

Abstract
Fenofibrate, a well-known normolipidemic drug, has been shown to exert strong anticancer effects against tumors of neuroectodermal origin including glioblastoma. Although some pharmacokinetic studies were performed in the past, data are still needed about the detailed subcellular and tissue distribution of fenofibrate (FF) and its active metabolite, fenofibric acid (FA), especially in respect to the treatment of intracranial tumors. We used high performance liquid chromatography (HPLC) to elucidate the intracellular, tissue and body fluid distribution of FF and FA after oral administration of the drug to mice bearing intracranial glioblastoma. Following the treatment, FF was quickly cleaved to FA by blood esterases and FA was detected in the blood, urine, liver, kidney, spleen and lungs. We have also detected small amounts of FA in the brains of two out of six mice, but not in the brain tumor tissue. The lack of FF and FA in the intracranial tumors prompted us to develop a new method for intracranial delivery of FF. We have prepared and tested in vitro biodegradable poly-lactic-co-glycolic acid (PLGA) polymer wafers containing FF, which could ultimately be inserted into the brain cavity following resection of the brain tumor. HPLC-based analysis demonstrated a slow and constant diffusion of FF from the wafer, and the released FF abolished clonogenic growth of glioblastoma cells. On the intracellular level, FF and FA were both present in the cytosolic fraction. Surprisingly, we also detected FF, but not FA in the cell membrane fraction. Electron paramagnetic resonance spectroscopy applied to spin-labeled phospholipid model-membranes revealed broadening of lipid phase transitions and decrease of membrane polarity induced by fenofibrate. Our results indicate that the membrane-bound FF could contribute to its exceptional anticancer potential in comparison to other lipid-lowering drugs, and advocate for intracranial delivery of FF in the combined pharmacotherapy against glioblastoma.
Keywords: glioblastoma, poly-lactic-co-glycolic acid, peroxisome proliferator activated receptor alpha, fenofibrate, drug delivery, membrane fluidity, hydrophobic barrier
INTRODUCTION
Invasiveness and metastatic dissemination characterize neuroectodermal tumors such as glioblastoma, neuroblastoma, medulloblastoma and melanoma. Despite the growing knowledge about their etiology and efforts to develop improved tools for early diagnosis and treatment, their invasive phenotype results in high mortality rates, especially among children and young adults. Brain tumors are particularly difficult to treat due to distinct anatomical and physiological traits of neural tissue and vasculature. The blood brain barrier (BBB) and blood-brain tumor barrier (BTB) represent the major obstacles that prevent chemotherapeutic agents from reaching intracranial tumors.
Several strategies have been developed to enhance BBB and BTB permeability via biochemical intervention. Carotid artery infusion with hyperosmotic (1.6 M) mannitol was shown to temporarily open BBB, by induction of endothelial cells shrinkage and tight junction disruption. However, the opening lasts less than 30 minutes, leaving a very narrow window for potential drug delivery (1, 2). Another strategy for more selective BTB opening involves the use of vasomodulators, most commonly bradykinin or nitric oxide donors, which are able to transiently (for 15–120 minutes) increase capillary permeability (3, 4). However, the use of bradykinin promotes glioma cell migration, invasion and tumor angiogenesis and this drug acts as a chemoattractant that guides glioma cells to the blood vessels (5, 6), thus increasing the aggressiveness of the tumor. An alternative effective way to overcome BBB and simultaneously reduce systemic toxicity exerted by chemotherapeutic drugs was reported by Henry Brem and his team in 1993, in a proof-of-concept paper describing interstitial chemotherapy through bischloroethylnitrosourea (BCNU) intracranial delivery in biodegradable or nonbiodegradable wafers. The BCNU wafers were tested in rats bearing 9L gliosarcoma intracranial tumors and the treated animals survived significantly longer than the control animals (receiving a placebo wafer) or those that received intraperitoneal BCNU (7). This concept of a direct delivery of chemotherapeutic agent to a tumor bed served as a basis for development of Gliadel® wafers, which have been used against recurrent glioblastoma tumors since 1996 (8).
Despite the moderate increase in the mean survival of glioblastoma patients with implanted BCNU wafers, Gliadel® has not fully fulfilled the hopes for effective treatment (8, 9). A recent report on randomized control trials performed so far, “demonstrated a significant survival benefit for Gliadel® only after adjustment for prognostic factors and the prospective cohort study reported no survival benefit for Gliadel® as compared with a historical control group” (9). A number of unanswered questions still remain about how to improve current treatments or develop new, more effective multi-drug targeted regimens against glial tumors. Progress in basic research provides the frames for the development of new therapeutic directions that employ modulation of tumor-associated inflammatory responses, autophagy and application of bioactive phytochemicals (10–12).
We present herein results that support a new approach to glioblastoma treatment with fenofibrate (FF), a widely used hypolipidemic drug with an exceptional anticancer potential. The main advantages of using FF as a supportive oncotherapeutic agent include its recently revealed broad anticancer activities, low systemic toxicity, and good tolerance even in chronic administration. Our recent studies have demonstrated that FF effectively induces: 1) cell cycle arrest, 2) blockage of migration and invasiveness, and 3) apoptotic death in various tumor cells of neuroectodermal origin (13–17). In spite of these very promising results, we still do not understand how FF is so effective in targeting tumor cells while providing low systemic toxicity (18).
Fenofibrate was launched for hypercholesterolemia treatment in 1974 but its mechanism of action remained unknown until the discovery of a novel member of the steroid hormone receptor superfamily, called peroxisome proliferator activated receptor alpha (PPARα) by Issemann and Green (19). This report was preceded by studies demonstrating that fibrates had a strong impact on the genes responsible for fatty acid oxidation and lipoprotein metabolism (20, 21). Fenofibrate, or more accurately its active metabolite, fenofibric acid (FA), (Fig. 1) acts as an agonist of transcription factor PPARα. PPARα transactivates a broad variety of genes involved in lipid and glucose metabolism (22–24). Other drugs that share the common structural aromatic motif with FF, such as gemfibrozil or prinixic acid (Wy-14,643) (Fig. 1), are known to be potent PPARα agonists (25). However, in contrast to FF, these latter two compounds are not processed by cellular enzymes and activate PPARα directly.
Fig. 1.

Chemical structures of fenofibrate (FF), fenofibric acid (FA), gemfibrozil and Wy-14,643 (pirinixic acid).
Interestingly, at least some effects of FF that contribute to its anticancer activities seem to be PPARα independent. These effects include inhibition of mitochondrial respiration and alterations in plasma membrane fluidity (26–28). Importantly, FF has been included in new anti-angiogenic therapeutic regimens of combined chemotherapy with etoposide, cyclophosphamide, thalidomide and celecoxib against progressive and relapsed cancer that aim to reduce tumor neovascularization (29–31).
In this report we show HPLC-based analyses of FF pharmacokinetics in cell culture and in various organs and body fluids from animals after oral delivery. Because oral administration did not provide the access of FF or FA to brain tumor tissue, we also present a method for potential intracranial delivery in which FF is incorporated in a wafer made of the poly-lactate-co-glycolate (PLGA) that can be placed directly in the cavity after the brain tumor resection in a similar fashion to Gliadel® (7). Additionally, we demonstrate that FF but not FA is present in the membrane fraction isolated from the glioblastoma cells exposed to this lipid-lowering drug. Using electron paramagnetic resonance (EPR) and spin-labeled liposomes, we further show that the membrane-bound FF significantly alters lipid phase transitions and decreases membrane polarity. This unique property of FF could contribute to its exceptional anticancer potential in comparison to other PPARα agonists or to metformin, which all have been previously reported to have putative anticancer potential (32).
MATERIALS AND METHODS
Cell culture
Human glioblastoma cell line LN-229 (ATCC# CRL-2611) monolayer cultures were maintained in DMEM supplemented with 50 U/ml penicillin, 50 ng/ml streptomycin, and 10% fetal bovine serum (FBS) at 37°C and 5% CO2 atmosphere. For HPLC analyses, the cells were seeded in 100 mm cell culture dishes and cultured in the presence of 10% FBS supplemented with FF (Sigma Aldrich, USA, at final concentration of 50 μM diluted from DMSO stock). The medium and cellular samples were collected after 6, 10, 24, 48 and 72 hours of incubation. Membrane and cytosolic fractions were prepared from the control (DMSO) and FF treated LN-229 cultures were prepared using Membrane Fractionation kit (Abcam, UK), according to the manufacturer’s protocol. Briefly, cells (1,000,000) were trypsinized and centrifuged. The pellet was resuspended in 150 μl of Buffer A and subjected to further steps as described in the protocol. The final membrane fraction (0.3 mL) was collected as a supernatant after centrifugation, then an aliquot of 100 μl was subjected to HPLC analysis (described below).
