

Abstract
Post-traumatic stress disorder (PTSD) is characterized by re-experiencing of a traumatic event, avoidance of trauma-associated stimulation, general changes in mood and cognition, and hyper arousal symptoms. Cyclooxygenase is involved in the production of prostaglandins and thromboxanes, and its inducible form cyclooxygenase-2(COX-2), an important mediator of cell injury in inflammation, is primarily expressed in leukocytes and brain cells. The present study investigated the expression of COX-2 in the hippocampi of rats with PTSD and evaluated the effect of COX-2 inhibition on PTSD. Adult male Wistar rats were randomly divided into three groups: Control (n=20), PTSD (n=20) and intervention group (PTSD+COX-2 inhibitor treatment, n=20). The expression of COX-2 was detected by immunohistochemistry, reverse transcription-quantitative polymerase chain reaction and western blotting. Terminal deoxynucleotidyl transferase mediated dUTP nick end labeling staining was used to observe the apoptosis of rat hippocampal neurons. Tumor necrosis factor α (TNF-α), interleukin (IL)-6 and prostaglandin E2 (PGE2) levels were analyzed by ELISA. Nitric oxide (NO) was detected using the Griess test. The behavioral and cognitive function of rats in the PTSD group was significantly decreased compared with the control group, while the behavioral and cognitive function of rats in the intervention group were improved. The COX-2 mRNA and protein expression levels in hippocampi of rats in the PTSD group were higher than in the control and intervention group. The apoptosis of hippocampus in rats with PTSD was significantly increased compared with the control group and following treatment with COX-2 inhibitor, apoptosis was decreased. In addition, compared with the control group and intervention group, the levels of TNF-α, IL-6, PGE2 and NO in hippocampi of rats were increased in the PTSD group. The present study indicated that COX-2 may be involved in the pathogenesis of PTSD, and inhibition of its expression serves a neuroprotective role in hippocampi of PTSD rats.
Keywords: post-traumatic stress disorder, cyclooxygenase-2, tumor necrosis factor α, interleukin-6, nitric oxide, prostaglandin E2
Introduction
Post-traumatic stress disorder (PTSD) is an abnormal mental reaction to severe stress factors, such as major disasters, war, terrorist attacks, traffic accidents and abuse, and has a high incidence overall (1,2). Clinical and community-based studies of PTSD among the elderly have identified elevated rates of anxiety, mood variation and substance use disorders which typically develop after PTSD (3–5). Such cognitive or behavioral changes are frequently ignored or underestimated during initial hospitalization, when severe psychological reactions or obvious physical injuries are presented (6). Recent studies have demonstrated that PTSD is dependent on environmental factors, which stimulate changes in the expression of biological susceptibility genes and associated proteins; however, but the specific mechanism remains to be elucidated (7,8). Current PTSD treatment includes antidepressant medication and psychotherapy, but a significant number of patients remain refractory to the treatment mostly due to substance use disorders, general medical illnesses and suicides (9–11).
Cyclooxygenase-2 (COX-2) is a rate-limiting enzyme in prostaglandin E2 (PGE2) synthesis. The expression of COX-2, an important mediator of cell injury in inflammation, is induced by cytokines and inhibited by glucocorticoids (12). It was reported that the expression of COX-2 could also be induced by neuronal excitation and increased intracellular calcium. COX-2 also serves an important role in the pathophysiology of neuronal death in ischemia and a variety of neurodegenerative diseases (13,14). The expression of the immediate early gene COX-2 is induced by vulnerable CA1 neurons. Several studies demonstrated that treatment with a COX-2selective inhibitor can decrease the damage following temporary focal ischemia (15,16). Therefore, inhibition of COX-2 expression might reduce the oxidative stress-induced damage to nerve cells.
Although it has been known for decades that cyclooxygenase inhibitors ameliorate brain injury (12), whether inhibition of COX-2 can have a positive effect on the symptoms of PTSD remain to be elucidated. In the present study, changes in COX-2 expression in rats with PTSD treated with COX-2 inhibitor were detected. Celecoxib, a first selective COX-2 inhibitor, is widely used in clinical practice and research (17–19). The apoptosis of rat hippocampi and levels of inflammatory factors, such as tumor necrosis factor α (TNF-α), interleukin (IL)-6, prostaglandin E2 (PGE2) and nitric oxide (NO), were used to evaluate the effect of COX-2 inhibition on PTSD. The objective of the present study was to reveal the mechanism of COX-2 in the pathogenesis of PTSD, and the therapeutic potential of COX-2 inhibition was investigated.
Materials and methods
Experimental animals
A total of 60 healthy, male Wistar rats (2–3 months, 150–200 g) were obtained from experimental animal center (Tongji Medical College, Huazhong University of Science and Technology) and acclimatized in the laboratory for 2 weeks prior to the experimental manipulation. Rats were reared in a cage kept at 25±3°C in a normal atmosphere and a 12 h light/dark cycle. Rats had free access to dry pellets and water with intermittent feeding of green fodder. Rats were randomly divided into three groups: Control, (n=20), PTSD (n=20) and intervention (PTSD+COX-2 inhibitor treatment, n=20). A PTSD model was established by single prolonged stress (SPS) as previously described by Liberzon et al (20,21). SPS was induced in three stages: Rats were restrained for 24 h, forced to swim for 20 min and administered ether anesthesia. All protocols involving animals were approved by the Ethics Committee for Experimental Animals (Wuhan No. 1 Hospital, Wuhan, China).
Rats in the control and PTSD groups were administered 0.9% normal saline and in the intervention group rats were treated with 25 mg/(kgd) COX-2 inhibitor celecoxib (Pfizer, Inc., New York, NY, USA) through a nasogastric gavage. Samples were collected after treatment for 2 weeks.
