

Abstract
Two prescient 1953 publications set the stage for the elucidation of a novel endocrine system: Schatzmann’s report that cardiotonic steroids (CTSs) are all Na+ pump inhibitors, and Szent-Gyorgi’s suggestion that there is an endogenous “missing screw” in heart failure that CTSs like digoxin may replace. In 1977 I postulated that an endogenous Na+ pump inhibitor acts as a natriuretic hormone and simultaneously elevates blood pressure (BP) in salt-dependent hypertension. This hypothesis was based on the idea that excess renal salt retention promoted the secretion of a CTS-like hormone that inhibits renal Na+ pumps and salt reabsorption. The hormone also inhibits arterial Na+ pumps, elevates myocyte Na+ and promotes Na/Ca exchanger-mediated Ca2+ gain. This enhances vasoconstriction and arterial tone—the hallmark of hypertension. Here I describe how those ideas led to the discovery that the CTS-like hormone is endogenous ouabain (EO), a key factor in the pathogenesis of hypertension and heart failure. Seminal observations that underlie the still-emerging picture of the EO-Na+ pump endocrine system in the physiology and pathophysiology of multiple organ systems are summarized. Milestones include: 1) cloning the Na+ pump isoforms and physiological studies of mutated pumps in mice; 2) discovery that Na+ pumps are also EO-triggered signaling molecules; 3) demonstration that ouabain, but not digoxin, is hypertensinogenic; 4) elucidation of EO’s roles in kidney development and cardiovascular and renal physiology and pathophysiology; 5) discovery of “brain ouabain”, a component of a novel hypothalamic neuromodulatory pathway; and 6) finding that EO and its brain receptors modulate behavior and learning.
Keywords: calcium, endogenous ouabain, hypertension, Na/Ca exchange, sodium pump
A new idea is the most quickly acting antigen known to science.
—Wilfred Trotter, FRS
Origins
In 1977, Professors Paul Horowicz and Paul De Weer (my Washington University colleague), the founding editor and associate editor, respectively, of AJP Cell Physiology, invited me to write a review article for their new journal. Paul De Weer, an Na+ pump expert, and I had discussed my ideas about the role of Na+ pumps and a postulated Na+ pump inhibitor/natriuretic hormone in the pathogenesis of hypertension. The present article is a personal account of the origin of those ideas and the developments in the field since my hypothesis was published in the journal’s first volume (29); Table 1 provides a timeline.
Table 1.
Milestones in elucidating the endogenous ouabain-Na+ pump endocrine system
| Year | Key Investigator | Discovery | Reference |
|---|---|---|---|
| 1785 | Withering | Digitalis is a therapy for heart failure (HF) | (301) |
| 1861 | Kirk | Discovery that S. kombé seed juice (contains ouabain) is cardiotonic | (182, 204) |
| 1888 | Arnaud | Isolation (from ouabaio tree bark) and naming of ouabain | (8, 139) |
| 1953 | Schatzmann | Cardiotonic steroids (CTS) inhibit Na+ pumps | (255) |
| 1953 | Szent-Gyorgi | Digitalis may replace a missing endogenous compound | (274) |
| 1957 | Skou | Na+ pump is an Na-K-ATPase (NKA) | (264) |
| 1967 | Dahl | A humoral factor contributes to salt-dependent hypertension | (50) |
| 1968 | Reuter | Na/Ca exchanger (NCX) discovery; | (246) |
| 1968/9 | Baker/Blaustein | NCX discovery: “missing link” explains the cardiotonic effect of CTSs | (13, 14) |
| 1973 | Reuter/Blaustein | NCX is present in arteries and is postulated to modulate blood pressure (BP) | (245) |
| 1975 | Bricker | Natriuretic hormone (“third factor”) is a NKA inhibitor | (72) |
| 1976 | Haddy | Na+ pump-endogenous CTS-hypertension hypothesis | (113) |
| 1977 | Blaustein | Na+ pump-NCX-endogenous CTS-hypertension hypothesis | (29) |
| 1978 | Brody | Sustained hypertension requires the hypothalamic AV3V region | (39) |
| 1982 | Hamlyn/Blaustein | A circulating NKA inhibitor correlates with BP in humans | (123) |
| 1983 | Atkinson | Low (≤10–7 M) ouabain concentrations trigger cell proliferation | (10) |
| 1984 | Erdmann | Rat cardiac myocytes express ~90% low (α1) and ~10% high (α2) ouabain affinity Na+ pumps | (299) |
| 1985–7 | Lingrel/Shull | NKA is cloned; 3 catalytic (α) subunit isoforms (α1–α3) identified | (259, 260) |
| 1989 | Philipson | Ouabain upregulates NCX1 expression in cardiomyocytes | (287) |
| 1989 | Delva | Plasma ouabain-like compound (OLC) is elevated in pregnancy and more so in preeclampsia | (54) |
| 1991 | Hamlyn/Upjohn group/Blaustein | An endogenous CTS in humans identified as “ouabain” (EO) | (118) |
| 1992 | Gottlieb/Hamlyn | Plasma EO correlates inversely with cardiac index in HF patients | (104) |
| 1993 | Haddy/Pamnani/Hamlyn | Ouabain (chronic treatment) causes hypertension in rats | (314) |
| 1993 | Hamlyn/Manunta | Digoxin doesn’t induce hypertension, but antagonizes ouabain’s effect | (196) |
| 1993/4 | Hamlyn | Adrenal glands synthesize and secrete EO | (36, 162) |
| 1994 | EP and CE Gomez-Sanchez | Active anti-ouabain immunization attenuates DOCA-salt hypertension | (101) |
| 1994 | Leenen | “Brain ouabain” mediates salt-sensitive hypertension | (133) |
| 1995 | Hollenberg | Fab fragments (Digibind) attenuate DOCA + salt hypertension in rats | (156) |
| 1995 | Leenen | “Brain ouabain” mediates sympathetic hyperactivity in HF | (169) |
| 1995 | Rossi/Manunta/Hamlyn Hamilton | ~50% of patients with essential hypertension or aldosterone-producing adenomas have elevated plasma EO that correlates with mean BP | (252) |
| 1996–8 | Askari/Xie | Ouabain binding to NKA activates protein kinase (PK) signaling cascades | (135, 153, 231) |
| 1997–8 | Defaye/Perrin | [14C]rhamnose is incorporated into EO in bovine adrenocortical cells | (217) |
| 1997–8 | Bianchi/Ferrari | Synthesis of rostafuroxin (PST2238), an ouabain antagonist but not NKA inhibitor, attenuates ouabain-induced hypertension | (79, 241) |
| 1997–8 | Blaustein/Juhaszova | α2 Na+ pumps and NCX1 colocalize at PLasmERosomes, and help regulate local [Ca2+]CYT and Ca2+ signaling | (33, 144, 145) |
| 2000 | Xie/Askari | Ouabain activates PK cascades independent of changes in [Na+] or [Ca2+] | (181) |
| 2005 | Lingrel | ACTH-hypertension requires α2 NKA ouabain binding sites and EO | (60) |
| 2006 | Hamlyn/Manunta/Hamilton | Plasma EO in humans is elevated by both high salt and salt depletion | (191) |
| 2008 | Lingrel/Lorenz | Digibind prevents natriuresis promoted by high ouabain affinity renal α1 NKA* | (184) |
| 2009 | Lichtstein | Anti-ouabain antibodies promote adrenal cortex, not medulla, hypertrophy | (213) |
| 2009 | Heiny/Lingrel | α2 NKA ouabain binding sites modulate skeletal muscle fatigue* | (231) |
| 2010 | Aperia | Ouabain rescues fetal kidney development in malnourished pregnant rats* | (174) |
| 2010 | Golovina | Arterial Ca2+ transporters are upregulated in ouabain- and salt-sensitive hypertension | (238, 322) |
| 2010 | Leenen | A novel hypothalamic slow neuromodulatory pathway [Ang II-AT1R-aldosterone-MR-ENaC-EO-(α2) Na+ pumps] sustains BP in hypertension* | (167) |
| 2011 | Lorenz/Lingrel | Pressure overload-induced cardiac hypertrophy and dysfunction requires ouabain-sensitive Na+ pumps and EO | (296) |
| 2011 | Van Huysse/Lingrel | NaCl-induced hypertension requires α2 NKA ouabain binding sites and EO | (286) |
| 2011 | Vorhees/Williams/Lingrel | Mice with ouabain-resistant α2 NKA exhibit specific behavioral abnormalities and learning impairment* | (254) |
| 2012 | Hamlyn | MS shows EO is elevated in pregnant rats; detection of two EO isomers in rat plasma | (138) |
| 2013 | Golovina/Hamlyn | Ouabain, not digoxin, upregulates arterial Ca2+ transporters and elevates BP via a Src-dependent mechanism; first evidence that an ion transporter can act as a biased hormone receptor | (323) |
| 2013 | Manunta/Gottlieb/Bianchi/Hamlyn | Preoperative plasma EO predicts acute kidney injury (AKI) in cardiac surgery patients; renal function is impaired in ouabain-hypertensive rats | (22) |
| 2014 | Ferrandi/Ferrari | Rostafuroxin attenuates ouabain-induced high BP and kidney injury | (78) |
| 2014 | Leenen/Hamlyn/Blaustein | A hypothalamic slow neuromodulatory pathway regulates plasma EO | (121) |
| 2015 | Lichtstein | In pregnant rats, anti-ouabain antibodies reduce fetal birth weight, and impair fetal kidney and liver development but increase cardiac growth* | (65) |
| 2017 | Scavone | α2 NKA silencing abolishes the anti-inflammatory and anti-apoptotic effects of nanomolar ouabain in cultured glial cells | (150) |
| 2018 | Blaustein | α2 NKA ouabain binding sites modulate basal BP* | Table 3 |
These data demonstrate that the EO-α2 Na+ pump endocrine system influences normal physiology by modulating, for example: 1) blood pressure; 2) skeletal muscle exercise endurance; 3) fetal renal, hepatic, and cardiac growth and development; 4) natriuresis (ouabain-sensitive α1 Na+ pumps); 5) long-term central sympathetic drive; and 6) behavior and learning.
Two key early influences were my experiences treating heart failure (HF) patients with cardiotonic steroids (CTSs, e.g., digoxin) as a medical student and intern, and my 18 months in Daniel Tosteson’s laboratory studying the Na+ pump, where I routinely used inhibition by the CTS ouabain (pronounced, “wä′bān”) (255) to measure pump activity. In 1958 I first met Tosteson who told me that “the Na-K-ATPase (NKA, or Na+ pump) has just been discovered (264). You’d better hurry up and work on it before everything about the pump is found out and there’s nothing left to do”–—so I went to work in his laboratory on a stipend from an NIH training grant. I’m still working on the pump 60 years later (43)!
My research and clinical experience with ouabain led me to ponder the question of how a pump inhibitor could enhance cardiac contraction (the positive inotropic effect) and thereby benefit patients with HF. The story of how I fortuitously found the answer by discovering the Na/Ca exchanger (NCX) (14), the “missing link,” while studying squid axon Na+ pumps as an NIH Special Fellow in Cambridge and Plymouth, England, is described elsewhere (28, 35). When we discovered the NCX, my coworker Peter Baker recommended I read Luttgau and Niedergerke’s paper on Na+-Ca2+ antagonism in cardiac muscle (189), which I did when a gale caused a brief hiatus in the squid supply. I immediately realized that Luttgau and Niedergerke had obtained indirect evidence of NCX in the heart. With the CTS conundrum on my mind I had an epiphany (28): Pump inhibition by CTS elevates the cytosolic Na+ concentration ([Na+]CYT) and, via NCX, the cytosolic Ca2+ concentration ([Ca2+]CYT). This in turn promotes cardiac contraction (i.e., the positive inotropic action). It was truly a “Eureka! moment.”
I spent a mini-sabbatical in Harald Reuter’s laboratory in Bern, Switzerland, in 1971 [Harald independently and simultaneously discovered NCX in 1966 (35, 246)]. We found that arterial smooth muscle (ASM) also expresses an NCX (35, 245) but, whereas NCX was readily accepted as a novel Ca2+ transport mechanism in mammalian cardiac muscle and squid nerve, the smooth muscle evidence encountered considerable skepticism (e.g., Refs. 211, 265, and see Refs. 28, 35). The original manuscript was rejected by Nature, Science, and Experientia and was eventually published in a nonrefereed journal (245) because Harald was invited to speak at a Royal Society conference. It wasn’t until NCX could be measured by molecular methods that the presence of NCX in smooth muscle became widely accepted (212, 266), but I can’t be too critical of skeptics because I, too, am often slow to accept novel ideas (30).
In the Discussion section of our 1973 article Harald and I speculated that ASM NCX might not only help to control vascular tone, but might also play a role in blood pressure (BP) regulation and the pathogenesis of hypertension (245). Although the original statement was undocumented, I continued to ponder this idea and started to search for evidence in the literature. This required me to spend a great deal of time in the library as there was no internet or PubMed. I also tried to reason from the viewpoint of comparative physiology. For example, I had studied muscles that contract tonically, namely, barnacle muscle (253) and ASM. Also, cardiac ventricular muscle is specialized so that it normally sustains [Ca2+]CYT above the contraction threshold for many, many milliseconds so that sufficient force can develop to contract the entire ventricle during each systole. I was intrigued by the possibility that localized (sub-plasma membrane, sub-PM) elevation of [Na+]CYT (i.e., [Na+]sub-PM) might help to slow NCX-mediated Ca2+ extrusion and contribute to tonic contraction. In our 1973 paper (245) we postulated a “privileged communication” between the extracellular fluid and the sarcoplasmic reticulum (SR), involving the PM NCX (and, presumably, PM Na+ pumps and the tiny intervening space between the PM and SR). A similar idea, the ASM “superficial buffer-barrier”, was later proposed by Van Breemen (285). These views were bolstered by evidence for local Na+ gradients in heart (298) and ASM (235) and by discoveries about the subcellular localization of Na+ pumps and NCX in PM microdomains (see The Plot Thickens—A Lot, below). This concept differs from the more widely distributed “fuzzy space” (165) that has recently been questioned (90, 187).1
The Pump, the Exchanger, and a Hypothesis
By the 1960s the role of dietary salt in the pathogenesis of essential hypertension (49) and its treatment with natriuretic agents (83) were well established, but the underlying mechanisms were unknown. In the late ’60s and early ’70s Guyton and his coworkers focused on the key role of the kidneys. They proposed that salt retention and plasma volume expansion led via arterial “autoregulation” to increased vasoconstriction and total peripheral vascular resistance (TPR) and thus to elevated BP (47, 108, 109). [Recall that BP = CO × TPR, where CO = cardiac output; if CO is constant, BP is directly proportional to TPR (18).] This “autoregulation theory” and the role of abnormal “pressure natriuresis” (115, 206) has dominated thinking on the mechanism of salt-dependent hypertension for the past half-century (47, 114, 207) although several groups have challenged this view (159, 209, 225). One critical problem is that “autoregulation” is a phenomenon, not a mechanism, as Nelson and colleagues showed by discovering specific mechanisms that regulate cerebral blood flow (183). Another problem is that the fundamental mechanisms that underlie the direct enhancement of systemic artery Ca2+ signaling, contractility, and tone in various forms of hypertension, including salt-dependent hypertension (178, 237, 238, 317), have been ignored. Further, brain mechanisms play a key role in hypertension (2, 38, 39, 143, 166, 197) and, when these central mechanisms are blocked, salt-induced hypertension can be averted (100, 286, 293) without evidence that they affect the renal and hemodynamic alterations postulated to trigger “autoregulation.”
I jumped into this abyss in 1977 with my AJP Cell Physiology article (29). One of its core concepts was the idea that the functional-coupling of Na+ pumps and NCX provides a unique connection between Na+ metabolism and ASM cell Ca2+ regulation and, thus, a link to arterial contraction, TPR, and BP. While scouring the literature for relevant clinical data, I found a number of reports that raised the possibility that a natriuretic hormone, or “third factor,” might be involved (37, 45, 54). Some studies even suggested that this factor was an “active sodium transport” (i.e., Na+ pump) inhibitor (72, 80) [the senior author, Neal Bricker, had taught me renal pathophysiology and clinical nephrology when I was a medical student!]. I leaped at the idea and wove a seemingly logical (but rather naïve) story about how salt retention, by an unknown mechanism, triggers the secretion of an endogenous Na+ pump inhibitor (i.e., a ouabain-like compound or “OLC”). I was unaware that Szent-Gyorgi had postulated an endogenous CTS 24 years earlier (274), and that Dahl had already shown that a “humoral agent” was instrumental in “salt” hypertension in rats (50). The OLC would be expected to: 1) inhibit Na+ pumps in the kidneys, thereby promoting natriuresis to compensate for the salt retention; and 2) inhibit Na+ pumps in arterial myocytes, thereby promoting Ca2+ entry via NCX [see Fig. 1, taken from the original article (29)]. The myocyte Ca2+ retention should, of course, increase arterial tone, TPR and BP. In fact Mason and Braunwald had shown that local ouabain infusion increases forearm vascular resistance in humans (198). Numerous novel findings have demonstrated that the model in Fig. 1 is overly simplistic. Nevertheless, the concepts of functional Na+ pump-NCX coupling and an OLC with vasotonic (and cardiotonic) actions have now been verified, and many of the details have been filled in.
Fig. 1.

Block diagram (1977 version) of Ca2+ regulation in arterial smooth muscle myocytes used to explain the hypothesis that Na+ pumps, Na+/Ca2+ exchangers (NCX) and an endogenous ouabain-like compound (OLC) help regulate myocyte Ca2+ and arterial tone (29). Some key Na+ and Ca2+ transport pathways are shown: 1) ouabain-sensitive Na+ pump; 2) NCX operating in the Ca2+ extrusion mode, although we now know that NCX usually operates in the Ca2+ entry mode in arteries with tone (31, 319); 3) sarcoplasmic reticulum (SR) Ca2+ pump (SERCA). Most of the Na+ and Ca2+ entry is through plasma membrane voltage-gated channels (k1 and k2, respectively) and Ca2+ is released from the SR through ryanodine receptors or inositol trisphosphate receptors (k3). This diagram anteceded the discovery of Na+ pump isoforms and PLasmERosomes. [Modified from Blaustein (29).]
My 1977 paper (29) was published while I was abroad. Upon my return, my laboratory colleagues (at Washington University Medical School in St. Louis, MO) all stopped what they were doing to watch me enter my office at the back of the laboratory. They wanted to see my astonishment when I opened the door and found my desk buried under a mound of several thousand reprint request cards (the article has been cited >1,700 times). Interest in those ideas was soon reflected in de Wardener and MacGregor’s review, “Dahl’s hypothesis that a saluretic substance (plays a) role in essential hypertension” (55), and in Kaplan’s reference to my “biochemical hypothesis” (147).
The Hunt for the OLC (the Holy Spirit)
While my AJP Cell article was in press, Haddy and Overbeck (113) also proposed that an OLC is a critical player in the pathogenesis of salt-sensitive hypertension. They, however, suggested that the critical effects of pump inhibition were reduced electrogenic Na+ pump current and consequent myocyte depolarization (112), but this mechanism cannot work in the steady state. Partial pump block will cause a transient decline in Na+ transport (and pump current), but when [Na+]CYT rises, pumped Na+ efflux must recover and must equal Na+ influx in the steady state to avoid continued net gain of Na+ (24). In other words, chronically, partially inhibited pumps operate with a higher [Na+]CYT but unchanged pump current.
These proposals about the role of an OLC in hypertension spurred interest in purifying and identifying this compound. One consequence was that a postdoctoral applicant, John Hamlyn, arrived at my office in 1981, shortly after I moved to the University of Maryland School of Medicine in Baltimore. He told me he wanted to purify this new hormone. I thought to myself, “Sure, you and who else?” Nevertheless, he had some good ideas so I agreed to take him on and let him try, perhaps swayed a bit by the fact that he grew up in the ’60s in Plymouth, UK, the site of our NCX discovery. Little did I—or any of us—realize how terribly difficult this quest for the holy spirit would be.
Most opportunely, shortly after John’s arrival I was introduced to Avinoam Kowarski and Bruce Hamilton, two clinical endocrinology colleagues who were studying hypertension and had collected plasmas from a number of patients and controls. They agreed to collaborate and provided us with de-identified plasmas, and John devised a direct but complex biochemical assay to measure Na+-K+-ATPase (NKA) inhibitory activity in the plasmas. On several occasions I hovered over John’s shoulder as he performed the assay, and I marveled at his skill; it left me with no doubt about the quality of his data. My mentors, Howard Schneiderman, Dan Tosteson, David Goldman and Alan Hodgkin (26) had imbued me with the need to perform meticulous experiments so there could be no question about their validity. John exemplified this approach.