Animal studies
Foxn1 nude mice (6 week old females, average body weight 25 g) were purchased from Harlan Laboratories (Houston TX, USA). The mice were maintained in groups of 5 animals/cages in a pathogen-free environment, with 12 hours light/dark cycles and free access to water and standard chow. Animals bearing intracranial human glioblastoma (LN-229) were given micronized FF (Lipanthyl 200M, Fournier, France, 50 mg/kg/day) by the oral gavage. The dose was chosen after consideration of relevance to human studies and assuming that oral dose of 45 mg/kg/day is equivalent to 1 × MRHD (maximal recommended human dose, (mg per m2 of body surface area)) for rats and 0.7 × MRHD for mice, according to FDA Approved Drug Product database (http://www.accessdata.fda.gov/scripts/cder/drugsatfda/). The treatment started 7 days after tumor cell implantation. Following 10 days of daily drug administration the animals were euthanized according to the standard ethically accepted procedures, and the following body fluids/organs were collected: blood, urine, liver, kidneys, spleen, heart, lungs, intact brain and intracranial tumor tissue. These tissues were subjected to sample preparation for the HPLC analysis (see below). All experiments were performed according to the Guide for the Care and Use of Laboratory Animals and local bioethical committee procedures at Louisiana State University Health Science Center (IACUC approval # 2902).
Sample preparation
Blood plasma, cell culture media, cellular and tissue lysates, and subcellular fractions were deproteinized by the addition of 150 μl of acetonitrile to 150 μl of sample, mixed well and centrifuged (15,000 g, 5 min). Urine and other samples that did not contain protein were centrifuged as above. In cell culture experiments, subconfluent monolayer cultures were washed in phosphate buffered saline (PBS) twice, then cells were scraped and lysed in 2% sodium dodecyl sulphate (SDS) in PBS. The lysates were sonicated on ice and centrifuged (15,000 g, 5 min). Finally, 150 μl of the supernatant was mixed with the equal volume of acetonitrile, filtered through 0.22 μm centrifuge filter (Sigma) and analyzed by high performance liquid chromatography (HPLC).
High performance liquid chromatography
All data were obtained from the Agilent Technologies 1100 apparatus equipped with a line degasser, binary pump (high pressure mixer), autosampler, column thermostat and Diode Array Detector (DAD). The YMCBase, 3 μm 4.6 × 150 mm analytical column was used and the solvent was 50 mM acetic acid in 60% acetonitrile (in water). Flow rate was set to 1 ml/min, elution was isocratic, column temperature was 20°C, and 5 μl of filtered (0.22 μm) sample was injected. DAD wavelength was set to 285 nm. Representative HPLC chromatograms that show detection of FF and FA in biological samples are shown in Fig. 2.
Fig. 2.

Detection of fenofibrate (FF) and fenofibric acid (FA) by high performance liquid chromatography [HPLC; Agilent Technologies 1100 with on line degasser, binary pump, auto-sampler, 3 μm, 4.6 × 150 mm column YMCBase 3 μm, 4.6 × 150 mm column (octyl silane C8 chemically bonded to totally porous silica particles), thermostat, and diode array detector (DAD)]. The sample separation parameters: solvent a: 50 mM acetic acid in water, solvent b: acetonitrile, isocratic 60%; flow rate 1ml/min; temp 20°C; injection fraction 5 μl; detection: DAD at 285 nm. (Panel a): HPLC chromatogram of FF and FA standards; (panel b): FF 50 μM solution in human blood just after mixing (time 0); (panel c): the same solution as in B after 4 hours incubation at 37°C; (panel d): HPLC analysis of urine from the patient who takes fenofibrate regularly (200 mg of micronized fenofibrate daily). FA and FF retention times are 3.904 ± 0.004 min and 10.436 ± 0.084 min, respectively. Insets: UV-Vis absorption spectra for FF and FA showing absorption maxima at 285 nm.
Western blot
The purity of the cytosolic and membrane fractions prepared from the LN-229 cell lysates was checked by the detection of protein markers characteristic for these fractions, namely N-cadherin (rabbit monoclonal antibody from Cell Signaling Technology, USA) for the membrane fraction and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, mouse monoclonal antibody from Fitzgerald Industries, USA) for the cytosolic fraction. Sample preparation and immunoblotting were performed according to standard procedures described in our previous publication (17).
Preparation of poly(glycolide-co-lactide) wafers containing fenofibrate
The fabrication consisted of two-layered porous biodegradable poly(glycolide-co-lactide) (PLGA) films, as described previously by our laboratories (33, 34). In these wafers, FF (Sigma Aldrich, USA) was incorporated into the bottom layer and sealed with a second PLGA layer without drug, as shown in Fig. 3a. The bottom layer was solvent cast and the sealing layer was prepared by spin-coating. An 8 mm circular piece of teflon, used as the substrate, was rinsed with 95% ethanol to remove any surface contaminants. To prepare the coating solution, 1.3% (w/v) of FF was first dissolved in methylene chloride followed by addition of 12.5% (w/v) of the PLGA (Resomer RG 506, 80 kDa) polymer to the solvent. The solution was vortex-mixed to ensure homogeneity. The appropriate volume (75 μL) of the solution containing PLGA and the drug (~107 mg drug/g polymer) was solvent cast onto the teflon at room temperature. Constant humidity was maintained (~50–60% relative humidity) while the solvent was slowly evaporated to produce a breath figure pattern on the film. The breath figure technique is a facile method of producing regular pores in the polymer film that enhance its drug delivery properties (35–37). The drug-loaded film (~150 microns thick) was dried for a day before fabricating the second layer. To coat a second layer, 50 μL of 15% (w/v) PLGA solution in methylene chloride (Resomer RG 504, 38–50 kDa) was spin coated at 1000 rpm for 25 s. This created a very thin film with an average thickness of 20 microns that was intended to be a seal over the first layer. Again, the second layer was cast in ~50–60% relative humidity to create pores on the top layer (SEM photomicrograph is shown in Fig. 3b). All fabricated samples were UV-sterilized for an hour and stored until cell clonogenic assays.
Fig. 3.
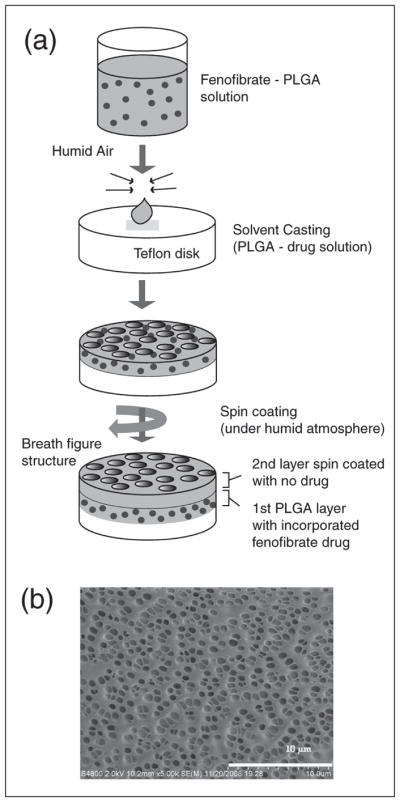
(Panel a): Schematic description of the preparation of poly-lactic-co-glycolic acid (PLGA) wafers containing fenofibrate. (Panel b): SEM image of highly porous PLGA film cast using breath figure methodology. SEM images were prepared as described in (33, 34).
Clonogenic assay
LN-229 cells were plated at the clonal density (1 × 103 cells per 35 mm dish or well in a 6-well plate) in the DMEM-based growth medium. The cells were exposed to the investigated compounds (FF, Sigma Aldrich; gemfibrozil, Sigma Aldrich; Wy-14,643, Cayman Chemicals, USA and metformin, Fluka; and MK-886, Calbiochem, Merck Millipore, USA,) or to 1 mg of FF incorporated in the PLGA wafer during the 12 days of incubation. Control cells were treated with the vehicle (DMSO). At the end of each experiment, the cells were fixed and stained in the 0.25% crystal violet solution in methanol, air dried and the colonies were counted. All the conditions were tested in duplicate and each experiment was performed at least three times.
