

Abstract
Background
There is a paucity of data about the mechanisms by which sacubitril/valsartan (also known as LCZ696) improves outcomes in patients with heart failure. Specifically, the effects of sacubitril/valsartan on vascular function and NO bioavailability have not been investigated. We hypothesized that sacubitril/valsartan therapy increases circulating NO levels and improves vascular function in the setting of heart failure.
Methods and Results
Male spontaneously hypertensive rats underwent myocardial ischemia/reperfusion surgery to induce heart failure and were followed for up to 12 weeks with serial echocardiography. Rats received sacubitril/valsartan (68 mg/kg), valsartan (31 mg/kg), or vehicle starting at 4 weeks after reperfusion. At 8 or 12 weeks of reperfusion, animals were euthanized and tissues were collected for ex vivo analyses of NO bioavailability, aortic vascular reactivity, myocardial and vascular histology, and cardiac molecular assays. Left ventricular structure and function were improved by both valsartan and sacubitril/valsartan compared with vehicle. Sacubitril/valsartan resulted in superior cardiovascular benefits, as evidenced by sustained improvements in left ventricular ejection fraction and end‐diastolic pressure. Ex vivo vascular function, as measured by aortic vasorelaxation responses to acetylcholine and sodium nitroprusside, was significantly improved by valsartan and sacubitril/valsartan, with more sustained improvements afforded by sacubitril/valsartan. Furthermore, myocardial NO bioavailability was significantly enhanced in animals receiving sacubitril/valsartan therapy.
Conclusions
Sacubitril/valsartan offers superior cardiovascular protection in heart failure and improves vascular function to a greater extent than valsartan alone. Sacubitril/valsartan‐mediated improvements in cardiac and vascular function are likely related to increases in NO bioavailability and explain, in part, the benefits beyond angiotensin receptor blockade.
Keywords: heart failure, myocardial fibrosis, myocardial infarction, renin angiotensin system, vascular function
Subject Categories: Heart Failure, Endothelium/Vascular Type/Nitric Oxide, Fibrosis, Vascular Biology, Animal Models of Human Disease
Clinical Perspective
What Is New?
This study is one of the first to describe a mechanism of action distinct from renin‐angiotensin‐aldosterone system inhibition in the new class of angiotensin receptor–neprilysin inhibitors that includes sacubitril/valsartan (also known as LCZ696).
Sacubitril/valsartan improves vascular function and preserves endothelium‐dependent vasodilatory responses during heart failure (HF) with reduced ejection fraction.
Sacubitril/valsartan increases NO bioavailability, which affords the previously described vascular protection and additional cardioprotective effects that are important during HF with reduced ejection fraction.
What Are the Clinical Implications?
This study provides insight into the poorly understood mechanisms of action of a new class of HF therapy that may aid in the understanding, development, and adoption of this treatment strategy.
In addition to patients with HF with reduced ejection fraction, sacubitril/valsartan may be of therapeutic benefit in patients affected by other diseases associated with vascular dysfunction, such as arterial stiffness, cerebrovascular disease, and HF with preserved ejection fraction.
Introduction
Heart failure (HF) is one of the leading causes of morbidity and mortality in the United States.1 Many of the newest pharmacological strategies used to treat HF rely on the modulation of endogenous neurohormonal systems that are maladaptive during HF progression.2 These changes include sympathetic nervous system and renin‐angiotensin‐aldosterone system overactivity with concomitant suppression of the natriuretic peptide (NP) system.3, 4, 5
The PARADIGM‐HF (Prospective Comparison of ARNI [Angiotensin Receptor–Neprilysin Inhibitor] With ACEI [Angiotensin‐Converting–Enzyme Inhibitor] to Determine Impact on Global Mortality and Morbidity in Heart Failure) trial demonstrated the superiority of the first in class angiotensin receptor–neprilysin inhibitor (ARNi), sacubitril/valsartan, over that of the stand‐alone renin‐angiotensin‐aldosterone system inhibition afforded by enalapril. ARNi therapy combines renin‐angiotensin‐aldosterone system inhibition via angiotensin receptor blockade (ARB) with augmentation of the NP system through inhibition of neprilysin (NEPi), a peptidase enzyme responsible for degradation of NPs.6 ARNi therapy represents a major step forward in HF therapy; however, introduction into clinical use has been met with skepticism because of a perceived lack of evidence.7, 8, 9 Preclinical studies involving animal models have demonstrated reductions in blood pressure,10 hypertrophy,11 fibrosis,11, 12 and ischemic brain damage.13 However, additional studies are important to further our understanding of precise mechanisms by which ARNi therapy protects the failing heart.
Cardiovascular diseases, including HF, are characterized by reductions in endothelium‐dependent vasodilation and vascular function.14, 15, 16 These decreases are associated with reduction in NO bioavailability and increases in systemic oxidative stress that occur during HF.17 There is a strong correlation between the reductions in vascular function associated with endothelial dysfunction and the pathophysiological changes that occur in cardiovascular disease.18, 19 The endothelium has also been suggested as a potential therapeutic target in HF.20, 21 NO plays a crucial role in the protection afforded by the endothelium, and NO bioavailability is important in overall cardiovascular health.22 The benefits of increasing NO bioavailability in cardiovascular disease include vasodilation; anti‐inflammatory, antiplatelet, antiproliferative, and antiapoptotic effects; reduced oxidative stress; and improved cardiac contractility.23, 24
In this study, we hypothesized that treatment with an ARNi would augment NO bioavailability, improve vascular function, attenuate adverse left ventricular (LV) remodeling, and improve cardiac function.
Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Experimental Animals
Male spontaneously hypertensive rats, at 18 to 20 weeks of age, were used in all experiments (Charles River Laboratories). Animals were pair housed in an animal facility with an automated 12‐hour light/dark cycle. Rodent chow (Harlan) and tap water were provided ad libitum. There was no significant difference in average body weight between groups through the length of the study (Figure S1A). All experimental protocols were approved by the Louisiana State University Health Sciences Center Institute for Animal Care and Use Committee and were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Myocardial Ischemia and Reperfusion
Rats were anesthetized using 3.0% isoflurane supplemented with 100% oxygen in an induction chamber. The chest area was shaved and covered with a chemical depilatory crème for 1 minute to ensure complete removal of fur. The animals were subsequently intubated and placed on a surgical table (Indus Instruments) and connected to a rodent ventilator (model 835; Harvard Apparatus) set to 85 breaths per minute at a tidal volume of 3.5 mL with 100% oxygen supplemented with 1.5% isoflurane. The surgical table contains ECG leads and a monitor, along with a homoeothermic heating element. The exposed skin was sterilized with 3 rounds of 70% isopropyl alcohol, followed by betadine, ending with betadine, and then covered with a sterile fenestrated surgical drape. A median sternotomy was performed just lateral to the sternum at the level of the second and third ribs to expose the chest cavity. The left anterior descending artery was visualized and ligated proximally using a 7‐0 Prolene suture mated to a BV‐1 needle (Ethicon). Animals were subjected to 45 minutes of ischemia, followed by reperfusion for up to 12 weeks. The chest cavity was closed in 3 layers using 4‐0 Vicryl suture mated to an FS‐1 needle (Ethicon), after which the animals were recovered. All animals received prophylactic antibiotic therapy with cefazolin and buprenorphine for pain.
A total of 83 animals were included in the present study after accounting for animal deaths and exclusions based on troponin values. The animals were randomly assigned to a treatment (vehicle, valsartan, or sacubitril/valsartan) and a treatment cohort (8 or 12 weeks). Ultimately, 14 animals were enrolled in each group, with the exception of the 12‐week sacubitril/valsartan group, in which 13 animals were enrolled. All of the 8‐week cohort animals were used in molecular analyses; however, only predetermined subsets of the 12‐week cohort animals were used in different molecular analyses.
Plasma Troponin Levels
Tail vein blood was collected at 4 hours after reperfusion to obtain plasma. Cardiac troponin I was measured using an ELISA kit optimized for rat plasma (Life Diagnostics). Troponin was measured in all 100 animals enrolled in the study (n=13–14). There was no significant difference in baseline circulating cardiac troponin I between groups (Figure S1B).