A pre-experiment was performed to select the optimum celecoxib concentration for treatment. A total of 9 rats from the PTSD group were divided into three groups and received celecoxib in dose of 15, 25 or 35 mg/(kgd) respectively via intraperitoneal route for one week, as previously described (22). COX-2 mRNA expression levels in all groups were detected by reverse transcription-quantitative polymerase chain reaction (RT-qPCR).
Behavioral detection
A total of four incandescent lamps (60 W) were placed in each of the four corners and served as an indoor light source for a behavioral laboratory, at a constant illumination of 150–300 Lux. In order to ensure clear observation of rat activity, the lights did not directly irradiate the detection equipment. The outdoor light was blocked by shading curtains. The open-field (OF), elevated plus maze (EPM) and Morris water maze (MWM) tests used in the present study are widely used procedures for examining the behavioral effects of anxiety (23,24). Horizontal movement distance and residence time are important indices of the open field test, while the open arm entry times and open arm residence times are indices for the EPM test. The results of the Morris water maze test were evaluated based on the escape latency. The open field test was performed as previously described by Choleris et al (25), the EPMtest was performed as previously described by Walf and Frye (26) and the Morris water maze test was performed as previously described by Morris (27). The primary method for data collection was a video-tracking system, which automatically detected and recorded the horizontal movement distance and residence time.
Immunohistochemical staining
Glass slides were pre-heated in an oven at 65°C for 1 h. Paraffin-embedded sections were dewaxed, rehydrated and treated with 3% hydrogen peroxide for quenching of endogenous peroxidase activity at room temperature. Sections were incubated with a rabbit anti-rat polyclonal COX-2 antibody (1:1,000; cat. no. BA0738, Boster Biological Technology, Pleasanton, CA, USA) overnight at 4°C. The spatial localization of COX-2 was visualized by incubation with mouse IgG horseradish peroxidase (HRP)-conjugated secondary antibody (1:10,000; cat. no. PV-9000; ZSGB-Bio; OriGene Technologies, Inc., Beijing, China) for 1 h at room temperature. A 3,3′-diaminobenzidine tetrahydrochloride (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) chromogenic reagent was added dropwise until the nucleus had stained brown. After wards, the sections were rinsed with PBS, counterstained with hematoxylin for 3 min at room temperature, dehydrated with graded ethanol (75, 85, 95% and anhydrous ethanol) and xylene and mounted with Entellan (cat. no. 1.07960.0500; Merck KGaA). Sections were observed under a light microscope, six visual fields were selected and the mean number of positive cells was counted.
RNA extraction and RT-qPCR
Total RNA from rat hippocampi was extracted using TRIzol reagent (Takara Biotechnology Co., Ltd., Dalian, China) and detected with an ultraviolet spectrophotometer and agarose-gel electrophoresis. For each sample, 1 µg RNA was reverse-transcribed to obtain first-strand cDNA using the PrimeScript® RT Reagent kit with a gDNA Eraser (Takara Biotechnology Co., Ltd.) according to the manufacturer's protocol. Expression levels of COX-2were analyzed using RT-qPCR. The primers were designed and synthesized by TsingKe Biological Company (Wuhan, China). The primer specific to COX-2 was: Forward, 5′-TCGCTGTGCCTGATGATTG-3′ and reverse, 5′-TCGCTTATGATCTGTCTTG-3′; and a primer specific to the internal control β-actin was: Forward, 5′-TGACGTGGACATCCGCAAAG-3′ and reverse, 5′-CTGGAAGGTGGACAGCGAGG-3′. Each reaction mixture (20 µl total volume) contained 10 µl of 2× SYBR Premix Ex Taq (Takara Biotechnology Co., Ltd.), 0.4 µmol/l each forward and reverse primer and 0.2±0.02 µg cDNA template. The thermocycling conditions were: 95°C for 30 sec, followed by 40 cycles at 95°C for 5 sec, 58°C for 20 sec and 72°C for 20 sec. The relative transcription levels of genes were calculated using the 2−∆∆Cq method (28). The quantitation cycle (Cq) was determined for each reaction, and the Cq values for each gene of interest were normalized to the endogenous control gene β-actin. The quantification of target and reference genes was evaluated using standard curves and the ratio between the target and reference gene represented the relative expression levels of target gene. For each group three technical replicates of each measurement were obtained.
Western blot analysis
Western blot analysis was performed to determine the expression of COX-2. Rat hippocampi were homogenized and the total protein was extracted. The protein concentration was determined using a Bicinchoninic Acid kit (Bio-Swamp, Wuhan, China). Equal amounts of protein (30 µg) were separated by 10% SDS-PAGE and then transferred onto polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA). Membranes were blocked for 2 h at room temperature with 5% skimmed milk in Tris-buffered saline (20 mmol/l Tris, 500 mmol/l NaCl and 0.05% Tween 20). Subsequently, the membrane was incubated with anti-COX-2 antibody (1:100; cat. no. ab52237; Abcam, Cambridge, UK) overnight at 4°C. An anti-β-actin antibody (1:10,000, cat. no. ab227387, Abcam) was selected as an internal reference. The membranes were washed with Tris-buffered saline and incubated with a rabbit anti-goat secondary antibody (1:10,000, cat. no. E030130, EarthOx, LLC, Millbrae, CA, USA) for 2 h at room temperature. Immunoreactivity was visualized by colorimetric reaction using enhanced chemiluminescence substrate buffer (EMD Millipore, Billerica, MA, USA). Membranes were scanned with a Gel DocEZ imager (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and bands were quantified using Quantity One software version 5.0 (Bio-Rad Laboratories, Inc.).