Liposome preparation for electron paramagnetic resonance analysis
L-α-phosphatidylcholine, dimyristoyl (DMPC) and TEMPO-PC (T-PC) spin label were purchased from Avanti Polar Lipids (USA), and other spin labels (5-SASL and 16-SASL) were from Sigma (Germany). T-PC, 5-SASL and 16-SASL have a nitroxide free radical moiety attached to the polar headgroup, or to the 5th or 16th carbon atom in the alkyl chain, respectively. Information can thus be obtained from three different regions of the membrane: the water-membrane interface, the region close to the polar headgroups or the membrane center. The membranes used in this work were multilamellar liposomes of DMPC containing 0, 2.5 or 5 mol% of FF and 1 mol% of lipid spin label, prepared according to (38). All compounds were dissolved in chloroform, which was then evaporated under stream of nitrogen. The formed lipid film was then put under vacuum for at least 12 hours. The dried lipids were suspended in 10 mM borate buffer (pH 9.0) and vortexed. The multilamellar liposome suspension was centrifuged at 14,000 rpm, for 15 min at 4°C, and the pellet was used for EPR measurements.
Electron paramagnetic resonance measurements of membrane fluidity and polarity
EPR measurements were performed using a Bruker EMX spectrometer equipped with a temperature control unit. Suspended multilamellar liposomes were placed in a gas permeable capillary (i.d. 0.9 mm) made of Teflon and located inside the EPR dewar insert in a resonant cavity of the spectrometer. The sample was thoroughly deoxygenated with nitrogen gas, which was also used for temperature control. In case of DMPC phase transition measurements, the temperature was changed in the range 25 to 20°C (cooling experiments) and the spectra were recorded every 0.5°C (within the region of phase transition (24–23°C), every 0.2°C). For polarity measurements the samples were frozen to –150°C. The parameter obtained from the EPR spectra of spin labels in frozen membranes, Azz (z-component of the hyperfine interaction tensor), depends only on the polarity of the surroundings (38).
RESULTS
Fenofibrate pharmacokinetics in cell culture
Our cell culture experiments have demonstrated that LN-229 human glioblastoma cells take up FF from culture medium, and that intracellular esterases (most likely carboxylesterases or arylesterases (39, 40)) metabolize FF to FA, which subsequently accumulates in the medium. The results in Fig. 4 show the time course of FF to FA exchange between cells and culture media. In cells exposed to 50 μM FF, the drug accumulates quickly inside the cells reaching the maximal concentration at 24 h, and then its concentration gradually decreases (Fig. 4a). In contrast, FA intracellular levels are much lower and constant during the course of experiment (Fig. 4a). In the medium, FA levels continuously increase, which together with the observed low intracellular FA levels, indicate that FA is continuously produced inside the cells and is released to the medium (Fig. 4b). However, it is also possible that FF may be processed to FA outside the cell by secreted esterases. To test this alternative interpretation, we collected conditioned medium from the LN-229 cells cultured for 72 hours. This conditioned medium was filtered (0.22 μm pore size filter), and 50 μM FF standard was added for 24 hours at 37°C. Since we did not detect any traces of FA after this procedure (data not shown), we concluded that the filtered conditioned medium had no esterase activity and the FA detected in Fig. 4b was most likely released from the cells treated with FF.
Fig. 4.

Fenofibrate (FF) and fenofibric acid (FA) concentrations in whole cell lysates (a) and cell culture media (b) from the monolayer culture of human glioblastoma cell line, LN-229 were treated with a single dose of fenofibrate (50 μM) FF and FA were determined at the indicated time points using as described in the materials and methods section. Data represent average values from 2 experiments in triplicate (n=6) ± S.D. (S.D. values for some points are too small to be visible).
Subcellular distribution of fenofibrate
Anticancer effects of PPARα agonists, FF in particular, have been postulated by several laboratories, including ours (14–16, 41–44). In addition, some anticancer effects of FF are suspected to be PPAR-independent, involving the antiangiogenic effects on the tumor stroma (13, 41). It has been also reported that FF may have cholesterol-like effects on biological membranes (28). Here we demonstrate, for the first time, the detection of FF in the membrane fraction isolated from human glioblastoma cells, LN-229 (Fig. 5a). In these experiments, the cells were subjected to subcellular fractionation following the treatment with 50 μM FF for 24 hours. HPLC analysis of the membrane fraction showed a single peak with a retention time that identified it as FF (data shown in Fig. 6). The membrane fraction isolated from control, DMSO-treated cells, was completely negative (data not shown). To further verify that the obtained peak corresponded to FF, the fraction was spiked with FF just before HPLC separation. This procedure again generated a single peak that reflects quantitatively the sum of detected membrane FF and the FF spike (data not shown). When the cytosolic fraction was analyzed, it generated two distinct peaks, one which corresponded to FA and a second that corresponded to unprocessed FF (Fig. 6). The data in Fig. 5a show the quantitative analysis of FF and FA in cytosolic (cyto) and membrane (mem) fractions isolated from 1 × 106 LN-229 cells treated with 50 μM FF for 24 hours. In membrane fractions, we detected 1.68 nmol of FF in a sample aliquot of 100 μl, which corresponds to 5.04 nmol of FF in total membrane fraction isolated from 106 cells; the levels of FA were below our limit of detection (~0.01 nmol for both FF and FA per 100 μl of sample). Cytosolic fractions contained 7.20 nmol of FF and 1.71 nmol of FA per 106 cells. The purity of analyzed subcellular fractions was determined by Western blot analysis in which GAPDH served a cytosolic marker, and N-cadherin as a membrane marker, respectively (Fig. 5b). This novel finding indicates that only FF partitions into biological membranes. This suggests that in addition to the PPARα-mediated metabolic effects induced by FA, FF itself could also contribute to the anticancer activity of the drug via membrane-mediated processes. To test the hypothesis that FF incorporated within the lipid bilayer alters their properties, we performed the series of experiments with spin labeling of model DMPC membranes.
Fig. 5.

Fenofibrate (FF) and its metabolite, fenofibric acid (FA), in cell membrane and cytosolic fractions isolated from FF treated human glioblastoma cell line, LN-229. (Panel a): quantitative analysis of FF and FA in membrane (mem) and cytosolic (cyto) fractions isolated from LN-229 cells exposed to 50 μM fenofibrate (FF50) for 24 hours. Concentrations of FF and FA were calculated from the corresponding calibration curves, and are expressed in nmols of FF and FA per 1 × 106 cells. Data represent average values from three separate measurements with standard deviation. (Panel b): Western blot analysis demonstrating purity of cytosolic (Cyto) and membrane (Mem) fractions in which N-cadherin and GAPDH were use as membrane and cytosolic markers, respectively.
Fig. 6.

HPLC-based detection of fenofibrate and its metabolite, fenofibric acid, in cell membrane (panel a) and cystolic fraction (panel b) isolated from fenofibrate treated human glioblastoma cell line, LN-229. Under these chromatographic conditions (see legend to Fig. 2), fenofibrate was eluted at 10.4 minutes and fenofibric acid at 3.9 min. Insets in (a) and (b) represent the unique UV-Vis absorbance spectra of the obtained peaks corresponding to fenofibrate and fenofibric acid, respectively.
Effects of fenofibrate on membrane properties
The mol% of fenofibrate relative to phospholipid in the membranes of LN-229 cells was estimated from values of phospholipid content in cell lysates reported for a LM mouse fibroblast cell line (45). In these previous studies, the total phospholipid in a cellular lysate ranged from 332–410 nmol/mg protein, with an average of 364.5 nmol/mg protein. Based on this average value for phospholipid content and back-calculating to the amount of cellular protein present in the total membrane fraction from 1 × 106 cells (0.3672 mg), we estimated the total phospholipid in this fraction to be ~160 nmols and the mol% of fenofibrate relative to phospholipid to be ~3.8% (5.04 nmol FF/160.37 nmol phospholipid × 100). While we acknowledge that such a calculation could vary 2–4-fold for different cultured cell lines, this mol% calculation was used to guide our choice of 2.5 and 5 mol% fenofibrate in the EPR experiments described below.