Echocardiography
Biweekly echocardiography was performed serially on the 12‐week cohort animals starting before myocardial ischemia/reperfusion (MI/R) and through the end of the protocol. A Vevo2100 (Fujifilm VisualSonics) rodent echocardiography system connected to an MS250 13‐ to 24‐MHz probe was used to determine cardiovascular function and structure parameters. Animals were anesthetized again using 3.0% isoflurane for induction and maintained via nose cone administration of 1.0% to 1.5% isoflurane. An ECG‐gated kilohertz visualization image was obtained by summation of 300 cardiac cycles. The ECG‐gated kilohertz visualization provided a B‐mode image, which was used for LV ejection fraction determinations; and regenerated M‐mode images, which were used for determination of LV end‐diastolic diameter (LVEDD) and LV end‐systolic diameter. Body weight was also collected when echocardiography measures were obtained. Echocardiography was performed on all animals enrolled in the 12‐week cohort (n=14 for vehicle and valsartan, and n=13 for sacubitril/valsartan).
Administration of Valsartan and Sacubitril/Valsartan
Starting at 4 weeks after reperfusion, animals began receiving daily therapy consisting of vehicle (water), valsartan (31 mg/kg), or sacubitril/valsartan (also known as LCZ696; 68 mg/kg). Drug was administered at 8 am every morning via oral gavage. Treatment assignment was based on plasma troponin levels and baseline cardiovascular function. Euthanized animals received their final dose the day of euthanasia. Valsartan and sacubitril/valsartan were provided by Novartis Pharmaceuticals (Novartis).
Tissue Collection
Animals were euthanized before MI/R, after 4 weeks of reperfusion, after 8 weeks of reperfusion, or after 12 weeks of reperfusion. At the time of euthanasia, echocardiography was performed, pressure measurements were made, blood was collected in K2 EDTA tubes and spun down to obtain plasma, thoracic aorta was collected and stored in cold Krebs‐Henseleit solution, and tissues were obtained and flash frozen for future studies. In addition to flash freezing, a midventricular myocardial specimen and the left kidney were placed in 10% zinc‐buffered formalin for fixation. Weights were also obtained for heart and lung specimens to measure hypertrophy and pulmonary edema, respectively. Tissues were obtained for all animals enrolled in the study (n=13–14). Tissue weights were measured for a subset of animals in the 8‐ and 12‐week cohorts (Figure S2) to ensure adequate preservation of tissue samples (n=7–9).
Blood and LV Pressure Measurements
Animals were anesthetized using methods previously described and subjected to carotid catheterization. A 1.6F catheter with a high‐fidelity pressure transducer probe on the tip was inserted into the carotid and advanced into the LV. Pressure was measured for 1 minute to obtain LV pressure parameters, including LV end‐diastolic pressure (LVEDP), isovolumetric relaxation parameter τ, and cardiac contractility index. After measurement of LV pressures, the catheter was removed from the ventricle and blood pressure was measured at the proximal aorta. Animals were euthanized immediately after pressure determinations. Pressure measurements were performed in all animals enrolled in the study (n=13–14).
NP Measurement
Fresh‐frozen plasma was used in peptide measurements. Brain NP (BNP) and bradykinin were measured using an ELISA kit, per the manufacturer's directions (Phoenix Pharmaceuticals). NT‐proBNP (N‐terminal pro‐B‐type NP) was measured using an ELISA kit, per the manufacturer's directions (MyBioSource). NP levels were measured for all animals enrolled in the study (n=13–14).
Renal Neprilysin Activity
Kidney cortex tissue was collected at the time of euthanasia and immediately flash frozen in liquid nitrogen. Kidney protein was isolated by homogenization of 100 mg of renal cortex in radioimmunoprecipitation assay buffer supplemented with Halt protease and phosphatase inhibitors (ThermoFisher). Isolated protein homogenate (300 µg) was incubated with the fluorogenic peptide N‐dansyl‐d‐Ala‐Gly‐p‐nitro‐Phe‐Gly (Sigma‐Aldrich) and captopril (Sigma‐Aldrich) at concentrations of 500 μmol/L and 100 mmol/L, respectively. This was done in 250 μL of 50 mmol/L Tris‐HCl, pH 7.4, for 30 minutes at 37°C, followed by heat inactivation of neprilysin by incubating for 5 minutes at 100°C. Samples were spun for 10 minutes at 5000 RCF, and 200 μL of supernatant was collected for fluorometric analysis with excitation at 342 nm and emission at 562 nm within 5 minutes of acquisition.25, 26 Renal neprilysin activity was measured for all animals enrolled in the study, and exclusions were made afterwards using the Grubbs' test for outliers (n=11–14).
Histological Analysis
Fixed short‐axis midventricular myocardial samples were embedded in paraffin, divided into sections, and stained with Masson's trichrome and Picrosirius red. Picrosirius red–stained specimens were analyzed using fluorescent confocal microscopy, with collagen volume fraction determined by ImageJ (National Institutes of Health). Masson's trichrome–stained specimens were analyzed in a blinded manner, and fibrosis was scored on the basis of histopathological findings, including percentage fibrosis and infarct expansion. Fibrosis scoring ranged from 0 (no evidence of fibrosis) to 5 (significant fibrosis). Histological analysis was performed on all animals enrolled in the study (n=13–14).
Vascular Function
Thoracic aorta was collected at euthanasia and divided into 4 0.5‐cm sections in ice‐cold Krebs‐Henseleit solution. Rings were placed in a vascular reactivity system composed of tissue chambers perfused with warm normal Krebs‐Henseleit solution and pressure transducer systems (Radnoti). Rings were allowed to equilibrate for 1 hour at a preload of 1.5 g. Rings were subsequently contracted with 1 μmol/L phenylephrine, and relaxation profiles were obtained for acetylcholine (1 nmol/L to 10 μmol/L) and sodium nitroprusside (SNP; 1 nmol/L to 10 μmol/L). Vascular function analysis was performed on a predetermined subset of animals per cohort (n=8 for 8‐week cohort, and n=6–10 for 12‐week cohort).
Vascular Compliance
Vascular compliance was determined on the basis of the parameters of stroke volume, as determined by 2‐dimensional echocardiography at the time of euthanasia and pulse pressure, which is defined as the difference between aortic systolic pressure and diastolic pressure measured by arterial pressure catheter.27, 28 Vascular compliance analysis was performed on all animals enrolled in the study (n=13–14).
Nitrite Measurement
Nitrite content of plasma and myocardial specimens was made using an ENO‐30 ion chromatography system with a PC‐PAK precolumn and an NO‐PAK separation column (Eicom), as previously described.29 The mobile phase was composed of 10% methanol with 0.15 mol/L NaCl‐NH4Cl and 0.5 g/L tetrasodium EDTA supplied at a flow of 0.33 mL/min. The color‐generating Griess reagent, composed of an equal mixture of 1.25% HCl containing 0.5 g/L sulfanilamide and 10% methanol containing 0.25 g/L N‐naphthylethylenediamine, was delivered at a combined flow rate of 0.1 mL/min. Nitrite bioavailability was measured for all animals enrolled in the study, and exclusions were made afterwards using the Grubbs' test for outliers (n=10–14).
Western Blot
Flash‐frozen tissue was homogenized in radioimmunoprecipitation assay buffer supplemented with Halt protease and phosphatase inhibitor cocktail (Thermo‐Fisher) and pepstatin‐A (Sigma‐Aldrich). Protein content was determined using a BCA protein assay (Thermo‐Fisher). Immunoblotting used specific antibodies for endothelial NO synthase (eNOS; BD Biosciences), phosphorylated Ser1177 eNOS (Cell Signaling Technologies), protein kinase G (PKG; Cell Signaling Technologies), GAPDH (Santa Cruz), and α‐tubulin (Santa Cruz). Western blot analysis was performed on a predetermined subset of animals in the 12‐week cohort (n=8).
Gene Expression Assays
RNA was isolated from LV tissue using Trizol reagent extraction, followed by RNA precipitation. RNA was quantified using Gen 5 software (BioTek), and cDNA was synthesized using I‐script cDNA synthesis kit (Bio‐Rad). Quantitative polymerase chain reaction was performed using TaqMan primers (Life Technologies) and the StepOnePlus RT‐PCR system (AppliedBiosystems). Genes probed included NOS1, NOS2, NOS3, CBS, CSE, 3MST, COL1, COL3, MMP2, MMP9, TIMP1, TIMP2, IL6, TGFβ, ANP, BNP, CNP, NEP, NPR1, NPR2, NPR3, as well as GAPDH, TUBA1A, and 18s for housekeeping. 2ΔΔCt was used to determine the relative gene expression changes. Gene expression analysis was performed on all animals enrolled in the study, and exclusions were made afterwards using mRNA quality analysis and the Grubbs' test for outliers (n=10–14).