Terminal deoxynucleotidyl transferase mediated dUTP nick end labeling (TUNEL) staining
Paraffin-embedded sections of rat hippocampi were obtained. TUNEL assay was performed using a TUNEL kit (Boster Biological Technology, Pleasanton, CA, USA) according to the manufacturer's protocol. The sections were sealed with natural gum. Apoptotic cells were observed under a light microscope (magnification, ×400) and counted in 6 fields of vision. The apoptosis index was expressed as the number of apoptotic cells within 1 mm2, and the apoptosis of rat hippocampal neurons was defined as the number of apoptotic neurons/total number of neurons ×100%.
ELISA
The levels of IL-6, TNFα and PGE2 in rat hippocampi were evaluated by an ELISA assay. The supernatant of 10% rat hippocampal homogenate was extracted. IL-6 (cat. no. RA20607), TNF-α (cat. no. RA20035) and PGE2ELISA kits (cat. no. RA20013) were obtained from Bio-Swamp (Wuhan, China) and the assay was carried out according to the manufacturer's protocol.
NO detection
The NO content in rat hippocampi was detected using the Griess test (29). The Nitric Oxide Assay kit (cat. no. S0021, Beyotime Institute of Biotechnology, Shanghai, China) was used according to the manufacturer's protocol. The hippocampal tissue was treated with S3090 lysate (Beyotime Institute of Biotechnology) prior to detection.
Statistics analysis
All data are expressed as the mean ± standard deviation. Statistical differences were analyzed by one-way using SPSS (version 18.0, SPSS, Inc., Chicago, IL, USA). Duncan's test was used as a post-hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
Behavioral changes in rats
In the pre-experiment, in the 25 mg/(kgd) celecoxib treatment group, the COX-2 mRNA expression level was lower than in rats administered 15 and 35 mg/(kgd) celecoxib (Fig. 1). Therefore, 25 mg/(kgd) celecoxib treatment was selected for the following experiments.
Figure 1.
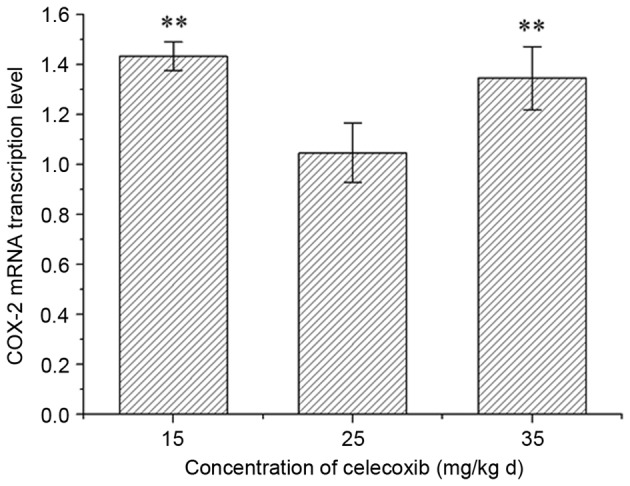
COX-2 mRNA expression levels in post-traumatic stress disorder rats treaded with the COX-2 inhibitor celecoxib. Data are expressed as the mean ± standard deviation (n=3). **P<0.01 vs. the 25 mg/(kgd) group. COX-2, cyclooxygenase-2.
As presented in Fig. 2, behavioral changes were observed in rats with PTSD and in rats from the intervention group, compared with the control group. The alertness, anxiety and environmental adaptability of rats were evaluated by the OF test. The results of the OF test demonstrated that the horizontal movement distance of rats within 5 min in the PTSD and intervention groups were significantly decreased compared with the control group (P<0.01 or P<0.05) and the horizontal movement distance in intervention group was greater than in the PTSD group (P<0.05) (Fig. 2A). The residence time of rats in the PTSD group was increased significantly compared with the control group (P<0.05), but in the intervention group the residence time was decreased significantly compared with the PTSD group (P<0.05; Fig. 2B).
Figure 2.

Behavioral changes in rats with PTSD, and the intervention group rats. (A) Horizontal movement distance and (B) residence time of rats within 5 min of the open-field test. (C) Percentage of open arm entry times and the open arm residence time of rats were observed during an EPM task. (D) The escape latency in the Morris water maze task. Data are expressed as the mean ± standard deviation (n=10). *P<0.05, **P<0.01 vs. the control group; #P<0.05 vs. the PTSD group. PTSD, post-traumatic stress disorder; EPM, elevated plus maze.
The anxiety levels of rats were evaluated by EPM and compared with the control group. The percentage of open arm entry times and the percentage of open arm residence time of rats in the PTSD group were decreased significantly (P<0.01 or P<0.05). Compared with the PTSD group, the percentage of open arm entry times and open arm residence time of rats were increased in the intervention group (P<0.05; Fig. 2C).
The escape latency of rats was detected by MWM and the results are presented in Fig. 2D. Compared with the control group, the escape latency was increased in the PTSD group (P<0.05) and decreased compared with the PTSD group (P<0.05).
COX-2 levels in rat hippocampi
The COX-2 levels in rat hippocampi were evaluated by immunohistochemical staining. In the control group, low or no COX-2 expression was observed (Fig. 3A). Compared with the control, the level of COX-2 expression in the PTSD group was increased significantly (Fig. 3B). In the intervention group, the COX-2 level was lower than that of the PTSD group (Fig. 3C).
Figure 3.