Parameters characterizing the main phase transition of lipids, such as the transition temperature (TM) and width (dT1/2) reflect the membrane physical state and are very sensitive to perturbations caused by some exogenous molecules. The main phase transition of DMPC membranes was monitored by observing the amplitude of the central line of the EPR spectra of 5 and 16-SASL (Ao). Fig. 7 shows the temperature dependence of Ao for DMPC membranes containing 0, 2.5 and 5 mol% of FF. These data demonstrate that FF shifts the TM to lower temperatures and significantly broadens the phase transition in a concentration-dependent manner. The values of phase transition parameters obtained from these plots are presented in Tables 1 and 2. These tables also report additional data obtained from EPR spectra of spin labels (the order parameter S for 5- and 16-SASL and correlation times τ2B and τ2C for 16-SASL), which reflect the degree of membrane fluidity. The effects of FF on these parameters clearly depend on temperature. At 20°C (below the TM of DMPC) a decrease in both S and correlation times is observed in the presence of FF, indicating a more fluid membrane system, whereas at 25°C (above Tm of DMPC) a slight increase of these parameters indicates that lipids are more ordered in the presence of FF. Another important property of membranes is the polarity at its different regions, and this can be measured by EPR in frozen samples, since at −150°C a spectral parameter 2AZ depends only on local polarity surrounding the nitroxide moiety (38). Fig. 8 demonstrates hydrophobicity profiles obtained for DMPC membranes in the absence and presence of FF (2.5 or 5 mol%). FF clearly raises the hydrophobicity barrier within the membrane (at positions C5 and C16), but does not affect the polarity in the membrane-water interface. The effect is stronger in the region close to the polar headgroups (C5) than in the membrane center (C16).
Fig. 7.

Normalized amplitude of the central peak of the EPR spectra of 5-SASL (a) and 16-SASL (b) plotted as a function of temperature (cooling experiments) in DMPC bilayer. Experiments were performed in the absence and presence of FF (2.5 mol% and 5 mol%). TM and dT1/2 were obtained by fitting sigmoidal curves using Origin software.
Table 1.
Values of Tm, dT1/2 and order parameter S obtained from EPR spectra of 5-SASL in DMPC membranes in the absence and presence of 2.5 or 5 mol% of FF.
| DMPC 5-SASL | Tm [°C] | dT1/2 [°C] | S | |
|---|---|---|---|---|
| 20 [°C] | 25 [°C] | |||
| control | 23.199±0.042 | 0.352±0.036 | 0.729 | 0.622 |
| 2.5 mol % FF | 22.547±0.055 | 0.589±0.057 | 0.723 | 0.626 |
| 5 mol % FF | 20.711±0.519 | 1.446±0.260 | 0.703 | 0.630 |
Table 2.
Values of Tm, dT1/2, order parameter S and correlation times obtained from EPR spectra of 16-SASL in DMPC membranes in the absence and presence of 2.5 or 5 mol% of FF.
| DMPC 16-SASL | Tm [°C] | dT1/2 [°C] | S | τ2B [ns] | τ2C [ns] | |||
|---|---|---|---|---|---|---|---|---|
| 20°C | 25°C | 20°C | 25°C | 20°C | 25°C | |||
| control | 23.703±0.018 | 0.176±0.015 | 0.248 | 0.123 | 2.31 | 1.19 | 3.83 | 1.32 |
| 2.5 mol % FF | 23.198±0.338 | 0.348±0.287 | 0.255 | 0.119 | 2.17 | 1.18 | 3.60 | 1.29 |
| 5 mol % FF | 22.400±0.044 | 0.618±0.047 | 0.224 | 0.126 | 1.84 | 1.23 | 2.80 | 1.35 |
Fig. 8.

Hydrophobicity profiles (2Az) across the DMPC membrane. Profiles were obtained for the membranes without additions and after addition of 2.5 mol% or 5 mol% of FF. Upward changes (lower values of the 2Az parameter) indicate increase in hydrophobicity. Approximate location of the nitroxide moieties of spin labels are indicated by arrows. The numbers with stars for n-SASLs indicate that these SASLs are intercalated in the right half of the bilayer, but the nitroxide attached to C16 may pass through the center of the bilayer and stay in the other leaflet of the membrane. For more details see (38).
Fenofibrate distribution in tissues following oral administration in mice
Considering possible anticancer application of FF, we decided to evaluate tissue distribution of FF and FA in mice after oral administration of 50 mg/kg/day of micronized FF. The results of the HPLC analysis are reported in the Table 3. Importantly, we did not detect FF in any of the analyzed tissues. The FA was detected in the blood plasma, urine, liver, kidneys, heart, spleen and lungs of the treated mice. In addition, we have detected very small amounts of FA in the intact brain tissue in two out of six mice treated with FF; however, LN-229 cells growing intracranially in these animals were completely negative. Our results are in close agreement with the previously published data presenting the tissue distribution of 14C-radiolabelled FF following oral administration in rats (46). The authors confirmed the 14C presence in liver, kidney, guts, lung, heart, spleen, testis, skin and epididymal fat, but almost none in brain and eyes. Radioisotope detection is much more sensitive than HPLC method, so this is strong evidence that neither FF nor its metabolites cross blood brain barrier at the doses tested. Similar results were obtained by Deplanque and coauthors (47), who estimated that fenofibrate permeation rate in an in vitro BBB model was very low, indicating that this drug can cross BBB but extremely slowly. Taken together, these data indicated that oral administration of FF may result in very limited uptake in intracranial tumors and that uptake is dependent on the dose.
Table 3.
HPLC-based quantification of fenofibrate (FF) and fenofibric acid (FA) in different tissues and body fluids of mice fed with micronized fenofibrate (50 mg/kg/day) over a period of four weeks. 1mean ±S.D.
| Tissue (mg)/body fluid (ml) | 1FA (nmol/mg or/ml) | 1FF (nmol/mg or/ml) |
|---|---|---|
| Liver (n=6) | 11.35 ± 3.9 | 0 |
| Kidneys (n=6) | 1.5 ± 0.8 | 0 |
| Brain (n=5) | 0.06 ± 0.1 | 0 |
| Heart (n=4) | 1.31 ± 1.4 | 0 |
| Lungs (n=6) | 0.36 ± 04 | 0 |
| Spleen (n=4) | 1.33 ± 1.1 | 0 |
| Intracranial tumor (n=6) | 0 | 0 |
| Blood (n=6) | 3.33 ± 1.6 | 0 |
| Urine (n=5) | 5.34 ± 2.4 | 0 |
Poly-lactic-co-glycolic acid wafers for fenofibrate delivery to the tumor site
Aggressive glial tumors are usually subjected to a surgical excision, which is rarely complete, and frequently patients experience tumor recurrence. Therefore, a direct delivery of the drug into the cavity that is formed after tumor resection could inhibit the glioblastoma cells that remain in the bed of the brain tissue. The only controlled delivery system on the market, Gliadel®, is a chemotherapeutic agent that cause side effects, including a significantly higher risk of increased intracranial hypertension (48). FF, in contrast, acts as neuroprotectant in ischemic stroke and decreases cerebral infarct volume and brain tissue injuries (47, 49); it is therefore much less likely to produce adverse effects and might be even beneficial. We have consequently developed a drug delivery system that would slowly release FF upon contact with cerebrospinal fluid. We employed a porous nanostructured poly-lactic-co-glycolic acid (PLGA) polymer matrix as shown in Fig. 3b. PLGA is a biodegradable, FDA approved polymer useful for both hydrophobic and hydrophilic drug delivery applications (35).
The use of highly porous PLGA matrix incorporated FF enabled slow release of the drug to the surrounding fluid with an increasing rate that is a consequence of drug diffusion and wafer erosion (33, 34). The results in Fig. 9 show that FF-loaded PLGA matrix (PLGA/FF) submerged into the cell culture medium began to release FF after 36 h, and the concentration of the drug constantly increased, reaching almost 4 μM after 4 days of the exposure. To determine whether the FF released from the PLGA matrix was still active against glioblastoma cells, we compared PLGA/FF with a single dose of 50 μM FF using the LN-229 clonogenic assay. The results in Fig. 9b demonstrated that the FF continuously released from the PLGA matrix inhibited clonal growth of LN-229 cell even more potently than a single dose of 50 μM FF. This could be attributed to a constant release of the active drug from the matrix, which seems to have an advantage over a single addition of FF. This is most likely because FF is quickly converted to FA, which has significantly lower anticancer activity in comparison to the unprocessed FF. The results in Fig. 9c show cumulative concentrations of FF and FA in culture media collected from the clonogenic assay, in which the PLGA/FF wafer was continuously present for 12 days. The data confirm the abundant presence of FF, which reached concentration of almost 30 μM at day 12 of the clonogenic assay. Conversely, we have detected only very low levels of FA in the same medium, which could be explained by a very low number of the tumor cells (capable of FF deesterification) due to FF-induced inhibition of their clonogenic growth.
