapy with plasminogen (human). (A) Left eye lesion for patient 2 at day 0, prior to administration of plasminogen (human). The subject received plasminogen (human) at an IV dose of 6.6 mg/kg every second day. (B) Complete resolution of the left eye lesion after 4 weeks of treatment with plasminogen (human).
A total of 7 patients had 10 nonvisible lesions at baseline, with sites including nasopharynx, bronchus, kidney, colon, cervix, and vagina (Table 3); of these, 7 lesions in 4 patients were assessed by the investigator at the time of the interim analysis. All 7 nonvisible lesions resolved or improved by week 24, with 2 (nasopharynx and bronchus) resolved by week 4, 2 (colon and vagina) resolved and 2 (bronchus and renal) improved by week 12, and 1 (cervix) resolved by week 24. For the latter bronchus lesion, spirometry testing showed >75% improvement in FEV1 between baseline (46.7%) and week 4 (91.4%), week 8 (89.0%), and week 12 (89.3%).
No new lesions were observed, and no lesions recurred in patients as of the data cutoff date for this interim analysis. Moreover, none of the patients required surgical intervention or initiation of adjunctive therapy for their disease.
CGI-I scores were either much or very much improved in all 14 patients, and quality-of-life scores were excellent in all but 1 patient. The remaining patient (patient 14) had a quality-of-life score of 5 at week 12; this is the same patient who had extensive disease burden and low plasminogen activity (<5%) at baseline. Further treatment is planned for this patient.
Safety
Plasminogen (human) was well tolerated in both children and adults at the time of the interim analysis. There were no deaths or serious adverse events, and no patient permanently discontinued treatment due to an adverse event. Two patients had adverse events of severe intensity, including 1 patient with a history of anxiety who had a cluster of severe adverse events (nausea, fatigue, arthralgia, back pain, dizziness, paresthesia, and flushing) after her 20th infusion that coincided with an episode of anxiety temporally related to a motor vehicle accident involving her child. The severe adverse events resolved after temporary treatment discontinuation without re-emergence with restart of therapy. The other patient had severe back pain that resolved 3 days later. Headache and nasopharyngitis were the most common adverse events in both children and adults (Table 4).
Table 4.
Adverse events in the safety population
| Adverse event* | Patient population, n (%) | ||
|---|---|---|---|
| Adults (n = 9) | Children (n = 5) | Combined (N = 14) | |
| Headache | 4 (44) | 2 (40) | 6 (43) |
| Nasopharyngitis | 3 (33) | 2 (40) | 5 (36) |
| Abdominal pain, upper | 1 (11) | 3 (60) | 4 (29) |
| Nausea | 3 (33) | 1 (20) | 4 (29) |
| Diarrhea | 2 (22) | 1 (20) | 3 (21) |
| Cough | 0 | 3 (60) | 3 (21) |
| Rhinorrhea | 2 (22) | 1 (20) | 3 (21) |
| Dyspepsia | 1 (11) | 1 (20) | 2 (14) |
| Vomiting | 1 (11) | 1 (20) | 2 (14) |
| Sinusitis | 2 (22) | 0 | 2 (14) |
| Urinary tract infection | 1 (11) | 1 (20) | 2 (14) |
| Epistaxis | 2 (22) | 0 | 2 (14) |
| Back pain | 2 (22) | 0 | 2 (14) |
| Pain in extremity | 2 (22) | 0 | 2 (14) |
| Hematuria | 2 (22) | 0 | 2 (14) |
| Dysmenorrhea | 1 (11) | 1 (20) | 2 (14) |
Adverse events were coded by version 18.0 of the Medical Dictionary for Regulatory Activities using preferred terms.
Shown are events that occurred in ≥2 patients as of the data cutoff date for the interim analysis, regardless of the relationship to the study drug.
Several patients had adverse events (eg, epistaxis, hematuria, and dysmenorrhea) and/or laboratory abnormalities (eg, blood in urine and elevated d-dimer levels) consistent with physiologic study drug activity, specifically increased fibrinolytic capacity with lesion dissolution. Minor bleeding events of hemorrhage (eye, skin, uterine, and vaginal), epistaxis, hematuria, cervical, and oral discharge occurred in a few patients in areas near the lesion sites or associated with urinary excretion. No additional analyses were performed on any of these secretions. These findings are consistent with abnormal urinalysis findings of blood and protein in urine, which became macroscopic with urinary tract lesion lysis. Gross hematuria was not persistent or continuous.
