

The overall survival of patients with chronic lymphocytic leukemia (CLL) has improved over the last decades mainly due to advances in the understanding of the disease biology and the introduction of novel therapeutic approaches.1 In this retrospective study we investigated trends in overall survival in subgroups of cases defined by genetic and immunogenetic features with the aim of addressing the question whether advances in chemoimmunotherapy had a uniform impact across all CLL patients. We found that such advances have translated into prolonged overall survival in all prognostic subgroups examined except those carrying TP53 abnormalities, as expected, but also those assigned to stereotyped subsets #1 and #2, which are generally devoid of such gene aberrations. This latter finding, reported here for the first time, indicates the need for alternative treatment options for these patients.
A milestone in the management of CLL was the introduction of combined chemoimmunotherapy, in particular the fludarabine-cyclophosphamide-rituximab (FCR) regimen.2 FCR is the gold standard first-line treatment for medically fit CLL patients except those carrying aberrations of the TP53 gene (TP53abs: i.e. deletion of chromosome 17p, del(17p) and/or TP53 mutations) who should be managed using signaling inhibitors.3 Additional options, consisting of different combinations of chemotherapeutic agents, anti-CD20 antibodies, signaling inhibitors and the BCL2 inhibitor venetoclax hold promise for further improvement of patients’ care.4
The remarkable efficacy of signaling inhibitors in CLL can be considered as in vivo evidence of the critical role of the B-cell receptor immunoglobulin in disease ontogeny and evolution.5 This is further supported by the fact that the somatic hypermutation status of the clonotypic immunoglobulin heavy variable (IGHV) gene segregates CLL cases into two categories with markedly different outcomes: cases with no or limited somatic hypermutation load (germline identity ≥98%, “unmutated CLL”, U-CLL), who generally have an aggressive disease course, in contrast to cases with a germline identify <98% (“mutated CLL”, M-CLL) who usually have a more indolent disease.5
Moreover, CLL patients can be assigned to specific subgroups, termed stereotyped subsets, each characterized by a distinctive variable heavy complementarity determining region 3 (VH CDR3) within the B-cell receptor immunoglobulin, which is shared between cases in each stereotyped subset.6 The two largest stereotyped subsets are subset #1 (clan I IGHV genes/IGKV1(D)-39, U-CLL), representing 2.2–2.5% of all cases of CLL and 5% of U-CLL, and subset #2 (IGHV3-21/IGLV3-21), the largest overall, representing approximately 3% of all CLL and comprising both U-CLL and mostly M-CLL.7 We have previously reported that patients assigned to subsets #1 and #2 have a short time-to-first-treatment, similar to that of patients harboring TP53abs, even though ~80% and ~95% of subset #1 and #2 cases, respectively, lack such aberrations.6,8
In the present study we explored survival trends based on the date of primary treatment in a cohort of 3504 patients who had received at least one line of treatment (Online Supplementary Tables S1 and S2), focusing on subgroups of patients with particular biomarker profiles including those belonging to stereotyped subsets #1 and #2. The present series was consolidated within the context of a multicenter collaboration of 15 institutions from nine countries in Europe and the USA. The clinicobiological data were retrieved from the local registry of each institution. Information regarding gender, age at the time of primary treatment, as well as immunogenetic features was available for all patients, while fluorescence in situ hybridization data were available for 1857 (53%) patients. Details regarding the molecular analyses are provided in the Online Supplementary Material. The study was approved by the local ethics review committee in each participating center.
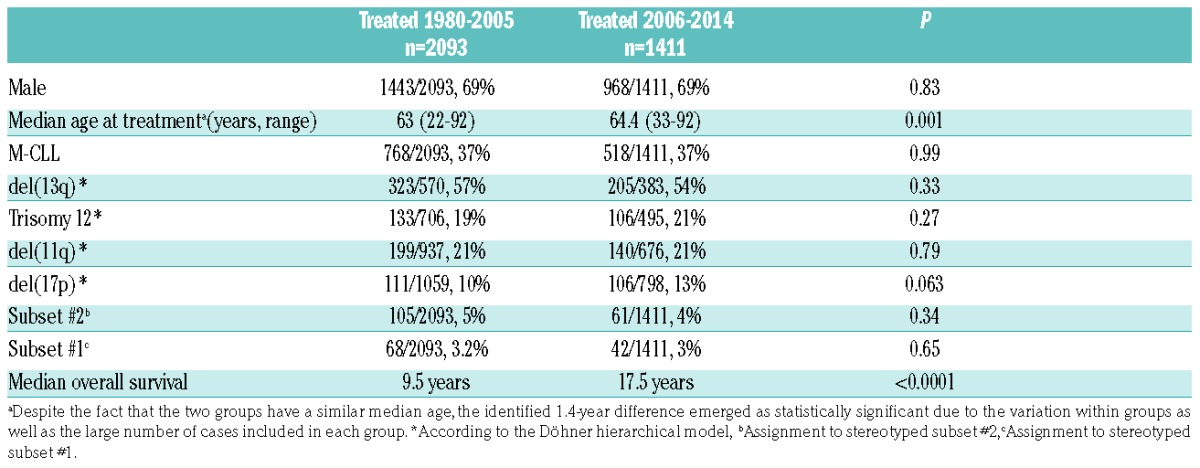
The evaluated patients received primary treatment between May 1980 and February 2014 and were stratified into two groups based on the date of this treatment; group A (n=2093) received primary treatment before 2006 and group B (n=1411) received primary treatment after January 1, 2006 (Table 1). The cut-off dates of January 2006 and February 2014 were chosen as they mark, respectively, the introduction of chemoimmunotherapy into clinical practice9 and, the date of the USA Food and Drug Administration approval for the use of ibrutinib in CLL.