Fig. 9.

Evaluation of PLGA wafer containing 1 mg of fenofibrate (FF). (Panel a): HPLC-based measurement of FF release to the culture media, after submerging the PLGA wafer containing 1 mg of FF. The analysis was performed in the absence of cells. (Panel b): Clonogenic growth of LN-229 cells in the presence of PLGA wafer containing 1 mg of FF (PLGA/FF); empty wafer (PLGA); and FF in DMSO applied as a single 50 μM dose (FF 50 μM). (Panel c): FF and FA concentrations in LN-229 cells cultured in the presence of PLGA/FF wafer for 12 days. (Panel d): Clonogenic assay in LN-229 cells treated with different PPARα agonists and metformin: DMSO (vehicle), FF - fenofibrate 50 μM; Met - metformin 50 μM; Gem - gemfibrozil 50 μM; Wy - Wy14,643 50 μM. (Panel e): Clonogenic assay in LN-229 treated with FF (25 μM), PPARα inhibitor MK-886 (10 μM) or both compounds together. Cells were treated with the indicated compounds for 12 days; medium with the drugs was changed every second day.
Since, FF-induced inhibition of glioblastoma clonogenic growth is indeed quite remarkable, we decided to compare FF with other known drugs that are postulated to have a similar anticancer activity, including other agonists of PPARα, gemfibrozil, Wy-14,643, and metformin, an anti-diabetic drug that is believed to induce energetic stress in cancer cells (32). The results in Fig. 9d show that FF is far more potent against LN-229 cells when directly compared to gemfibrozil, Wy-14,643 and metformin (all used at 50 μM, and with the addition of a fresh aliquot for each drug every second day). The fact, that those very specific PPARα agonists are not capable of reproducing the FF effects suggests a receptor-independent mechanism. Further evidence for an independent mechanism is presented in Fig. 9e, which shows the results from the clonogenic assay performed in the presence of a selective PPARα inhibitor, MK-886 (50). MK-886 treatment did not rescue the LN-229 cells from FF-induced growth retardation. Unexpectedly, the cells treated with MK-886 and FF at the same time experienced even stronger growth arrest and seemed to have the proliferation blocked completely (Fig. 9e). These results also suggest that PPARα-independent mechanism is a strong contributing factor in this FF-mediated inhibition of LN-229 cells, and that direct interaction between FF and cellular membranes (Fig. 5a) could be involved.
DISCUSSION
As previously reported, FF exerts strong antiproliferative, antimetastatic and proapoptotic activities towards various tumors of neuroectodermal origin, including glioblastoma, melanoma and medulloblastoma (13–17). This is a very interesting finding for a drug that originally was used for normalizing plasma lipid and lipoprotein profiles in patients with hypercholesterolemia. The potent anticancer activity of FF has gained much attention and led to its incorporation within clinically applied drug regimens for patients with aggressive, recurrent brain malignancies, childhood primitive neuroectodermal tumors (PNETs) and leukemias. These regimens include COMBAT (combined oral metronomic biodifferentiating antiangiogenic treatment) and other metronomic antiangiogenic therapies (29, 51, 52). Metronomic chemotherapy is defined as chronic administration of chemotherapeutic and cytostatic drugs at relatively low doses to minimize toxicity and acute side effects (53). Importantly, this treatment scheme omits the drug-free recovery periods that usually lead to the tumor growth acceleration (30). Antiangiogenic multidrug metronomic regimens combining bevacizumab, thalidomide, celecoxib, etoposide, cyclophosphamide and FF (30) or antiangiogenic differentiating regimen that include temozolomide, etoposide, celecoxib, vitamin D, FF and retinoic acid (29), are well tolerated and produce encouraging effects in pediatric patients with aggressive brain tumors. These benefits include increased 2-year survival, good overall response to the treatment and only minor side effects. Apart from being a treatment option for patients with brain malignancies, FF has been shown to exert neuroprotective effects in traumatic brain injuries and ischemic stroke (47). In animal models of ischemic stroke that involve temporal middle cerebral artery occlusion and subsequent reperfusion, mice pretreated with FF had significantly decreased cerebral infarct volume in the cortex and reduced oxidative stress in the brain tissue (47). These effects have been attributed to the antiinflammatory and antioxidative activity of PPARα, since FF had no effect on ischemic/reperfusion injury in PPARα −/− mice. Of note, in this study the FF/FA concentration in the brain was not actually measured. To assess FF penetration to brain tissue, the authors employed a blood-brain barrier (BBB) in vitro model that consisted of bovine capillary endothelial cells and rat astrocytes cocultured in the cell culture vessels with inserts (47). The estimated BBB permeability coefficient for FA was very low (0.68 × 10−3 cm/min) and similar to that of sucrose, so the authors concluded that this molecule crosses BBB at a very slow rate (47). Therefore, we might assume that the FA concentration in the brain might teeter on the edge of detection limit of HPLC method. Another interesting study that recognized a neuroprotective potential of FF tried to address the problem of poor access to the brain tissue through BBB by developing FF and FA loaded PLGA microparticles for intracranial delivery (54). The drug-containing microparticles were injected intracranially to rats prior to the stroke induction. That procedure allowed the injection of 10 μl of the suspension, which due to the relatively low drug release rate (estimated not to exceed 0.004 % daily, (54)) limited the drug accessibility area. It is remarkable that in this study, FF, in contrast to FA, significantly reduced cortical infarct volume, despite its much lower solubility (54). In the light of our results, this might be attributed to better solubility of FF in the neuronal plasma membranes. It cannot be excluded that the neuroprotective activity of FF and the lack of such for FA could be associated with the PPARα independent, direct interactions of FF with biological membranes.
The study by Gamerdinger and colleagues (28) presented the evidence that FF influenced the membrane fluidity in the manner similar to cholesterol, and particularly increased the long-chain fatty acid order that resulted in a thicker and more rigid membrane. Indeed, the results reported herein with EPR and model liposome membranes showed that FF affects membrane fluidity and polarity in the manner similar to cholesterol, and at even lower concentrations. The main phase transition of DMPC is strongly affected by FF, which results in a membrane less ordered below TM, and more ordered above TM. This is also reflected by the changes of order parameter S and correlation times (Table 2 and 3). The TM of DMPC is significantly shifted to lower temperatures by FF, and at higher drug concentration, no phase transition is observed at all, especially with 5-SASL. Similar effects were observed in DMPC membranes after adding nonsteroid anti-inflammatory drugs (NSAIDs) (55). Membranes of such properties are typical for natural physiological conditions where sudden fluidity changes (like from gel to liquid phase at TM) do not take place. Also, the polarity decreases observed in the presence of FF in the membrane center (monitored by 16-SASL), and even to a higher degree, in the region close to the lipid head groups (5-SASL), are important. It was shown (38) that unsaturated lipid chains, as well as the presence of cholesterol, decrease membrane polarity (creating a hydrophobicity barrier). The level of unsaturated phospholipids and cholesterol:phospholipid ratio also decline during progressive carcinogenesis, thus rendering the membranes more fluid (56). Addition of FF to such membranes could counteract and compensate for these alterations and facilitate reestablishing the hydrophobicity barrier, which is critical to maintaining physiological homeostasis in healthy cells.
Stronger effects of FF on membrane properties were observed close to the head group region of the membrane, which suggests that FF is located predominantly in the region close to the membrane surface. At the membrane-water interface, however, the effect is not significant (i.e., only a slight increase in head group mobility was observed by means of T-PC (data not shown)), contrary to the strong effect of cholesterol in this region (57). On the other hand, the observed decrease of TM in the presence of FF may be caused by the disorganization of the membrane in the region of the polar head groups (58).