When all the results had been collected, John and I sat in my office and plotted the BP as a function of NKA inhibitory activity. The results were almost too good to believe: although the signals were generally small and there was a fair amount of scatter, there appeared to be a virtually linear correlation between BP and NKA inhibitory activity (Fig. 2)! The paper was published in Nature (123), accompanied by a “News and Views” commentary (96). Our article whetted everyone’s appetite and intensified the search for the endogenous OLC. In fact, three major pharmaceutical companies contacted me about the possibility of a collaboration. Scientists at the Upjohn Company (Kalamazoo, MI), led by Jerry Zinn and Donald DuCharme, proposed a joint effort, and I decided to work with them (an excellent choice in retrospect, as our Upjohn colleagues were wonderful people, and excellent scientists and colleagues). The decision to work with Upjohn was cemented when NIH (National Heart, Lung, and Blood Institute, NHLBI) turned down our two grant applications to purify the OLC. The rationale was (to paraphrase the Summary Statements) “Everybody else is doing this so why do we need another group in the field?” and “Everyone who has tried this has failed, so why waste money on anyone else?”
Fig. 2.

Mean arterial pressure (MAP) is directly correlated with plasma Na+-K+-ATPase (NKA) inhibitory activity. Data are from 20 normotensive (green and blue circles) and 26 hypertensive (red circles) subjects. Green circles indicate discrete single plasma samples; red and blue circles indicate plasma samples drawn slowly over 6 hours (“integrated samples”). The regression line is a least-squares fit to the data; the correlation between MAP and % NKA inhibition is highly significant (r = 0.73; P < 0.0005). Details are given in Ref. 123. [Modified from Hamlyn et al. (123) with permission from Macmillan Publishers Ltd., copyright 1982.]
During the next decade, Upjohn funded and participated in our research as follows: I was tasked with procuring, literally, tons of plasma. A good source was blood banks at our university and other nearby large medical institutions that performed plasmapheresis. John worked out the initial steps of extraction and purification on large, normal phase chromatographic columns, but the latter proved unsatisfactory. Accordingly, subsequent work was done on smaller, reverse-phase HPLC (high-pressure liquid chromatography) columns that gave far superior resolution and reliability. At every step, the eluates were carefully screened for NKA inhibitory activity, and the most promising, high-activity samples were saved for further analysis. Once false-positive had been excluded and the location of relevant biological activities was understood, the isolation steps worked out in Baltimore were refined and scaled up for batch processing at Upjohn.
The Competition Heats Up
In 1987 I attended a meeting in Switzerland and stopped off to visit Reuter in Bern. Harald asked me if I had seen the latest issue of Biochemical and Biophysical Research Communications. I hadn’t, so he escorted me to the library, retrieved the issue from the rack, and opened it to an article by Tamura and Inagami (278). I skimmed through the report in which they claimed to have purified an endogenous NKA inhibitor of molecular mass 336 Da, with “striking similarity to… ouabain,” and was alarmed. I immediately phoned Hamlyn in Baltimore. He hadn’t seen the article either, so I started to read it to him. Before I’d gotten very far, he interrupted me: “They missed it!” John, who has an exceptional understanding of steroid chemistry, recognized that they, too, had seen what appeared to be the same bioactive polar material eluting from their columns, but that they had been led astray by a co-eluting contaminant. When I returned to Baltimore, I sought John out. He wasn’t in his office, but an 8½ × 11 inch photocopy of an Inagami portrait was hanging from each of his bookcases—and from each of the eye-level shelves in his laboratory. As John explained, “I want to know who my competition is.”
In fact, there was no shortage of competition. In 1988, a prominent Tokyo group, Goto and Yamada, described a bioactive material in dog plasma that “closely resembles ouabain” (103). Their extracts, while impure, almost certainly contained OLC, but they, too, were thrown off the scent by a contaminant that had an estimated mass of only 343 Da; ouabain’s mass is 585 Da. The following year, another very active group, Hallaq and Haupert in Boston, described the ouabain-like positive inotropic effects of a partially purified material from bovine hypothalamus (116). Clearly, the key to winning the “race” was to put sufficient pure material in the hands of an analytical chemist who made a living by identifying small molecules.
A New Twist—and an Astonishing Answer
By 1989 we had 0.8 μg of a very highly enriched OLC with an inhibitory action on NKA indistinguishable from that of plant ouabain (120). At this point we were close, but not yet there. The OLC was purified by a factor of ~10 billion but wasn’t completely pure, and the 0.8 μg were insufficient for the best mass spectrometers (MS) at the time. We needed several micrograms of pure material to be sure not to get caught with a contaminant. Fred Mandel at Upjohn suggested employing the NKA, itself, as an affinity ligand. A slurry of NKA was used to bind up the OLC; after washing to remove unbound contaminants, the OLC was eluted from the NKA. Two more rounds of HPLC were sufficient to obtain pure OLC, which was submitted to the excellent Upjohn analytical chemistry facility for MS.
Don Ducharme phoned a couple of weeks later to tell me that they had identified the structure: “Are you sitting down? You’d better sit down. You won’t believe this. It’s ouabain!” It took him some time to convince me that he wasn’t joking (“Okay, Don, now really, what’s the structure?”). I immediately went to John’s office to inform him (he was then an assistant professor). John was just as incredulous; he phoned Don back for confirmation. The chemistry and mechanism of action had indicated that the endogenous molecule was very “ouabain-like”; nevertheless, we were all stunned that it was “indistinguishable from ouabain” (Fig. 3A) (118). This manuscript, too, had a rough time. It was submitted to Nature, and although one referee offered to write a “News and Views” commentary, another had “a gut feeling that (endogenous ouabain, EO) is an artifact.” Nature therefore rejected the manuscript so I consulted Professor Joseph Hoffman, a respected leader in the Na+ pump field, who communicated it to Proceedings of the National Academy of Sciences (PNAS; 118). Our report merited an editorial, “Welcome to ouabain,” in The Lancet (68); the original presentation to the American Heart Association High Blood Pressure Council was reported in The New York Times (7). John and our Upjohn colleagues also published full details of the EO purification and mass spectroscopy (119, 188, 199). The PNAS article and the earlier Nature (123) and AJP Cell (29) publications were all included among “The Top 100 Cited Scientific Reports… on Hypertension Research“ published in the past century (221). Regrettably, a change in leadership at Upjohn led the company to abandon our project shortly after the discovery of EO. Our Upjohn collaborators, Don DuCharme, Jim Ludens, Fred Mandel, and Rod Mathews, were dismayed because they described the EO study as “the most exciting project” in their careers at the company.
Fig. 3.
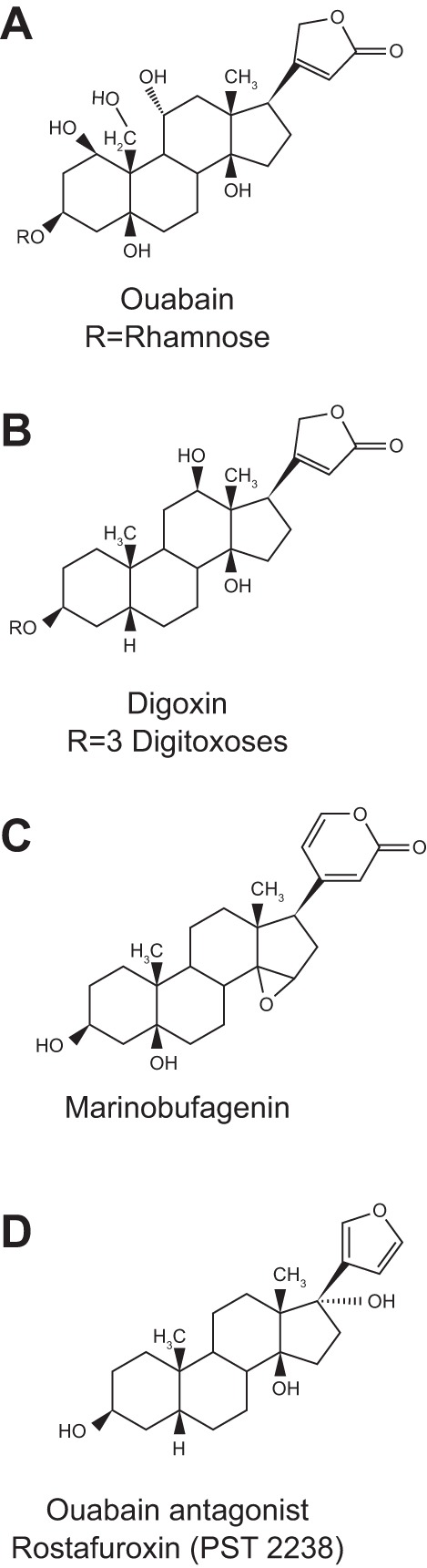
Molecular structures of ouabain (A), digoxin (B), marinobufagenin (C), and rostafuroxin (D). The sugars in ouabain and digoxin are indicated by “Rs.” [Reproduced from Schoner and Scheiner-Bobis (257).]
The skepticism voiced by some colleagues when we announced the result was understandable (a few thought I’d gone off the deep end). After all, ouabain had been used in Africa as an arrow poison for hunting and warfare since the 3rd century (215). Why would anyone have expected ouabain to be a normal human hormone? It seems weird, but doesn’t this simply reflect our prejudice because we learned that ouabain is a poison more than a century before discovering that it is a hormone?
Confirmation—Despite a Few Vocal Naysayers
Within a decade six groups on three continents had verified by MS and NMR spectroscopy that human, bovine, and rodent materials (plasma, urine, adrenals, and hypothalamic tissue) contain EO (Table 2A); the highest concentrations were found in the adrenals (118). A few outspoken investigators were unable to detect EO in human plasma and claimed that EO did not exist [Table 2C; (172)]. Regrettably, this distracted many researchers and clinicians outside the immediate field who could not evaluate or, more often, were unaware of the actual data. In the face of so much positive analytical data, including published MS and NMR spectra of EO (Table 2A), very rigorous experiments are needed to prove its absence. Could so many fine investigators have found the same artifact in specimens from different species? A more likely explanation is that the authors listed in Table 2C may have not detected EO because they did not replicate the original protocols; e.g., one used homogenized placenta (126) rather than plasma, and the others (12, 59, 173) relied on immunoassays. In contrast, many of those in Table 2A used the native receptor: i.e., they bioassayed eluates for NKA binding and/or inhibitory activity at each stage of HPLC separation (e.g., refs. 118, 120, 188, 232, 256, 276).
Table 2.
Analytic detection of EO in mammals
| Senior/Key Author(s) (Location) | Year | Source | Material | Analytical Method | Reference |
|---|---|---|---|---|---|
| A. Positive detection of EO in mammals (published spectra/MS and NMR data) | |||||
| 1. Hamlyn/Blaustein/DuCharme (Baltimore, MD/Kalamazoo, MI) | 1991 | Human | Plasma | MS | (118) |
| 2. Mathews/DuCharme/Hamlyn (Kalamazoo/Baltimore) | 1991 | Human | Plasma | MS | (199) |
| 3. Inagami/Tamura (Nashville, TN) | 1994 | Rat | Urine | MS, NMR | (276) |
| 4. Doris (Lubbock, TX) | 1994 | Bovine | Adrenal | ——* | (59) |
| 5. Defaye/Perrin (Grenoble, France) | 1997 | Bovine | Adrenal | MS | (232) |
| 6. Schoner/Schneider (Giessen, Germany) | 1998 | Bovine | Adrenal | MS, NMR | (256) |
| 7. Nakanishi/Kawamura/Haupert (New York, NY/Boston, MA) | 1999 | Bovine | Hypothalamus | NMR | (149) |
| 8. Takahashi/Komiyama (Tokyo, Japan) | 2000 | Human | Plasma | MS | (155) |
| 9. Manunta/Hamlyn/Pitzalis (Milan, Italy/Baltimore) | 2005 | Human | Plasma | MS | (234) |
| 10. Hamlyn/Manunta (Baltimore/Milan) | 2006 | Human | Plasma | MS | (191) |
| 11. Hamlyn/Jacobs (Baltimore/Milan) | 2012 | Rat | Plasma | MS | (138) |
| 12. Hamlyn/Leenen/Blaustein (Baltimore/Ottawa, Canada) | 2014 | Rat | Plasma | MS | (121) |
| 13. Hamlyn/Blaustein (Baltimore) | 2016 | Rat/Mouse | Plasma | MS | (117) |
| B. Identification of EO isomers (published spectra) | |||||
| 1. Hamlyn/Jacobs (Baltimore) | 2012 | Rat | Plasma | MS | (138) |
| 2. Hamlyn/Leenen (Baltimore/Ottawa) | 2014 | Rat | Plasma | MS | (121) |
| C. Did not identify EO in human samples | |||||
| 1. Doris (Lubbock, TX) | 1994 | Human | Plasma | RIA† | (59) |
| 2. Nicholls/Lewis (Christchurch, New Zealand) | 1994 | Human | Plasma | ELISA† | (173) |
| 3. Hilton (London, UK) | 1996 | Human | Placenta | MS | (126) |
| 4. Vogeser/Baecher (Munich) | 2014 | Human | Plasma | MS‡ | (12, 289) |
Identified by elution pattern.
Not a definitive analytical method.
The MS trace signal is broken at ∼5.0 min in the original publication (Fig. 2, upper spectrum) (12), but an ion current at that position is shown in spectra from two patients later published in a letter to the editor (289) (and see Ref. 117 online supplement Fig. S7). That ion current had the same mass/charge ratio as ouabain but eluted 0.3 min ahead of ouabain.
Ouabain, also called “g-strophanthin,” is an unusual steroid. It is highly polar and therefore can be challenging to separate by column chromatography, especially when it is present in a complex matrix such as plasma. Hamlyn recently identified two ouabain isomers in rodent plasma that are absent from commercial (plant) ouabain, one even more polar than ouabain [Table 2B; (121, 138)]. This is additional evidence that EO is of animal origin. In fact, one group that was unable to detect EO in human plasma (12) did observe an ion currrent peak in their MS-MS product ion spectra that eluted shortly ahead of ouabain ((289) and see footnote to Table 2 line C4). That ion current peak contained a product ion with the same mass/charge ratio as ouabain, and we have therefore suggested that it might correspond to the more polar of two ouabain isomers (27, 117) previously observed in rat plasma by multistage MS (121, 138).
Is EO the Only Endogenous CTS in Mammals?
The OLC has also been called a “digitalis-like compound” (CTSs obtain from plants of the genus, Digitalis) or DLC. As discussed in the next section, however, digitalis-like, very hydrophobic compounds (e.g., digoxin; Fig. 3B) do not mimic the unusually polar ouabain-like CTSs (from flora of the genera Strophanthus and Acokanthera) in some of their key actions. To date, there is negligible evidence that DLCs are endogenous in mammals. Plant ouabain comes from the bark of the Somali “ouabaio” tree, Acokanthera ouabaio (alt., A. schimperi), hence the name (“waabaayo” is “arrow poison” in Somali), and from the seeds of the flowering woody vines, Strophanthus gratus and Strophanthus kombé. The cardiotonic, pulse-slowing effect of juice from S. kombé seeds, which contains ouabain, was discovered by the British botanist, John Kirk, in 1861 (182, 204). Ouabain was first identified chemically and named by the French chemist, Arnaud, in 1888 (8, 139).
Another related CTS, marinobufagenin (MBG; Fig. 3C), a bufadienolide first identified in amphibia (15), has also been linked to hypertension (73, 74), and elevated levels of MBG and another bufadienolide, telocinobufagin, have been found in patients in renal failure (154). The linkage to hypertension is based mainly on immunologic (versus analytic) identification and on immuno-neutralization by “Digibind” or “DigiFab,” commercial anti-digoxin Fab fragments with much higher affinity for ouabain and digoxin than for MBG (239, 240). Also, MBG binds preferentially to α1 Na+ pumps (74, 75, 295). Here I focus on α2 Na+ pumps and EO (see The Plot Thickens – a Lot for a discussion of Na+ pump α-subunit isoforms). Many of the immuno-neutralization studies mentioned below indicate that the main endogenous CTS binding site ligand is an OLC. Nevertheless, there are likely to be other related endogenous Na+ pump ligands such as the two ouabain isomers mentioned above (121, 138).
An oft-stated concern about EO is that the biosynthetic pathway is unknown (172), but this is not entirely correct. The adrenals are an important source of EO because they have high concentrations (118, 256, 277). Adrenalectomy markedly reduces plasma EO in rats (118), and patients with adrenal insufficiency have very low circulating EO levels (268). Also, prolonged immuno-neutralization of EO with custom anti-ouabain antibodies increases adrenal cortical, but not medullary, weight by about 40%, suggesting feedback enhancement of EO production (213). There are several reports of hypertensive patients with adrenocortical tumors associated with excessive production of EO or endogenous “DLC” (190, 252, 258). Further, plasma aldosterone and EO are co-elevated in patients with essential hypertension (279). Also, excision of the adrenocortical tumors (some may even be “ouabainomas”; e.g., Ref. 190) markedly reduced plasma EO or DLC and attenuated the hypertension (190, 252, 258). (In this case, as in many other reports, “DLC” is simply used as the generic term for an endogenous substance that binds with high affinity to anti-digoxin fab fragments, rather than a reference to an entity chemically identified as “digitalis-like.”]
Primary cultured bovine adrenocortical cells exhibit net synthesis of EO (162, 232), as do the adrenals of awake dogs (36). Perrin and colleagues (232) showed that the biosynthesis of ouabain, like aldosterone, begins with cholesterol side chain cleavage to form pregnenolone, which is then converted to progesterone. The more distal steps where the pathway diverges from that of aldosterone synthesis, including the addition of the unsaturated five-member lactone ring and the hexose, l-rhamnose (ouabain is a rhamnoside; Fig. 3A), still need to be clarified. The observation that addition of rhamnose to the medium more than doubled the synthesis of ouabain, but not cortisol, by primary cultured bovine adrenocortical cells implies that coupling the sugar to the steroid is one of the late steps in EO biosynthesis (232). Regrettably, the research needed to identify the distal EO biosynthetic steps is unlikely to be performed in today’s funding climate, but that is surely not evidence that EO doesn’t exist. It is fascinating to contemplate the likelihood that the biosynthetic pathways for animal and plant ouabain evolved independently, and to consider how similar/different the pathways are.
The Proof of the Pudding Is in the Tasting
If EO is the Na+ pump endogenous ligand, and if it does promote the elevation of BP, as Haddy and Overbeck (113) and I (29) had postulated, there is a direct test: infuse ouabain into animals to elevate the plasma level and determine if it raises BP. Accordingly, Hamlyn, Haddy, and their colleagues showed that simply administering ouabain to normal outbred rats for 6–8 weeks causes hypertension (314). This landmark observation has now been replicated in numerous laboratories (reviewed in Refs. 92, 117). Because ouabain does not always raise BP in outbred rats, a few investigators have mistakenly concluded that the ouabain-hypertension link is unimportant (92). This view ignores the evidence that the susceptibility to ouabain-induced hypertension is genetically determined. As Hamlyn and colleagues showed, by inbreeding the least and the most ouabain-sensitive rats, one can generate a highly ouabain-resistant strain and one that is exquisitely ouabain-sensitive within just three generations (6).
Based on the knowledge that all CTSs are pump inhibitors (255) with a cardiotonic action (291), one might have anticipated that the influence of ouabain on BP depends exclusively on its inhibition of Na+ pumps and consequent NCX-mediated elevation of cytosolic Ca2+ [as postulated (29) and see Ref. 14]. In this case, all CTSs, which are also vasotonic (198, 283), would be expected to have a similar hypertensinogenic effect. When Hamlyn and his post-doc, Paolo Manunta, infused digoxin and related compounds into rats for 5 weeks, however, the BP did not rise (193, 196), even though these compounds, too, are vasotonic (267, 283). In fact, when digoxin was infused into ouabain-hypertensive rats, the BP fell; i.e., digoxin behaved like an ouabain antagonist (193, 196). Despite these novel findings, the original report (193) was initially rejected for publication as a full-length article. It was only published 7 years later, after another report had verified the antagonistic effect of digoxin on ouabain-induced and salt-dependent hypertension (131). Subsequently, Lichtstein and colleagues corroborated the ouabain-digoxin antagonism. They showed that in anesthetized guinea pigs given a toxic dose of digoxin, and in isolated cardiac muscle treated with toxic concentrations of digoxin or bufalin, low-dose ouabain attenuated the cardiotoxicity (214). Also, we later observed ouabain-digoxin antagonism in isolated arteries and in primary cultured neurons (267). In retrospect, these results should have been anticipated because digoxin has been used clinically for more than two centuries (301) without any evidence that it is hypertensinogenic. Some of the clinically beneficial effects of Digitalis steroids might even be explained by (unrecognized) ouabain-digoxin antagonism.