Immunohistochemical staining for COX-2 in rat hippocampi (magnification, ×200). (A) Control group COX-2 was not expressed or expressed at a very low level. (B) Post-traumatic stress disorder group COX-2 expression was visibly enhanced and mainly localized to the cytoplasm and membranes of keratinocytes in rat hippocampi. (C) Intervention group COX-2 level was decreased following treatment with COX-2 inhibitor. COX-2, cyclooxygenase-2.
Transcription level of COX-2 mRNA in rat hippocampi
The mRNA level of COX-2 in rat hippocampi was evaluated by RT-qPCR (Fig. 4). Compared with the control group, COX-2 mRNA levels were increased significantly in the PTSD group (P<0.01) and intervention group (P<0.05). Compared with the PTSD group, COX-2 levels in the intervention group were decreased (P<0.05).
Figure 4.
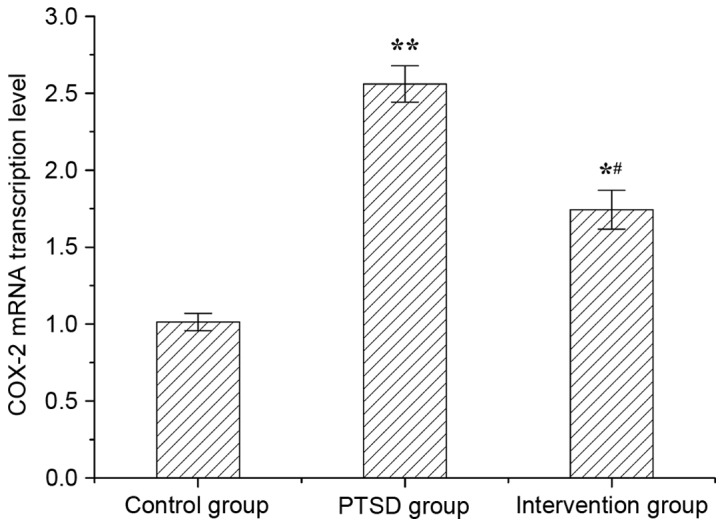
mRNA expression levels of COX-2 in rat hippocampi. Data are expressed as the mean ± standard deviation (n=6). *P<0.05, **P<0.01 vs. the control group; #P<0.05 vs. the PTSD group. COX-2, cyclooxygenase-2; PTSD, post-traumatic stress disorder.
Protein level of COX-2 in rat hippocampi
The protein level of COX-2 in rat hippocampi is presented in Fig. 5. In the PTSD group, the protein expression level of COX-2 was higher than in the control group (P<0.01). However, compared with the PTSD group, the expression level of protein COX-2 was lower in the intervention group (P<0.05).
Figure 5.
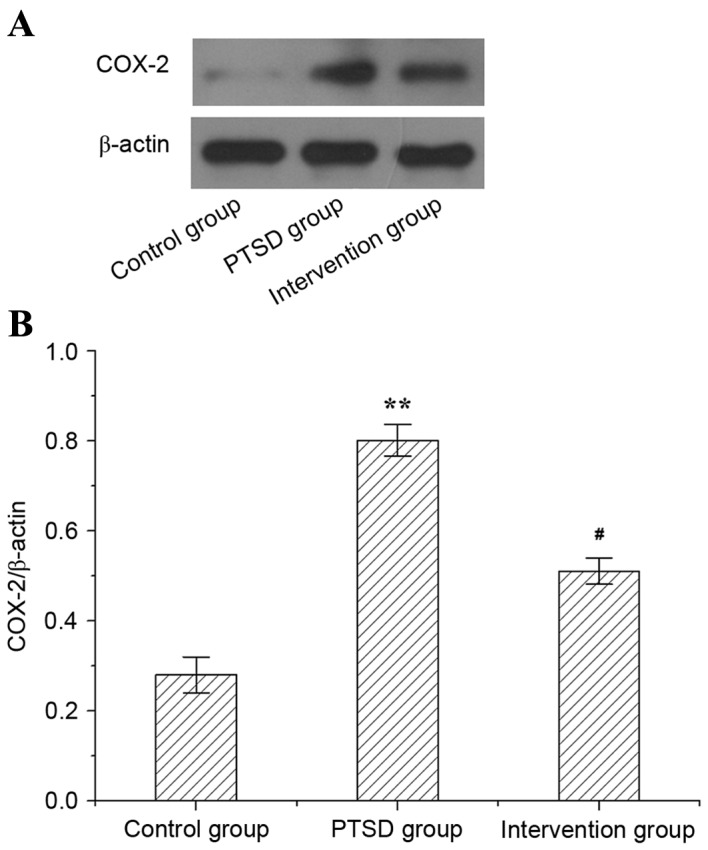
Western blot analysis of COX-2 protein expression levels in rat hippocampi. (A) Western blot visualization of COX-2 protein expression levels in the experimental and control groups. (B) Quantitative analysis of COX-2/β-actin levels in rat hippocampi. Data are expressed as the mean ± standard deviation (n=3). **P<0.01 vs. the control group; #P<0.05 vs. the PTSD group. COX-2, cyclooxygenase-2; PTSD, post-traumatic stress disorder.
Apoptosis of rat hippocampi
The apoptosis of rat hippocampi was evaluated by TUNEL staining and the results are presented in Fig. 6. In the PTSD group and the intervention group, the TUNEL staining was positive. Compared with the control group, the apoptosis in the PTSD group increased significantly (Fig. 6B). However, apoptosis in the intervention group was decreased significantly compared with the PTSD group (Fig. 6C). The apoptotic rate in the PTSD group and the intervention group were higher than in the control group (P<0.01; Fig. 6D). Compared with the PTSD group, the apoptotic rate in the intervention group decreased significantly (P<0.01; Fig. 6D).
Figure 6.