In addition, there were no clinically significant findings for vital signs or viral testing. Antiplasminogen antibodies were not detected in any patient.
Discussion
Replacement therapy with human Glu-plasminogen consistently increased plasminogen activity levels and improved the clinical symptoms of children and adults with congenital plasminogen deficiency. Upon administration, physiological levels of plasminogen activity were achieved and maintained for ∼24 hours after the first dose and for ∼48 hours after the week 12 dose. During this period, mean plasminogen activity AUC0-t increased ∼1.5-fold. Moreover, target trough plasminogen activity levels were achieved and sustained across the course of the treatment period. The rare instances where trough levels were not maintained were due to changes in infusion intervals (patients 1, 2, 10, 12, 13, and 14; Table 2), emphasizing the importance of maintaining the prescribed dosing intervals. These findings coincided with overall clinical success in the resolution or improvement of lesions associated with congenital plasminogen deficiency.
Most patients in this study had significant disease history and burden. All but 1 patient had LC, with most patients having multisystem manifestations. In most patients, the first clinical manifestation occurred during infancy or early childhood. This finding is consistent with the literature3,5,19 and may partly be associated with a higher general susceptibility to external irritants (eg, dust, local infections, small foreign bodies, and minor injuries after scratching) and higher vulnerability of the mucous membranes at this early age.5 Moreover, such irritants and/or vulnerability may initiate or serve to perpetuate local inflammation and formation of ligneous lesions on other mucous membranes.5 Once triggered, the clinical manifestations of congenital plasminogen deficiency can persist for several months to years, the duration likely dependent on the endogenous plasminogen levels and/or susceptibility for further injury.5 In this study, prior clinical manifestations ranged in duration from 1 to 42 years, with the most extensive disease histories observed in patients with plasminogen activity <5%.
Study participant’s disease histories also provide important insight into the limitations of prior treatment options. Although surgical removal of pseudomembranes provides an acute treatment effect, its effectiveness is limited by lesion regrowth likely triggered by the procedure itself.1,5 This finding is most evident in patients who had undergone multiple surgical procedures prior to study enrollment. One patient with a plasminogen activity level <5% had a history of at least 18 conjunctival surgeries and at least 43 laryngoscopies and/or bronchoscopies to remove airway lesions. Other treatment options (plasminogen-containing and non–plasminogen-containing eye drops and/or systemic fresh frozen plasma infusion) appeared less than optimally effective, as disease burden recurred or persisted despite their use.
In the literature, clinical success was observed in several patients with LC who achieved plasminogen activity trough levels 7% to 20% above baseline following systemic Lys-plasminogen.7,18-20 The concept of plasminogen replacement therapy as a treatment of clinical manifestations of congenital plasminogen deficiency is supported by these historical reports. This study evaluated the native circulating and longer half-life form of plasminogen, Glu-plasminogen. The drug substance is derived from human plasma collected from healthy North American donors through a process that incorporates purification, concentration, filtration, pH, and buffer exchange steps, as well as affinity chromatography, solvent/detergent treatment, and nanofiltration to remove viruses.
Despite the extensive morbidity of enrolled patients, a significant clinical effect was evident as early as 4 weeks as 14 visible lesions in the conjunctiva and gingiva resolved by at least 90% in size (100% overall success rate), with 12 of these lesions disappearing. Of the 2 remaining visible lesions, 1 resolved at week 8 and 1 was no longer measurable at week 12 (this lesion resolved at week 24); these lesions occurred in a young child whose baseline disease burden was the largest among all patients assessed in the interim analysis. Moreover, 9 lesions showed improvement by week 4. This clinical effect persisted through 12 weeks and for all of these lesions through 24 weeks (for those patients who had 24-week data as of the data cutoff date of the interim analysis). Improvement was also seen for the 8 manifestations of abnormal wound healing. A similar effect was also observed for other disease manifestations, as 7 nonvisible lesions in the nasopharynx, bronchus, kidney, colon, cervix, and vagina either completely resolved or improved. Moreover, in a patient with chronic bronchial lesions, spirometry testing demonstrated >75% improvement in FEV1 from week 4 to week 12. It is important to note that this patient had persistent variable intrathoracic airways obstruction on the flow volume loop likely due to chronic tracheal injury, highlighting the need for treatment at an early age to avoid the permanent consequences of this disease. The sustainability of this clinical effect without recurrence of lesions or emergence of new lesions through 48 weeks of treatment is currently being investigated in the ongoing study.