The implications of these straightforward observations are profound! First, because both digoxin and ouabain similarly inhibit the Na+ pump (267, 310), this inhibition alone cannot account for ouabain’s hypertensinogenic effect, but an explanation wasn’t recognized until a decade later (see The NKA Is More Than a Cation Pump, below). Second, these data demonstrate that the CTS-Na+ pump interactions for digoxin and ouabain are not identical; they support the view that the endogenous ligand is very “ouabain-like” and not “digoxin-like.” Third, the results suggest that digoxin or a digoxin-like molecule might be a therapeutically useful antihypertensive. In fact, digoxin has been used to treat hypertension (1). (During a luncheon following a seminar I gave at Johns Hopkins University three years ago, a post-doc from India told me that his mother, who had hypertension, had been treated with digoxin. “What happened?” I asked. “Her blood pressure came down” was his reply.) This concept was quickly recognized by others and led Bianchi and colleagues to develop rostafuroxin (PST2238; Fig. 3D), a derivative of digitoxigenin (241) that displaces [3H]ouabain from NKA at concentrations that don’t inhibit the ATPase (79). Rostafuroxin antagonizes ouabain-induced hypertension in rats (79) but may have too low an affinity for the pump (267) to be useful in unselected patients with essential hypertension (269). This drug, however, effectively lowers BP in hypertensive patients who carry specific isomorphisms (161). Nevertheless, a more effective EO antagonist is obviously needed.
In a prescient article, Manunta, Hamilton, and Hamlyn (192) identified some of the structural features of ouabain that are required for its hypertensinogenic effect. They showed that the aglycone ouabagenin is as effective as ouabain in inducing hypertension in the rat. Surprisingly, modifications of the normally unsaturated lactone ring—i.e., saturation, conformational constraint, or opening of the ring—strengthen the hypertensinogenic effect even though these alterations greatly weaken the ATPase inhibitory activity by increasing the EC50 almost 100-fold. This, too, indicates that the hypertensinogenic effect is not tightly linked to the Na+ pump inhibitory activity but instead suggests that the effect is more closely related to EO-activated signaling mechanisms (see The NKA Is More Than a Cation Pump).
An alternative test of the EO-hypertension hypothesis is to neutralize the circulating EO in hypertensive models (or humans). Indeed, immuno-neutralization of EO with Digibind significantly lowers BP in rats with DOCA-salt (156) or reduced renal mass (146) hypertension and prevents ACTH-induced BP elevation in mice (60). By the late 1990s the link between ouabain/EO and hypertension was already well documented in humans (195, 233, 252) and rodents—despite a few awkward and as yet unexplained observations such as why only about half of essential hypertensives and patients with aldosterone-producing adenomas have substantially elevated EO levels (252). Nonetheless, plasma EO correlates directly with BP in essential hypertensives and their offspring (194, 233).
The Plot Thickens—a Lot (and One Type of Na+ Pump Isn’t Enough)
In 1985 Lingrel and Shull cloned the Na+ pump catalytic (α) subunit which contains the transport machinery and ouabain binding site (259). By the early ’90s they had cloned all 4 α-subunit isoforms (α1–α4) and the small, glycosylated mammalian β-subunit isoforms (β1 and β2) (23, 180). The pumps are αβ dimers, and β is a chaperone that is required for maturation, intracellular routing, and activity of the pump, including ouabain binding (124). The pump complex may also include a small regulatory “FXYD family” protein, sometimes called the γ-subunit (91). Importantly, all cells express pumps with an α1-subunit, usually the predominant pumps [80–90% of total pumps in the heart (19, 299)]. Most cells also express pumps with an α2- or α3-subunit [the remainder (19, 141, 299)]. In addition to α1 pumps, for example, all types of muscle (cardiac, skeletal, and smooth), glial cells, and hippocampal neurons also express α2 Na+ pumps, while most neurons express α3 pumps (179, 203). The α1 pumps are “housekeepers” that maintain the low [Na+]CYT in bulk cytosol and mediate net Na+ transport across epithelia (201). So what do the α2 and α3 Na+ pumps do?
While studying α-subunit distribution in ASM cells and neurons, we serendipitously discovered that the pumps with α2- or α3-subunits were confined to PM microdomains that were closely apposed to “junctional” SR or endoplasmic reticulum (S/ER) (144, 145). The NCX colocalized with α2 or α3 in these microdomains at PM-S/ER junctions, which we have called “PLasmERosomes” (33). In contrast, α1 pumps were more ubiquitously distributed in the PM (144). This was the first experimental support for our conjecture that there is preferential communication between the PM and S/ER at the PLasmERosomes (245). Further, the unusually low affinity of rodent α1 for CTSs (218) implicated α2 and/or α3 pumps and NCX in the cellular action of low-dose CTSs (9, 33).
Does EO Directly Affect Cardiovascular Function?
Crucially, Lingrel’s Na+ pump-cloning studies opened the door to genetic alteration of the pumps to facilitate the study of α isoform function. Mutations included the generation of mice with ouabain-resistant (R) α2 pumps [α1R/Rα2R/R mice (48, 61)] and R-α2 mice with ouabain-sensitive (S) α1 pumps (α1S/Sα2R/R, or “SWAP” mice (63)), to determine the role of the ouabain binding site (179). The α1R/Rα2R/R mice, with two amino acids normally found only in ouabain-resistant rodent α1 substituted into the α2 ouabain binding site (48), apparently have normal Na+-K+-ATPase activity (61), but they exhibit a baseline cardiovascular phenotype. Telemetry studies show that baseline mean BP (MBP) is 4–10 mm Hg higher in awake α1R/Rα2R/R than wild-type (WT) α1R/Rα2S/S mice (Table 3). Also, pregnant α1R/Rα2R/R dams have about 10 mm Hg lower systolic BP than do pregnant WT controls (228). Thus, the α2 ouabain binding site and, presumably, its endogenous ligand play a role in normal basal BP regulation.
Table 3.
Baseline MBP in awake WT (α1R/Rα2S/S) mice and ouabain-resistant α2 (α1R/Rα2R/R) mice measured by telemetry; ΔMBP is the difference between the two
| Study | α1R/Rα2S/S (WT) MBP, mm Hg | α1R/Rα2R/R MBP, mm Hg | ΔMBP, mm Hg | Reference |
|---|---|---|---|---|
| 1.* | 104.0 ± 2.6 (11)† | 108.2 ± 3.2 (10) | 4.2 | (185) |
| 2.‡ | 115 ± 3 (8) | 125 ± 5 (10) | 10 | (286) |
| 3a.§ | 121 ± 8 (4) | 128 ± 3 (5) | 7 | (168) |
| 3b.§ | 115 ± 6 (5) | 125 ± 4 (7) | 10 | (168) |
| 4.§ | 97.8 ± 0.8 (7) | 108.2 ± 0.8 (7) | 10.4 | (43) |
Values are means ± SE. WT, wild type; MBP, mean blood pressure.
Mice were “paired for age, weight and sex.”
Numbers of mice shown in parentheses.
Mouse sex not specified. §Male mice; 3a = Experiment no. 1 and 3b = Experiment no. 2. WT, wild type; MBP, mean blood pressure.
The α1R/Rα2R/R mice are resistant to hypertension induced by ouabain, ACTH, elevated intracerebroventricular (icv) NaCl, and high dietary NaCl (60, 62, 168, 185, 286). Even angiotensin (Ang) II-high salt hypertension, a common hypertension model, apparently requires S-α2 pumps for full expression (25, 187a). Trans-aortic constriction (TAC)-induced cardiac hypertrophy also is delayed/attenuated in these mice despite the TAC-induced BP elevation (296). Conversely, SWAP (α1S/Sα2R/R) mice, which have 5–10 times as many S-pumps as WT mice, exhibit a smaller BP rise but accelerated TAC-induced hypertrophy and HF (25, 296). In addition, ACTH-induced and salt-induced hypertension and TAC-induced cardiac hypertrophy, but not TAC-induced pressure overload, are all markedly attenuated by immuno-neutralization of EO (60, 168, 176, 286, 296). Also, active anti-ouabain immunization with an ouabain conjugate attenuates hypertension in salt-sensitive Dahl rats (101). Collectively, these seminal studies demonstrate that EO and its receptor site participate in the pathogenesis of hypertension, cardiac hypertrophy, and HF. Moreover, the SWAP mouse data may be relevant to humans and other non-rodent mammals that have a α1S/Sα2S/S Na+ pump genotype.
I first learned of the ACTH-hypertension results when Jerry Lingrel presented them in a symposium in 2004 honoring Joe Hoffman on his retirement. Joe, who was sitting in the front row, turned around and gave me a “thumbs-up” when Jerry finished speaking. It was welcome support for the EO-Na+ pump-hypertension hypothesis, when many had anticipated a negative result.
There is a fly in the ointment, however. The α1R/Rα2R/R mice apparently can develop DOCA-salt hypertension despite the ouabain resistance (186). The discrepancy between this result and the attenuation of DOCA-salt hypertension in rats by immuno-neutralization of EO with Digibind (156) needs to be resolved. Also, the apparent paradox that α1R/Rα2R/R mice have elevated basal BP (Table 3) yet are unable to develop several forms of hypertension requires an explanation.
The NKA Is More Than a Cation Pump
Several other unexpected and astonishing observations that revolutionized our conception of the CTS-Na+ pump endocrine mechanism were made in the 1990s. One is the discovery by Xie and Askari and colleagues that ouabain binding to Na+ pumps activates protein kinase (PK) cascades and cell signaling apparently independent of active cation transport (110, 135, 153, 181, 231, 305). The two pathways that have been identified, epidermal growth factor receptor (EGFR)/Src/MEK/ERK1/2 and PI3K1A/PDK/Akt (87, 111, 175, 303), are activated by ouabain in a variety of cell types, including cardiomyocytes, ASM cells, and kidney tubule cells. Ouabain signaling also generates reactive oxygen species (ROS) (307). Interestingly, ouabain may either activate (150) or suppress (53, 160) some signaling pathway-mediated responses, perhaps indicating that low EO concentrations are necessary, but higher concentrations are detrimental. Also, how the PK signaling pathways are linked to the Na+ pumps and activated by EO is still unsettled (87, 242, 292, 320). Some investigators have suggested or implied that the activation of PK signaling is mediated by ouabain concentrations below those needed to inhibit cation transport (148, 224, 261). This would be surprising, however, because there is evidence for only a single high-affinity binding site on each αβ dimer, and X-ray crystallography shows that it lies within the cation transport channel (163, 308). Another possibility is that Na+ pumps function as tetramers of the αβ protomer, i.e. (αβ)4 (267).
At first I was quite skeptical that the pump did anything other than pump cations. After all, I had lived with that classical concept for about 40 years, and it’s hard to teach on old dog a new trick (30). Nonetheless, the discovery that prolonged ouabain, but not digoxin, treatment triggers PK-mediated ion transporter reprogramming, including upregulation of NCX1 both in vivo and in vitro in ASM cells (237, 238, 323) and cardiomyocytes (32) was the clincher. Our initial report was rejected by Journal of Physiology with the claim that EO doesn’t exist (yes, it’s getting monotonous!), but was published in AJP Heart and Circulatory Physiology (238) with an accompanying “Editorial Focus” (94). Importantly, here was a possible explanation for the fact that digoxin inhibits NKA and is vasotonic but does not raise BP: The PK-mediated upregulation of NCX1 and enhancement of ouabain-induced, NCX-mediated Ca2+ entry at PLasmERosomes may be crucial for BP elevation (see Ref. 31). In a striking turnabout, Journal of Physiology recently invited and published our review of the evidence that α2 and its ouabain binding site are pivotal in cardiovascular physiology and pathophysiology (31).
“Brain Ouabain”—Really?
Another unanticipated development in the mid-90s was Frans Leenen’s discovery of the role of “brain ouabain” in the pathogenesis of hypertension (129, 130, 132). This followed earlier reports of an OLC in the hypothalamus (275, 282). But why was brain EO necessary, as Leenen’s experiments clearly suggested (130), if most of the circulating EO comes from the adrenal cortex (36, 118, 268) where it is synthesized (162), and if circulating EO/ouabain has a direct vasotonic effect (9, 238, 317, 318)? How did brain ouabain fit into the “big picture” if circulating EO directly augments Ca2+ signaling and activates the arterial (and cardiac) PK-dependent Ca2+ transporter reprogramming? Despite these key, unresolved questions, Leenen, and Hamlyn and I, proceeded on our separate ways for more than a decade without trying to reconcile our different viewpoints. Then, in 2010 Leenen invited me to participate in a satellite conference he organized in conjunction with the International Society on Hypertension’s Vancouver meeting. Following our divergent presentations he asked me if there wasn’t some way to reconcile our differing views. For the first time this got me to think seriously about the dilemma (30). I invited Frans to Baltimore, and John and I initiated a collaboration with him. This led to a jointly authored review in which we postulated that Leenen’s novel hypothalamic pathway, involving “brain EO” (88), helps regulate adrenal secretion of EO and circulating EO levels (34). We then embarked on a collaborative study that tested this hypothesis (121). Specifically, we found that icv infusion of low-dose Ang II elevated BP and plasma EO and upregulated arterial expression of NCX1 and SERCA type 2 (SERCA2). All these effects were blocked by icv infusion of an aldosterone synthase inhibitor or a mineralocorticoid receptor (MR) blocker (121). Yet despite all this evidence for a novel EO-Na+ pump endocrine mechanism, grant applications, including the renewal of our Program Project Grant (which generated much of these data) and a new collaborative program with Leenen, were rejected. For example, despite two enthusiastic reviews of one application, the primary reviewer stated (Summary Statement) that “It is intuitive that these... (central nervous system, CNS) pathways likely exist… Hence, the innovation is modest at best.” Anything is obvious when you’re given the answer [in this case, the preliminary data that supported a novel explanation for the paradoxical finding that high salt and salt restriction both elevate plasma EO (191)].
EO: a “Missing Screw” in Congestive Heart Failure?
In his 1953 monograph, Szent-Gyorgi suggested that in HF there might be a “missing screw” that exogenous CTSs are able to replace (274). Therefore, following the discovery of EO, Hamlyn, Manunta, and Stephen Gottlieb, a University of Maryland cardiologist, measured EO levels in HF patients. The results were clear: EO was elevated, not reduced (as implied by Szent-Gyorgi), in HF patients, and the level correlated inversely with cardiac index (104). Was this already a hint of EO-digoxin antagonism? A later corroborative study from Milan demonstrated that EO correlates directly with the extent of cardiac hypertrophy in patients with end stage renal disease (270). In these patients and in patients undergoing cardiopulmonary bypass surgery (22) the highest EO levels were associated with the most severe cardiac disease and the worst clinical outcomes (263).
The NIH (NHLBI) funded the Digitalis Investigative Group (DIG) clinical trial (1990 to 1998), a randomized digoxin/placebo test on 7,788 HF patients, to investigate the effectiveness of digoxin therapy [https://clinicaltrials.gov/ (58)]. The presence of EO and occurrence of ouabain-digoxin antagonism surely raise fundamental questions about the interpretation and significance of this trial’s outcome (93, 229).
During the past 15 years it has become apparent that the pathogenesis of hypertension and HF involves renin-Ang-aldosterone system (RAAS)-mediated activation of the same central pathways that drive sympathetic input to the arteries and heart (Fig. 4) (57, 226, 227, 300, 313, 321). Thus, in retrospect it is not surprising that Leenen found that “brain ouabain” participates in the same CNS mechanisms that enhance sympathetic nerve activity (SNA) in HF (169) and hypertension (129, 130, 132). After all, similar agents, including Ang-converting enzyme (ACE) inhibitors, Ang receptor blockers (ARBs), and MR blockers, are all effective therapies for both hypertension and HF (42, 140, 157, 170). Furthermore, the progression from uncontrolled hypertension to cardiac hypertrophy and then HF is well documented (46, 128, 290, 316). Additionally, as noted above (The Plot Thickens), the cardiac hypertrophy that results from TAC-induced pressure overload in mice also depends in part on the α2 Na+ pump ouabain binding site and on EO (25, 296). Indeed, in SWAP mice with S-α1 Na+ pumps the progression to HF is accelerated (296). This is a further manifestation of the EO-ouabain-sensitive Na+ pump-hypertension-HF relationship.
Fig. 4.

The centrally controlled, parallel sympathetic nerve and slow neurohumoral pathways that regulate both arterial and cardiac function, and participate in the pathogenesis of hypertension and heart failure (HF). Angiotensin II (Ang II) and high dietary salt (↑[Na+]) are convergent signals that act via hypothalamic Ang-type 1 receptors (AT1R) to activate central nervous system (CNS) sympathetic pathways; the role of the kidneys in the retention of salt has been omitted for the sake of simplicity. The increased sympathetic nerve activity (SNA) promotes vasoconstriction and increases cardiac rate and contractile force. Prolonged stimulation of hypothalamic AT1R also activates a novel neurohumoral pathway (box at top right) that includes aldosterone (Aldo), mineralocorticoid receptors (MR), epithelial Na+ channels (ENaC), endogenous ouabain (EO), and α2 Na+ pumps. This hypothalamic pathway feeds back (green line, “+”) to modulate Ang II-activated SNA and also promotes adrenal secretion of EO, triggered by, e.g., ACTH, adrenal SNA, and/or Ang II. The elevated plasma EO acutely inhibits α2 Na+ pumps in both the heart and arteries, and the rise in intracellular Na+ rapidly induces Na/Ca exchanger (NCX)-mediated Ca2+ gain, and cardiotonic and vasotonic effects. Prolonged plasma EO elevation also activates an α2 Na+ pump-associated protein kinase cascade (e.g., EGFR/Src/MEK/ERK-mediated) that increases cardiac and arterial NCX expression, and arterial sarcoplasmic reticulum (SR) Ca2+ pump (SERCA2) expression. In arteries with tone, NCX normally favors Ca2+ entry and helps to sustain cytosolic Ca2+ ([Ca2+]CYT) above contraction threshold. The EO-induced NCX and SERCA2 upregulation enhances Ca2+ signaling and helps the very modestly increased SNA to increase vascular tone and resistance and elevate blood pressure. In the heart, NCX promotes Ca2+ extrusion during diastole, but prolonged α2 pump inhibition by EO reduces the Na+ gradient driving force so that [Na+]CYT and diastolic [Ca2+]CYT are both elevated; consequently, cardiac relaxation is slow and/or incomplete. Also, cardiac SERCA2 expression is usually reduced in HF (perhaps due to the high EO), as are SR Ca2+ stores and [Ca2+]CYT transients, and systolic function is impaired. The diastolic dysfunction and attenuated cardiac contraction and stroke volume contribute to HF. These cellular mechanisms are discussed further in a recent review (31). [Modified from Blaustein et al. (31) with permission.]
Initially, I questioned the EO-HF link, so John Hamlyn and I explored the effects of coronary occlusion in mice. We found that the resultant HF is associated with elevated plasma EO and enhanced expression of both cardiac and arterial NCX1 (32). This upregulation of NCX1 is seen in HF (219, 249, 271) as well as in hypertension (237, 322, 323) (see The NKA Is More Than a Cation Pump). However, whereas SERCA2 expression in ASM is significantly upregulated as in several hypertension models (237, 238, 322), cardiac SERCA2 is decreased in the HF mice (32) as others have observed in human and experimental HF (219, 271). Not surprisingly, the signaling mechanisms appear to be cell type-specific.
How Does the Brain Talk to the Heart and Arteries?
The preceding discussion about the cardiovascular consequences of interfering with the EO-Na+ pump mechanism raises critical questions about how this system works. In other endocrine systems, either absence or excess of the hormone can have pathophysiological consequences as in, for example, hypo- and hyperthyroidism. This is also the case for the EO-Na+ pump system; furthermore, the pathophysiological consequences are not always “all-or-none.” For example, ouabain resistance in mice delays/attenuates TAC-induced cardiac hypertrophy but does not prevent it (296; see also Ref. 25). Also, transient or modest elevation of EO may be beneficial (e.g., the cardiotonic effect), but greater and more sustained elevation may have pathological consequences (hypertension, cardiac hypertrophy, and HF). How can we explain these seeming contradictions?