TUNEL staining analysis of the apoptosis in rat hippocampi (magnification, ×400). (A) The control group was negative for the TUNEL staining. (B) PTSD group, the TUNEL staining was positive and the apoptosis was increased compared with the control group. (C) Intervention group, TUNEL staining was positive. (D) The apoptosis rate in rat hippocampi from different groups. Data are expressed as the mean ± standard deviation (n=6). **P<0.01 vs. the control group; ##P<0.01 vs. the PTSD group. TUNEL, terminal deoxynucleotidyl transferase mediated dUTP nick end labeling; PTSD, post-traumatic stress disorder.
Levels of TNF-α, IL-6, PGE2and NO in rat hippocampi
Levels of TNF-α, IL-6, PGE2 and NO in rat hippocampi are presented in Fig. 7. In the PTSD group, the levels of TNF-α, IL-6, PGE2 and NO were higher than in the control group (P<0.01). The levels of TNF-α and IL-6 in the intervention group were higher than in the control group (P<0.05, P<0.01), but lower than in the PTSD group (P<0.01; Fig. 7A). Compared with the PTSD group, the levels of PGE2 and NO in the intervention group were increased significantly (P<0.01; Fig. 7B).
Figure 7.
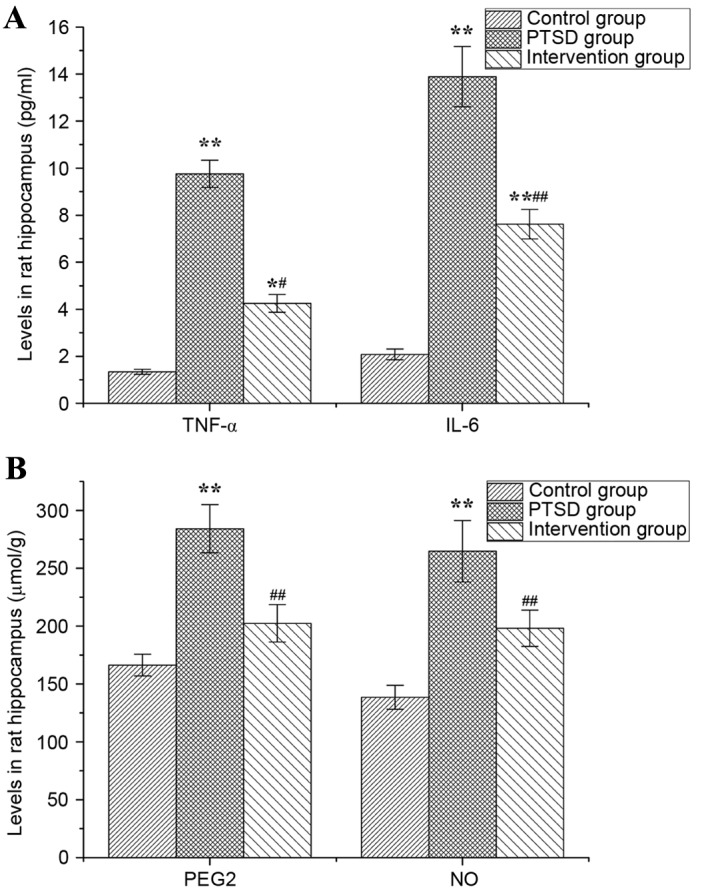
Levels of TNF-α, IL-6, PGE2 and NO in rat hippocampi. The levels of (A) TNF-α and IL-6, and (B) PGE2 and NO. Data are expressed as the mean ± standard deviation (n=6). *P<0.05, **P<0.01 vs. the control group; #P<0.05, ##P<0.01 vs. the PTSD group. TNF-α, tumor necrosis factor-α; IL, interleukin; PGE2, prostaglandin E2; NO, nitric oxide; PTSD, post-traumatic stress disorder.
Discussion
Although the morbidity of PTSD has been reported multiple times, the pathogenesis of the disease remains to be elucidated. PTSD can significantly reduce the quality life of patients, causing psychiatric and physical comorbidity (30). Research over the past two decades has focused on identifying the neurocircuitry mediating the core clinical symptoms of PTSD, in attempt to develop optimal, effective interventions for PTSD (31). The hypothalamic-pituitary-adrenal (HPA) axis, sympathetic and parasympathetic nervous system and 5-hydroxytryptamine (5-HT) are the main regulators of the stress response, emotion and arousal of an organism (32). Intense external stimulation might induce over-excitement of HPA, which can lead to an increased cytokine secretion (33). It has been reported that transmembrane proteins can transport TNF-α and IL-6 into the central nervous system through active transport, causing a series of inflammatory injuries to the central nervous system (34,35). Increased stress-associated protein expression is a key pathogenic symptom of PTSD. In the present study, the levels of TNF-α and IL-6 in rats with PTSD were increased significantly, indicating that TNF-α and IL-6 may serve an important role in the pathogenesis of PTSD. The increase in TNF-α and IL-6 expression in rats with PTSD was prevented by the inhibition of COX-2, which suggested that a feedback loop might exist between COX-2 and inflammation. Stress can stimulate the immune system to release pro-inflammatory factors. IL-6 in plasma of patients with depression was positively correlated with the severity of their clinical symptoms (36). Therefore, the authors of the present study hypothesized that excessive inflammatory cytokine expression can lead to secondary intracranial injury and resulting pathological changes associated with PTSD.