Statistical Analysis
Data are expressed as mean±SEM unless specified otherwise. Statistical differences were determined using 1‐way ANOVA when comparing single time points. Two‐way ANOVA with repeated measures using multiple comparisons and a Bonferroni post‐test correction was used for echocardiography and analyses requiring multiple time point comparisons; significance was only reported if there was an interaction between time and treatment. Significance was attained when P<0.05.
Results
Sacubitril/Valsartan (LCZ696) Improves Cardiovascular Function and Structure
LV ejection fraction was significantly reduced after MI/R and continued to decline in the vehicle treatment group for the entirety of the study, along with significant and sustained dilation of the LV (Figure 1A through 1C).
Figure 1.
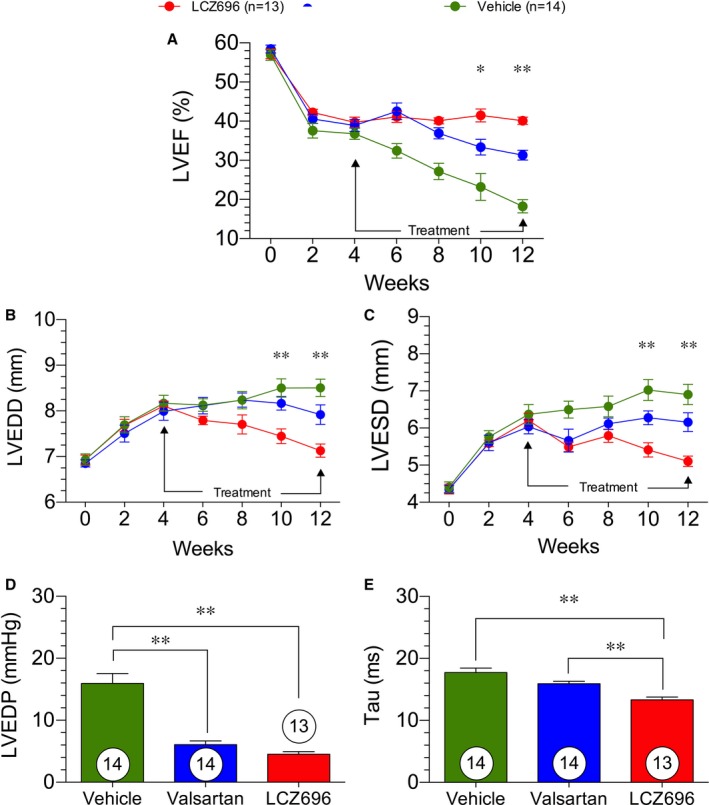
Cardiovascular function and structure in heart failure. Cardiovascular structure and function was measured by 2‐dimensional serial echocardiography at baseline and every 2 weeks after acute myocardial infarction for a period of 12 weeks. Left ventricular ejection fraction (LVEF; %; A), left ventricular end‐diastolic diameter (LVEDD; mm; B), left ventricular end‐systolic diameter (LVESD; mm; C), left ventricular end‐diastolic pressure (LVEDP) at 12 weeks (D), and τ at 12 weeks (E). The figure legend indicates treatment groups and number of animals in each group. *P<0.05, **P<0.01 for the difference between valsartan and sacubitril/valsartan (also known as LCZ696).
At 4 weeks after reperfusion, treatment with valsartan and sacubitril/valsartan was initiated; by week 6 weeks after reperfusion, both valsartan‐ and sacubitril/valsartan‐treated animals showed significant improvements in cardiovascular function compared with vehicle. We did not observe any significant difference among the treatment groups through the first 4 weeks of treatment (ie, weeks 6–10). Through the first 4 weeks of treatment, LVEDD was significantly reduced in the sacubitril/valsartan group compared with the vehicle and the valsartan groups. There were no differences in LVEDD between the valsartan and vehicle groups. In contrast, LV end‐systolic diameter in the valsartan‐ and sacubitril/valsartan‐treated groups were both significantly reduced when compared with the group of animals receiving vehicle.
At 8 to 12 weeks after reperfusion, the sacubitril/valsartan‐treated group exhibited sustained improvements in cardiovascular function, whereas the valsartan group experienced reductions in LV ejection fraction coupled with increased LV dilation at end systole. At 8 to 12 weeks after MI/R, sacubitril/valsartan treatment was superior to valsartan and vehicle in terms of LV ejection fraction, LVEDD, and LV end‐systolic diameter.
LVEDP was elevated in the vehicle group to levels suggestive of HF after MI/R30 (Figure 1D and 1E). Treatment with valsartan or sacubitril/valsartan resulted in a significant reduction in LVEDP at 12 weeks after MI/R. These data suggest that either treatment reduces LV congestion during HF. The isovolumetric relaxation constant τ was significantly reduced with sacubitril/valsartan treatment compared with either vehicle and valsartan treatment at 12 weeks after reperfusion. These data suggest that treatment with an ARNi significantly improves LV relaxation and LV hemodynamic strain.
Although valsartan afforded short‐term preservation of cardiovascular function and structure, sacubitril/valsartan treatment resulted in sustained preservation of cardiovascular function from 6 to 12 weeks after MI/R and reversed the adverse LV remodeling that occurred before initiation of treatment. The reductions in LVEDP and τ are also suggestive of improved cardiovascular function of the LV and reduced severity of HF. Furthermore, the improvements in τ by sacubitril/valsartan compared with valsartan suggest a possible diastolic function improvement afforded by the combination drug compared with the ARB alone.
Sacubitril/Valsartan Increases Circulating NPs While Reducing Their Production Through Inhibition of Neprilysin
Myocardial ischemia and reperfusion resulting in HF led to an increased production of NT‐proBNP that was processed to mature bioactive BNP at both 8 and 12 weeks after reperfusion, with significant elevation at 12 weeks (Figure 2A and 2B). The production of NT‐proBNP was significantly lower in the sacubitril/valsartan group than in both vehicle and valsartan groups. Although NT‐proBNP, an index of BNP production, was reduced, the levels of mature BNP were significantly greater in the sacubitril/valsartan group at 12 weeks (Figure 2C and 2D). To understand whether this effect was attributable to neprilysin inhibition, neprilysin activity was measured at both the 8‐ and 12‐week time point (Figure 2G and 2H). Renal neprilysin activity was significantly attenuated at both time points, confirming the efficacy of the sacubitril moiety of sacubitril/valsartan to inhibit the endopeptidase. Circulating bradykinin levels were unchanged at 8 weeks after acute myocardial infarction, but increased significantly at 12 weeks after reperfusion in the sacubitril/valsartan group (Figure 2E and 2F). There were no significant differences between the vehicle and valsartan groups in terms of circulating NP levels.
Figure 2.
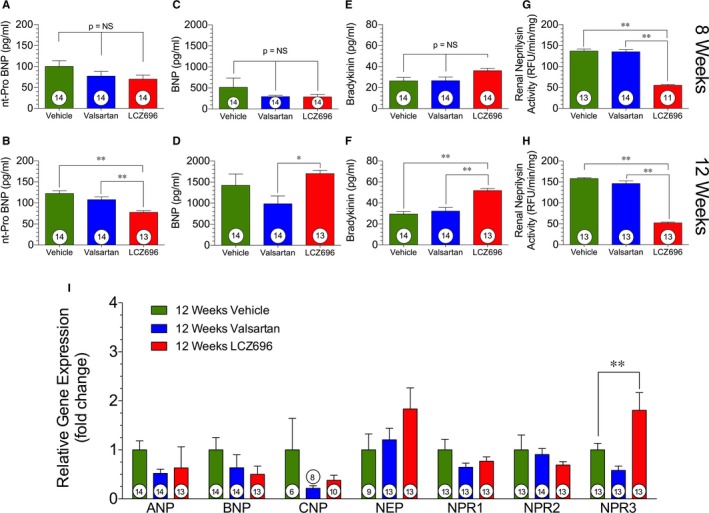
Natriuretic peptides and neprilysin activity. Circulating natriuretic peptide levels, including brain natriuretic peptide (BNP), at 8 and 12 weeks after myocardial ischemia/reperfusion (4 and 8 weeks after valsartan or sacubitril/valsartan [also known as LCZ696] therapy, respectively). A, NT‐proBNP (N‐terminal pro‐B‐type natriuretic peptide; pg/mL) at 8 weeks. B, NT‐proBNP (pg/mL) at 12 weeks. C, BNP (ng/mL) at 8 weeks. D, BNP (ng/mL) at 12 weeks. E, Bradykinin levels (pg/mL) at 8 weeks. F, Bradykinin levels (ng/mL) at 12 weeks. G, Neprilysin activity as measured in relative fluorescence units (RFU/min per mg) at 8 weeks.. H, Neprilysin activity (RFU/min per mg) at 12 weeks. I, Myocardial gene expression at 12 weeks. Animal numbers are shown in white circles. NS indicates not significant. *P<0.05, **P<0.01.