The key role of the sympathetics in cardiovascular control is widely accepted, but it seems logical that other mechanisms also contribute to these functions that are so vital for survival. In this regard the EO-Na+ pump system operates in conjunction with and in parallel to other mechanisms, especially the sympathetic nervous system (Fig. 4). Whereas α1R/Rα2R/R mice have only a modest baseline BP phenotype (Table 3; see The Plot Thickens), α-adrenoceptor blockade markedly reduces arterial tone (302, 315) and lowers BP (105, 220). Enhanced sympathetic drive in hypertension and HF is maintained at least in part by activation of a novel, slow neuromodulatory pathway in the hypothalamus (Fig. 4, top right corner), one component of which is locally secreted (paracrine) “brain EO” (88, 121, 300). Interruption of this pathway by lesioning the anteroventral third ventricle (AV3V) could explain why AV3V lesions prevent the maintenance of established hypertension (39). The neuromodulatory pathway feeds forward via Ang II onto the brainstem sympathetic system that drives both the heart and arteries (171). In addition to modulating SNA, the slow pathway regulates plasma EO, perhaps via SNA to the adrenals and Ang II (31, 121). This endocrine mechanism serves as a parallel pathway to the sympathetics in influencing cardiac and vascular signaling (Fig. 4; see also Ref. 31). Block of the neuromodulatory pathway therefore attenuates hypertension and HF by both central and peripheral mechanisms (121, 134) and has obvious implications for hypertension and HF therapy (166).
Circulating EO acts directly on the heart and arteries in two ways. First, by inhibiting α2 Na+ pumps, EO exerts acute cardiotonic and vasotonic actions [Fig. 4, bottom half); (31)]. Second, by activating PK cascades, EO triggers reprogramming of Ca2+ transporter protein expression exemplified by increased NCX1 in both the heart and arteries, and increased arterial but decreased cardiac SERCA2 (see “chronic” pathways in Fig. 4, bottom). In arteries with tone, because NCX1 normally mediates net Ca2+ entry, the upregulation of both NCX1 and SERCA2 contributes to enhanced Ca2+ signaling and increased tone and fosters hypertension. In the heart, however, although NCX1 normally mediates Ca2+ extrusion, primarily, the combination of Na+ pump inhibition and increased NCX1 expression tends to promote net Ca2+ gain, and thereby increases ventricular “stiffness” and reduces diastolic dilation (Fig. 4; bottom right) (86, 95, 200, 236). Also, the decreased SERCA2 expression reduces Ca2+ storage and signaling (20, 21, 273, 280). Thus the elevated plasma EO initially contributes to enhanced Ca2+ signaling and cardiac workload, and to hypertrophy by a sustained cardiotonic effect as well as the protein reprogramming. But, if EO elevation is greatly prolonged, and especially if the EO level continues to rise, this will eventually increase cardiac stiffness, reduce Ca2+ signaling, and cause HF (additional details in Ref. 31). The key role of EO and α2 Na+ pumps is indicated by the results of studies on TAC-induced cardiac hypertrophy. Contrary to the dogma that cardiac stretch directly induces the hypertrophy (223, 297, 309), pressure overload-induced hypertrophy is attenuated/delayed when EO is immuno-neutralized, or its receptor is knocked out or mutated to an ouabain-resistant form (248, 296).
Is EO Exclusively a Cardiovascular Hormone?
Ouabain-sensitive Na+ pumps are expressed in all tissues. Therefore it is appropriate to ask whether ouabain-resistant mutants and EO immuno-neutralization induce dysfunction in organ systems other than the cardiovascular system. An affirmative answer would help validate the EO-Na+ pump endocrine concept. The CNS might, however, be protected from circulating EO by the blood-brain barrier, which polar EO does not permeate—except at the circumventricular organs, which lack the barrier. Nevertheless, as discussed next, several groups explored the idea that brain neurons and/or astrocytes might express an EO-dependent phenotype.
EO is a mood modulator.
Following up on their earlier studies demonstrating learning and behavior alterations in heterozygous mice with a single null mutation in α2 (or α3) Na+ pumps (210), Lingrel and coworkers discovered that α1R/Rα2R/R mice have a behavioral phenotype: They exhibit decreased locomotion in a novel environment, a blunted acoustic startle reflex, enhanced response to methamphetamine (an indirect sympathomimetic), and impaired egocentric learning (254). Importantly, these specific behavioral and memory deficits, which are likely related to α2 Na+ pump expression in hippocampal pyramidal neurons and/or brain astrocytes, differ from those in heterozygous α2 mice (α2S/−) (254). The α2S/− mice, which have a null mutation in one α2 allele and express only half the normal complement of S-α2 pumps, exhibit increased anxiety, decreased locomotor activity, and impaired spatial learning (137, 210). In fact, a difference would be expected because α2S/− is equivalent to elevation of the EO level (i.e., greater inhibition of α2) but reduction of EO-activated signaling, while α2R/R is equivalent to elimination of EO. Thus the α2 ouabain binding site and, presumably, EO are associated with specific brain functions.
Low-dose icv ouabain infusion induces mania-like behavior in rats (69, 70). Also, immuno-neutralization of EO with icv anti-ouabain antibodies attenuates amphetamine-induced hyperactivity (another mania model) and is antidepressive in the Flinders Sensitive Line of genetically depressed rats (97, 127). An important caveat to these experiments, however, is that most neurons express (S) α3 Na+ pumps which have a different behavioral phenotype from α2. For example, α3 heterozygotes (α3S/−) have spatial learning and memory deficits, and increased locomotor activity and locomotor response to methamphetamine (210). Heterozygous Mishkin mice, with an inactivating point mutation in one α3 gene (44), also display mania-like behavior resembling that of bipolar patients in the manic phase, with enhanced exploratory locomotion in a novel environment and increased sensitivity to amphetamine. These symptoms are treatable with mood-stabilizing drugs such as lithium and valproic acid, and with rostafuroxin (151). Because ouabain infusion and EO immuno-neutralization should affect both α2 and α3 pumps, the results of those tests support the view that both EO and its binding sites are important for behavior and learning, but they do not distinguish between the roles of the α2 and α3 Na+ pumps. [Regrettably, no α3R/R mouse has yet been generated.]
The source of the EO that regulates the neurons involved in behavior and learning is unknown. Circulating EO might influence hypothalamic neurons in the circumventricular organs that participate in cardiovascular control (294), if only as feedback to help regulate the plasma EO level. However, EO-regulated neuronal activity in other areas of the brain surely require local EO synthesis. This circumstantial evidence that EO is synthesized and secreted in the brain complements Leenen’s findings (“Brain Ouabain”—Really?).
EO and α2 modulate skeletal muscle fatigue.
In skeletal muscle, α2, not α1, Na+ pumps are the most prevalent, and the α1:α2 ratio is approximately 1:6 (125), but this can vary depending on fiber type and conditions. Most skeletal muscle α2 pumps are, however, located in intracellular membranes, and exercise training and insulin promote the trafficking of α2 pumps to the PM (89, 136, 272).
EO and the α2 ouabain binding site modulate exercise endurance. α1R/Rα2R/R mice have fewer exercise failures on a treadmill than do WT mice (243). Conversely, mice with targeted knockout of skeletal muscle α2 Na+ pumps (SkM-α2−/− mice) fatigue faster, i.e., they have more treadmill failures than do WT mice (244). Further, the increased fatigability of isolated extensor digitorum longus muscle from SkM-α2−/− mice is mimicked by treating WT extensor digitorum longus muscle with ouabain (244). Thus, reduction of EO binding enhances, and inhibition or knockout of α2 pumps curtails exercise endurance.
EO is a stress hormone that modulates neuroinflammatory responses.
Numerous reports demonstrate that EO is a stress hormone. Plasma EO is elevated not only in hypertension, and cardiac hypertrophy and failure, but following cardiac surgery (263), in renal failure (122), in ectopic corticotropin syndrome (102), in normal human and rodent pregnancy and even more so in preeclampsia (56, 138), following intense exercise (17), and during salt depletion as well as dietary salt excess (191).
Many reports indicate that low dose ouabain/EO is a neuroprotective and antiapoptotic agent whose effects appear to be mediated by the Ca2+-dependent nuclear transcription factor, NF-κB (4, 53, 148, 175, 261). Several in vivo and in vitro studies also show that ouabain/EO has an anti-inflammatory action (53, 160, 224). On the other hand, some in vivo and in vitro studies indicate that ouabain/EO can activate proinflammatory responses (148, 150, 152). For example, in primary cultured glia, knockout of α2 Na+ pumps by siRNA prevents activation of the NF-kB-mediated inflammatory response to lipopolysaccharides (150). This is clearly a “hot area”, and new studies are needed to elucidate how EO can elicit both pro- and anti-inflammatory responses. Is this simply a concentration-dependent effect (as seems unlikely) or are other factors involved?
EO is a growth factor: role in the kidneys.
The preceding observations demonstrate that the actions of EO are widespread. Additionally, several groups have shown that low nanomolar ouabain/EO is a growth factor in a variety of tissues (10, 11, 66, 98, 217, 257). For example, ouabain promotes cyst growth and cystic fluid secretion in renal tissue from patients with an autosomal dominant polycystic disease mutation, apparently via the EGFR/Src/MEK/ERK signaling pathway (142, 216).
But is EO a growth factor in vivo? To determine the role of EO in pregnant rats, Lichtstein and colleagues immuno-neutralized the EO by intraperitoneal infusion with anti-ouabain antibodies. This treatment (vs. control IgG) significantly retards fetal development and reduces kidney and liver growth (65). Conversely, prolonged infusion of ouabain in rats increases heart and kidney weights, and these effects are abrogated by rostafuroxin (77). In vitro, immuno-neutralization of EO in growth medium containing fetal bovine serum attenuates the growth of several types of primary cells in culture (65, 66). Further, Aperia and coworkers report that malnourishment of pregnant rats by feeding a low-protein diet impairs fetal kidney growth, and that subcutaneous infusion of low-dose ouabain in protein-restricted dams protects fetal development (174). In vitro, nanomolar ouabain sustains kidney development and attenuates apoptosis of renal epithelial cells and podocytes (40, 174, 175). These latter effects appear to be due to upregulation of NF-κB (174).
It has been suggested that activation of signaling cascades by low-dose (nanomolar) ouabain in rodents is mediated by α1 pumps (174, 304), but this is at odds with the finding that ouabain-induced hypertension, which apparently depends on this signaling (31), is averted in α1R/Rα2R/R mice (62). Thus, the low-dose (nanomolar) ouabain/EO-induced effects on rodent kidneys needs to be reconciled with reports that R-α1 is the only Na+ pump α isoform detected in this tissue (71, 202). Others, however, suggest that very low levels of α2 and/or α3 are also expressed in some rat renal tubule segments (5, 76); also, α3 has been observed in rabbit kidneys in which S-α1 is the predominant isoform (16). This issue may be settled by the aforementioned report that in mouse glial cells, which express R-α1 (80–90% of pumps) and S-α2 Na+ pumps (99), silencing α2 with siRNA prevents ouabain from activating PK signaling and the neuroinflammatory pathway (150). Thus, as in rodent hearts (25, 31) and glial cells, a tiny minority of pumps (the S-α2 or S-α3) might mediate the signaling actions of EO in rodent kidneys. Nevertheless, given these examples of EO acting as a growth factor in the kidneys and liver, and in view of the broad distribution of ouabain-sensitive Na+ pumps, it seems that additional examples of this action are likely to be identified in other tissues. Clearly, we have much to learn about this aspect of EO function.
If the ouabain/EO level is too high, however, the hormone can stimulate generation of ROS and trigger apoptosis (66, 158, 284, 306, 307). Manunta and colleagues found that acute kidney injury, which may occur (e.g.) as a serious complication of cardiopulmonary bypass surgery (250, 251), is associated with high levels of circulating EO (22, 262). In rats with ouabain-induced hypertension or renal ischemia-reperfusion injury, renal podocyte nephrin, a key component of the glomerular filtration barrier, and creatinine clearance are reduced and proteinuria is increased. All these signs of impaired barrier function are prevented or reversed by treating the rats with rostafuroxin (78, 288).
Is EO a natriuretic agent?
In 1977 I suggested that the endogenous Na+ pump inhibitor should be a natriuretic as well as a hypertensinogenic agent (29). But, is there evidence that the low concentrations in the circulation directly inhibit Na+ reabsorption by the kidneys? During intravenous (iv) infusion of anti-ouabain antibodies (passive immunization) in normal rats, a transient anti-natriuresis (i.e., salt retention) was observed a day after starting the EO immuno-neutralization while BP remained normal (213). This suggests that EO may be a natriuretic agent, although the mechanism is unclear because virtually all the Na+ pumps responsible for Na+ reabsorption in the rodent renal tubule epithelium are R-α1 (202). The transience of the effect is expected with constant dietary salt because, following a brief net gain of Na+ and attendant anions, a new steady state should soon be achieved so that Na+ intake is once again balanced by output.
In an isolated case report, an 11-year-old patient who developed hyponatremia due to salt wasting following the removal of a large brain tumor was treated with iv Digibind; this reduced the natriuresis and corrected the hyponatremia (205). Here, too, the mechanism for the anti-natriuresis was not resolved but, in this case, could have been due to immuno-neutralization of nanomolar EO that normally binds to α1 Na+ pumps in the human kidney tubule epithelium. Furthermore, saline loading enhances natriuresis in SWAP mice, and the fact that this effect is attenuated by infusion of Digibind (184), indicates that EO is natriuretic.
In sum, the data from mutant mice and from EO immuno-neutralization studies are powerful evidence that EO, and α2 and α3 Na+ pumps, are components of a major endocrine/paracrine system with critical cardiovascular, renal, and CNS influences (31, 179). This endocrine system has been largely overlooked, perhaps in part because of the insistence by some that “endogenous ouabain is not ouabain” (Ref. 172; see Confirmation—Despite a Few Vocal Naysayers). In practice, however, the exact structure of EO seems secondary to validating the concept of an EO-Na+ pump endocrine system.
Ouabain: a Therapeutic Agent?
Circulating EO, like most hormones, is expected to have a normal physiological concentration range. Plasma levels that are too high or too low are pathophysiological as summarized in this review: e.g., elevated EO leads to reduced cardiac index (104) and increased left ventricular hypertrophy (234), and low EO impairs fetal development (65, 174). In the latter case, ouabain would appear to be the ideal replacement therapy [recall Szent-Gyorgi’s “missing screw” (274)] provided that the normal plasma level and the appropriate dosage are known.
While they are equipotent (64, 267, 311, 312), ouabain acts much more rapidly than digoxin (81) and the absorption (only ≈2–2.5% vs. ≈70–80% for digoxin) and bioavailability of orally administered ouabain (106) are far less than for digoxin (247). The latter difference is reflected in the recommended oral (or sublingual) dosage (6–12 mg for ouabain vs. 0.375–0.5 mg for digoxin) (84, 106, 107). Ouabain was therefore recommended primarily for acute iv administration in heart failure emergencies with an oral digitalis preparation then used to maintain the cardiotonic effect (84). Ever since 1885 when Fraser popularized it as a cardiotonic agent (82, 204) some physicians have advocated use of oral ouabain (85). Digitalis preparations are, however, far more widely prescribed, and ouabain is no longer approved for clinical use in the US, perhaps because iv EO may cause sudden death (3). [After all, ouabain has been used as an arrow poison for centuries because of its proven efficacy (215)!] Ouabain’s apparently narrow therapeutic window and its low and variable absorption rate are fundamental concerns. Moreover, there has been no controlled clinical trial of oral ouabain efficacy; all evidence is anecdotal. To address these concerns we need: 1) a convenient EO assay; 2) solid information about the normal plasma EO level under a variety of conditions (as noted, EO is normally elevated during intense exercise and during pregnancy); and 3) a well-controlled clinical trial including plasma EO measurements before and during therapy.
From a Na+-Hypertension Link to a Novel Endocrine System: Summary and Conclusions
This article summarizes the origins and development of ideas and accrual of knowledge about a newly recognized endocrine system that has been hidden in plain sight. During the past 25 years numerous ground-breaking discoveries about Na+ pumps and CTSs, and their influences on cardiovascular, renal, and brain physiology and pathophysiology have helped to elucidate the EO-Na+ pump endocrine system. The mammalian Na+ pump is a most unusual hormone receptor because it functions both as a cation transporter that does not require the hormone for activity (although some isoforms can be modulated by EO), and as an EO-activated signal transducer that appears to function independently of the cation transport. The α-subunit is actually an ancient protein: it is found in Archea and bacteria as well as eukaryotes. Indeed, hydra, nematode, and fruit fly NKA α-subunits have substantial sequence homology and tertiary structure similarity to the human NKA α (208, 222, 281). The invertebrate β- and human β-subunit sequences are, however, more distinct (222). Nevertheless, ouabain also inhibits the hydra and nematode NKAs (41, 51, 52). Thus, CTS binding activity has been conserved through evolution despite the occasional appearance of ouabain resistant pumps, as in rodents and insects (51), but the significance of CTS binding in invertebrates and lower vertebrates is unknown. It should be very instructive to learn when and how the endogenous CTS-Na+ pump couple evolved into a hormone-receptor complex. When, during evolution, did EO first appear? And when did EO-activated cell signaling evolve?
The functional consequences of interfering with the EO-Na+ pump (hormone-receptor) interaction, either by mutation of the ligand binding site to an ouabain-resistant form or by immuno-neutralization of EO, demonstrate that EO and Na+ pumps are an endocrine/paracrine system with very broad impact. The idea that an endogenous Na+ pump inhibitor/CTS is a vasotonic agent that plays a role in the pathogenesis of hypertension has been validated (31), but the mechanism is far more complicated than originally envisioned (29). This is exemplified by the multitude of unexpected, dogma-defying findings (see Table 1). These include: cloning of the Na+ pump α and β subunit isoforms; discovery that hormone ouabain (EO) is synthesized in the brain as well as the adrenals, and that circulating EO is regulated by a novel neurohumoral pathway in the hypothalamus; discovery that EO and brain Na+ pumps modulate behavior and learning; identification of the ouabain-Na+ pump interaction as an activator of PK cascades and cell signaling; recognition that the actions of ouabain-like and digoxin-like CTSs are not identical, and that DLCs do not activate PK cascades; discovery that ouabain/EO can be both pro- and anti-inflammatory; and recognition that EO is a stress hormone and a growth factor and is essential for normal fetal development. It is difficult to grasp the full scope of this novel humoral system with all of its complexity and myriad actions.
The past 40 years have been an exciting roller coaster ride, but our odyssey is far from complete. We undoubtedly will learn much more about EO as a hormone when a specific antagonist becomes available, as happened with the RAAS when ARBs like losartan were developed. We also need a convenient commercial assay for EO. There are still numerous questions to answer and controversies to resolve, starting with elucidation of the steps in the synthesis of EO from progesterone. A major concern is whether funds will be available for the requisite studies that utilize standard methods and are not very “glitzy.” Because of a research funding squeeze the US is starting to lose its leadership in the EO-Na+ pump field despite its obvious clinical relevance, just as research on this novel endocrine system is picking up in Canada, Israel, Italy, Japan, Sweden, and the UK.
The EO-Na+ pump system influences the physiology of most, if not all, organs in the body. The complexity of this system is illustrated by the role of EO and Na+ pumps in hypertension (Fig. 4), from whence I began with a far more simplistic view (Fig. 1). Too often we lose sight of Page’s prescient insight that hypertension is a mosaic (230). Multiple factors (genetic, environmental, immunologic, renal, neural, endocrine, cardiac, and vascular) are essential contributors to the BP elevation—even when a single cause such as a monogenic defect in salt transport (177) can be identified as the initiating element. A plethora of components and interacting pathways that influence BP have been elucidated, but they are generally studied one bit at a time, and they can’t all be “essential.” Which ones are really important? It appears that we need a new, more quantitative approach, a new way to juxtapose all the pieces to determine how these elements all work together to regulate BP. It’s time for the engineer-biologists: we need to learn how to “fix a radio” (164). Here is the essence and future of (integrative) physiology!
The history of progress with the EO-Na+ pump endocrine system calls to mind T. H. Huxley’s admonition (letter to C. Kingsley, 1860): “Sit down before fact as a little child, be prepared to give up every preconceived notion, follow humbly wherever and to whatever abysses nature leads, or you shall learn nothing.”—but be sure of the “facts.”
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.P.B. drafted, edited and revised the manuscript, and approved the final version.