The physiological role of plasminogen replacement therapy for the dissolution of mucosal lesions was supported by laboratory evidence of increased fibrinolysis in 3 patients with clinically significant elevations in d-dimer temporally related to clinical response (reduction or resolution of lesions). All 3 patients had plasminogen activity <5% and large disease burden at baseline. Similar d-dimer elevations were reported in the Lys-plasminogen studies.7,19 Dissolution of mucosal lesions may result in minor bleeding and/or tissue sloughing. Adverse events such as epistaxis, hematuria, or dysmenorrhea and/or laboratory abnormalities such as blood in urine also provide evidence of the physiologic activity of plasminogen (human). Such events were observed in our study.
Plasminogen (human) was well tolerated at the time of the interim analysis, as no patient experienced a serious adverse event or adverse event that resulted in permanent discontinuation of study drug. Headache and nasopharyngitis were the most common adverse events in both children and adults.
In conclusion, this study provides the critical first evidence of the clinical utility of ongoing replacement therapy with human Glu-plasminogen for the treatment of children and adults with congenital plasminogen deficiency. Upon administration, physiological levels of plasminogen activity are achieved that temporally coincide with clinical efficacy and improved disease management. This product represents a pivotal breakthrough in the treatment of this very rare coagulation deficiency and an important therapeutic advance for affected patients who have suffered under the burden of their disease due to lack of an available efficacious therapy.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the patients and their families, study coordinators, and support staff. The authors also thank Maria Rubinacci for managing the study, Christine Pan for statistical support, Angela Styhler for manuscript preparation, and Prometic Biotherapeutics Inc.
The study was designed, performed, and sponsored by Prometic Biotherapeutics, Inc. in consultation with A.D.S. Prometic also provided Glu-plasminogen free to participants.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: P.L., J.E.M., and A.D.S. designed the study; A.D.S., C.N., B.M.H., and P.M.S. enrolled and followed patients in the study; N.T. coordinated the study; J.M.P. was the responsible Medical Officer for the study; K.T. performed the PK analysis for plasminogen; A.D.S. and G.R.A. wrote the first draft; and all authors provided critical revisions, gave final approval, and agreed to be accountable for the work.
Conflict-of-interest disclosure: A.D.S. and C.N. received nonfinancial (clinical research) and other support (speaker fee, consulting, and travel) from Prometic, whose drug is studied in this work, and Kedrion. N.T. received nonfinancial support (clinical research) from Prometic and Kedrion. B.M.H. received personal fees and nonfinancial support from Shire, Novo Nordisk, and Octapharma AG. P.L., J.E.M., J.M.P., K.T., and G.R.A. received personal fees from Prometic Life Sciences, Inc. or its affiliates, outside the submitted work. In addition, Prometic has a patent number pending. P.M.S. declares no competing financial interests.
Correspondence: Amy D. Shapiro, Indiana Hemophilia and Thrombosis Center, 8326 Naab Rd, Indianapolis, IN 46260; e-mail: ashapiro@ihtc.org.