Table 1.
Main clinicobiological features of cases treated before and after 2006.
Associations regarding categorical variables were assessed using the χ2 test. Overall survival was measured from the date of diagnosis until the date of last follow-up or death, in order to minimize potential bias due to the longer follow up of the patients treated before 2006. Survival curves were constructed with the Kaplan-Meier method, and the log-rank test was used to determine statistically significant differences between survival proportions. All tests were two-sided and statistical significance was defined as a P-value <0.05. Statistical analysis was performed using the Statistica Software v.10·0 (StatSoftInc, Tulsa, OK, USA).
Group A (1980–2005) and group B (2006–2014) had similar basic demographics, immunogenetic features and cytogenetic profiles (Table 1). However, the overall survival of group A was significantly (P<0.0001) inferior compared to that of group B [median overall survival: 9.5 years (95% confidence interval [CI]: 0.1–17.1) versus 17.5 years (CI: 0.1–17.9) in groups A and B respectively, P<0.0001] (Figure 1A). This superior outcome of group B patients was evident across subgroups defined by age, gender, somatic hypermutation status, del(11q), trisomy 12 and del(13q) (P<0.05 for all comparisons to the corresponding group A subgroups) (Figure 1, Online Supplementary Figures S1 and S2).
Figure 1.

Overall survival for patients with chronic lymphocytic leukemia in the present cohort. (A) Inferior overall survival (OS) for cases treated between 1980–2005 (blue line) versus cases treated between 2006–2014 (red line). (B) Inferior OS for all U-CLL cases treated between 1980–2005. (C–E) No improvement in OS over time for patients carrying del(17p) (C) or patients belonging to subset #1 (D) or subset #2 (E). (F) No improvement in OS over time for cases belonging to subset #2 even after excluding del(17p) cases.
In contrast, no increase in overall survival was seen over time for cases with del(17p) [median overall survival: 7.7 years (95% CI: 0.1–18.1) versus 5.2 years (95% CI: 0.1–10.1) in groups A and B respectively, P=0.61] (Figure 1C), which is not unexpected given the documented low efficacy of chemo(immuno)therapy in patients with TP53abs.2 Notably, a similar lack of improvement in overall survival was observed for cases assigned to subset #1 [median overall survival: 6.6 years (95% CI: 0.1–8.5) versus 8.3 years (95% CI: 0.1–15.1) in groups A and B respectively, P=0.31] and subset #2 [median overall survival: 7.3 years (95% CI: 0.1–10.3) versus 10.7 years (95% CI: 0.1–16.4) in groups A and B respectively, P=0.14] (Figure 1D,E). Survival differences between groups A and B remained non-significant for subsets #1 and #2, even when cases positive for del(17p) were excluded from the analysis (P=0.94 and P=0.95, respectively) (Figure 1F, Online Supplementary Figure S3).
TP53abs represent the only predictive biomarker affecting the treatment choice in CLL,3 but not all chemorefractory cases carry TP53abs. Instead, emerging evidence highlights other genomic aberrations that may complete the puzzle of chemorefractoriness.10 The present study goes beyond genomic aberrations, highlighting a notable lack of improvement in overall survival over the last 35 years for patients belonging to stereotyped subsets #1 and #2, despite the refinement of chemo(immuno)therapy regimens. Admittedly, despite this evidence, caution is warranted since, due to the retrospective nature of our study, the evaluated patients had received different therapeutic regimens rather than a uniform treatment, thus necessitating further investigation before definitive conclusions can be drawn.
Obviously, it would be reasonable to ask whether the genomic landscape of these subsets per se might explain their noted clinical aggressiveness. This question could not be addressed systematically in the present study due to missing information, especially concerning recurrent gene mutations. Nonetheless, based on the literature, subset #1 exhibits a rather diverse genomic landscape,8 hence rendering it difficult to draw definitive conclusions regarding the potential impact of each single individual abnormality. In contrast, subset #2 frequently shows del(13q) and del(11q) (in up to 54% and 24% cases, respectively), as well as enrichment for SF3B1 and ATM mutations (frequency ~45% and 26%, respectively), which might reasonably be considered as contributing to the clinical aggressiveness of mutant cases.8,11 Notably, however, subset #2 cases lacking SF3B1 mutations have an equally aggressive clinical course as mutant cases, implying that the dismal outcome of subset #2 is more closely linked to its unique clonotypic antigen receptor rather than a particular genomic aberration.8 In line with this, del(13q) or del(11q) did not have an impact on overall survival within subset #2 cases of our study (Online Supplementary Figure S4).