ACKNOWLEDGMENTS
This review is in memory of Daniel C. Tosteson who planted the seed, and for John M. Hamlyn who found the spirit and Ellen B. Blaustein whose support and encouragement helped the seed flourish and the spirit soar.
I thank my numerous trainees, laboratory associates and collaborators, many of whom are cited here, who contributed to this story, and my granddaughter, Madeleine Cahn, whose thoughtful questions and careful grammar and punctuation editing helped me to clarify this presentation. I also thank the National Institutes of Health (National Heart, Lung, and Blood Institute and National Institute of Neurological Disorders and Stroke) and the American Heart Association (just completed: 15GRNT24940022) for many years of research support.
Glossary
- α1R/Rα2R/R
Ouabain-resistant (R-) α2 Na+ pump mice
- α2S/− (α3S/−)
Heterozygous α2 (α3) Na+ pump mice
- ACE
Angiotensin-converting enzyme
- Ang
Angiotensin
- ARB
Ang receptor blocker
- ASM
Arterial smooth muscle
- BP
Blood pressure
- CNS
Central nervous system
- CO
Cardiac output
- CTS
cardiotonic steroid
- CYT
Cytosolic
- DLC
Digitalis-like compound
- ELISA
Enzyme-linked immunosorbent assay
- EO
Endogenous ouabain
- HF
Heart failure
- HPLC
High pressure liquid chromatography
- icv
Intracerebroventricular
- iv
Intravenous
- MBP
Mean BP
- MBG
Marinobufagenin
- MR
Mineralocorticoid receptor
- MS
Mass spectroscopy
- NCX
Na/Ca exchanger
- NKA
Na+-K+-ATPase (Na+ pump)
- NMR
Nuclear magnetic resonance
- PK
Protein kinase
- PM
Plasma membrane
- RAAS
Renin-angiotensin-aldosterone system
- RIA
Radioimmunoassay
- ROS
Reactive oxygen species
- SERCA
Sarco-/endoplasmic reticulum Ca2+ pump
- SNA
Sympathetic nerve activity
- SR
Sarcoplasmic reticulum
- SWAP (α1S/Sα2R/R)
R-α2 Na+ pump mice with ouabain-sensitive (S-) α1 Na+ pumps
- TAC
Trans-aortic constriction
- TPR
Total peripheral resistance
- WT
Wild type (α1R/Rα2S/S) mice.
Footnotes
Glossary of abbreviations appears at the end of the article.
REFERENCES
- 1.Abarquez RF., Jr Digitalis in the treatment of hypertension. A prelminary report. Acta Med Philipp 3: 161–170, 1967. [PubMed] [Google Scholar]
- 2.Abboud FM. The sympathetic system in hypertension. State-of-the-art review. Hypertension 4: 208–225, 1982. [PubMed] [Google Scholar]
- 3.Abelmann WH. Acute hypertensive effect of digitalis glycosides. Chest 63: 2–3, 1973. doi: 10.1378/chest.63.1.2. [DOI] [PubMed] [Google Scholar]
- 4.Abushik PA, Sibarov DA, Eaton MJ, Skatchkov SN, Antonov SM. Kainate-induced calcium overload of cortical neurons in vitro: dependence on expression of AMPAR GluA2-subunit and down-regulation by subnanomolar ouabain. Cell Calcium 54: 95–104, 2013. doi: 10.1016/j.ceca.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahn KY, Madsen KM, Tisher CC, Kone BC. Differential expression and cellular distribution of mRNAs encoding α- and β-isoforms of Na+-K+-ATPase in rat kidney. Am J Physiol Renal Physiol 265: F792–F801, 1993. [DOI] [PubMed] [Google Scholar]
- 6.Aileru AA, De Albuquerque A, Hamlyn JM, Manunta P, Shah JR, Hamilton MJ, Weinreich D. Synaptic plasticity in sympathetic ganglia from acquired and inherited forms of ouabain-dependent hypertension. Am J Physiol Regul Integr Comp Physiol 281: R635–R644, 2001. [DOI] [PubMed] [Google Scholar]
- 7.Altman LK. “Scientists link hormone similar to poison to high blood pressure”. The New York Times. September 15, 1990.
- 8.Arnaud M. Sur la matière cristallisér active des flèsches empoisonnées des şomalis, estraite du bois d’Ouabaio [On the crystalized active substance of Somali arrow poison extracted from the Ouabaio tree]. Compt Rend Acad Sci 107: 1011–1014, 1888. [Google Scholar]
- 9.Arnon A, Hamlyn JM, Blaustein MP. Ouabain augments Ca2+ transients in arterial smooth muscle without raising cytosolic Na+. Am J Physiol Heart Circ Physiol 279: H679–H691, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Atkinson MJ, Cade C, Perris AD. Sodium and ouabain induce proliferation of rat thymic lymphocytes via calcium- and magnesium- dependent reactions. Cell Calcium 4: 1–12, 1983. doi: 10.1016/0143-4160(83)90044-1. [DOI] [PubMed] [Google Scholar]
- 11.Aydemir-Koksoy A, Abramowitz J, Allen JC. Ouabain-induced signaling and vascular smooth muscle cell proliferation. J Biol Chem 276: 46605–46611, 2001. doi: 10.1074/jbc.M106178200. [DOI] [PubMed] [Google Scholar]
- 12.Baecher S, Kroiss M, Fassnacht M, Vogeser M. No endogenous ouabain is detectable in human plasma by ultra-sensitive UPLC-MS/MS. Clin Chim Acta 431: 87–92, 2014. doi: 10.1016/j.cca.2014.01.038. [DOI] [PubMed] [Google Scholar]
- 13.Baker PF, Blaustein MP. Sodium-dependent uptake of calcium by crab nerve. Biochim Biophys Acta 150: 167–170, 1968. doi: 10.1016/0005-2736(68)90023-0. [DOI] [PubMed] [Google Scholar]
- 14.Baker PF, Blaustein MP, Hodgkin AL, Steinhardt RA. The influence of calcium on sodium efflux in squid axons. J Physiol 200: 431–458, 1969. doi: 10.1113/jphysiol.1969.sp008702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barbier M, Shroter H, Meyer K, Schindler O, Reichstein T. Die Bufogenine des Paratoidensekrets von Bufo marinus (L.) Schneider [Bufagenin in the parotid secretion of Bufo marinus (L.) Schneider] Helv Chim Acta 42: 2486–2506, 1959. doi: 10.1002/hlca.19590420720. [DOI] [Google Scholar]
- 16.Barlet-Bas C, Arystarkhova E, Cheval L, Marsy S, Sweadner K, Modyanov N, Doucet A. Are there several isoforms of Na,K-ATPase α subunit in the rabbit kidney? J Biol Chem 268: 11512–11515, 1993. [PubMed] [Google Scholar]
- 17.Bauer N, Müller-Ehmsen J, Krämer U, Hambarchian N, Zobel C, Schwinger RH, Neu H, Kirch U, Grünbaum EG, Schoner W. Ouabain-like compound changes rapidly on physical exercise in humans and dogs: effects of β-blockade and angiotensin-converting enzyme inhibition. Hypertension 45: 1024–1028, 2005. doi: 10.1161/01.HYP.0000165024.47728.f7. [DOI] [PubMed] [Google Scholar]
- 18.Berne RM, Levy MN. Cardiovascular Physiology. St. Louis, MO: Mosby, 2001, p. 115–153. [Google Scholar]
- 19.Berry RG, Despa S, Fuller W, Bers DM, Shattock MJ. Differential distribution and regulation of mouse cardiac Na+/K+-ATPase α1 and α2 subunits in T-tubule and surface sarcolemmal membranes. Cardiovasc Res 73: 92–100, 2007. doi: 10.1016/j.cardiores.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 20.Bers DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 21: 380–387, 2006. doi: 10.1152/physiol.00019.2006. [DOI] [PubMed] [Google Scholar]
- 21.Bers DM, Despa S. Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J Pharmacol Sci 100: 315–322, 2006. doi: 10.1254/jphs.CPJ06001X. [DOI] [PubMed] [Google Scholar]
- 22.Bignami E, Casamassima N, Frati E, Lanzani C, Corno L, Alfieri O, Gottlieb S, Simonini M, Shah KB, Mizzi A, Messaggio E, Zangrillo A, Ferrandi M, Ferrari P, Bianchi G, Hamlyn JM, Manunta P. Preoperative endogenous ouabain predicts acute kidney injury in cardiac surgery patients. Crit Care Med 41: 744–755, 2013. doi: 10.1097/CCM.0b013e3182741599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol Renal Physiol 275: F633–F650, 1998. [DOI] [PubMed] [Google Scholar]
- 24.Blausstein MP. What is the link between vascular smooth muscle sodium pumps and hypertension? Clin Exp Hypertens 3: 173–182, 1981. doi: 10.3109/10641968109037175. [DOI] [PubMed] [Google Scholar]
- 25.Blaustein MP. How does pressure overload cause cardiac hypertrophy and dysfunction? High ouabain affinity cardiac Na+ pumps are crucial. Am J Physiol Heart Circ Physiol 313: H919–H930, 2017. doi: 10.1152/ajpheart.00131.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blaustein MP. It was the best of times – A postdoctoral experience in the UK in the 1960s. Physiology News Autumn 2016: 32–35, 2016. [Google Scholar]
- 27.Blaustein MP. Letter to the Editor concerning Baecher et al. (Clin Chim Acta 2014;431:87-92). Clin Chim Acta 448: 248–249, 2015. doi: 10.1016/j.cca.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 28.Blaustein MP. Livin’ with NCX and lovin’ it: a 45 year romance. Adv Exp Med Biol 961: 3–15, 2013. doi: 10.1007/978-1-4614-4756-6_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blaustein MP. Sodium ions, calcium ions, blood pressure regulation, and hypertension: a reassessment and a hypothesis. Am J Physiol Cell Physiol 232: C165–C173, 1977. [DOI] [PubMed] [Google Scholar]
- 30.Blaustein MP. Why isn’t endogenous ouabain more widely accepted? Am J Physiol Heart Circ Physiol 307: H635–H639, 2014. doi: 10.1152/ajpheart.00404.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blaustein MP, Chen L, Hamlyn JM, Leenen FH, Lingrel JB, Wier WG, Zhang J. Pivotal role of α2 Na+ pumps and their high affinity ouabain binding site in cardiovascular health and disease. J Physiol 594: 6079–6103, 2016. doi: 10.1113/JP272419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blaustein MP, Chen L, Song H, Leenen FHH, Hamlyn JM. How does the brain talk to the arteries and heart? (Abstract). FASEB J 29: 984.3, 2015. doi: 10.1096/fj.1530-6860. [DOI] [Google Scholar]
- 33.Blaustein MP, Juhaszova M, Golovina VA. The cellular mechanism of action of cardiotonic steroids: a new hypothesis. Clin Exp Hypertens 20: 691–703, 1998. doi: 10.3109/10641969809053247. [DOI] [PubMed] [Google Scholar]
- 34.Blaustein MP, Leenen FH, Chen L, Golovina VA, Hamlyn JM, Pallone TL, Van Huysse JW, Zhang J, Wier WG. How NaCl raises blood pressure: a new paradigm for the pathogenesis of salt-dependent hypertension. Am J Physiol Heart Circ Physiol 302: H1031–H1049, 2012. doi: 10.1152/ajpheart.00899.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blaustein MP, Reuter H. A tale of three cities: the discovery of sodium/calcium exchange and some of its cardiovascular implications. Physiology News Autumn 2014: 32–37, 2014. [Google Scholar]
- 36.Boulanger BR, Lilly MP, Hamlyn JM, Laredo J, Shurtleff D, Gann DS. Ouabain is secreted by the adrenal gland in awake dogs. Am J Physiol Endocrinol Metab 264: E413–E419, 1993. [DOI] [PubMed] [Google Scholar]
- 37.Bricker NS, Schmidt RW, Favre H, Fine L, Bourgoignie JJ. On the biology of sodium excretion: the search for a natriuretic hormone. Yale J Biol Med 48: 293–303, 1975. [PMC free article] [PubMed] [Google Scholar]
- 38.Brody MJ. Central nervous system and mechanisms of hypertension. Clin Physiol Biochem 6: 230–239, 1988. [PubMed] [Google Scholar]
- 39.Buggy J, Fink GD, Haywood JR, Johnson AK, Brody MJ. Interruption of the maintenance phase of established hypertension by ablation of the anteroventral third ventricle (AV3V) in rats. Clin Exp Hypertens 1: 337–353, 1978-1979. doi: 10.3109/10641967809068612. [DOI] [PubMed] [Google Scholar]
- 40.Burlaka I, Nilsson LM, Scott L, Holtbäck U, Eklöf AC, Fogo AB, Brismar H, Aperia A. Prevention of apoptosis averts glomerular tubular disconnection and podocyte loss in proteinuric kidney disease. Kidney Int 90: 135–148, 2016. doi: 10.1016/j.kint.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Canfield VA, Xu KY, D’Aquila T, Shyjan AW, Levenson R. Molecular cloning and characterization of Na,K-ATPase from Hydra vulgaris: implications for enzyme evolution and ouabain sensitivity. New Biol 4: 339–348, 1992. [PubMed] [Google Scholar]
- 42.Chatterjee K. Congestive heart failure: what should be the initial therapy and why? Am J Cardiovasc Drugs 2: 1–6, 2002. doi: 10.2165/00129784-200202010-00001. [DOI] [PubMed] [Google Scholar]
- 43.Chen L, Lingrel J.B., Hamlyn J.M., Blaustein M.P.. Angiotensin-salt hypertension requires ouabain-sensitive Na+ pumps (Abstract). Hypertension 70, Suppl 1: AP331, 2017. [Google Scholar]
- 44.Clapcote SJ, Duffy S, Xie G, Kirshenbaum G, Bechard AR, Rodacker Schack V, Petersen J, Sinai L, Saab BJ, Lerch JP, Minassian BA, Ackerley CA, Sled JG, Cortez MA, Henderson JT, Vilsen B, Roder JC. Mutation I810N in the α3 isoform of Na+,K+-ATPase causes impairments in the sodium pump and hyperexcitability in the CNS. Proc Natl Acad Sci USA 106: 14085–14090, 2009. doi: 10.1073/pnas.0904817106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clarkson EM, Raw SM, de Wardener HE. Two natriuretic substances in extracts of urine from normal man when salt-depleted and salt-loaded. Kidney Int 10: 381–394, 1976. doi: 10.1038/ki.1976.124. [DOI] [PubMed] [Google Scholar]
- 46.Cody RJ. Hypertensive heart disease and heart failure. Curr Opin Cardiol 10: 450–457, 1995. doi: 10.1097/00001573-199509000-00003. [DOI] [PubMed] [Google Scholar]
- 47.Coleman TG, Guyton AC. Hypertension caused by salt loading in the dog. 3. Onset transients of cardiac output and other circulatory variables. Circ Res 25: 153–160, 1969. doi: 10.1161/01.RES.25.2.153. [DOI] [PubMed] [Google Scholar]
- 48.Croyle ML, Woo AL, Lingrel JB. Extensive random mutagenesis analysis of the Na+/K+-ATPase alpha subunit identifies known and previously unidentified amino acid residues that alter ouabain sensitivity--implications for ouabain binding. Eur J Biochem 248: 488–495, 1997. doi: 10.1111/j.1432-1033.1997.00488.x. [DOI] [PubMed] [Google Scholar]
- 49.Dahl LK, Heine M, Tassinari L. Effects of chronic excess salt ingestion. Role of genetic factors in both DOCA-salt and renal hypertension. J Exp Med 118: 605–617, 1963. doi: 10.1084/jem.118.4.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dahl LK, Knudsen KD, Heine M, Leitl G. Effects of chronic excess salt ingestion. Genetic influence on the development of salt hypertension in parabiotic rats: evidence for a humoral factor. J Exp Med 126: 687–699, 1967. doi: 10.1084/jem.126.4.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dalla S, Swarts HG, Koenderink JB, Dobler S. Amino acid substitutions of Na,K-ATPase conferring decreased sensitivity to cardenolides in insects compared to mammals. Insect Biochem Mol Biol 43: 1109–1115, 2013. doi: 10.1016/j.ibmb.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 52.Davis MW, Somerville D, Lee RY, Lockery S, Avery L, Fambrough DM. Mutations in the Caenorhabditis elegans Na,K-ATPase alpha-subunit gene, eat-6, disrupt excitable cell function. J Neurosci 15: 8408–8418, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Vasconcelos DI, Leite JA, Carneiro LT, Piuvezam MR, de Lima MR, de Morais LC, Rumjanek VM, Rodrigues-Mascarenhas S. Anti-inflammatory and antinociceptive activity of ouabain in mice. Mediators Inflamm 2011: 912925, 2011. doi: 10.1155/2011/912925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Wardener HE. Natriuretic hormone. Clin Sci Mol Med 53: 1–8, 1977. [DOI] [PubMed] [Google Scholar]
- 55.de Wardener HE, MacGregor GA. Dahl’s hypothesis that a saluretic substance may be responsible for a sustained rise in arterial pressure: its possible role in essential hypertension. Kidney Int 18: 1–9, 1980. doi: 10.1038/ki.1980.104. [DOI] [PubMed] [Google Scholar]
- 56.Delva P, Capra C, Degan M, Minuz P, Covi G, Milan L, Steele A, Lechi A. High plasma levels of a ouabain-like factor in normal pregnancy and in pre-eclampsia. Eur J Clin Invest 19: 95–100, 1989. [DOI] [PubMed] [Google Scholar]
- 57.DiBona GF. Central angiotensin modulation of baroreflex control of renal sympathetic nerve activity in the rat: influence of dietary sodium. Acta Physiol Scand 177: 285–289, 2003. doi: 10.1046/j.1365-201X.2003.01074.x. [DOI] [PubMed] [Google Scholar]
- 58.Digitalis Investigation Group The effect of digoxin on mortality and morbidity in patients with heart failure. N Engl J Med 336: 525–533, 1997. doi: 10.1056/NEJM199702203360801. [DOI] [PubMed] [Google Scholar]
- 59.Doris PA, Jenkins LA, Stocco DM. Is ouabain an authentic endogenous mammalian substance derived from the adrenal? Hypertension 23: 632–638, 1994. doi: 10.1161/01.HYP.23.5.632. [DOI] [PubMed] [Google Scholar]
- 60.Dostanic-Larson I, Van Huysse JW, Lorenz JN, Lingrel JB. The highly conserved cardiac glycoside binding site of Na,K-ATPase plays a role in blood pressure regulation. Proc Natl Acad Sci USA 102: 15845–15850, 2005. doi: 10.1073/pnas.0507358102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dostanic I, Lorenz JN, Schultz JJ, Grupp IL, Neumann JC, Wani MA, Lingrel JB. The α2 isoform of Na,K-ATPase mediates ouabain-induced cardiac inotropy in mice. J Biol Chem 278: 53026–53034, 2003. doi: 10.1074/jbc.M308547200. [DOI] [PubMed] [Google Scholar]
- 62.Dostanic I, Paul RJ, Lorenz JN, Theriault S, Van Huysse JW, Lingrel JB. The α2-isoform of Na-K-ATPase mediates ouabain-induced hypertension in mice and increased vascular contractility in vitro. Am J Physiol Heart Circ Physiol 288: H477–H485, 2005. doi: 10.1152/ajpheart.00083.2004. [DOI] [PubMed] [Google Scholar]
- 63.Dostanic I, Schultz JJ, Lorenz JN, Lingrel JB. The α 1 isoform of Na,K-ATPase regulates cardiac contractility and functionally interacts and co-localizes with the Na/Ca exchanger in heart. J Biol Chem 279: 54053–54061, 2004. doi: 10.1074/jbc.M410737200. [DOI] [PubMed] [Google Scholar]
- 64.Dutta S. Cardiac uptake and binding of cardiac glycosides. In: Cardiac Glycosides Part II: Pharmakokinetics and Clinical Pharmacology, edited by Greeff K. Berlin: Springer-Verlag, 1981, p. 141–168. doi: 10.1007/978-3-642-68166-0_7. [DOI] [Google Scholar]
- 65.Dvela-Levitt M, Cohen-Ben Ami H, Rosen H, Ornoy A, Hochner-Celnikier D, Granat M, Lichtstein D. Reduction in maternal circulating ouabain impairs offspring growth and kidney development. J Am Soc Nephrol 26: 1103–1114, 2015. doi: 10.1681/ASN.2014020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dvela M, Rosen H, Ben-Ami HC, Lichtstein D. Endogenous ouabain regulates cell viability. Am J Physiol Cell Physiol 302: C442–C452, 2012. doi: 10.1152/ajpcell.00336.2011. [DOI] [PubMed] [Google Scholar]