Analysis of myocardial ANP, BNP, and CNP gene expression at 12 weeks after reperfusion revealed no significant differences among treatment groups (Figure 2I). However, there were trends for reduced expression of BNP and CNP by the sacubitril/valsartan treatment compared with vehicle therapy. There was a significant elevation of NPR3 expression in the myocardium; NPR3 encodes the clearance receptor responsible for binding and degrading NPs.31, 32, 33, 34 The elevation of NPR3 expression suggests that the increases in circulating NPs leads to an upregulation of the endogenous mechanisms to remove them.
Sacubitril/Valsartan and Valsartan Reduce Fibrosis in the Infarct Border Zone
Representative images of myocardial infarct size and infarct border zone expansion are depicted in Figure 3A through 3C. We evaluated the extent of myocardial fibrosis and infarct border zone expansion in the valsartan and sacubitril/valsartan treatment groups compared with vehicle therapy at 12 weeks after myocardial infarction (Figure 3). We failed to observe any significant differences in interstitial and perivascular fibrosis in the myocardium (Figure 3D and 3E) among any of the study groups. We did, however, observe significant reductions in infarct border zone expansion in animals treated with either valsartan or sacubitril/valsartan (Figure 3D) compared with vehicle. The reduction in collagen volume fraction in both the valsartan and sacubitril/valsartan groups was ≈2‐fold (Figure 3F). There was a significant reduction of collagen 3 expression and a trend for reductions in collagen 1 expression in sacubitril/valsartan compared with vehicle therapy (Figure 3G). There was also a significant reduction of tissue inhibitor of metalloproteinases 2 and transforming growth factor‐β in both valsartan and sacubitril/valsartan treatment groups compared with vehicle. Taken together, this suggests an overall reduction in collagen deposition and decreased extracellular matrix remodeling in the infarct border zone after treatment with valsartan or sacubitril/valsartan.
Figure 3.
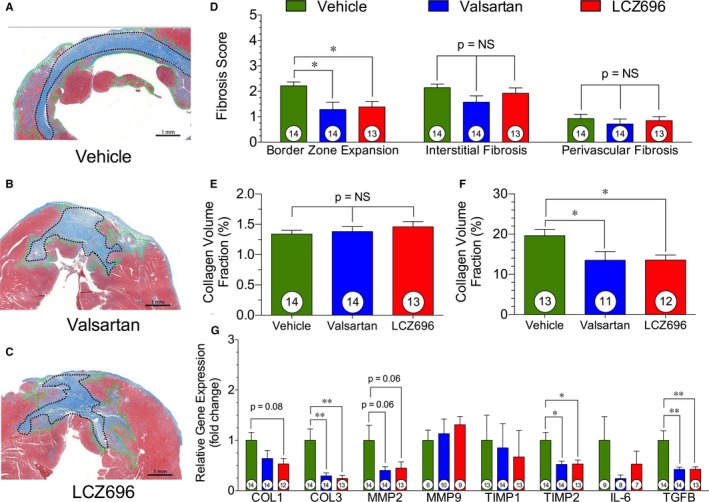
Myocardial fibrosis. Myocardial fibrosis at 12 weeks after acute myocardial infarction. A, Representative photomicrographs of infarct border zone expansion, with infarct scar outlined in black and border zone outlined in green in a vehicle heart. B, Representative image of infarct and infarct border zone expansion, with infarct scar outlined in black and border zone outlined in green in a valsartan‐treated heart. C, Representative image of sacubitril/valsartan (also known as LCZ696) heart infarct. Infarct scar is outlined in black, and the infarct border zone is outlined in green. D, Myocardial fibrosis scoring of infarct border zone expansion, interstitial fibrosis, and perivascular fibrosis. E, Quantification of collagen content in the infarct border zone. F, Quantification of collagen content in the myocardial interstitum. G, Expression of fibrosis genes in the myocardium immediately adjacent to the scar. Animal numbers are shown in white circles within their respective columns. NS indicates not significant. *P<0.05, **P<0.01.
Sacubitril/Valsartan and Valsartan Improve Endothelium‐Independent and Endothelium‐Dependent Vasodilation Responses
Vascular reactivity of isolated aortic vascular rings to SNP are depicted in Figure 4. Aorta from valsartan‐ and sacubitril/valsartan‐treated animals had better relaxation responses to SNP at both 8 and 12 weeks after reperfusion (Figure 4A and 4C). These improvements in the relaxation curve occurred starting at 10 nmol/L for both valsartan and sacubitril/valsartan. The improvement in relaxation to SNP after valsartan and sacubitril/valsartan was reflected in significant reductions in EC50 at both time points (8 and 12 weeks) between the valsartan and sacubitril/valsartan treatment group and animals receiving vehicle (Figure 4B and 4D). There were no differences between valsartan and sacubitril/valsartan therapy in terms of maximal relaxation or EC50 at any time point nor was there a difference in SNP maximal relaxation response from 8 to 12 weeks (Figure 4E). This suggests that both valsartan and sacubitril/valsartan provided a similar benefit in terms of endothelium‐independent vascular reactivity to SNP.
Figure 4.
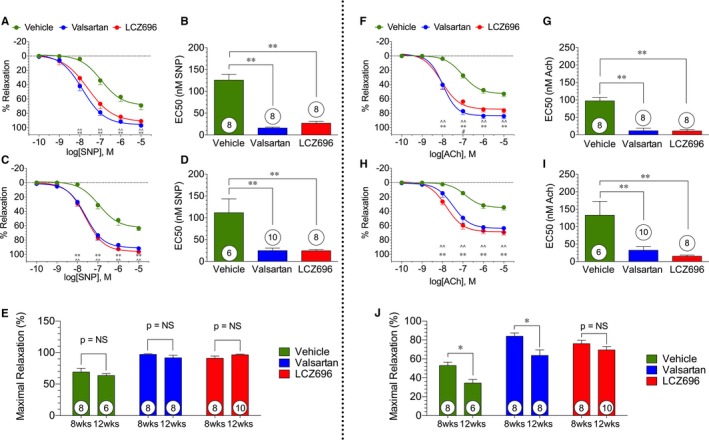
Sodium nitroprusside (SNP)– and acetylcholine (ACh)‐mediated aortic vasorelaxation. SNP‐ and ACh‐mediated vasorelaxation of isolated aortic vascular rings at 8 and 12 weeks after myocardial ischemia/reperfusion/ (ie, 4 and 8 weeks of valsartan or sacubitril/valsartan [also known as LCZ696] therapy). A, Dose‐response curve to SNP administration at 8 weeks with nonlinear curve fitted. B, EC 50 for SNP‐mediated vasorelaxation at 8 weeks. C, Dose‐response curve to SNP administration at 12 weeks with nonlinear curve fitted. D, EC 50 for SNP‐mediated vasorelaxation at 12 weeks. E, Maximal relaxation response to SNP at 8 and 12 weeks. F, Concentration‐response curve to ACh administration at 8 weeks with nonlinear curve fitted. G, EC 50 for ACh‐mediated vasorelaxation at 8 weeks. H, Concentration‐response curve to ACh administration at 12 weeks with nonlinear curve fitted. I, EC 50 for ACh‐mediated vasorelaxation at 12 weeks. J, Maximal relaxation response to ACh at 8 and 12 weeks. The number of animals in each group is shown in white circles within their respective columns. NS indicates not significant. Key: ^ indicates a significant difference between vehicle and valsartan; *, a difference between vehicle and sacubitril/valsartan; #, a difference between valsartan and sacubitril/valsartan; single notation, P<0.05; double notation, P<0.01.