Recent studies support that patients with M-CLL treated with FCR achieve long-lasting responses, often with no detectable minimal residual disease, thus in contrast with U-CLL cases,12–14 prompting consideration of whether somatic hypermutation status should be used for making treatment decisions in medically fit patients with CLL. Along this line, our study implies that other immunogenetic features in addition to, but also beyond, somatic hypermutation status i.e. B-cell receptor immunoglobulin stereotypy, may predict inferior responses to chemo(immuno)therapy, regardless of genomic aberrations, further highlighting the significance of comprehensive immunogenetic analysis in CLL.15 Consequently, it could be argued that alternative options should be considered for subset #1 and #2 patients in the context of prospective trials. However, given the inherent limitations of retrospective analysis, subgroup analyses based on prospective clinical studies with targeted agents are warranted to further inform such a change in treatment regimens for these subsets.
Supplementary Material
Acknowledgments
The authors thank Stavroula Smerla, Eva Koravou, Evangelia Mouchtaropoulou and Diane Hatzioannou for their technical support with data assessment and definitions.
Footnotes
Funding: this work was supported in part by the Swedish Cancer Society, the Swedish Research Council, the Lion’s Cancer Research Foundation, the Marcus Börgström Foundation and Selander’s Foundation, Uppsala; H2020 “AEGLE, An analytics framework for integrated and personalized healthcare services in Europe” by the EU; H2020 “MEDGENET, Medical Genomics and Epigenomics Network” (No.692298) by the EU; GCH-CLL: funded by the General Secretariat for Research and Technology (GSRT) of Greece and the Italian Ministry of Health (MoH); IMI2 “HARMONY”, funded by the EU; project CEITEC 2020 (LQ1601) by MEYS-CZ, project AZV-MH-CZ 15-30015A-4/2015.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.da Cunha-Bang C, Simonsen J, Rostgaard K, Geisler C, Hjalgrim H, Niemann CU. Improved survival for patients diagnosed with chronic lymphocytic leukemia in the era of chemo-immunotherapy: a Danish population-based study of 10455 patients. Blood Cancer J. 2016;6(11):e499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376(9747):1164–1174. [DOI] [PubMed] [Google Scholar]
- 3.Hallek M. Chronic lymphocytic leukemia: 2015 update on diagnosis, risk stratification, and treatment. Am J Hematol. 2015;90(5):446–460. [DOI] [PubMed] [Google Scholar]
- 4.Lamanna N, O’Brien S. Novel agents in chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program. 2016;2016(1):137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fabbri G, Dalla-Favera R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat Rev Cancer. 2016;16(3):145–162. [DOI] [PubMed] [Google Scholar]
- 6.Baliakas P, Hadzidimitriou A, Sutton LA, et al. Clinical effect of stereotyped B-cell receptor immunoglobulins in chronic lymphocytic leukaemia: a retrospective multicentre study. Lancet Haematol. 2014;1(2):e74–84. [DOI] [PubMed] [Google Scholar]
- 7.Baliakas P, Agathangelidis A, Hadzidimitriou A, et al. Not all IGHV3–21 chronic lymphocytic leukemias are equal: prognostic considerations. Blood. 2015;125(5):856–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sutton LA, Young E, Baliakas P, et al. Different spectra of recurrent gene mutations in subsets of chronic lymphocytic leukemia harboring stereotyped B-cell receptors. Haematologica. 2016;101(8):959–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zoellner AK, Hohler T, Fries S, et al. Altered treatment of chronic lymphocytic leukemia in Germany during the last decade. Ann Hematol. 2016;95(6):853–861. [DOI] [PubMed] [Google Scholar]
- 10.Rosenquist R, Bea S, Du MQ, Nadel B, Pan-Hammarstrom Q. Genetic landscape and deregulated pathways in B-cell lymphoid malignancies. J Intern Med. 2017;282(5):371–394. [DOI] [PubMed] [Google Scholar]
- 11.Jeromin S, Haferlach C, Dicker F, Alpermann T, Haferlach T, Kern W. Differences in prognosis of stereotyped IGHV3-21 chronic lymphocytic leukaemia according to additional molecular and cytogenetic aberrations. Leukemia. 2016;30(11):2251–2253. [DOI] [PubMed] [Google Scholar]
- 12.Rossi D, Terzi-di-Bergamo L, De Paoli L, et al. Molecular prediction of durable remission after first-line fludarabine-cyclophosphamide-rituximab in chronic lymphocytic leukemia. Blood. 2015;126(16): 1921–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer K, Bahlo J, Fink AM, Goede V, et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2016;127(2):208–215. [DOI] [PubMed] [Google Scholar]
- 14.Thompson PA, Tam CS, O’Brien SM, et al. Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood. 2016;127(3):303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenquist R, Ghia P, Hadzidimitriou A, et al. Immunoglobulin gene sequence analysis in chronic lymphocytic leukemia: updated ERIC recommendations. Leukemia. 2017;31(7):1477–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.