References
- 1.Mehta R, Shapiro AD. Plasminogen deficiency. Haemophilia. 2008;14(6):1261-1268. [DOI] [PubMed] [Google Scholar]
- 2.Schuster V, Hügle B, Tefs K. Plasminogen deficiency. J Thromb Haemost. 2007;5(12):2315-2322. [DOI] [PubMed] [Google Scholar]
- 3.Tefs K, Gueorguieva M, Klammt J, et al. Molecular and clinical spectrum of type I plasminogen deficiency: A series of 50 patients. Blood. 2006;108(9):3021-3026. [DOI] [PubMed] [Google Scholar]
- 4.Bateman JB, Pettit TH, Isenberg SJ, Simons KB. Ligneous conjunctivitis: an autosomal recessive disorder. J Pediatr Ophthalmol Strabismus. 1986;23(3):137-140. [DOI] [PubMed] [Google Scholar]
- 5.Schuster V, Seregard S. Ligneous conjunctivitis. Surv Ophthalmol. 2003;48(4):369-388. [DOI] [PubMed] [Google Scholar]
- 6.Kayikcioglu F, Bulbul D, Celiker S, Kucukali T. Ligneous inflammation of the cervix: a case report. J Reprod Med. 2005;50(10):801-804. [PubMed] [Google Scholar]
- 7.Schott D, Dempfle CE, Beck P, et al. Therapy with a purified plasminogen concentrate in an infant with ligneous conjunctivitis and homozygous plasminogen deficiency. N Engl J Med. 1998;339(23):1679-1686. [DOI] [PubMed] [Google Scholar]
- 8.Babcock MF, Bedford RF, Berry FA. Ligneous tracheobronchitis: an unusual cause of airway obstruction. Anesthesiology. 1987;67(5):819-821. [PubMed] [Google Scholar]
- 9.Klammt J, Kobelt L, Aktas D, et al. Identification of three novel plasminogen (PLG) gene mutations in a series of 23 patients with low PLG activity. Thromb Haemost. 2011;105(3):454-460. [DOI] [PubMed] [Google Scholar]
- 10.Tait RC, Walker ID, Conkie JA, Islam SI, McCall F. Isolated familial plasminogen deficiency may not be a risk factor for thrombosis. Thromb Haemost. 1996;76(6):1004-1008. [PubMed] [Google Scholar]
- 11.Castellino FJ, Ploplis VA. Structure and function of the plasminogen/plasmin system. Thromb Haemost. 2005;93(4):647-654. [DOI] [PubMed] [Google Scholar]
- 12.Collen D, Ong EB, Johnson AJ. Human plasminogen: in vitro and in vivo evidence for the biological integrity of NH2-terminal glutamic acid plasminogen. Thromb Res. 1975;7(4):515-529. [DOI] [PubMed] [Google Scholar]
- 13.Twining SS, Wilson PM, Ngamkitidechakul C. Extrahepatic synthesis of plasminogen in the human cornea is up-regulated by interleukins-1alpha and -1beta. Biochem J. 1999;339(Pt 3):705-712. [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Seiffert D, Fowler BJ, et al. Plasminogen has a broad extrahepatic distribution. Thromb Haemost. 2002;87(3):493-501. [PubMed] [Google Scholar]
- 15.Takada A, Takada Y. Physiology of plasminogen: with special reference to activation and degradation. Haemostasis. 1988;18(Suppl 1):25-35. [DOI] [PubMed] [Google Scholar]
- 16.Caputo R, Pucci N, Mori F, Secci J, Novembre E, Frosini R. Long-term efficacy of surgical removal of pseudomembranes in a child with ligneous conjunctivitis treated with plasminogen eyedrops. Thromb Haemost. 2008;100(6):1196-1198. [PubMed] [Google Scholar]
- 17.Tabbara KF. Prevention of ligneous conjunctivitis by topical and subconjunctival fresh frozen plasma. Am J Ophthalmol. 2004;138(2):299-300. [DOI] [PubMed] [Google Scholar]
- 18.Kraft J, Lieb W, Zeitler P, Schuster V. Ligneous conjunctivitis in a girl with severe type I plasminogen deficiency. Graefes Arch Clin Exp Ophthalmol. 2000;238(9):797-800. [DOI] [PubMed] [Google Scholar]
- 19.Mingers AM, Philapitsch A, Zeitler P, Schuster V, Schwarz HP, Kreth HW. Human homozygous type I plasminogen deficiency and ligneous conjunctivitis. APMIS. 1999;107(1):62-72. [DOI] [PubMed] [Google Scholar]
- 20.Pergantou H, Likaki D, Fotopoulou M, Katsarou O, Xafaki P, Platokouki H. Management of ligneous conjunctivitis in a child with plasminogen deficiency. Eur J Pediatr. 2011;170(10):1333-1336. [DOI] [PubMed] [Google Scholar]
- 21.Busner J, Targum SD. The clinical global impressions scale: applying a research tool in clinical practice. Psychiatry (Edgmont). 2007;4(7):28-37. [PMC free article] [PubMed] [Google Scholar]
- 22.Spearing MK, Post RM, Leverich GS, Brandt D, Nolen W. Modification of the Clinical Global Impressions (CGI) Scale for use in bipolar illness (BP): the CGI-BP. Psychiatry Res. 1997;73(3):159-171. [DOI] [PubMed] [Google Scholar]
- 23.American Chronic Pain Association. Quality of life scale: a measure of function for people with pain. Developed by Penney Cowan and Nicole Kelly. 2003. Available at https://www.theacpa.org/uploads/documents/Life_Scale_3.pdf. Accessed 1 July 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.