The observed alterations in physical parameters of model membranes suggest that FF is likely to change properties of plasma membranes in the treated cells. Increased order above the TM temperature, which is in the physiological range, may affect activity of signal transduction molecules residing in the membranes, such as phospholipase Cγ, protein kinase C (PKC) or trimeric G proteins. All these proteins play crucial role in triggering the signaling cascades from the membrane bound growth factor receptors to the nucleus. Classical PKC isoenzymes (α, βI, βII, γ) localized in the membranes are subsequently activated by both diacylglycerol (DAG) and Ca2+ ions and amplify the signaling cascade through upregulation of Ras/Raf-MEK and Erk pathway that controls cell survival, proliferation and invasion (59). PKC activity depends on protein - lipid interactions within membranes and fluid, less ordered membranes with high propensity to non-lamellar hexagonal (HII) phase formation strongly favor the PKC activation (60). PKC activity is regulated by the membrane composition, which determines the size of polar head group space and regional bilayer curvature, showing a preference for a narrow range of optimal content of some phospholipid species, such as phosphatidylethanolamine (61). Phosphatidylethanolamine, similarly to PKC activator DAG, is a cone shaped molecule with a small polar head group. Membrane regions rich in this species are less “packed” and possess larger head group spacing, as well as a higher hydration level, which creates preferred microenvironment for PKC (61–63). Slater and coworkers studying in detail the influence of membrane microenvironment on PKC activity concluded that “the narrow range over which activation of the enzyme can be achieved indicates that the enzyme may be highly sensitive to bilayer modifications that lead to changes in head group spacing. Such modifications include the presence of membrane perturbents such as drugs, alcohol and anesthetics (61). Fenofibrate may be regarded as such a substance. As our results depicted in Fig. 7 and 8 show, FF increases hydrophobicity, particularly in the vicinity of membrane surface (5-SASL), which is likely to affect activity of lipid-submerged protein, such as PKC.
PKC isoenzymes are important oncogenes that boost up the proliferation in various tumors, especially high-grade gliomas (59, 64). Therefore, it is conceivable that FF-induced alterations towards more ordered membranes downregulate PKC signaling and triggers growth arrest in LN-229 cells. This statement can be further supported by our previous observations that FF potently inhibits phosphorylation of PKC down-stream effectors, such as Erk1/2 in various cancer cells of neuroectodermal origin (16, 65). Some reports show that clofibrate and FF attenuate PKC activation in endothelin-1 induced animal model of cardiac hypertrophy, which alleviates the symptoms of this pathology (66, 67). FF and FA have also been observed to inhibit PKC enzymatic activity that drives platelet activation and aggregation (68). Interestingly, in this case the effect was entirely PPARα dependent and involved direct interaction of PPARα with PKC (69). To make the situation even more complex, it was revealed that PKC both stimulates PPARα expression and directly phosphorylates PPARα protein on Ser179 and Ser230, which enhances transactivation properties of this receptor (70).
The results of clonogenic assays performed on LN-229 cells treated with potent PPARα agonists Wy-14,643 and gemfibrozil and PPARα inhibitor MK-886 present the next rationale suggesting that the strong anti-proliferative and anti-invasive effects of FF against glioblastoma cells are largely PPARα independent. Wy-14,643 or gemfibrozil are not able to reproduce the growth suppressive effect of FF (Fig. 8d), and PPARα inhibition by MK-886 could not reverse it (Fig. 8e). Conversely, MK-886 administered together with FF showed synergistic growth inhibitory effect (Fig. 8e). Logical explanation is that the FF accumulated in the cellular membranes is neither converted into the PPARα ligand, FA, nor interacts with the nuclear receptor.
In conclusion, it is likely that membrane-bound and PPARα-independent action of FF is an important factor, which advocates for the use of this drug in glioblastoma patients after tumor resection. In this study we present the rationales for future clinical trials with the FF- loaded PLGA matrices (wafers) to be placed intracranially in the cavity that remains after glioma resection, similarly to Gliadel® wafers. This approach, combined with standard chemotherapy or metronomic treatment, could limit both the danger of recurrence and reduce severe toxicity and in consequence lead to substantial improvement in the prognosis for the patients. PLGA is both biocompatible with brain tissue and biodegradable, which means that the drug release is driven by both diffusion through the polymer and erosion (hydrolysis) of the carrier (54, 71). Both diffusion and erosion require a liquid environment, which is present in the tumor resection area. Such PLGA-based systems have already been tested for carmustine, temozolomide and paclitaxel delivery for glioma treatment (72, 73). One of PLGA’s advantages is its relatively high hydrophobicity, which enables efficient encapulation of nonpolar drugs such as FF. Slow release of drug from the hydrophobic PLGA matrix might also favor drug distribution into the lipid rich neuronal tissue.
Acknowledgments
K.R. is supported by R01 CA095518, P20 GM103501 and LSUHSC startup funds. M.G. is supported by the Foundation for Polish Science - POMOST Program co-financed by the European Union within European Regional Development Fund. D.A.B. acknowledges support from the Louisiana Board of Regents grant #LEQSF(2010–12)RD-B-05 and V.T.J. received support from the Department of Defense under Grant No. W81XWH-10-1-0377.
Footnotes
Conflict of interests: Tulane University has filed a provisional patent on the drug delivery system described herein with D.A. Blake, V.T. John and R.S. Ayyala as the inventors. Other authors declare none.
References
- 1.Doolittle ND, Miner ME, Hall WA, et al. Safety and efficacy of a multicenter study using intraarterial chemotherapy in conjunction with osmotic opening of the blood-brain barrier for the treatment of patients with malignant brain tumors. Cancer. 2000;88:637–647. doi: 10.1002/(sici)1097-0142(20000201)88:3<637::aid-cncr22>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 2.Ikeda M, Nagashima T, Bhattacharjee AK, Kondoh T, Kohmura E, Tamaki N. Quantitative analysis of hyperosmotic and hypothermic blood-brain barrier opening. Acta Neurochir Suppl. 2003;86:559–563. doi: 10.1007/978-3-7091-0651-8_114. [DOI] [PubMed] [Google Scholar]
- 3.Black KL, Ningaraj NS. Modulation of brain tumor capillaries for enhanced drug delivery selectively to brain tumor. Cancer Control. 2004;11:165–173. doi: 10.1177/107327480401100304. [DOI] [PubMed] [Google Scholar]
- 4.Yin D, Wang X, Konda BM, et al. Increase in brain tumor permeability in glioma-bearing rats with nitric oxide donors. Clin Cancer Res. 2008;14:4002–4009. doi: 10.1158/1078-0432.CCR-07-1826. [DOI] [PubMed] [Google Scholar]
- 5.Lu DY, Leung YM, Huang SM, Wong KL. Bradykinin-induced cell migration and COX-2 production mediated by the bradykinin B1 receptor in glioma cells. J Cell Biochem. 2010;110:141–150. doi: 10.1002/jcb.22520. [DOI] [PubMed] [Google Scholar]
- 6.Montana V, Sontheimer H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J Neurosci. 2011;31:4858–4867. doi: 10.1523/JNEUROSCI.3825-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tamargo RJ, Myseros JS, Epstein JI, Yang MB, Chasin M, Brem H. Interstitial chemotherapy of the 9L gliosarcoma: controlled release polymers for drug delivery in the brain. Cancer Res. 1993;53:329–333. [PubMed] [Google Scholar]