We also evaluated vascular relaxation responses of isolated thoracic aorta to acetylcholine at 8 and 12 weeks after MI/R, and these data are presented in Figure 4. Vascular relaxation curves (% relaxation) are presented in Figure 4F (8 weeks) and 4H (12 weeks). Vascular relaxation to acetylcholine was significantly improved after treatment with valsartan or sacubitril/valsartan at both 8 and 12 weeks after reperfusion. EC50 (nmol/L) data for acetylcholine are summarized in Figure 4G (8 weeks) and 4I (12 weeks) for all study groups. The improvement in relaxation was reflected in a significant reduction in EC50 at both the 8‐ and 12‐week time points between valsartan and sacubitril/valsartan treatment compared with vehicle therapy. Data for maximal relaxation responses to acetylcholine are shown in Figure 4J. Interestingly, we observed significant decreases in the maximal relaxation response to acetylcholine between 8 and 12 weeks in the vehicle and valsartan groups, suggesting that vascular injury is progressive over time in this HF with reduced ejection fraction (HFrEF) model. There was a significant improvement in maximal relaxation for the valsartan and sacubitril/valsartan groups at both time points when compared with the vehicle group. At both 8 and 12 weeks, there was, however, no difference between valsartan and sacubitril/valsartan therapy in terms of maximal relaxation to acetylcholine.
We did not observe a time‐dependent change in SNP relaxation (Figure 4E). In the acetylcholine‐treated groups, the vehicle and valsartan groups exhibited reductions in the maximal relaxation responses (Figure 4I) and in increases in the EC50 from 8 to 12 weeks. There was, however, no change in maximal relaxation responses or EC50 in the sacubitril/valsartan group among these time points. These data suggest that sacubitril/valsartan is superior to valsartan in terms of vascular endothelial protection.
Sacubitril/Valsartan and Valsartan Improve Vascular Compliance
We also measured the in vivo aortic vascular compliance using LV stroke volume and aortic pulse pressure. These data are presented for both 8 and 12 weeks after myocardial infarction in Figure 5.
Figure 5.
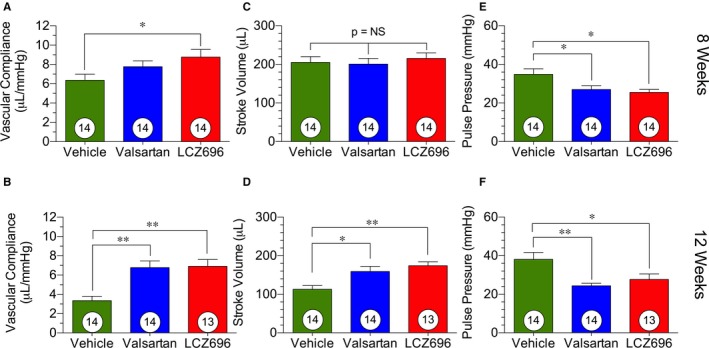
Vascular compliance. Vascular compliance, stroke volume, and pulse pressure at 8 and 12 weeks after myocardial ischemia/reperfusion, corresponding to 4 and 8 weeks of therapy, respectively. A, Vascular compliance at 8 weeks. B, Vascular compliance at 12 weeks. C, Stroke volume at 8 weeks. D, Stroke volume at 12 weeks. E, Pulse pressure at 8 weeks. F, Pulse pressure at 12 weeks. Animal numbers are shown in white circles. LCZ696 indicates sacubitril/valsartan; and NS, not significant. *P<0.05, **P<0.01.
Treatment with sacubitril/valsartan improved vascular compliance beginning at 8 weeks after reperfusion, which corresponds to 4 weeks of treatment (Figure 5A). This improvement was attributable to the reductions in pulse pressure in these animals. At 12 weeks after reperfusion, the improvement in vascular compliance was seen in both the valsartan‐ and sacubitril/valsartan‐treated animals (Figure 5B), with an equal improvement in both groups compared with vehicle therapy. This improvement was attributable to the combined effects of a significantly increased stroke volume and significantly reduced pulse pressure (Figure 5C through 5F). This suggests that the benefits afforded by both treatments are attributable to both reductions in arterial stiffness, as reflected in improved pulse pressure, and improved cardiovascular function, as reflected in improved stroke volume. More important, we did not observe a difference in blood pressure reduction between valsartan and sacubitril/valsartan treatment (Figure S3). There was also no significant difference between molecular markers of hypertensive activity between valsartan‐ and sacubitril/valsartan‐treated animals (Figure S4).
Sacubitril/Valsartan Improves NO Bioavailability
We measured both plasma and myocardial nitrite levels (μmol/L) at 8 and 12 weeks after reperfusion as an index of NO bioavailability (Figure 6). Valsartan and sacubitril/valsartan both increased circulating plasma nitrite compared with vehicle treatment at 8 weeks, with increases maintained at 12 weeks for sacubitril/valsartan therapy (Figure 6A and 6B). Despite the observed increases in plasma nitrite at 8 weeks after valsartan and sacubitril/valsartan treatment, only sacubitril/valsartan treatment increased myocardial nitrite at 12 weeks after reperfusion (Figure 6C and 6D). The increase in NO stores within the myocardium is especially important given the cardioprotective effects NO can exert and the decreases in NO known to occur in HF.35, 36, 37
Figure 6.
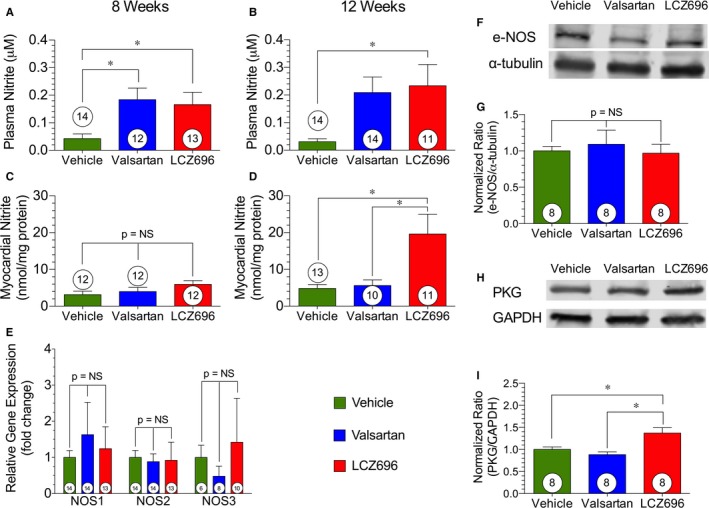
NO bioavailability. NO bioavailability and production at 8 and 12 weeks after myocardial ischemia/reperfusion. A, Plasma nitrite levels at 8 weeks. B, Plasma nitrite levels at 12 weeks. C, Myocardial nitrite levels at 8 weeks. D, Myocardial nitrite levels at 12 weeks. E, Myocardial gene expression of endothelial NO synthase (eNOS)–producing enzymes at 12 weeks. F, Representative immunoblot of total eNOS at 12 weeks with α‐tubulin as a housekeeping control. G, Normalized ratio of myocardial expression of eNOS compared with control α‐tubulin at 12 weeks. H, Representative immunoblot of protein kinase G (PKG) at 12 weeks with GAPDH as a housekeeping protein control. I, Normalized ratio of myocardial expression of PKG compared with control GAPDH at 12 weeks. Animal numbers are shown in white circles within their respective columns. LCZ696 indicates sacubitril/valsartan; and NS, not significant. *P<0.05.
Myocardial eNOS gene expression (Figure 6E) and protein levels (Figure 6F and 6G) of endogenous NO synthesizing enzymes were unchanged among groups. However, sacubitril/valsartan therapy increased protein levels of PKG, a downstream effector of NO34 (Figure 6H and 6I), providing further evidence of increased NO signaling after sacubitril/valsartan treatment in HF.
Discussion
Sacubitril/valsartan received Food and Drug Administration approval for use in HFrEF in May 2015 after the groundbreaking results of the PARADIGM‐HF clinical trial.6 Shortly after its approval, groups, including the American Heart Association, the American College of Cardiology, the Heart Failure Society of America, and the European Society of Cardiology, recommended the use of sacubitril/valsartan in their guidelines for treatment of acute and chronic HF.38, 39 The guidelines were made on the basis of significant clinical evidence in support of sacubitril/valsartan over angiotensin‐converting enzyme inhibitor, as seen in a large randomized controlled clinical trial.6 The clinical trial was halted early because of the significant benefit afforded to patients; however, there were serious limitations to the study that have been described at length in many opinion articles.40, 41 There are limited publicly available preclinical data describing the precise mechanisms by which LCZ protects the failing heart. The present study provides a direct comparison between sacubitril/valsartan and valsartan in a clinically relevant in vivo animal model of HFrEF. This head‐to‐head comparison allows the elucidation of how NEPi augments and improves on angiotensin receptor antagonism alone.