Studies have demonstrated that COX-2 expression is abnormal in patients with depression (36,37). In the present study, rats with PTSD were treated with COX-2 inhibitor celecoxib which alleviated their anxiety and cognitive function. In rats with PTSD, the level of COX-2 was higher than in the control group, while it was decreased significantly following treatment with celecoxib. The above observations suggest that the enhanced expression of COX-2 is involved in the pathogenesis of PTSD and COX-2 is a potential target for PTSD treatment. Celecoxib serves an important role in the treatment of PTSD through the inhibition of the expression of COX-2. The results of the present study are consistent with previous studies, in which inhibition of COX-2 production lead to a reduction in a variety of stress-induced behavioral pathologies in mice (38). The protein level of COX-2 is closely associated with inflammation in vivo (39,40). The present study confirmed that the increase in the abundance of pro-inflammatory factors is associated with an increased COX-2 expression and causes up-regulation of PGE2, NO and other products of oxidative stress. The mechanism of cell apoptosis induced by oxidative stress has been confirmed (41,42). PEG2 is one of the causative factors of apoptosis. Takadera et al (43) demonstrated that apoptosis was observed following treatment of mouse neurons with PGE2 for 48 h and that PGE2 induced apoptosis in a dose-dependent manner via activation of cysteine protein kinase. Li et al (44) reported that COX-2/PGE2 signaling serves a pivotal role in the accumulation and function of myeloid-derived suppressor cells following traumatic stress and that COX-2 blockade inhibits accumulation and function of myeloid-derived suppressor cells and restores T cell response following traumatic stress. In the present study, the level of PEG2 in hippocampi of rats with PTSD was increased and following intervention with celecoxib, the PEG2 level was down-regulated. These results of the present study indicated that celecoxib intervention can inhibit COX-2 production and induce a protective effect on rat hippocampi.
NO, as a highly reactive free radical, can induce free radical chain reaction with ONOO−, which can directly inhibit mitochondrial respiratory chain and cause damage to DNA. Previous studies have shown that activation of NF-κB and up-regulation of the inducible nitric oxide synthase can lead to the synthesis of NO causing tissue injury (45). In the present study, NO expression in hippocampi of rats with PTSD was increased significantly compared with the control group but in the intervention group NO levels were lower than in the PTSD group, indicating that COX-2 inhibition may protect the nervous system by reducing the expression of NO and its downstream pathway.
PTSD as a stress disorder disease and COX-2 may serve an important role in its pathogenesis. In the present study, the levels of TNF-α, IL-6 and COX-2 in the hippocampi of rats with PTSD were increased significantly compared with healthy rats, which is consistent with the PTSD pathology detected by imaging. Levels of proinflammatory cytokines were increased by stress stimuli. Up-regulation of COX-2 was involved in the oxidative stress and its downstream products such as NO and PGE2 were elevated causing neuronal injury and apoptosis. Oxidative stress responses are a common pathway for multiple intracellular signal transduction pathways and drug therapy has been shown to significantly reduce the symptoms of PTSD (46). Therefore, exploring the pathogenesis of PTSD-related signal transduction pathways can contribute to the development of new drugs and clinical treatments.
In conclusion, COX-2 can induce inflammation and cell apoptosis in rats and promote the development of PTSD. COX-2 inhibition can decrease the levels of TNF-α, IL-6, PEG2 and NO in the hippocampi of rats with PTSD and reduce the apoptosis of cells in this tissue. COX-2 inhibition reduces the prevalence of oxidative stress products and apoptosis, and may in the future serve an important role in the clinical research and treatment of PTSD.
Acknowledgements
This study was supported by the grant from Nature Science Foundation of Hubei Province (grant no. 2015CFB694).
References
- 1.Jones KD, Young T, Leppma M. Mild traumatic brain injury and posttraumatic stress disorder in returning Iraq and Afghanistan War Veterans: Implications for assessment and diagnosis. J Counsel Dev. 2010;88:372–376. doi: 10.1002/j.1556-6678.2010.tb00036.x. [DOI] [Google Scholar]
- 2.Olatunji BO, Cisler JM, Tolin DF. Quality of life in the anxiety disorders: A meta-analytic review. Clin Psychol Rev. 2007;27:572–581. doi: 10.1016/j.cpr.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 3.Pietrzak RH, Goldstein RB, Southwick SM, Grant BF. Psychiatric comorbidity of full and partial posttraumatic stress disorder among older adults in the United States: Results from wave 2 of the National Epidemiologic Survey on Alcohol and Related Conditions. Am J Geriatr Psychiatry. 2012;20:380–390. doi: 10.1097/JGP.0b013e31820d92e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen H, Kaplan Z, Koresh O, Matar MA, Geva AB, Zohar J. Early post-stressor intervention with propranolol is ineffective in preventing posttraumatic stress responses in an animal model for PTSD. Eur Neuro Psychopharmacol. 2011;21:230–240. doi: 10.1016/j.euroneuro.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 5.Rauch SA, Morales KH, Zubritsky C, Knott K, Oslin D. Posttraumatic stress, depression, and health among adults in primary care. Am J Geriatr Psychiatry. 2006;14:316–324. doi: 10.1097/01.JGP.0000199382.96115.86. [DOI] [PubMed] [Google Scholar]