- 68.Editorial Welcome to ouabain–a new steroid hormone. Lancet 338: 543–544, 1991. doi: 10.1016/0140-6736(91)91104-3. [DOI] [PubMed] [Google Scholar]
- 69.El-Mallakh RS, El-Masri MA, Huff MO, Li XP, Decker S, Levy RS. Intracerebroventricular administration of ouabain as a model of mania in rats. Bipolar Disord 5: 362–365, 2003. doi: 10.1034/j.1399-5618.2003.00053.x. [DOI] [PubMed] [Google Scholar]
- 70.El-Mallakh RS, Harrison LT, Li R, Changaris DG, Levy RS. An animal model for mania: preliminary results. Prog Neuropsychopharmacol Biol Psychiatry 19: 955–962, 1995. doi: 10.1016/0278-5846(95)00123-D. [DOI] [PubMed] [Google Scholar]
- 71.Farman N, Corthesy-Theulaz I, Bonvalet JP, Rossier BC. Localization of α-isoforms of Na+-K+-ATPase in rat kidney by in situ hybridization. Am J Physiol Cel Physiol 260: C468–C474, 1991. [DOI] [PubMed] [Google Scholar]
- 72.Favre H, Hwang KH, Schmidt RW, Bricker NS, Bourgoignie JJ. An inhibitor of sodium transport in the urine of dogs with normal renal function. J Clin Invest 56: 1302–1311, 1975. doi: 10.1172/JCI108206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fedorova OV, Kolodkin NI, Agalakova NI, Lakatta EG, Bagrov AY. Marinobufagenin, an endogenous alpha-1 sodium pump ligand, in hypertensive Dahl salt-sensitive rats. Hypertension 37: 462–466, 2001. doi: 10.1161/01.HYP.37.2.462. [DOI] [PubMed] [Google Scholar]
- 74.Fedorova OV, Shapiro JI, Bagrov AY. Endogenous cardiotonic steroids and salt-sensitive hypertension. Biochim Biophys Acta 1802: 1230–1236, 2010. doi: 10.1016/j.bbadis.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fedorova OV, Talan MI, Agalakova NI, Lakatta EG, Bagrov AY. Endogenous ligand of α(1) sodium pump, marinobufagenin, is a novel mediator of sodium chloride-dependent hypertension. Circulation 105: 1122–1127, 2002. doi: 10.1161/hc0902.104710. [DOI] [PubMed] [Google Scholar]
- 76.Féraille E, Barlet-Bas C, Cheval L, Rousselot M, Carranza ML, Dreher D, Arystarkhova E, Doucet A, Favre H. Presence of two isoforms of Na, K-ATPase with different pharmacological and immunological properties in the rat kidney. Pflugers Arch 430: 205–212, 1995. doi: 10.1007/BF00374651. [DOI] [PubMed] [Google Scholar]
- 77.Ferrandi M, Molinari I, Barassi P, Minotti E, Bianchi G, Ferrari P. Organ hypertrophic signaling within caveolae membrane subdomains triggered by ouabain and antagonized by PST 2238. J Biol Chem 279: 33306–33314, 2004. doi: 10.1074/jbc.M402187200. [DOI] [PubMed] [Google Scholar]
- 78.Ferrandi M, Molinari I, Rastaldi MP, Ferrari P, Bianchi G, Manunta P. Rostafuroxin protects from podocyte injury and proteinuria induced by adducin genetic variants and ouabain. J Pharmacol Exp Ther 351: 278–287, 2014. doi: 10.1124/jpet.114.217133. [DOI] [PubMed] [Google Scholar]
- 79.Ferrari P, Torielli L, Ferrandi M, Padoani G, Duzzi L, Florio M, Conti F, Melloni P, Vesci L, Corsico N, Bianchi G. PST2238: a new antihypertensive compound that antagonizes the long-term pressor effect of ouabain. J Pharmacol Exp Ther 285: 83–94, 1998. [PubMed] [Google Scholar]
- 80.Fine LG, Bourgoignie JJ, Hwang KH, Bricker NS. On the influence of the natriuretic factor from patients with chronic uremia on the bioelectric properties and sodium transport of the isolated mammalian collecting tubule. J Clin Invest 58: 590–597, 1976. doi: 10.1172/JCI108505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Forester W, Lewis RP, Weissler AM, Wilke TA. The onset and magnitude of the contractile response to commonly used digitalis glycosides in normal subjects. Circulation 49: 517–521, 1974. doi: 10.1161/01.CIR.49.3.517. [DOI] [PubMed] [Google Scholar]
- 82.Fraser TR. The action and uses of digitalis and its substitutes, with special reference to Strophanthus (hispidus?). BMJ 2: 904–910, 1885. doi: 10.1136/bmj.2.1298.904. [DOI] [Google Scholar]
- 83.Freis ED. Mechanism of the antihypertensive effects of diuretics. Possible role of salt in hypertension. Clin Pharmacol Ther 1: 337–344, 1960. doi: 10.1002/cpt196013337. [DOI] [PubMed] [Google Scholar]
- 84.Friedberg CK. Diseases of the Heart. Philadelphia, PA: Saunders, 1956, p. 256–263. [Google Scholar]
- 85.Fürstenwerth H. Ouabain – the insulin of the heart. Int J Clin Pract 64: 1591–1594, 2010. doi: 10.1111/j.1742-1241.2010.02395.x. [DOI] [PubMed] [Google Scholar]
- 86.Gaasch WH. Congestive heart failure in patients with normal left ventricular systolic function: a manifestation of diastolic dysfunction. Herz 16: 22–32, 1991. [PubMed] [Google Scholar]
- 87.Gable ME, Abdallah SL, Najjar SM, Liu L, Askari A. Digitalis-induced cell signaling by the sodium pump: on the relation of Src to Na+/K+-ATPase. Biochem Biophys Res Commun 446: 1151–1154, 2014. doi: 10.1016/j.bbrc.2014.03.071. [DOI] [PubMed] [Google Scholar]
- 88.Gabor A, Leenen FH. Central neuromodulatory pathways regulating sympathetic activity in hypertension. J Appl Physiol (1985) 113: 1294–1303, 2012. doi: 10.1152/japplphysiol.00553.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Galuska D, Kotova O, Barrès R, Chibalina D, Benziane B, Chibalin AV. Altered expression and insulin-induced trafficking of Na+-K+-ATPase in rat skeletal muscle: effects of high-fat diet and exercise. Am J Physiol Endocrinol Metab 297: E38–E49, 2009. doi: 10.1152/ajpendo.90990.2008. [DOI] [PubMed] [Google Scholar]
- 90.Garcia A, Liu CC, Cornelius F, Clarke RJ, Rasmussen HH. Glutathionylation-dependence of Na+-K+-pump currents can mimic reduced subsarcolemmal Na+ diffusion. Biophys J 110: 1099–1109, 2016. doi: 10.1016/j.bpj.2016.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Geering K. FXYD proteins: new regulators of Na-K-ATPase. Am J Physiol Renal Physiol 290: F241–F250, 2006. doi: 10.1152/ajprenal.00126.2005. [DOI] [PubMed] [Google Scholar]
- 92.Ghadhanfar E, Al-Bader M, Turcani M. Wistar rats resistant to the hypertensive effects of ouabain exhibit enhanced cardiac vagal activity and elevated plasma levels of calcitonin gene-related peptide. PLoS One 9: e108909, 2014. doi: 10.1371/journal.pone.0108909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gheorghiade M, Pitt B. Digitalis Investigation Group (DIG) trial: a stimulus for further research. Am Heart J 134: 3–12, 1997. doi: 10.1016/S0002-8703(97)70100-5. [DOI] [PubMed] [Google Scholar]
- 94.Giachini FR, Tostes RC. Does Na+ really play a role in Ca2+ homeostasis in hypertension? Am J Physiol Heart Circ Physiol 299: H602–H604, 2010. doi: 10.1152/ajpheart.00542.2010. [DOI] [PubMed] [Google Scholar]
- 95.Given BD, Lee TH, Stone PH, Dzau VJ. Nifedipine in severely hypertensive patients with congestive heart failure and preserved ventricular systolic function. Arch Intern Med 145: 281–285, 1985. doi: 10.1001/archinte.1985.00360020109018. [DOI] [PubMed] [Google Scholar]
- 96.Glynn IM, Rink TJ. Hypertension and inhibition of the sodium pump: a strong link but in which chain? Nature 300: 576–577, 1982. doi: 10.1038/300576a0. [DOI] [PubMed] [Google Scholar]
- 97.Goldstein I, Lax E, Gispan-Herman I, Ovadia H, Rosen H, Yadid G, Lichtstein D. Neutralization of endogenous digitalis-like compounds alters catecholamines metabolism in the brain and elicits anti-depressive behavior. Eur Neuropsychopharmacol 22: 72–79, 2012. doi: 10.1016/j.euroneuro.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 98.Golomb E, Hill MR, Brown RG, Keiser HR. Ouabain enhances the mitogenic effect of serum in vascular smooth muscle cells. Am J Hypertens 7: 69–74, 1994. doi: 10.1093/ajh/7.1.69. [DOI] [PubMed] [Google Scholar]
- 99.Golovina VA, Song H, James PF, Lingrel JB, Blaustein MP. Na+ pump α2-subunit expression modulates Ca2+ signaling. Am J Physiol Cell Physiol 284: C475–C486, 2003. doi: 10.1152/ajpcell.00383.2002. [DOI] [PubMed] [Google Scholar]
- 100.Gomez-Sanchez EP, Gomez-Sanchez CE. Effect of central infusion of benzamil on Dahl S rat hypertension. Am J Physiol Heart Circ Physiol 269: H1044–H1047, 1995. [DOI] [PubMed] [Google Scholar]
- 101.Gomez-Sanchez EP, Gomez-Sanchez CE, Fort C. Immunization of Dahl SS/jr rats with an ouabain conjugate mitigates hypertension. Am J Hypertens 7: 591–596, 1994. doi: 10.1093/ajh/7.7.591. [DOI] [PubMed] [Google Scholar]
- 102.Goto A, Yamada K, Hazama H, Uehara Y, Atarashi K, Hirata Y, Kimura K, Omata M. Ouabainlike compound in hypertension associated with ectopic corticotropin syndrome. Hypertension 28: 421–425, 1996. doi: 10.1161/01.HYP.28.3.421. [DOI] [PubMed] [Google Scholar]
- 103.Goto A, Yamada K, Ishii M, Yoshioka M, Ishiguro T, Eguchi C, Sugimoto T. Presence of digitalis-like factor in mammalian plasma. Biochem Biophys Res Commun 152: 322–327, 1988. doi: 10.1016/S0006-291X(88)80717-4. [DOI] [PubMed] [Google Scholar]
- 104.Gottlieb SS, Rogowski AC, Weinberg M, Krichten CM, Hamilton BP, Hamlyn JM. Elevated concentrations of endogenous ouabain in patients with congestive heart failure. Circulation 86: 420–425, 1992. doi: 10.1161/01.CIR.86.2.420. [DOI] [PubMed] [Google Scholar]
- 105.Graham RM, Oates HF, Stoker LM, Stokes GS. Alpha blocking action of the antihypertensive agent, prazosin. J Pharmacol Exp Ther 201: 747–752, 1977. [PubMed] [Google Scholar]
- 106.Greeff K, Wirth KE. Pharmakokinetics of Strophanthus glycosides. In: Cardiac Glycosides Part II: Pharmakokinetics and Clinical Pharmacology, edited by Greeff K. Berlin: Springer -Verlag, 1981, p. 57–85. doi: 10.1007/978-3-642-68166-0_3. [DOI] [Google Scholar]
- 107.Grosse-Brockhoff F, Peters U. Clinical indications and choice of cardiac glycosides, clinical conditions influencing glycoside effect. In: Cardiac Glycosides Part II: Pharmakokinetics and Clinical Pharmacology, edited by Greeff K. Berlin: Springer-Verlag, 1981, p. 239–274. doi: 10.1007/978-3-642-68166-0_12. [DOI] [Google Scholar]
- 108.Guyton AC. Blood pressure control–special role of the kidneys and body fluids. Science 252: 1813–1816, 1991. doi: 10.1126/science.2063193. [DOI] [PubMed] [Google Scholar]
- 109.Guyton AC, Coleman TG, Cowley AVJ Jr, Scheel KW, Manning RDJ Jr, Norman RAJ Jr. Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med 52: 584–594, 1972. doi: 10.1016/0002-9343(72)90050-2. [DOI] [PubMed] [Google Scholar]
- 110.Haas M, Askari A, Xie Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem 275: 27832–27837, 2000. [DOI] [PubMed] [Google Scholar]
- 111.Haas M, Wang H, Tian J, Xie Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J Biol Chem 277: 18694–18702, 2002. doi: 10.1074/jbc.M111357200. [DOI] [PubMed] [Google Scholar]
- 112.Haddy F, Pamnani M, Clough D. The sodium-potassium pump in volume expanded hypertension. Clin Exp Hypertens 1: 295–336, 1978-1979. doi: 10.3109/10641967809068611. [DOI] [PubMed] [Google Scholar]
- 113.Haddy FJ, Overbeck HW. The role of humoral agents in volume expanded hypertension. Life Sci 19: 935–947, 1976. doi: 10.1016/0024-3205(76)90284-8. [DOI] [PubMed] [Google Scholar]
- 114.Hall JE. Renal dysfunction, rather than nonrenal vascular dysfunction, mediates salt-induced hypertension. Circulation 133: 894–906, 2016. doi: 10.1161/CIRCULATIONAHA.115.018526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hall JE, Guyton AC, Brands MW. Pressure-volume regulation in hypertension. Kidney Int Suppl 55: S35–S41, 1996. [PubMed] [Google Scholar]
- 116.Hallaq HA, Haupert GT Jr. Positive inotropic effects of the endogenous Na+/K+-transporting ATPase inhibitor from the hypothalamus. Proc Natl Acad Sci USA 86: 10080–10084, 1989. doi: 10.1073/pnas.86.24.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hamlyn JM, Blaustein MP. Endogenous ouabain: recent advances and controversies. Hypertension 68: 526–532, 2016. doi: 10.1161/HYPERTENSIONAHA.116.06599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, Mathews WR, Ludens JH. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci USA 88: 6259–6263, 1991. doi: 10.1073/pnas.88.14.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hamlyn JM, Harris DW, Clark MA, Rogowski AC, White RJ, Ludens JH. Isolation and characterization of a sodium pump inhibitor from human plasma. Hypertension 13: 681–689, 1989. doi: 10.1161/01.HYP.13.6.681. [DOI] [PubMed] [Google Scholar]
- 120.Hamlyn JM, Harris DW, Ludens JH. Digitalis-like activity in human plasma. Purification, affinity, and mechanism. J Biol Chem 264: 7395–7404, 1989. [PubMed] [Google Scholar]
- 121.Hamlyn JM, Linde CI, Gao J, Huang BS, Golovina VA, Blaustein MP, Leenen FH. Neuroendocrine humoral and vascular components in the pressor pathway for brain angiotensin II: a new axis in long term blood pressure control. PLoS One 9: e108916, 2014. doi: 10.1371/journal.pone.0108916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hamlyn JM, Manunta P. Endogenous cardiotonic steroids in kidney failure: a review and an hypothesis. Adv Chronic Kidney Dis 22: 232–244, 2015. doi: 10.1053/j.ackd.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hamlyn JM, Ringel R, Schaeffer J, Levinson PD, Hamilton BP, Kowarski AA, Blaustein MP. A circulating inhibitor of (Na+ + K+)ATPase associated with essential hypertension. Nature 300: 650–652, 1982. doi: 10.1038/300650a0. [DOI] [PubMed] [Google Scholar]
- 124.Hasler U, Wang X, Crambert G, Béguin P, Jaisser F, Horisberger JD, Geering K. Role of β-subunit domains in the assembly, stable expression, intracellular routing, and functional properties of Na,K-ATPase. J Biol Chem 273: 30826–30835, 1998. doi: 10.1074/jbc.273.46.30826. [DOI] [PubMed] [Google Scholar]
- 125.He S, Shelly DA, Moseley AE, James PF, James JH, Paul RJ, Lingrel JB. The α1- and α2-isoforms of Na-K-ATPase play different roles in skeletal muscle contractility. Am J Physiol Regul Integr Comp Physiol 281: R917–R925, 2001. [DOI] [PubMed] [Google Scholar]
- 126.Hilton PJ, White RW, Lord GA, Garner GV, Gordon DB, Hilton MJ, Forni LG, McKinnon W, Ismail FM, Keenan M, Jones K, Morden WE. An inhibitor of the sodium pump obtained from human placenta. Lancet 348: 303–305, 1996. doi: 10.1016/S0140-6736(96)02257-X. [DOI] [PubMed] [Google Scholar]
- 127.Hodes A, Rosen H, Deutsch J, Lifschytz T, Einat H, Lichtstein D. Endogenous cardiac steroids in animal models of mania. Bipolar Disord 18: 451–459, 2016. doi: 10.1111/bdi.12413. [DOI] [PubMed] [Google Scholar]
- 128.Hollenberg NK. Management of hypertension: considerations involving cardiovascular risk reduction. J Cardiovasc Pharmacol 15, Suppl 5: S73–S78, 1990. doi: 10.1097/00005344-199000155-00011. [DOI] [PubMed] [Google Scholar]
- 129.Huang BS, Harmsen E, Yu H, Leenen FH. Brain ouabain-like activity and the sympathoexcitatory and pressor effects of central sodium in rats. Circ Res 71: 1059–1066, 1992. doi: 10.1161/01.RES.71.5.1059. [DOI] [PubMed] [Google Scholar]
- 130.Huang BS, Huang X, Harmsen E, Leenen FH. Chronic central versus peripheral ouabain, blood pressure, and sympathetic activity in rats. Hypertension 23: 1087–1090, 1994. doi: 10.1161/01.HYP.23.6.1087. [DOI] [PubMed] [Google Scholar]
- 131.Huang BS, Kudlac M, Kumarathasan R, Leenen FH. Digoxin prevents ouabain and high salt intake-induced hypertension in rats with sinoaortic denervation. Hypertension 34: 733–738, 1999. doi: 10.1161/01.HYP.34.4.733. [DOI] [PubMed] [Google Scholar]
- 132.Huang BS, Leenen FH. Both brain angiotensin II and “ouabain” contribute to sympathoexcitation and hypertension in Dahl S rats on high salt intake. Hypertension 32: 1028–1033, 1998. doi: 10.1161/01.HYP.32.6.1028. [DOI] [PubMed] [Google Scholar]
- 133.Huang BS, Leenen FH. Brain “ouabain” mediates the sympathoexcitatory and hypertensive effects of high sodium intake in Dahl salt-sensitive rats. Circ Res 74: 586–595, 1994. doi: 10.1161/01.RES.74.4.586. [DOI] [PubMed] [Google Scholar]
- 134.Huang BS, Zheng H, Tan J, Patel KP, Leenen FH. Regulation of hypothalamic renin-angiotensin system and oxidative stress by aldosterone. Exp Physiol 96: 1028–1038, 2011. doi: 10.1113/expphysiol.2011.059840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Huang L, Li H, Xie Z. Ouabain-induced hypertrophy in cultured cardiac myocytes is accompanied by changes in expression of several late response genes. J Mol Cell Cardiol 29: 429–437, 1997. doi: 10.1006/jmcc.1996.0320. [DOI] [PubMed] [Google Scholar]
- 136.Hundal HS, Marette A, Mitsumoto Y, Ramlal T, Blostein R, Klip A. Insulin induces translocation of the α2 and β1 subunits of the Na+/K+-ATPase from intracellular compartments to the plasma membrane in mammalian skeletal muscle. J Biol Chem 267: 5040–5043, 1992. [PubMed] [Google Scholar]
- 137.Ikeda K, Onaka T, Yamakado M, Nakai J, Ishikawa TO, Taketo MM, Kawakami K. Degeneration of the amygdala/piriform cortex and enhanced fear/anxiety behaviors in sodium pump α2 subunit (Atp1a2)-deficient mice. J Neurosci 23: 4667–4676, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jacobs BE, Liu Y, Pulina MV, Golovina VA, Hamlyn JM. Normal pregnancy: mechanisms underlying the paradox of a ouabain-resistant state with elevated endogenous ouabain, suppressed arterial sodium calcium exchange, and low blood pressure. Am J Physiol Heart Circ Physiol 302: H1317–H1329, 2012. doi: 10.1152/ajpheart.00532.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jacobs WA, Bigelow NM. Ouabain or g-strophanthin. J Biol Chem 96: 647–658, 1932. [Google Scholar]
- 140.James PA, Oparil S, Carter BL, Cushman WC, Dennison-Himmelfarb C, Handler J, Lackland DT, LeFevre ML, MacKenzie TD, Ogedegbe O, Smith SC Jr, Svetkey LP, Taler SJ, Townsend RR, Wright JT Jr, Narva AS, Ortiz E. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 311: 507–520, 2014. doi: 10.1001/jama.2013.284427. [DOI] [PubMed] [Google Scholar]