In the present study, we found that treatment with sacubitril/valsartan results in better preservation of cardiac structure and LV function in comparison to valsartan. This preservation was similar between the ARB alone, valsartan, and the ARB and NEPi combination, sacubitril/valsartan, through the first 4 weeks of treatment (ie, 8 weeks after myocardial infarction). Interestingly, this functional benefit was sustained out to 12 weeks after reperfusion with sacubitril/valsartan therapy in comparison to valsartan. The cardiac structural changes that occurred in response to treatment were different between valsartan and sacubitril/valsartan: valsartan improved LV end‐systolic diameter similarly to sacubitril/valsartan but was more similar to vehicle therapy in terms of LVEDD. Although there was a similar attenuation of LVEDP increases afforded by both treatments, sacubitril/valsartan was superior to valsartan in terms of isovolumetric relaxation constant τ. One possible explanation for this finding is a decrease in interstitial fibrosis; however, we did not observe significant differences in myocardial interstitial fibrosis between valsartan therapy and sacubitril/valsartan therapy throughout the study protocol. Therefore, the improvement in LVEDD and τ may suggest that the benefits of NEPi beyond those offered by valsartan in the later phase of LV dysfunction may be attributable to a reduction in preload caused by enhanced natriuresis, improved vascular compliance, or increases in NO bioavailability and, therefore, improved diastolic function of the ventricle.
Despite increases in mature NPs, there was not a similar increase in NT‐proBNP because neprilysin only degrades BNP and not NT‐proBNP. Pro‐BNP is cleaved by corin into the inactive NT‐proBNP and mature active BNP‐32.42, 43 We believe the presence of increased BNP in the sacubitril/valsartan‐treated animals is suggestive of NEPi whereas reduced NT‐proBNP is likely attributable to decreased cardiomyocyte stress and, thus, reduced BNP transcription. The benefits of NPs are multidimensional and include reduced hemodynamic load, antifibrotic effects, antihypertrophic effects, vasodilatory effects, and others.44, 45 Given the strong effect of NPs on these parameters, we next investigated the vascular function of the subject animals.
The pathological significance of endothelial dysfunction, arterial stiffness, decreased vascular function, and reduced NO bioavailability has been well characterized and studied in the context of HF.46, 47 The interplay of these factors is extremely important, with healthy endothelium being central to overall cardiovascular health.18 Valsartan and sacubitril/valsartan treatment both improved vascular reactivity, with improved vasorelaxation responses to both SNP and acetylcholine. We also observed improved vascular compliance in sacubitril/valsartan‐treated animals. This improvement in vascular compliance was attributable to both increased stroke volume, reflective of improved LV function, and decreased pulse pressure, reflective of decreased arterial stiffness.
At 4 and 8 weeks after therapy with valsartan and sacubitril/valsartan, both agents equally benefitted SNP‐mediated vascular relaxation. However, there was not a significant deterioration over this span, suggesting that injury to the vascular smooth muscle in the aorta is completed by 8 weeks after reperfusion. In contrast, although acetylcholine‐mediated endothelium‐dependent vasorelaxation was similar among both treatment groups, there was a time‐dependent effect, suggesting further endothelial damage as HF progressed over time. In the valsartan treatment group, we observed that the maximal relaxation response was improved at both time points compared with vehicle therapy and that there was an overall deterioration in this response over time. However, at 8 and 12 weeks after reperfusion, there was no decrease in maximal relaxation in response to acetylcholine in the sacubitril/valsartan treatment group. This suggests that part of the mechanism of action of sacubitril/valsartan‐mediated cardioprotection in HFrEF is via sustained protection of the vascular endothelium.
To further understand how sacubitril/valsartan treatment protects the failing heart, we measured NO bioavailability in circulation and in the myocardium. NO is both a key mediator and marker of endothelial function48, 49, 50 and a well‐established cardioprotective factor in acute myocardial infarction and HF.35, 36, 37 We did observe increased circulating nitrite levels, a storage form of NO, in both valsartan‐ and sacubitril/valsartan‐treated animals, which is likely attributable to reduced oxidative stress and reactive oxygen species and perhaps increased generation of NO via eNOS. Interestingly, myocardial nitrite was also significantly higher in the sacubitril/valsartan group, suggesting that sacubitril/valsartan therapy was able to augment NO synthesis in the myocardium in the setting of HF. There was no difference among any study groups in terms of myocardial transcription of eNOS or myocardial eNOS protein levels. However, we did observe an increase in PKG. Increases in NO lead to increased cGMP levels; increased cGMP levels activate PKG; PKG affects many of the physiological and cardioprotective signals that are attributed to NO.51, 52, 53 Increased NO bioavailability can lead to benefits, including angiogenesis, reduced oxidative stress and apoptosis, reductions in smooth muscle cell hypertrophy, antiplatelet effects, and antithrombotic effects.50, 51, 52, 53 It is also important to consider that NPs also increase cGMP through their interaction with the NPR1 and NPR2 receptors.31, 32 Furthermore, even the clearance receptor NPR3 may be able to increase cGMP through coupling with eNOS.32, 33 We observed an increase in the expression of NPR3 in the myocardium of sacubitril/valsartan‐treated animals that is likely a compensatory response to the increased circulating levels of NPs. Last, bradykinin is partially degraded by neprilysin, and we observed increases in circulating bradykinin in sacubitril/valsartan‐treated animals. Bradykinin can increase activity of eNOS and, therefore, also increase NO bioavailability.54, 55, 56, 57, 58 We believe that the increased NO bioavailability observed is attributable to the combination of increased circulating bradykinin and NPR3 expression, and the significant increases in PKG expression are attributable to both increased NO and increased cGMP via NPR1 and NPR2.
The ability of sacubitril/valsartan to increase NO bioavailability in the myocardium is a clinically important factor that distinguishes ARNi therapy from ARB therapy. Higher levels of NO can reduce the symptomatic burden of HF through angiogenesis and vasodilation as well as alter the pathological progression of HF through effects on oxidative stress and hypertrophy.59 This increase in NO bioavailability may be 1 of the unique mechanisms by which sacubitril/valsartan therapy was superior to ARB therapy in our study and superior to angiotensin‐converting enzyme inhibitor therapy in clinical studies.
In conclusion, our data demonstrate the sustained benefits of sacubitril/valsartan on LV function and remodeling in a clinically relevant in vivo model of HFrEF. We found that one of the cardioprotective benefits of sacubitril/valsartan beyond the benefits afforded by valsartan alone during HF is related to potent vascular protection. This protection was evidenced by improvements in endothelium‐dependent vasorelaxation and improved vascular compliance. This vascular protection is correlated with increased NO bioavailability in both the circulation and in the myocardium after treatment with sacubitril/valsartan. The increases in myocardial NO may also explain a previously unknown mechanism through which ARNi therapy leads to superior clinical outcomes compared with renin‐angiotensin‐aldosterone system inhibition alone. Future studies will be aimed at confirming these results in additional preclinical models of HF and in determining how increases in NP levels upregulate NO bioavailability and vascular function to improve LV performance.
Sources of Funding
This work was supported by an investigator‐initiated trial supported by Novartis Pharmaceuticals Corporation (LCZ696BUSNC15T to Lefer). This work was supported by grants from the National Heart, Lung, and Blood Institute (National Institutes of Health; 5R01HL092141, 5R01 HL093579, 1R01 HL11657, and 1U24 HL094373 to Lefer). We are also grateful for the generous funding from Louisiana State University Medical School Foundation and Louisiana State University Medical School Alumni Association.
Disclosures
None.
Supporting information
Figure S1. A, Body weight of treatment groups over 12 weeks and (B) Plasma circulating cardiac troponin‐I was measured at 4 hours post‐reperfusion as an index of acute myocardial cell death following myocardial ischemia. No significance was observed among groups.
Figure S2. Effect of treatment on heart weight at term sacrifice for both the 8 week cohort and 12 week cohort. A, heart weights at 8 weeks, (B) left ventricular (LV) weights at 8 weeks, (C) right ventricular (RV) weight at 8 weeks, (D) atrias weight at 8 weeks, (E) ratio of wet lung weight to dry lung weight at 8 weeks, (F) heart weights at 12 weeks, (G) LV weight at 12 weeks, (H) RV weight at 12 weeks, (I) atria weight at 12 weeks, and (J) ratio of wet lung weight to dry lung weight at 12 weeks. Numbers of animals in each are shown in white circles under their respective dot plots. *P<0.05, **P<0.01, P=NS (not statistically significant).