- 6.Knight JA, Taft CT. Assessing neuropsychological concomitants of trauma and PTSD. In: Wilson JP, Keane TM, editors. Assessing Psychological Trauma and PTSD. 2nd. The Guilford Press; New York, NY: 2004. pp. 344–388. [Google Scholar]
- 7.Neylan TC, Schadt EE, Yehuda R. Biomarkers for combat-related PTSD: Focus on molecular networks from high-dimensional data. Eur J Psychotraumatol. 2014;5:23938. doi: 10.3402/ejpt.v5.23938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moeller DR, Duffy JM, Goolsby AM, Gallimore JT. Use of a removable mandibular neuroprosthesis for the reduction of posttraumatic stress disorder (PTSD) and mild traumatic brain injury/PTSD/associated nightmares, headaches, and sleep disturbances. J Spec Oper Med. 2014;14:64–73. doi: 10.55460/MHVO-MN5Q. [DOI] [PubMed] [Google Scholar]
- 9.Mills KL, Teesson M, Ross J, Peters L. Trauma, PTSD, and substance use disorders: Findings from the Australian national survey of mental health and well-being. Am J Psychiatry. 2006;163:652–658. doi: 10.1176/ajp.2006.163.4.652. [DOI] [PubMed] [Google Scholar]
- 10.Cohen BE, Marmar CR, Neylan TC, Schiller NB, Ali S, Whooley MA. Posttraumatic stress disorder and health-related quality of life in patients with coronary heart disease: Findings from the heart and soul study. Arch Gen Psychiatry. 2009;66:1214–1220. doi: 10.1001/archgenpsychiatry.2009.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang HK, Bullman TA. Risk of suicide among US veterans after returning from the Iraq or Afghanistan war zones. JAMA. 2008;300:652–653. doi: 10.1001/jama.300.6.652. [DOI] [PubMed] [Google Scholar]
- 12.Nakayama M, Uchimura K, Zhu RL, Nagayama T, Rose ME, Stetler RA, Isakson PC, Chen J, Graham SH. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia; Proc Nati Acad Sci USA; 1998; pp. 10954–10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic-ischemic brain damage. Ann Neurol. 1986;19:105–111. doi: 10.1002/ana.410190202. [DOI] [PubMed] [Google Scholar]
- 14.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 15.Singh DP, Chopra K. Flavocoxid, dual inhibitor of cyclooxygenase-2 and 5-lipoxygenase, exhibits neuroprotection in rat model of ischaemic stroke. Pharmacol Biochem Behav. 2014;120:33–42. doi: 10.1016/j.pbb.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Yagami T, Koma H, Yamamoto Y. Pathophysiological roles of cyclooxygenases and prostaglandins in the central nervous system. Mol Neurobiol. 2016;53:4754–4771. doi: 10.1007/s12035-015-9355-3. [DOI] [PubMed] [Google Scholar]
- 17.Grösch S, Tegeder I, Niederberger E, Bräutigam L, Geisslinger G. COX-2 independent induction of cell cycle arrest and apoptosis in colon cancer cells by the selective COX-2 inhibitor celecoxib. FASEB J. 2001;15:2742–2744. doi: 10.1096/fj.01-0299fje. [DOI] [PubMed] [Google Scholar]
- 18.Altorki NK, Keresztes RS, Port JL, Libby DM, Korst RJ, Flieder DB, Ferrara CA, Yankelevitz DF, Subbaramaiah K, Pasmantier MW, Dannenberg AJ. Celecoxib, a selective cyclo-oxygenase-2 inhibitor, enhances the response to preoperative paclitaxel/carboplatin in early stage lung cancer. J Clin Oncol. 2003;21:2645–2650. doi: 10.1200/JCO.2003.07.127. [DOI] [PubMed] [Google Scholar]
- 19.Karim A, Tolbert DS, Hunt TL, Hubbard RC, Harper KM, Geis GS. Celecoxib, a specific COX-2 inhibitor, has no significant effect on methotrexate pharmacokinetics in patients with rheumatoid arthritis. J Rheumatol. 1999;26:2539–2543. [PubMed] [Google Scholar]
- 20.Liberzon I, Krstov M, Young EA. Stress-restress: Effects on ACTH and fast feedback. Psychoneuroendocrinology. 1997;22:443–453. doi: 10.1016/S0306-4530(97)00044-9. [DOI] [PubMed] [Google Scholar]
- 21.Liberzon I, López JF, Flagel SB, Vázquez DM, Young EA. Differential regulation of hippocampal glucocorticoid receptors mRNA and fast feedback: Relevance to post-traumatic stress disorder. J Neuroendocrinol. 1999;11:11–17. doi: 10.1046/j.1365-2826.1999.00288.x. [DOI] [PubMed] [Google Scholar]
- 22.Nadeem MN, Maqdoom M. Evaluation of anticonvulsant effect of celecoxib, a selective cyclooxygenase-2 inhibitor in experimentally induced convulsions in albino rats. Int J Basic Clin Pharmacol. 2016;5:1466–1470. doi: 10.18203/2319-2003.ijbcp20162454. [DOI] [Google Scholar]
- 23.Carola V, D'Olimpio F, Brunamonti E, Mangia F, Renzi P. Evaluation of the elevated plus-maze and open-field tests for the assessment of anxiety-related behaviour in inbred mice. Behav Brain Res. 2002;134:49–57. doi: 10.1016/S0166-4328(01)00452-1. [DOI] [PubMed] [Google Scholar]
- 24.Brandeis R, Brandys Y, Yehuda S. The use of the Morris Water Maze in the study of memory and learning. Int J Neurosci. 1989;48:29–69. doi: 10.3109/00207458909002151. [DOI] [PubMed] [Google Scholar]
- 25.Choleris E, Thomas AW, Kavaliers M, Prato FS. A detailed ethological analysis of the mouse open field test: Effects of diazepam, chlordiazepoxide and an extremely low frequency pulsed magnetic field. Neurosci Biobehav Rev. 2001;25:235–260. doi: 10.1016/S0149-7634(01)00011-2. [DOI] [PubMed] [Google Scholar]