- 141.James PF, Grupp IL, Grupp G, Woo AL, Askew GR, Croyle ML, Walsh RA, Lingrel JB. Identification of a specific role for the Na,K-ATPase α2 isoform as a regulator of calcium in the heart. Mol Cell 3: 555–563, 1999. doi: 10.1016/S1097-2765(00)80349-4. [DOI] [PubMed] [Google Scholar]
- 142.Jansson K, Nguyen AN, Magenheimer BS, Reif GA, Aramadhaka LR, Bello-Reuss E, Wallace DP, Calvet JP, Blanco G. Endogenous concentrations of ouabain act as a cofactor to stimulate fluid secretion and cyst growth of in vitro ADPKD models via cAMP and EGFR-Src-MEK pathways. Am J Physiol Renal Physiol 303: F982–F990, 2012. doi: 10.1152/ajprenal.00677.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Johnson AK, Buggy J, Fink GD, Brody MJ. Prevention of renal hypertension and of the central pressor effect of angiotensin by ventromedial hypothalamic ablation. Brain Res 205: 255–264, 1981. doi: 10.1016/0006-8993(81)90337-1. [DOI] [PubMed] [Google Scholar]
- 144.Juhaszova M, Blaustein MP. Distinct distribution of different Na+ pump alpha subunit isoforms in plasmalemma. Physiological implications. Ann NY Acad Sci 834: 524–536, 1997. doi: 10.1111/j.1749-6632.1997.tb52310.x. [DOI] [PubMed] [Google Scholar]
- 145.Juhaszova M, Blaustein MP. Na+ pump low and high ouabain affinity alpha subunit isoforms are differently distributed in cells. Proc Natl Acad Sci USA 94: 1800–1805, 1997. doi: 10.1073/pnas.94.5.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kaide J, Ura N, Torii T, Nakagawa M, Takada T, Shimamoto K. Effects of digoxin-specific antibody Fab fragment (Digibind) on blood pressure and renal water-sodium metabolism in 5/6 reduced renal mass hypertensive rats. Am J Hypertens 12: 611–619, 1999. doi: 10.1016/S0895-7061(99)00029-1. [DOI] [PubMed] [Google Scholar]
- 147.Kaplan NM. Clinical Hypertension (3rd ed.) Baltimore, MD: Williams & Wilkins, 1982. [Google Scholar]
- 148.Kawamoto EM, Lima LS, Munhoz CD, Yshii LM, Kinoshita PF, Amara FG, Pestana RR, Orellana AM, Cipolla-Neto J, Britto LR, Avellar MC, Rossoni LV, Scavone C. Influence of N-methyl-d-aspartate receptors on ouabain activation of nuclear factor-κB in the rat hippocampus. J Neurosci Res 90: 213–228, 2012. doi: 10.1002/jnr.22745. [DOI] [PubMed] [Google Scholar]
- 149.Kawamura A, Guo J, Itagaki Y, Bell C, Wang Y, Haupert GT Jr, Magil S, Gallagher RT, Berova N, Nakanishi K. On the structure of endogenous ouabain. Proc Natl Acad Sci USA 96: 6654–6659, 1999. doi: 10.1073/pnas.96.12.6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kinoshita PF, Yshii LM, Orellana AMM, Paixão AG, Vasconcelos AR, Lima LS, Kawamoto EM, Scavone C. Alpha 2 Na+,K+-ATPase silencing induces loss of inflammatory response and ouabain protection in glial cells. Sci Rep 7: 4894, 2017. doi: 10.1038/s41598-017-05075-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kirshenbaum GS, Clapcote SJ, Duffy S, Burgess CR, Petersen J, Jarowek KJ, Yücel YH, Cortez MA, Snead OC III, Vilsen B, Peever JH, Ralph MR, Roder JC. Mania-like behavior induced by genetic dysfunction of the neuron-specific Na+,K+-ATPase α3 sodium pump. Proc Natl Acad Sci USA 108: 18144–18149, 2011. doi: 10.1073/pnas.1108416108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kobayashi M, Usui-Kawanishi F, Karasawa T, Kimura H, Watanabe S, Mise N, Kayama F, Kasahara T, Hasebe N, Takahashi M. The cardiac glycoside ouabain activates NLRP3 inflammasomes and promotes cardiac inflammation and dysfunction. PLoS One 12: e0176676, 2017. doi: 10.1371/journal.pone.0176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kometiani P, Li J, Gnudi L, Kahn BB, Askari A, Xie Z. Multiple signal transduction pathways link Na+/K+-ATPase to growth-related genes in cardiac myocytes. The roles of Ras and mitogen-activated protein kinases. J Biol Chem 273: 15249–15256, 1998. doi: 10.1074/jbc.273.24.15249. [DOI] [PubMed] [Google Scholar]
- 154.Komiyama Y, Dong XH, Nishimura N, Masaki H, Yoshika M, Masuda M, Takahashi H. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin Biochem 38: 36–45, 2005. doi: 10.1016/j.clinbiochem.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 155.Komiyama Y, Nishimura N, Dong XH, Hirose S, Kosaka C, Masaki H, Masuda M, Takahashi H. Liquid chromatography mass spectrometric analysis of ouabainlike factor in biological fluid. Hypertens Res 23, Suppl: S21–S27, 2000. doi: 10.1291/hypres.23.Supplement_S21. [DOI] [PubMed] [Google Scholar]
- 156.Krep H, Price DA, Soszynski P, Tao QF, Graves SW, Hollenberg NK. Volume sensitive hypertension and the digoxin-like factor. Reversal by a Fab directed against digoxin in DOCA-salt hypertensive rats. Am J Hypertens 8: 921–927, 1995. doi: 10.1016/0895-7061(95)00181-N. [DOI] [PubMed] [Google Scholar]
- 157.Krum H, Driscoll A. Management of heart failure. Med J Aust 199: 334–339, 2013. doi: 10.5694/mja12.10993. [DOI] [PubMed] [Google Scholar]
- 158.Kulikov A, Eva A, Kirch U, Boldyrev A, Scheiner-Bobis G. Ouabain activates signaling pathways associated with cell death in human neuroblastoma. Biochim Biophys Acta 1768: 1691–1702, 2007. doi: 10.1016/j.bbamem.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 159.Kurtz TW, DiCarlo SE, Morris RC Jr. Logical issues with the pressure natriuresis theory of chronic hypertension. Am J Hypertens 29: 1325–1331, 2016. doi: 10.1093/ajh/hpw073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lancaster MC, Vegad JL. Suppression of the early inflammatory response in the sheep by strophanthin G. Nature 213: 840–841, 1967. doi: 10.1038/213840b0. [DOI] [PubMed] [Google Scholar]
- 161.Lanzani C, Citterio L, Glorioso N, Manunta P, Tripodi G, Salvi E, Carpini SD, Ferrandi M, Messaggio E, Staessen JA, Cusi D, Macciardi F, Argiolas G, Valentini G, Ferrari P, Bianchi G. Adducin- and ouabain-related gene variants predict the antihypertensive activity of rostafuroxin, part 2: clinical studies. Sci Transl Med 2: 59ra87, 2010. doi: 10.1126/scitranslmed.3001814. [DOI] [PubMed] [Google Scholar]
- 162.Laredo J, Hamilton BP, Hamlyn JM. Ouabain is secreted by bovine adrenocortical cells. Endocrinology 135: 794–797, 1994. doi: 10.1210/endo.135.2.8033829. [DOI] [PubMed] [Google Scholar]
- 163.Laursen M, Yatime L, Nissen P, Fedosova NU. Crystal structure of the high-affinity Na+K+-ATPase-ouabain complex with Mg2+ bound in the cation binding site. Proc Natl Acad Sci USA 110: 10958–10963, 2013. doi: 10.1073/pnas.1222308110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lazebnik Y. Can a biologist fix a radio?—Or, what I learned while studying apoptosis. Cancer Cell 2: 179–182, 2002. doi: 10.1016/S1535-6108(02)00133-2. [DOI] [PubMed] [Google Scholar]
- 165.Lederer WJ, Niggli E, Hadley RW. Sodium-calcium exchange in excitable cells: fuzzy space. Science 248: 283, 1990. doi: 10.1126/science.2326638. [DOI] [PubMed] [Google Scholar]
- 166.Leenen FH. Actions of circulating angiotensin II and aldosterone in the brain contributing to hypertension. Am J Hypertens 27: 1024–1032, 2014. doi: 10.1093/ajh/hpu066. [DOI] [PubMed] [Google Scholar]
- 167.Leenen FH. The central role of the brain aldosterone–“ouabain” pathway in salt-sensitive hypertension. Biochim Biophys Acta 1802: 1132–1139, 2010. doi: 10.1016/j.bbadis.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 168.Leenen FH, Hou X, Wang HW, Ahmad M. Enhanced expression of epithelial sodium channels causes salt-induced hypertension in mice through inhibition of the α2-isoform of Na+, K+-ATPase. Physiol Rep 3: e12383, 2015. doi: 10.14814/phy2.12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Leenen FH, Huang BS, Yu H, Yuan B. Brain ‘ouabain’ mediates sympathetic hyperactivity in congestive heart failure. Circ Res 77: 993–1000, 1995. doi: 10.1161/01.RES.77.5.993. [DOI] [PubMed] [Google Scholar]
- 170.Leenen FH, Ruzicka M, Floras JS. Central sympathetic inhibition by mineralocorticoid receptor but not angiotensin II type 1 receptor blockade: are prescribed doses too low? Hypertension 60: 278–280, 2012. doi: 10.1161/HYPERTENSIONAHA.112.197012. [DOI] [PubMed] [Google Scholar]
- 171.Leenen FHH, Blaustein MP, Hamlyn JM. Update on angiotensin II: new endocrine connections between the brain, adrenal glands and the cardiovascular system. Endocr Connect 6: R131–R145, 2017. doi: 10.1530/EC-17-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lewis LK, Yandle TG, Hilton PJ, Jensen BP, Begg EJ, Nicholls MG. Endogenous ouabain is not ouabain. Hypertension 64: 680–683, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03919. [DOI] [PubMed] [Google Scholar]
- 173.Lewis LK, Yandle TG, Lewis JG, Richards AM, Pidgeon GB, Kaaja RJ, Nicholls MG. Ouabain is not detectable in human plasma. Hypertension 24: 549–555, 1994. doi: 10.1161/01.HYP.24.5.549. [DOI] [PubMed] [Google Scholar]
- 174.Li J, Khodus GR, Kruusmägi M, Kamali-Zare P, Liu XL, Eklöf AC, Zelenin S, Brismar H, Aperia A. Ouabain protects against adverse developmental programming of the kidney. Nat Commun 1: 42, 2010. doi: 10.1038/ncomms1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Li J, Zelenin S, Aperia A, Aizman O. Low doses of ouabain protect from serum deprivation-triggered apoptosis and stimulate kidney cell proliferation via activation of NF-kappaB. J Am Soc Nephrol 17: 1848–1857, 2006. doi: 10.1681/ASN.2005080894. [DOI] [PubMed] [Google Scholar]
- 176.Li M, Wen C, Whitworth JA. Hemodynamic effects of the Fab fragment of digoxin antibody (digibind) in corticotropin (ACTH)-induced hypertension. Am J Hypertens 10: 332–336, 1997. doi: 10.1016/S0895-7061(96)00318-4. [DOI] [PubMed] [Google Scholar]
- 177.Lifton RP. Genetic dissection of human blood pressure variation: common pathways from rare phenotypes. Harvey Lect 100: 71–101, 2004–2005. [PubMed] [Google Scholar]
- 178.Linde CI, Karashima E, Raina H, Zulian A, Wier WG, Hamlyn JM, Ferrari P, Blaustein MP, Golovina VA. Increased arterial smooth muscle Ca2+ signaling, vasoconstriction, and myogenic reactivity in Milan hypertensive rats. Am J Physiol Heart Circ Physiol 302: H611–H620, 2012. doi: 10.1152/ajpheart.00950.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lingrel JB. The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Annu Rev Physiol 72: 395–412, 2010. doi: 10.1146/annurev-physiol-021909-135725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lingrel JB, Orlowski J, Shull MM, Price EM. Molecular genetics of Na,K-ATPase. Prog Nucleic Acid Res Mol Biol 38: 37–89, 1990. doi: 10.1016/S0079-6603(08)60708-4. [DOI] [PubMed] [Google Scholar]
- 181.Liu J, Tian J, Haas M, Shapiro JI, Askari A, Xie Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J Biol Chem 275: 27838–27844, 2000. [DOI] [PubMed] [Google Scholar]
- 182.Livingstone D, Livingstone C. Narrative of an Expedition to the Zambesi and Its Tributaries. London: Murray, 1865, p. 466. [Google Scholar]
- 183.Longden TA, Dabertrand F, Koide M, Gonzales AL, Tykocki NR, Brayden JE, Hill-Eubanks D, Nelson MT. Capillary K+-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat Neurosci 20: 717–726, 2017. doi: 10.1038/nn.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Loreaux EL, Kaul B, Lorenz JN, Lingrel JB. Ouabain-sensitive α1 Na,K-ATPase enhances natriuretic response to saline load. J Am Soc Nephrol 19: 1947–1954, 2008. doi: 10.1681/ASN.2008020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lorenz JN, Loreaux EL, Dostanic-Larson I, Lasko V, Schnetzer JR, Paul RJ, Lingrel JB. ACTH-induced hypertension is dependent on the ouabain-binding site of the α2-Na+-K+-ATPase subunit. Am J Physiol Heart Circ Physiol 295: H273–H280, 2008. doi: 10.1152/ajpheart.00183.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lorenz JN, Oshiro N, Loreaux EL, Lingrel JB. DOCA-salt hypertension does not require the ouabain-sensitive binding site of the α2 Na,K-ATPase. Am J Hypertens 25: 421–429, 2012. doi: 10.1038/ajh.2011.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lu FM, Hilgemann DW. Na/K pump inactivation, subsarcolemmal Na measurements, and cytoplasmic ion turnover kinetics contradict restricted Na spaces in murine cardiac myocytes. J Gen Physiol 149: 727–749, 2017. doi: 10.1085/jgp.201711780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187a.Lu J, Wang H-W, Ahmad M, Kreshtar-Jahromi M, Blaustein MP, Hamlyn J, Leenen FHH. Central and peripheral slow-pressor mechanisms contributing to angiotensin II-salt hypertension in rats. Cardiovasc Res doi: 10.1093/cvr/cvx214 [Epub ahead of print] 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ludens JH, Clark MA, DuCharme DW, Harris DW, Lutzke BS, Mandel F, Mathews WR, Sutter DM, Hamlyn JM. Purification of an endogenous digitalislike factor from human plasma for structural analysis. Hypertension 17: 923–929, 1991. doi: 10.1161/01.HYP.17.6.923. [DOI] [PubMed] [Google Scholar]
- 189.Lüttgau HC, Niedergerke R. The antagonism between Ca and Na ions on the frog’s heart. J Physiol 143: 486–505, 1958. doi: 10.1113/jphysiol.1958.sp006073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Manunta P, Evans G, Hamilton B, Gann D, Resau J, Hamlyn J. A new syndrome with elevated plasma ouabain and hypertension secondary to an adrenocortical tumor (Abstract). J Hypertens 10: S27, 1992. [Google Scholar]
- 191.Manunta P, Hamilton BP, Hamlyn JM. Salt intake and depletion increase circulating levels of endogenous ouabain in normal men. Am J Physiol Regul Integr Comp Physiol 290: R553–R559, 2006. doi: 10.1152/ajpregu.00648.2005. [DOI] [PubMed] [Google Scholar]
- 192.Manunta P, Hamilton BP, Hamlyn JM. Structure-activity relationships for the hypertensinogenic activity of ouabain: role of the sugar and lactone ring. Hypertension 37: 472–477, 2001. doi: 10.1161/01.HYP.37.2.472. [DOI] [PubMed] [Google Scholar]
- 193.Manunta P, Hamilton J, Rogowski AC, Hamilton BP, Hamlyn JM. Chronic hypertension induced by ouabain but not digoxin in the rat: antihypertensive effect of digoxin and digitoxin. Hypertens Res 23, Suppl: S77–S85, 2000. doi: 10.1291/hypres.23.Supplement_S77. [DOI] [PubMed] [Google Scholar]
- 194.Manunta P, Iacoviello M, Forleo C, Messaggio E, Hamlyn JM, Lucarelli K, Guida P, Romito R, De Tommasi E, Bianchi G, Rizzon P, Pitzalis MV. High circulating levels of endogenous ouabain in the offspring of hypertensive and normotensive individuals. J Hypertens 23: 1677–1681, 2005. doi: 10.1097/01.hjh.0000177049.38417.67. [DOI] [PubMed] [Google Scholar]
- 195.Manunta P, Stella P, Rivera R, Ciurlino D, Cusi D, Ferrandi M, Hamlyn JM, Bianchi G. Left ventricular mass, stroke volume, and ouabain-like factor in essential hypertension. Hypertension 34: 450–456, 1999. doi: 10.1161/01.HYP.34.3.450. [DOI] [PubMed] [Google Scholar]
- 196.Manunta P, Tyzack J, Hamilton BP, Hamlyn JM. Augmentation and antagonism of ouabain-induced hypertension. Hypertension 22: 432, 1993. [Google Scholar]
- 197.Mark AL. The sympathetic nervous system in hypertension: a potential long-term regulator of arterial pressure. J Hypertens Suppl 14: S159–S165, 1996. [PubMed] [Google Scholar]
- 198.Mason DT, Braunwald E, Karsh RB, Bullock FA. Studies on digitalis. X. Effects of ouabain on forearm vascular resistance and venous tone in normal subjects and in patients in heart failure. J Clin Invest 43: 532–543, 1964. doi: 10.1172/JCI104939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Mathews MR, DuCharme DW, Hamlyn JM, Harris DW. Mandel F, Clark MA, Ludens JH. Mass spectral characterization of an endogenous digitalis-like factor from human plasma. Hypertension 17: 930–935, 1991. doi: 10.1161/01.HYP.17.6.930. [DOI] [PubMed] [Google Scholar]
- 200.McCurdy DT, Palmer BM, Maughan DW, LeWinter MM. Myocardial cross-bridge kinetics in transition to failure in Dahl salt-sensitive rats. Am J Physiol Heart Circ Physiol 281: H1390–H1396, 2001. [DOI] [PubMed] [Google Scholar]
- 201.McDonough AA, Azuma KK, Lescale-Matys L, Tang MJ, Nakhoul F, Hensley CB, and Komatsu Y. Physiologic rationale for multiple sodium pump isoforms. Differential regulation of alpha 1 vs alpha 2 by ionic stimuli. Ann NY Acad Sci 671: 156–168, 1992. [DOI] [PubMed] [Google Scholar]
- 202.McDonough AA, Magyar CE, Komatsu Y. Expression of Na+-K+-ATPase α- and β-subunits along rat nephron: isoform specificity and response to hypokalemia. Am J Physiol Cell Physiol 267: C901–C908, 1994. [DOI] [PubMed] [Google Scholar]
- 203.McGrail KM, Phillips JM, Sweadner KJ. Immunofluorescent localization of three Na,K-ATPase isozymes in the rat central nervous system: both neurons and glia can express more than one Na,K-ATPase. J Neurosci 11: 381–391, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.McKenzie AG. The rise and fall of strophanthin. In: The History of Anesthesia, edited by Diz JC, Franco A, Bacon DR, Rupreht J, Alvarez J. Amsterdam: Elsevier, 2002, p. 95–100. [Google Scholar]
- 205.Menezes JC, Troster EJ, Dichtchekenian V. Digoxin antibody decreases natriuresis and diuresis in cerebral hemorrhage. Intensive Care Med 29: 2291–2296, 2003. doi: 10.1007/s00134-003-1955-0. [DOI] [PubMed] [Google Scholar]
- 206.Mizelle HL, Montani JP, Hester RL, Didlake RH, Hall JE. Role of pressure natriuresis in long-term control of renal electrolyte excretion. Hypertension 22: 102–110, 1993. doi: 10.1161/01.HYP.22.1.102. [DOI] [PubMed] [Google Scholar]
- 207.Montani JP, Van Vliet BN. Understanding the contribution of Guyton’s large circulatory model to long-term control of arterial pressure. Exp Physiol 94: 382–388, 2009. doi: 10.1113/expphysiol.2008.043299. [DOI] [PubMed] [Google Scholar]