- 8.Bota DA, Desjardins A, Quinn JA, Affronti ML, Friedman HS. Interstitial chemotherapy with biodegradable BCNU (Gliadel) wafers in the treatment of malignant gliomas. Ther Clin Risk Manag. 2007;3:707–715. [PMC free article] [PubMed] [Google Scholar]
- 9.Perry J, Chambers A, Spithoff K, Laperriere N. Gliadel wafers in the treatment of malignant glioma: a systematic review. Curr Oncol. 2007;14:189–194. doi: 10.3747/co.2007.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toton E, Lisiak N, Sawicka P, Rybczynska M. Beclin-1 and its role as a target for anticancer therapy. J Physiol Pharmacol. 2014;65:459–467. [PubMed] [Google Scholar]
- 11.Stagos D, Apostolou A, Poulios E, et al. Antiangiogenic potential of grape stem extract through inhibition of vascular endothelial growth factor expression. J Physiol Pharmacol. 2014;65:843–852. [PubMed] [Google Scholar]
- 12.Buldak RJ, Polaniak R, Buldak L, et al. Exogenous administration of visfatin affects cytokine secretion and increases oxidative stress in human malignant melanoma Me45 cells. J Physiol Pharmacol. 2013;64:377–385. [PubMed] [Google Scholar]
- 13.Drukala J, Urbanska K, Wilk A, et al. ROS accumulation and IGF-IR inhibition contribute to fenofibrate/PPARalpha -mediated inhibition of glioma cell motility in vitro. Mol Cancer. 2010;9:159. doi: 10.1186/1476-4598-9-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grabacka M, Placha W, Plonka PM, et al. Inhibition of melanoma metastases by fenofibrate. Arch Dermatol Res. 2004;296:54–58. doi: 10.1007/s00403-004-0479-y. [DOI] [PubMed] [Google Scholar]
- 15.Grabacka M, Placha W, Urbanska K, Laidler P, Plonka PM, Reiss K. PPAR gamma regulates MITF and beta-catenin expression and promotes a differentiated phenotype in mouse melanoma S91. Pigment Cell Melanoma Res. 2008;21:388–396. doi: 10.1111/j.1755-148X.2008.00460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urbanska K, Pannizzo P, Grabacka M, et al. Activation of PPARalpha inhibits IGF-I-mediated growth and survival responses in medulloblastoma cell lines. Int J Cancer. 2008;123:1015–1024. doi: 10.1002/ijc.23588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilk A, Urbanska K, Grabacka M, et al. Fenofibrate-induced nuclear translocation of FoxO3A triggers Bim-mediated apoptosis in glioblastoma cells in vitro. Cell Cycle. 2012;11:1–13. doi: 10.4161/cc.21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Najib J. Fenofibrate in the treatment of dyslipidemia: a review of the data as they relate to the new suprabioavailable tablet formulation. Clin Ther. 2002;24:2022–2050. doi: 10.1016/s0149-2918(02)80095-9. [DOI] [PubMed] [Google Scholar]
- 19.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 20.Reddy JK, Goel SK, Nemali MR, et al. Transcription regulation of peroxisomal fatty acyl-CoA oxidase and enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase in rat liver by peroxisome proliferators. Proc Natl Acad Sci USA. 1986;83:1747–1751. doi: 10.1073/pnas.83.6.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Staels B, van Tol A, Verhoeven G, Auwerx J. Apolipoprotein A-IV messenger ribonucleic acid abundance is regulated in a tissue-specific manner. Endocrinology. 1990;126:2153–2163. doi: 10.1210/endo-126-4-2153. [DOI] [PubMed] [Google Scholar]
- 22.Frederiksen KS, Wulff EM, Sauerberg P, Mogensen JP, Jeppesen L, Fleckner J. Prediction of PPAR-alpha ligand-mediated physiological changes using gene expression profiles. J Lipid Res. 2004;45:592–601. doi: 10.1194/jlr.M300239-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Muoio DM, Way JM, Tanner CJ, et al. Peroxisome proliferator-activated receptor-alpha regulates fatty acid utilization in primary human skeletal muscle cells. Diabetes. 2002;51:901–909. doi: 10.2337/diabetes.51.4.901. [DOI] [PubMed] [Google Scholar]
- 24.Ribet C, Montastier E, Valle C, et al. Peroxisome proliferator-activated receptor-alpha control of lipid and glucose metabolism in human white adipocytes. Endocrinology. 2010;151:123–133. doi: 10.1210/en.2009-0726. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez FJ, Peters JM, Cattley RC. Mechanism of action of the nongenotoxic peroxisome proliferators: role of the peroxisome proliferator-activator receptor alpha. J Natl Cancer Inst. 1998;90:1702–1709. doi: 10.1093/jnci/90.22.1702. [DOI] [PubMed] [Google Scholar]
- 26.Zhou S, Wallace KB. The effect of peroxisome proliferators on mitochondrial bioenergetics. Toxicol Sci. 1999;48:82–89. doi: 10.1093/toxsci/48.1.82. [DOI] [PubMed] [Google Scholar]
- 27.Brunmair B, Lest A, Staniek K, et al. Fenofibrate impairs rat mitochondrial function by inhibition of respiratory complex I. J Pharmacol Exp Ther. 2004;311:109–114. doi: 10.1124/jpet.104.068312. [DOI] [PubMed] [Google Scholar]
- 28.Gamerdinger M, Clement AB, Behl C. Cholesterol-like effects of selective cyclooxygenase inhibitors and fibrates on cellular membranes and amyloid-beta production. Mol Pharmacol. 2007;72:141–151. doi: 10.1124/mol.107.034009. [DOI] [PubMed] [Google Scholar]
- 29.Zapletalova D, Andre N, Deak L, et al. Metronomic chemotherapy with the COMBAT regimen in advanced pediatric malignancies: a multicenter experience. Oncology. 2012;82:249–260. doi: 10.1159/000336483. [DOI] [PubMed] [Google Scholar]
- 30.Peyrl A, Chocholous M, Kieran MW, et al. Antiangiogenic metronomic therapy for children with recurrent embryonal brain tumors. Pediatr Blood Cancer. 2012;59:511–517. doi: 10.1002/pbc.24006. [DOI] [PubMed] [Google Scholar]
- 31.Robison NJ, Campigotto F, Chi SN, et al. A phase II trial of a multi-agent oral antiangiogenic (metronomic) regimen in children with recurrent or progressive cancer. Pediatr Blood Cancer. 2013;61:636–642. doi: 10.1002/pbc.24794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pierotti MA, Berrino F, Gariboldi M, et al. Targeting metabolism for cancer treatment and prevention: metformin, an old drug with multi-faceted effects. Oncogene. 2013;32:1475–1487. doi: 10.1038/onc.2012.181. [DOI] [PubMed] [Google Scholar]
- 33.Ponnusamy T, Lawson LB, Freytag LC, Blake DA, Ayyala RS, John VT. In vitro degradation and release characteristics of spin coated thin films of PLGA with a “breath figure” morphology. Biomatter. 2012;2:77–86. doi: 10.4161/biom.20390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ponnusamy T, Yu H, John VT, Ayyala RS, Blake DA. A novel antiproliferative drug coating for glaucoma drainage devices. J Glaucoma. 2014;23:526–534. doi: 10.1097/IJG.0b013e318294869b. [DOI] [PubMed] [Google Scholar]
- 35.Makadia HK, Siegel SJ. Poly Lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel) 2011;3:1377–1397. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bunz UHF. Breath figures as a dynamic templating method for polymers and nanomaterials. Adv Mater. 2006;18:973–989. [Google Scholar]
- 37.Madej W, Budkowski A, Raczkowska J, Rysz J. Breath figures in polymer and polymer blend films spin-coated in dry and humid ambience. Langmuir. 2008;24:3517–3124. doi: 10.1021/la703363a. [DOI] [PubMed] [Google Scholar]
- 38.Subczynski WK, Wisniewska A, Yin JJ, Hyde JS, Kusumi A. Hydrophobic barriers of lipid bilayer membranes formed by reduction of water penetration by alkyl chain unsaturation and cholesterol. Biochemistry. 1994;33:7670–7681. doi: 10.1021/bi00190a022. [DOI] [PubMed] [Google Scholar]
- 39.Bahar FG, Ohura K, Ogihara T, Imai T. Species difference of esterase expression and hydrolase activity in plasma. J Pharm Sci. 2012;101:3979–3988. doi: 10.1002/jps.23258. [DOI] [PubMed] [Google Scholar]
- 40.Wong CC, Cheng KW, Xie G, et al. Carboxylesterases 1 and 2 hydrolyze phospho-nonsteroidal anti-inflammatory drugs: relevance to their pharmacological activity. J Pharmacol Exp Ther. 2012;340:422–432. doi: 10.1124/jpet.111.188508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Panigrahy D, Kaipainen A, Huang S, et al. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc Natl Acad Sci USA. 2008;105:985–990. doi: 10.1073/pnas.0711281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shigeto T, Yokoyama Y, Xin B, Mizunuma H. Peroxisome proliferator-activated receptor α and γ ligands inhibit the growth of human ovarian cancer. Oncol Rep. 2007;18:833–840. [PubMed] [Google Scholar]