Figure S3. Systemic blood pressure at the time of sacrifice for the 8‐weeks cohort and 12 weeks. A, Mean arterial blood pressure (mm Hg) at 8 weeks, (B) heart rate (beats per minute) at 8 weeks, (C) peak systolic arterial pressure (mm Hg) at 8 weeks, (D) plateau diastolic arterial pressure (mm Hg) at 8 weeks, (E) mean arterial pressure (mm Hg) at 12 weeks, (F) heart rate (beats per minute) at 12 weeks, (G) peak systolic arterial pressure at 12 weeks, and (H) plateau diastolic arterial pressure (mm Hg) at 12 weeks. Number of animals in each study group shown in white circles inside their respective columns. *P<0.05, **P<0.01. P=NS (not statistically significant).
Figure S4. Hypertension markers at 12 weeks post‐reperfusion (ie, corresponds to 8 weeks of treatment). A, Renal norepinephrine content, (B) plasma angiotensin II, and (C) plasma renin activity. Animal numbers are shown in white circles inside their respective columns. *P<0.05, **P<0.01, no significance bars indicate lack of statistical difference among groups.
Acknowledgments
We thank Craig Zibilich and Jean Carnal for invaluable assistance with technical aspects of the study; and Dr Luis Marrero and Laura Scott for expert assistance in preparing histological specimens.
(J Am Heart Assoc. 2018;7:e008268 DOI:10.1161/JAHA.117.008268.)29502102
References
- 1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jiménez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER III, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB. Heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation. 2016;133:e38–e360. [DOI] [PubMed] [Google Scholar]
- 2. Sacks CA, Jarcho JA, Curfman GD. Paradigm shifts in heart‐failure therapy: a timeline. N Engl J Med. 2014;371:989–991. [DOI] [PubMed] [Google Scholar]
- 3. Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure: physiology, pathophysiology, and clinical implications. J Am Coll Cardiol. 2009;54:1747–1762. [DOI] [PubMed] [Google Scholar]
- 4. Sayer G, Bhat G. The renin‐angiotensin‐aldosterone system and heart failure. Cardiol Clin. 2014;32:21–32. [DOI] [PubMed] [Google Scholar]
- 5. Volpe M, Carnovali M, Mastromarino V. The natriuretic peptides system in the pathophysiology of heart failure: from molecular basis to treatment. Clin Sci. 2015;130:57–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR. Angiotensin‐neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371:993–1004. [DOI] [PubMed] [Google Scholar]
- 7. Gradman AH. LCZ696: the next step in improving RAS inhibition? Curr Hypertens Rep. 2015;17:37. [DOI] [PubMed] [Google Scholar]
- 8. Pham AQ, Patel Y, Gallagher B. LCZ696 (angiotensin‐neprilysin inhibition): the new kid on the heart failure block? J Pharm Pract. 2015;28:137–145. [DOI] [PubMed] [Google Scholar]
- 9. Mauro G, Senni M. Sacubitril/valsartan (LCZ696) for the treatment of heart failure. Expert Rev Cardiovasc Ther. 2016;14:145–153. [DOI] [PubMed] [Google Scholar]
- 10. Kusaka H, Sueta D, Koibuchi N, Hasgawa Y, Nakagawa T, Lin B, Ogawa H, Kim‐Mitsuyama S. LCZ696, angiotensin II receptor‐neprilysin inhibitor, ameliorates high‐salt‐induced hypertension and cardiovascular injury more than valsartan alone. Am J Hypertens. 2015;28:1409–1417. [DOI] [PubMed] [Google Scholar]
- 11. von Lueder TG, Wang BH, Kompa AR, Huang L, Webb R, Jordaan P, Atar D, Krum H. Angiotensin receptor neprilysin inhibitor LCZ696 attenuates cardiac remodeling and dysfunction after myocardial infarction by reducing cardiac fibrosis and hypertrophy. Circ Heart Fail. 2015;8:71–78. [DOI] [PubMed] [Google Scholar]
- 12. Suematsu Y, Miura SI, Goto M, Matsuo Y, Arimura T, Kuwano T, Imaizumi S, Iwata A, Yahiro E, Saku K. LCZ696, an angiotensin receptor‐neprilysin inhibitor, improves cardiac function with the attenuation of fibrosis in heart failure with reduced ejection fraction in streptozotocin‐induced diabetic mice. Eur J Heart Fail. 2016;18:386–393. [DOI] [PubMed] [Google Scholar]
- 13. Bai HY, Mogi M, Nakaoka H, Kan‐no H, Tsukada K, Chisaka T, Wang XL, Kukida M, Shan BS, Yamauchi T. Pre‐treatment with LCZ696, an orally active angiotensin receptor neprilysin inhibitor, prevents ischemic brain damage. Eur J Pharmacol. 2015;762:293–298. [DOI] [PubMed] [Google Scholar]
- 14. Witman MAH, Fjeldstad AS, McDaniel J, Ives SJ, Zhao J, Barrett‐O'Keefe Z, Nativi JN, Stehlik J, Wray DW, Richardson RS. Vascular function and the role of oxidative stress in heart failure, heart transplant, and beyond. Hypertension. 2012;60:659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Katz SD, Biasucci L, Sabba C, Strom JA, Jondeau G, Galvao M, Solomon S, Nikolic SD, Forman R, LeJemtel TH. Impaired endothelium‐mediated vasodilation in the peripheral vasculature of patients with congestive heart failure. J Am Coll Cardiol. 1992;19:918–925. [DOI] [PubMed] [Google Scholar]
- 16. Drexler H, Hayoz D, Munzel T, Hornig B, Just H, Brunner HR, Zelis R. Endothelial function in chronic congestive heart failure. Am J Cardiol. 1992;69:1596–1601. [DOI] [PubMed] [Google Scholar]
- 17. Katz SD, Khan T, Zeballos GA, Mathew L, Potharlanka P, Knecht M, Whelan J. Decreased activity of the L‐arginine‐nitric oxide metabolic pathway in patients with congestive heart failure. Circulation. 1999;99:2113–2117. [DOI] [PubMed] [Google Scholar]
- 18. Marti CN, Gheorghiade M, Kalogeropoulos AP, Georgiopoulou VV, Quyyumi AA, Butler J. Endothelial dysfunction, arterial stiffness, and heart failure. J Am Coll Cardiol. 2012;60:1455–1469. [DOI] [PubMed] [Google Scholar]
- 19. Bank AJ, Lee PC, Kubo SH. Endothelial dysfunction in patients with heart failure: relationship to disease severity. J Card Fail. 2000;6:29–36. [DOI] [PubMed] [Google Scholar]
- 20. Tousoulis D, Charakida M, Stefanadis C. Inflammation and endothelial dysfunction as therapeutic targets in patients with heart failure. Int J Cardiol. 2005;100:347–353. [DOI] [PubMed] [Google Scholar]
- 21. Drexler H. Endothelium as a therapeutic target in heart failure. Circulation. 1998;98:2652–2655. [DOI] [PubMed] [Google Scholar]
- 22. Ignarro LJ. Nitric oxide as a unique signaling molecule in the vascular system: a historical overview. J Physiol Pharmacol. 2002;53:503–514. [PubMed] [Google Scholar]
- 23. Liu VW, Huang PL. Cardiovascular role of nitric oxide: a review of insights from nitric oxide synthase gene disrupted mice. Cardiovasc Res. 2008;77:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Polhemus DJ, Lefer DJ. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ Res. 2014;14:730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Florentin D, Sassi A, Roques BP. A highly sensitive fluorometric assay for “enkephalinase,” a neutral metalloendopeptidase that releases tyrosine‐glycine‐glycine from enkephalins. Anal Biochem. 1984;141:62–69. [DOI] [PubMed] [Google Scholar]
- 26. Polhemus DJ, Trivedi RK, Gao J, Li Z, Scarborough AL, Goodchild TT, Varner KJ, Xia H, Smart FW, Kapusta DR, Lefer DJ. Renal denervation protects the failing heart via inhibition of neprilysin activity in the kidney. J Am Coll Cardiol. 2017;70:2139–2153. [DOI] [PubMed] [Google Scholar]
- 27. Nakamura M, Sugawara S, Arakawa N, Nagano M, Shizuka T, Shimoda Y, Sakai T, Hiramori K. Reduced vascular compliance is associated with impaired endothelium‐dependent dilatation in the brachial artery of patients with congestive heart failure. J Card Fail. 2004;10:36–42. [DOI] [PubMed] [Google Scholar]
- 28. Borlaug BA, Kass DA. Ventricular‐vascular interaction in heart failure. Heart Fail Clin. 2008;4:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Polhemus DJ, Gao J, Scarborough AL, Trivedi RK, McDonough KH, Goodchild TT, Smart F, Kapusta DR, Lefer DJ. Radiofrequency renal denervation protects the ischemic heart via inhibition of GRK2 and increased nitric oxide signaling. Circ Res. 2016;119:470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mielniczuk LM, Lamas GA, Flaker GC, Mitchell G, Smith SC, Gersh BJ, Solomon SD, Moye LA, Rouleau JL, Rutherford JD, Pfeiffer MA. Left ventricular end‐diastolic pressure and risk of subsequent heart failure in patients following an acute myocardial infarction. Congest Heart Fail. 2007;13:209–214. [DOI] [PubMed] [Google Scholar]