- 26.Walf AA, Frye CA. The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat Protoc. 2007;2:322–328. doi: 10.1038/nprot.2007.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris R. Development of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 29.Han Y, Li X, Zhou S, Meng G, Xiao Y, Zhang W, Wang Z, Xie L, Liu Z, Lu H, Ji Y. 17ß-estradiol antagonizes the down-regulation of ERα/NOS-3 signaling in vascular endothelial dysfunction of female diabetic rats. PLoS One. 2012;7:e50402. doi: 10.1371/journal.pone.0050402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vermetten E, Vythilingam M, Southwick SM, Charney DS, Bremner JD. Long-term treatment with paroxetine increases verbal declarative memory and hippocampal volume in posttraumatic stress disorder. Biol Psychiatry. 2003;54:693–702. doi: 10.1016/S0006-3223(03)00634-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cisler JM, Bush K, James GA, Smitherman S, Kilts CD. Decoding the traumatic memory among women with PTSD: Implications for neurocircuitry models of PTSD and real-time fMRI neurofeedback. PLoS One. 2015;10:e0134717. doi: 10.1371/journal.pone.0134717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Babson KA, Feldner MT. Temporal relations between sleep problems and both traumatic event exposure and PTSD: A critical review of the empirical literature. J Anxiety Disord. 2010;24:1–15. doi: 10.1016/j.janxdis.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boals A, Hathaway LM. The importance of the DSM-IV E and F criteria in self-report assessments of PTSD. J Anxiety Disord. 2010;24:161–166. doi: 10.1016/j.janxdis.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 34.Bailey JN, Goenjian AK, Noble EP, Walling DP, Ritchie T, Goenjian HA. PTSD and dopaminergic genes, DRD2 and DAT, in multigenerational families exposed to the Spitak earthquake. Psychiatry Res. 2010;178:507–510. doi: 10.1016/j.psychres.2010.04.043. [DOI] [PubMed] [Google Scholar]
- 35.Carpenter LL, Gawuga CE, Tyrka AR, Lee JK, Anderson GM, Price LH. Association between plasma IL-6 response to acute stress and early-life adversity in healthy adults. Neuropsychopharmacology. 2010;35:2617–2623. doi: 10.1038/npp.2010.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson RA. Molecular clue to PTSD. PLoS Biol. 2015;13:e1002283. doi: 10.1371/journal.pbio.1002283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Müller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: Towards an integrated view of depression. Mol Psychiatry. 2007;12:988–1000. doi: 10.1038/sj.mp.4002006. [DOI] [PubMed] [Google Scholar]
- 38.Gamble-George JC, Baldi R, Halladay L, Kocharian A, Hartley N, Silva CG, Roberts H, Haymer A, Marnett LJ, Holmes A, Patel S. Cyclooxygenase-2 inhibition reduces stress-induced affective pathology. Elife. 2016;5(pii):e14137. doi: 10.7554/eLife.14137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao HM, Liu B, Zhang W, Hong JS. Novel anti-inflamatory therapy for Parkinson's disease. Trends Pharmacol Sci. 2003;24:395–401. doi: 10.1016/S0165-6147(03)00176-7. [DOI] [PubMed] [Google Scholar]
- 40.Hinz B, Brune K. Cyclooxygenaxe-2-10 years later. J Pharmacol Exp Ther. 2002;300:367–375. doi: 10.1124/jpet.300.2.367. [DOI] [PubMed] [Google Scholar]
- 41.Said RS, Badr AM, Nada AS, El-Demerdash E. Sodium selenite treatment restores long-lasting ovarian damage induced by irradiation in rats: Impact on oxidative stress and apoptosis. Reprod Toxicol. 2014;43:85–93. doi: 10.1016/j.reprotox.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 42.Nury T, Zarrouk A, Vejux A, Doria M, Riedinger JM, Delage-Mourroux R, Lizard G. Induction of oxiapoptophagy, a mixed mode of cell death associated with oxidative stress, apoptosis and autophagy, on 7-ketocholesterol-treated 158n murine oligodendrocytes: Impairment by α-tocopherol. Biochem Biophys Res Commun. 2014;446:714–719. doi: 10.1016/j.bbrc.2013.11.081. [DOI] [PubMed] [Google Scholar]
- 43.Takadera T, Yumoto H, Tozuka Y, Ohyashiki T. Prostaglandin E(2) induces caspase-dependent apoptosis in rat cortical cells. Neurosci Lett. 2002;317:61–64. doi: 10.1016/S0304-3940(01)02449-1. [DOI] [PubMed] [Google Scholar]
- 44.Li RJ, Liu L, Gao W, Song XZ, Bai XL, Li ZF. Cyclooxygenase-2 blockade inhibits accumulation and function of myeloid-derived suppressor cells and restores T cell response after traumatic stress. J Huazhong Univ Sci Technolog Med Sci. 2014;34:234–240. doi: 10.1007/s11596-014-1264-6. [DOI] [PubMed] [Google Scholar]
- 45.Morioka N, Inoue A, Hanada T, Kumagai K, Takeda K, Ikoma K, Hide I, Tamura Y, Shiomi H, Dohi T, Nakata Y. Nitric oxide synergistically potentiates interleukin-1 beta-induced increase of cyclooxygenase-2 mRNA levels, resulting in the facilitation of substance P release from primary afferent neurons: Involvement of cGMP-independent mechanisms. Neuropharmacology. 2002;43:868–876. doi: 10.1016/S0028-3908(02)00143-0. [DOI] [PubMed] [Google Scholar]
- 46.Puetz TW, Youngstedt SD, Herring MP. Effects of pharmacotherapy on combat-related PTSD, anxiety, and depression: A systematic review and meta-regression analysis. PLoS One. 2015;10:e0126529. doi: 10.1371/journal.pone.0126529. [DOI] [PMC free article] [PubMed] [Google Scholar]