- 43.Yokoyama Y, Xin B, Shigeto T, et al. Clofibric acid, a peroxisome proliferator-activated receptor alpha ligand, inhibits growth of human ovarian cancer. Mol Cancer Ther. 2007;6:1379–1386. doi: 10.1158/1535-7163.MCT-06-0722. [DOI] [PubMed] [Google Scholar]
- 44.Strakova N, Ehrmann J, Bartos J, Malikova J, Dolezel J, Kolar Z. Peroxisome proliferator-activated receptors (PPAR) agonists affect cell viability, apoptosis and expression of cell cycle related proteins in cell lines of glial brain tumors. Neoplasma. 2005;52:126–136. [PubMed] [Google Scholar]
- 45.Schroeder F, Perlmutter JF, Glaser M, Vagelos PR. Isolation and characterization of subcellular membranes with altered phospholipid composition from cultured fibroblasts. J Biol Chem. 1976;251:5015–5026. [PubMed] [Google Scholar]
- 46.Weil A, Caldwell J, Strolin-Benedetti M. The metabolism and disposition of fenofibrate in rat, guinea pig, and dog. Drug Metab Dispos. 1988;16:302–309. [PubMed] [Google Scholar]
- 47.Deplanque D, Gele P, Petrault O, et al. Peroxisome proliferator-activated receptor-alpha activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. J Neurosci. 2003;23:6264–6271. doi: 10.1523/JNEUROSCI.23-15-06264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Westphal M, Hilt DC, Bortey E, et al. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003;5:79–88. doi: 10.1215/S1522-8517-02-00023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stahel PF, Smith WR, Bruchis J, Rabb CH. Peroxisome proliferator-activated receptors: “key” regulators of neuroinflammation after traumatic brain injury. PPAR Res. 2008;2008:538141. doi: 10.1155/2008/538141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kehrer JP, Biswal SS, La E, et al. Inhibition of peroxisome-proliferator-activated receptor (PPAR)alpha by MK886. Biochem J. 2001;356:899–906. doi: 10.1042/0264-6021:3560899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Padovani L, Andre N, Gentet JC, et al. Reirradiation and concomitant metronomic temozolomide: an efficient combination for local control in medulloblastoma disease? J Pediatr Hematol Oncol. 2011;33:600–604. doi: 10.1097/MPH.0b013e3182331eaf. [DOI] [PubMed] [Google Scholar]
- 52.Sterba J, Valik D, Mudry P, et al. Combined biodifferentiating and antiangiogenic oral metronomic therapy is feasible and effective in relapsed solid tumors in children: single-center pilot study. Onkologie. 2006;29:308–313. doi: 10.1159/000093474. [DOI] [PubMed] [Google Scholar]
- 53.Lam T, Hetherington JW, Greenman J, Maraveyas A. From total empiricism to a rational design of metronomic chemotherapy phase I dosing trials. Anticancer Drugs. 2006;17:113–121. doi: 10.1097/00001813-200602000-00001. [DOI] [PubMed] [Google Scholar]
- 54.Klose D, Laprais M, Leroux V, et al. Fenofibrate-loaded PLGA microparticles: effects on ischemic stroke. Eur J Pharm Sci. 2009;37:43–52. doi: 10.1016/j.ejps.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 55.Manrique-Moreno M, Garidel P, Suwalsky M, Howe J, Brandenburg K. The membrane-activity of ibuprofen, diclofenac, and naproxen: a physico-chemical study with lecithin phospholipids. Biochim Biophys Acta. 2009;1788:1296–1303. doi: 10.1016/j.bbamem.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 56.Singh Kanwar S, Vaish V, Nath Sanyal S. Altered membrane lipid dynamics and chemoprevention by non-steroidal anti inflammatory drugs during colon carcinogenesis. Nutr Hosp. 2011;26:1141–1154. doi: 10.1590/S0212-16112011000500034. [DOI] [PubMed] [Google Scholar]
- 57.Ashikawa I, Yin JJ, Subczynski WK, Kouyama T, Hyde JS, Kusumi A. Molecular organization and dynamics in bacteriorhodopsin-rich reconstituted membranes: discrimination of lipid environments by the oxygen transport parameter using a pulse ESR spin-labeling technique. Biochemistry. 1994;33:4947–4952. doi: 10.1021/bi00182a025. [DOI] [PubMed] [Google Scholar]
- 58.Genis RB. The structure and properties of membrane lipids. In: Cantor CR, editor. Biomembranes Molecular Structure and Functions. New York: Springer Verlag; 1989. pp. 36–46. [Google Scholar]
- 59.do Carmo A, Balca-Silva J, Matias D, Lopes MC. PKC signaling in glioblastoma. Cancer Biol Ther. 2013;14:287–294. doi: 10.4161/cbt.23615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Escriba PV, Gonzalez-Ros JM, Goni FM, et al. Membranes: a meeting point for lipids, proteins and therapies. J Cell Mol Med. 2008;12:829–875. doi: 10.1111/j.1582-4934.2008.00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Slater SJ, Kelly MB, Taddeo FJ, Ho C, Rubin E, Stubbs CD. The modulation of protein kinase C activity by membrane lipid bilayer structure. J Biol Chem. 1994;269:4866–4871. [PubMed] [Google Scholar]
- 62.Bazzi MD, Youakim MA, Nelsestuen GL. Importance of phosphatidylethanolamine for association of protein kinase C and other cytoplasmic proteins with membranes. Biochemistry. 1992;31:1125–1134. doi: 10.1021/bi00119a022. [DOI] [PubMed] [Google Scholar]
- 63.Langner M, Kubica K. The electrostatics of lipid surfaces. Chem Phys Lipids. 1999;101:3–35. doi: 10.1016/s0009-3084(99)00052-3. [DOI] [PubMed] [Google Scholar]
- 64.da Rocha AB, Mans DR, Regner A, Schwartsmann G. Targeting protein kinase C: new therapeutic opportunities against high-grade malignant gliomas? Oncologist. 2002;7:17–33. doi: 10.1634/theoncologist.7-1-17. [DOI] [PubMed] [Google Scholar]
- 65.Grabacka M, Plonka PM, Urbanska K, Reiss K. Peroxisome proliferator-activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin Cancer Res. 2006;12:3028–3036. doi: 10.1158/1078-0432.CCR-05-2556. [DOI] [PubMed] [Google Scholar]
- 66.Yakubu MA, Nsaif RH, Oyekan AO. Peroxisome proliferator-activated receptor alpha activation-mediated regulation of endothelin-1 production via nitric oxide and protein kinase C signaling pathways in piglet cerebral microvascular endothelial cell culture. J Pharmacol Exp Ther. 2007;320:774–781. doi: 10.1124/jpet.106.104992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang Y, Zhang H, Shao Z, et al. Suppression of endothelin-1-induced cardiac myocyte hypertrophy by PPAR agonists: role of diacylglycerol kinase zeta. Cardiovasc Res. 2011;90:267–275. doi: 10.1093/cvr/cvq401. [DOI] [PubMed] [Google Scholar]
- 68.Ali FY, Armstrong PC, Dhanji AR, et al. Antiplatelet actions of statins and fibrates are mediated by PPARs. Arterioscler Thromb Vasc Biol. 2009;29:706–711. doi: 10.1161/ATVBAHA.108.183160. [DOI] [PubMed] [Google Scholar]
- 69.Ainsworth CR, Pamplin JC, Rn DA, Linfoot JA, Chung KK. A bedside communication tool did not improve the alignment of a multidisciplinary team’s goals for intensive care unit patients. J Crit Care. 2013;28:112.e7–112.e13. doi: 10.1016/j.jcrc.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 70.Blanquart C, Mansouri R, Paumelle R, Fruchart JC, Staels B, Glineur C. The protein kinase C signaling pathway regulates a molecular switch between transactivation and transrepression activity of the peroxisome proliferator-activated receptor alpha. Mol Endocrinol. 2004;18:1906–1918. doi: 10.1210/me.2003-0327. [DOI] [PubMed] [Google Scholar]
- 71.Sawyer AJ, Piepmeier JM, Saltzman WM. New methods for direct delivery of chemotherapy for treating brain tumors. Yale J Biol Med. 2006;79:141–152. [PMC free article] [PubMed] [Google Scholar]
- 72.Naraharisetti KP, Sheng Ong YB, Xie WJ, Yiu Lee KT, Wang CH, Sahinidis NV. In vivo performance of implantable biodegradable preparations delivering Paclitaxel and Etanidazole for the treatment of glioma. Biomaterials. 2007;28:886–894. doi: 10.1016/j.biomaterials.2006.09.044. [DOI] [PubMed] [Google Scholar]
- 73.Lee JS, An TK, Chae GS, et al. Evaluation of in vitro and in vivo antitumor activity of BCNU-loaded PLGA wafer against 9L gliosarcoma. Eur J Pharm Biopharm. 2005;59:169–175. doi: 10.1016/j.ejpb.2004.06.006. [DOI] [PubMed] [Google Scholar]