- 31. Kone BC. Molecular biology of natriuretic peptides and nitric oxide synthases. Cardiovasc Res. 2001;51:429–441. [DOI] [PubMed] [Google Scholar]
- 32. Murthy KS, Teng BQ, Jin JG, Makhlouf GM. G protein‐dependent activation of smooth muscle eNOS via natriuretic peptide clearance receptor. Am J Physiol. 1998;275:C1409–C1416. [DOI] [PubMed] [Google Scholar]
- 33. Anand‐Srivastava MB, Sehl PD, Lowe DG. Cytoplasmic domain of natriuretic peptide receptor‐C inhibits adenylyl cyclase: involvement of a pertussis toxin‐sensitive G protein. J Biol Chem. 1996;271:19324–19329. [DOI] [PubMed] [Google Scholar]
- 34. Hammond J, Balligand JL. Nitric oxide synthase and cyclic GMP signaling in cardiac myocytes: from contractility to remodeling. J Mol Cell Cardiol. 2012;52:330–340. [DOI] [PubMed] [Google Scholar]
- 35. Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G Sr, Gojon G Jr, Wang R, Karusula N, Nicholson CK, Calvert JW, Lefer DJ. H2S protects against pressure overload‐induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127:1116–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Donnarumma E, Ali MJ, Rushing AM, Scarborough AL, Bradley JM, Organ CL, Islam KN, Polhemus DJ, Evangelista S, Cirino G, Jenkins JS, Patel RA, Lefer DJ, Goodchild TT. Zofenopril protects against myocardial ischemia‐reperfusion injury by increasing nitric oxide and hydrogen sulfide bioavailability. J Am Heart Assoc. 2016;5:e003531 DOI: 10.1161/JAHA.116.003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bhushan S, Kondo K, Polhemus DJ, Otsuka H, Nicholson CK, Tao YX, Huang H, Georgiopoulou VV, Murohara T, Calvert JW, Butler J, Lefer DJ. Nitrite therapy improves left ventricular function during heart failure via restoration of nitric oxide‐mediated cytoprotective signaling. Circ Res. 2014;114:1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Colvin MM, Drazner MH, Filippatos G, Fonarow GC, Givertz MM, Hollenberg SM, Lindenfeld J, Masoudi FA, McBride PE, Peterson PN, Stevenson LW, Westlake C. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2016;134:e298. [DOI] [PubMed] [Google Scholar]
- 39. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, González‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37:2129–2200. [DOI] [PubMed] [Google Scholar]
- 40. Chen CH. Critical questions about PARADIGM‐HF and the future. Acta Cardiol Sin. 2016;32:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vardeny O, Claggett B, Packer M, Zile MR, Rouleau J, Swedberg K, Teerlink JR, Desai AS, Lefkowitz M, Shi V, McMurray JJ, Solomon SD. Efficacy of sacubitril/valsartan vs. enalapril at lower than target doses in heart failure with reduced ejection fraction: the PARADIGM‐HF trial. Eur J Heart Fail. 2016;18:1228–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McKie PM, Burnett JC. NT‐proBNP: the gold standard biomarker in heart failure. J Am Coll Cardiol. 2016;68:2437–2439. [DOI] [PubMed] [Google Scholar]
- 43. Zois NE, Bartels ED, Hunter I, Kousholt BS, Olsen LH, Goetze JP. Natriuretic peptides in cardiometabolic regulation and disease. Nat Rev Cardiol. 2014;11:403–412. [DOI] [PubMed] [Google Scholar]
- 44. Potter LR, Yoder AR, Flora DR, Antos LK, Dickey DM. Natriuretic peptides: their structures, receptors, physiological functions and therapeutic applications. Handb Exp Pharmacol. 2009;191:341–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee NS, Daniels LB. Current understanding of the compensatory actions of cardiac natriuretic peptides in cardiac failure: a clinical perspective. Card Fail Rev. 2016;2:14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bauersachs J, Widder JD. Endothelial dysfunction in heart failure. Pharmacol Rep. 2008;60:119–126. [PubMed] [Google Scholar]
- 47. Lam CS, Brutsaert DL. Endothelial dysfunction: a pathophysiological factor in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2012;60:1787–1789. [DOI] [PubMed] [Google Scholar]
- 48. Shantsila E, Wrigley BJ, Blann AD, Gill PS, Lip GY. A contemporary view on endothelial function in heart failure. Eur J Heart Fail. 2012;14:873–881. [DOI] [PubMed] [Google Scholar]
- 49. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. [DOI] [PubMed] [Google Scholar]
- 50. Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium‐derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84:9265–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Forstermann U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch. 2010;459:923–939. [DOI] [PubMed] [Google Scholar]
- 52. Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langston W, Patel RP, Yet SF, Wang X, Kevil CG, Gladwin MT, Lefer DJ. Cytoprotective effects of nitrite during in vivo ischemia reperfusion of the heart and liver. J Clin Invest. 2005;115:1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, Wang X, MacArthur PH, Shoja A, Raghavachari N, Calvert JW, Brookes PS, Lefer DJ, Gladwin MT. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204:2089–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, Maggi CA, Geppetti P, Ledda F. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J Clin Invest. 1994;94:2036–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101:2567–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Searles CD, Harrison DG. The interaction of nitric oxide, bradykinin, and the angiotensin II type 2 receptor: lessons learned from transgenic mice. J Clin Invest. 1999;104:1013–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hartman JC. The role of bradykinin and nitric oxide in the cardioprotective actions of ACE inhibitors. Ann Thorac Surg. 1995;60:789–792. [DOI] [PubMed] [Google Scholar]
- 58. Bae SW, Kim HS, Cha YN, Park YS, Jo SA, Jo I. Rapid increase in endothelial nitric oxide production by bradykinin is mediated by protein kinase A signaling pathway. Biochem Biophys Res Commun. 2003;306:981–987. [DOI] [PubMed] [Google Scholar]
- 59. Naseem KM. The role of nitric oxide in cardiovascular diseases. Mol Aspects Med. 2005;26:33–65. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. A, Body weight of treatment groups over 12 weeks and (B) Plasma circulating cardiac troponin‐I was measured at 4 hours post‐reperfusion as an index of acute myocardial cell death following myocardial ischemia. No significance was observed among groups.
Figure S2. Effect of treatment on heart weight at term sacrifice for both the 8 week cohort and 12 week cohort. A, heart weights at 8 weeks, (B) left ventricular (LV) weights at 8 weeks, (C) right ventricular (RV) weight at 8 weeks, (D) atrias weight at 8 weeks, (E) ratio of wet lung weight to dry lung weight at 8 weeks, (F) heart weights at 12 weeks, (G) LV weight at 12 weeks, (H) RV weight at 12 weeks, (I) atria weight at 12 weeks, and (J) ratio of wet lung weight to dry lung weight at 12 weeks. Numbers of animals in each are shown in white circles under their respective dot plots. *P<0.05, **P<0.01, P=NS (not statistically significant).
Figure S3. Systemic blood pressure at the time of sacrifice for the 8‐weeks cohort and 12 weeks. A, Mean arterial blood pressure (mm Hg) at 8 weeks, (B) heart rate (beats per minute) at 8 weeks, (C) peak systolic arterial pressure (mm Hg) at 8 weeks, (D) plateau diastolic arterial pressure (mm Hg) at 8 weeks, (E) mean arterial pressure (mm Hg) at 12 weeks, (F) heart rate (beats per minute) at 12 weeks, (G) peak systolic arterial pressure at 12 weeks, and (H) plateau diastolic arterial pressure (mm Hg) at 12 weeks. Number of animals in each study group shown in white circles inside their respective columns. *P<0.05, **P<0.01. P=NS (not statistically significant).
Figure S4. Hypertension markers at 12 weeks post‐reperfusion (ie, corresponds to 8 weeks of treatment). A, Renal norepinephrine content, (B) plasma angiotensin II, and (C) plasma renin activity. Animal numbers are shown in white circles inside their respective columns. *P<0.05, **P<0.01, no significance bars indicate lack of statistical difference among groups.