- 208.Morrill GA, Kostellow AB, Liu L, Gupta RK, Askari A. Evolution of the α-subunit of Na/K-ATPase from Paramecium to Homo sapiens: invariance of transmembrane helix topology. J Mol Evol 82: 183–198, 2016. doi: 10.1007/s00239-016-9732-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Morris RC Jr, Schmidlin O, Sebastian A, Tanaka M, Kurtz TW. Vasodysfunction that involves renal vasodysfunction, not abnormally increased renal retention of sodium, accounts for the initiation of salt-induced hypertension. Circulation 133: 881–893, 2016. doi: 10.1161/CIRCULATIONAHA.115.017923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Moseley AE, Williams MT, Schaefer TL, Bohanan CS, Neumann JC, Behbehani MM, Vorhees CV, Lingrel JB. Deficiency in Na,K-ATPase α isoform genes alters spatial learning, motor activity, and anxiety in mice. J Neurosci 27: 616–626, 2007. doi: 10.1523/JNEUROSCI.4464-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Murphy RA. Contraction of muscle cells. In: Physiology (2nd ed.), edited by Berne RM, Levy MN. St. Louis, MO: Mosby, 1988, p. 315–342. [Google Scholar]
- 212.Murphy RA. Smooth muscle. In: Physiology (3rd ed.), edited by Berne RM, Levy MN. St. Louis, MO: Mosby, 1993, p 309–324. [Google Scholar]
- 213.Nesher M, Dvela M, Igbokwe VU, Rosen H, Lichtstein D. Physiological roles of endogenous ouabain in normal rats. Am J Physiol Heart Circ Physiol 297: H2026–H2034, 2009. 10.1152/ajpheart.00734.2009. . [DOI] [PubMed] [Google Scholar]
- 214.Nesher M, Shpolansky U, Viola N, Dvela M, Buzaglo N, Cohen Ben-Ami H, Rosen H, Lichtstein D. Ouabain attenuates cardiotoxicity induced by other cardiac steroids. Br J Pharmacol 160: 346–354, 2010. doi: 10.1111/j.1476-5381.2010.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Neuwinger HD. African Ethnobotany: Poisons and Drugs. Weinheim, Germany: Chapman & Hall, 1996. [Google Scholar]
- 216.Nguyen AN, Jansson K, Sánchez G, Sharma M, Reif GA, Wallace DP, Blanco G. Ouabain activates the Na-K-ATPase signalosome to induce autosomal dominant polycystic kidney disease cell proliferation. Am J Physiol Renal Physiol 301: F897–F906, 2011. doi: 10.1152/ajprenal.00095.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Nguyen AN, Wallace DP, Blanco G. Ouabain binds with high affinity to the Na,K-ATPase in human polycystic kidney cells and induces extracellular signal-regulated kinase activation and cell proliferation. J Am Soc Nephrol 18: 46–57, 2007. doi: 10.1681/ASN.2006010086. [DOI] [PubMed] [Google Scholar]
- 218.O’Brien WJ, Lingrel JB, Wallick ET. Ouabain binding kinetics of the rat alpha two and alpha three isoforms of the sodium-potassium adenosine triphosphate. Arch Biochem Biophys 310: 32–39, 1994. doi: 10.1006/abbi.1994.1136. [DOI] [PubMed] [Google Scholar]
- 219.O’Rourke B, Kass DA, Tomaselli GF, Kääb S, Tunin R, Marbán E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ Res 84: 562–570, 1999. doi: 10.1161/01.RES.84.5.562. [DOI] [PubMed] [Google Scholar]
- 220.Oates HF, Graham RM, Stoker LM, Stokes GS. Haemodynamic effects of prazosin. Arch Int Pharmacodyn Ther 224: 239–247, 1976. [PubMed] [Google Scholar]
- 221.Oh YS, Galis ZS. Anatomy of success: the top 100 cited scientific reports focused on hypertension research. Hypertension 63: 641–647, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Okamura H, Yasuhara JC, Fambrough DM, Takeyasu K. P-type ATPases in Caenorhabditis and Drosophila: implications for evolution of the P-type ATPase subunit families with special reference to the Na,K-ATPase and H,K-ATPase subgroup. J Membr Biol 191: 13–24, 2003. doi: 10.1007/s00232-002-1041-5. [DOI] [PubMed] [Google Scholar]
- 223.Oparil S. Pathogenesis of ventricular hypertrophy. J Am Coll Cardiol 5, Suppl: 57B–65B, 1985. doi: 10.1016/S0735-1097(85)80528-3. [DOI] [PubMed] [Google Scholar]
- 224.Orellana AM, Kinoshita PF, Leite JA, Kawamoto EM, Scavone C. Cardiotonic steroids as modulators of neuroinflammation. Front Endocrinol (Lausanne) 7: 10, 2016. doi: 10.3389/fendo.2016.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Osborn JW, Averina VA, Fink GD. Current computational models do not reveal the importance of the nervous system in long-term control of arterial pressure. Exp Physiol 94: 389–396, 2009. doi: 10.1113/expphysiol.2008.043281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Osborn JW, Fink GD, Kuroki MT. Neural mechanisms of angiotensin II-salt hypertension: implications for therapies targeting neural control of the splanchnic circulation. Curr Hypertens Rep 13: 221–228, 2011. doi: 10.1007/s11906-011-0188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Osborn JW, Fink GD, Sved AF, Toney GM, Raizada MK. Circulating angiotensin II and dietary salt: converging signals for neurogenic hypertension. Curr Hypertens Rep 9: 228–235, 2007. doi: 10.1007/s11906-007-0041-3. [DOI] [PubMed] [Google Scholar]
- 228.Oshiro N, Dostanic-Larson I, Neumann JC, Lingrel JB. The ouabain-binding site of the α2 isoform of Na,K-ATPase plays a role in blood pressure regulation during pregnancy. Am J Hypertens 23: 1279–1285, 2010. doi: 10.1038/ajh.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Packer M. End of the oldest controversy in medicine. Are we ready to conclude the debate on digitalis? N Engl J Med 336: 575–576, 1997. doi: 10.1056/NEJM199702203360809. [DOI] [PubMed] [Google Scholar]
- 230.Page IH. The mosaic theory 32 years later. Hypertension 4: 177, 1982. doi: 10.1161/01.HYP.4.2.177. [DOI] [PubMed] [Google Scholar]
- 231.Peng M, Huang L, Xie Z, Huang WH, Askari A. Partial inhibition of Na+/K+-ATPase by ouabain induces the Ca2+-dependent expressions of early-response genes in cardiac myocytes. J Biol Chem 271: 10372–10378, 1996. doi: 10.1074/jbc.271.17.10372. [DOI] [PubMed] [Google Scholar]
- 232.Perrin A, Brasmes B, Chambaz EM, Defaye G. Bovine adrenocortical cells in culture synthesize an ouabain-like compound. Mol Cell Endocrinol 126: 7–15, 1997. doi: 10.1016/S0303-7207(96)03964-0. [DOI] [PubMed] [Google Scholar]
- 233.Pierdomenico SD, Bucci A, Manunta P, Rivera R, Ferrandi M, Hamlyn JM, Lapenna D, Cuccurullo F, Mezzetti A. Endogenous ouabain and hemodynamic and left ventricular geometric patterns in essential hypertension. Am J Hypertens 14: 44–50, 2001. doi: 10.1016/S0895-7061(00)01225-5. [DOI] [PubMed] [Google Scholar]
- 234.Pitzalis MV, Hamlyn JM, Messaggio E, Iacoviello M, Forleo C, Romito R, de Tommasi E, Rizzon P, Bianchi G, Manunta P. Independent and incremental prognostic value of endogenous ouabain in idiopathic dilated cardiomyopathy. Eur J Heart Fail 8: 179–186, 2006. doi: 10.1016/j.ejheart.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 235.Poburko D, Fameli N, Kuo KH, van Breemen C. Ca2+ signaling in smooth muscle: TRPC6, NCX and LNats in nanodomains. Channels (Austin) 2: 10–12, 2008. doi: 10.4161/chan.2.1.6053. [DOI] [PubMed] [Google Scholar]
- 236.Popovich MI, Kobets VA, Kostin SI, Kapelko VI. Myocardial alterations induced by prolonged noradrenaline administration in various doses. Clin Cardiol 15: 660–665, 1992. doi: 10.1002/clc.4960150909. [DOI] [PubMed] [Google Scholar]
- 237.Pulina MV, Zulian A, Baryshnikov SG, Linde CI, Karashima E, Hamlyn JM, Ferrari P, Blaustein MP, Golovina VA. Cross talk between plasma membrane Na+/Ca2+ exchanger-1 and TRPC/Orai-containing channels: key players in arterial hypertension. Adv Exp Med Biol 961: 365–374, 2013. doi: 10.1007/978-1-4614-4756-6_31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Pulina MV, Zulian A, Berra-Romani R, Beskina O, Mazzocco-Spezzia A, Baryshnikov SG, Papparella I, Hamlyn JM, Blaustein MP, Golovina VA. Upregulation of Na+ and Ca2+ transporters in arterial smooth muscle from ouabain-induced hypertensive rats. Am J Physiol Heart Circ Physiol 298: H263–H274, 2010. doi: 10.1152/ajpheart.00784.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Pullen MA, Brooks DP, Edwards RM. Characterization of the neutralizing activity of digoxin-specific Fab toward ouabain-like steroids. J Pharmacol Exp Ther 310: 319–325, 2004. doi: 10.1124/jpet.104.065250. [DOI] [PubMed] [Google Scholar]
- 240.Pullen MA, Harpel MR, Danoff TM, Brooks DP. Comparison of non-digitalis binding properties of digoxin-specific Fabs using direct binding methods. J Immunol Methods 336: 235–241, 2008. doi: 10.1016/j.jim.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 241.Quadri L, Bianchi G, Cerri A, Fedrizzi G, Ferrari P, Gobbini M, Melloni P, Sputore S, Torri M. 17 β-(3-furyl)-5 β-androstane-3 β, 14 β, 17 α-triol (PST 2238). A very potent antihypertensive agent with a novel mechanism of action. J Med Chem 40: 1561–1564, 1997. doi: 10.1021/jm970162e. [DOI] [PubMed] [Google Scholar]
- 242.Quintas LE, Pierre SV, Liu L, Bai Y, Liu X, Xie ZJ. Alterations of Na+/K+-ATPase function in caveolin-1 knockout cardiac fibroblasts. J Mol Cell Cardiol 49: 525–531, 2010. doi: 10.1016/j.yjmcc.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Radzyukevich TL, Lingrel JB, Heiny JA. The cardiac glycoside binding site on the Na,K-ATPase α2 isoform plays a role in the dynamic regulation of active transport in skeletal muscle. Proc Natl Acad Sci USA 106: 2565–2570, 2009. doi: 10.1073/pnas.0804150106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Radzyukevich TL, Neumann JC, Rindler TN, Oshiro N, Goldhamer DJ, Lingrel JB, Heiny JA. Tissue-specific role of the Na,K-ATPase α2 isozyme in skeletal muscle. J Biol Chem 288: 1226–1237, 2013. doi: 10.1074/jbc.M112.424663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Reuter H, Blaustein MP, Haeusler G. Na-Ca exchange and tension development in arterial smooth muscle. Philos Trans R Soc Lond B Biol Sci 265: 87–94, 1973. doi: 10.1098/rstb.1973.0011. [DOI] [PubMed] [Google Scholar]
- 246.Reuter H, Seitz N. The dependence of calcium efflux from cardiac muscle on temperature and external ion composition. J Physiol 195: 451–470, 1968. doi: 10.1113/jphysiol.1968.sp008467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Rietbrock N, Woodcock BG. Pharmakokinetics of digoxin and derivatives. In: Cardiac Glycosides Part II: Pharmakokinetics and Clinical Pharmacology , edited by Greeff K. Berlin: Springer-Verlag, 1981, p. 31–56. doi: 10.1007/978-3-642-68166-0_2. [DOI] [Google Scholar]
- 248.Rindler TN, Lasko VM, Nieman ML, Okada M, Lorenz JN, Lingrel JB. Knockout of the Na,K-ATPase α2-isoform in cardiac myocytes delays pressure overload-induced cardiac dysfunction. Am J Physiol Heart Circ Physiol 304: H1147–H1158, 2013. doi: 10.1152/ajpheart.00594.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Rodriguez JS, Velez Rueda JO, Salas M, Becerra R, Di Carlo MN, Said M, Vittone L, Rinaldi G, Portiansky EL, Mundiña-Weilenmann C, Palomeque J, Mattiazzi A. Increased Na+/Ca2+ exchanger expression/activity constitutes a point of inflection in the progression to heart failure of hypertensive rats. PLoS One 9: e96400, 2014. doi: 10.1371/journal.pone.0096400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Rosner MH, Okusa MD. Acute kidney injury associated with cardiac surgery. Clin J Am Soc Nephrol 1: 19–32, 2006. doi: 10.2215/CJN.00240605. [DOI] [PubMed] [Google Scholar]
- 251.Rosner MH, Portilla D, Okusa MD. Cardiac surgery as a cause of acute kidney injury: pathogenesis and potential therapies. J Intensive Care Med 23: 3–18, 2008. doi: 10.1177/0885066607309998. [DOI] [PubMed] [Google Scholar]
- 252.Rossi G, Manunta P, Hamlyn JM, Pavan E, De Toni R, Semplicini A, Pessina AC. Immunoreactive endogenous ouabain in primary aldosteronism and essential hypertension: relationship with plasma renin, aldosterone and blood pressure levels. J Hypertens 13: 1181–1191, 1995. doi: 10.1097/00004872-199510000-00013. [DOI] [PubMed] [Google Scholar]
- 253.Russell JM, Blaustein MP. Calcium efflux from barnacle muscle fibers. Dependence on external cations. J Gen Physiol 63: 144–167, 1974. doi: 10.1085/jgp.63.2.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Schaefer TL, Lingrel JB, Moseley AE, Vorhees CV, Williams MT. Targeted mutations in the Na,K-ATPase α 2 isoform confer ouabain resistance and result in abnormal behavior in mice. Synapse 65: 520–531, 2011. doi: 10.1002/syn.20870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Schatzmann HJ. Herzglykoside als Hemmstoffe für den aktiven Kalium- und Natriumtransport durch die Erythrocytenmembran [Cardiac glycosides as inhibitors of active potassium and sodium transport by erythrocyte membrane] Helv Physiol Pharmacol Acta 11: 346–354, 1953. [PubMed] [Google Scholar]
- 256.Schneider R, Wray V, Nimtz M, Lehmann WD, Kirch U, Antolovic R, Schoner W. Bovine adrenals contain, in addition to ouabain, a second inhibitor of the sodium pump. J Biol Chem 273: 784–792, 1998. doi: 10.1074/jbc.273.2.784. [DOI] [PubMed] [Google Scholar]
- 257.Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am J Physiol Cell Physiol 293: C509–C536, 2007. doi: 10.1152/ajpcell.00098.2007. [DOI] [PubMed] [Google Scholar]
- 258.Shimada T, Nagasaka Y, Ishibashi Y, Tsukihashi H, Murakami Y, Sano K, Tanabe K, Hiroyuki Y, Murakami R, Morioka S. A probable relationship between an endogenous digitalis-like substance and concentric cardiac hypertrophy in primary aldosteronism. Intern Med 38: 655–659, 1999. doi: 10.2169/internalmedicine.38.655. [DOI] [PubMed] [Google Scholar]
- 259.Shull GE, Schwartz A, Lingrel JB. Amino-acid sequence of the catalytic subunit of the (Na+ + K+)ATPase deduced from a complementary DNA. Nature 316: 691–695, 1985. doi: 10.1038/316691a0. [DOI] [PubMed] [Google Scholar]
- 260.Shull MM, Lingrel JB. Multiple genes encode the human Na+,K+-ATPase catalytic subunit. Proc Natl Acad Sci USA 84: 4039–4043, 1987. doi: 10.1073/pnas.84.12.4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Sibarov DA, Bolshakov AE, Abushik PA, Krivoi II, Antonov SM. Na+,K+-ATPase functionally interacts with the plasma membrane Na+,Ca2+ exchanger to prevent Ca2+ overload and neuronal apoptosis in excitotoxic stress. J Pharmacol Exp Ther 343: 596–607, 2012. doi: 10.1124/jpet.112.198341. [DOI] [PubMed] [Google Scholar]
- 262.Simonini M, Lanzani C, Bignami E, Casamassima N, Frati E, Meroni R, Messaggio E, Alfieri O, Hamlyn J, Body SC, Collard CD, Zangrillo A, Manunta P, Body SC, Daniel Muehlschlegel J, Shernan SK, Fox AA, David Collard C; CABG Genomics Investigators . A new clinical multivariable model that predicts postoperative acute kidney injury: impact of endogenous ouabain. Nephrol Dial Transplant 29: 1696–1701, 2014. doi: 10.1093/ndt/gfu200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Simonini M, Pozzoli S, Bignami E, Casamassima N, Messaggio E, Lanzani C, Frati E, Botticelli IM, Rotatori F, Alfieri O, Zangrillo A, Manunta P. Endogenous ouabain: an old cardiotonic steroid as a new biomarker of heart failure and a predictor of mortality after cardiac surgery. BioMed Res Int 2015: 714793, 2015. doi: 10.1155/2015/714793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Skou JC. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta 23: 394–401, 1957. doi: 10.1016/0006-3002(57)90343-8. [DOI] [PubMed] [Google Scholar]
- 265.Somlyo AP, Broderick R, Somlyo AV. Calcium and sodium in vascular smooth muscle. Ann NY Acad Sci 488: 228–239, 1986. doi: 10.1111/j.1749-6632.1986.tb46561.x. [DOI] [PubMed] [Google Scholar]
- 266.Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature 372: 231–236, 1994. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- 267.Song H, Karashima E, Hamlyn JM, Blaustein MP. Ouabain-digoxin antagonism in rat arteries and neurones. J Physiol 592: 941–969, 2014. doi: 10.1113/jphysiol.2013.266866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Sophocleous A, Elmatzoglou I, Souvatzoglou A. Circulating endogenous digitalis-like factor(s) (EDLF) in man is derived from the adrenals and its secretion is ACTH-dependent. J Endocrinol Invest 26: 668–674, 2003. doi: 10.1007/BF03347027. [DOI] [PubMed] [Google Scholar]
- 269.Staessen JA, Thijs L, Stolarz-Skrzypek K, Bacchieri A, Barton J, Espositi ED, de Leeuw PW, Dłużniewski M, Glorioso N, Januszewicz A, Manunta P, Milyagin V, Nikitin Y, Souček M, Lanzani C, Citterio L, Timio M, Tykarski A, Ferrari P, Valentini G, Kawecka-Jaszcz K, Bianchi G. Main results of the ouabain and adducin for Specific Intervention on Sodium in Hypertension Trial (OASIS-HT): a randomized placebo-controlled phase-2 dose-finding study of rostafuroxin. Trials 12: 13, 2011. doi: 10.1186/1745-6215-12-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Stella P, Manunta P, Mallamaci F, Melandri M, Spotti D, Tripepi G, Hamlyn JM, Malatino LS, Bianchi G, Zoccali C. Endogenous ouabain and cardiomyopathy in dialysis patients. J Intern Med 263: 274–280, 2008. doi: 10.1111/j.1365-2796.2007.01883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Studer R, Reinecke H, Bilger J, Eschenhagen T, Böhm M, Hasenfuss G, Just H, Holtz J, Drexler H. Gene expression of the cardiac Na+-Ca2+ exchanger in end-stage human heart failure. Circ Res 75: 443–453, 1994. doi: 10.1161/01.RES.75.3.443. [DOI] [PubMed] [Google Scholar]
- 272.Sweeney G, Klip A. Regulation of the Na+/K+-ATPase by insulin: why and how? Mol Cell Biochem 182: 121–133, 1998. doi: 10.1023/A:1006805510749. [DOI] [PubMed] [Google Scholar]
- 273.Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev 79: 215–262, 1999. [DOI] [PubMed] [Google Scholar]
- 274.Szent-Gyorgi A. Chemical Physiology of Contraction in Body and Heart Muscle. New York: Academic, 1953. [Google Scholar]
- 275.Takahashi H, Matsuzawa M, Okabayashi H, Suga K, Ikegaki I, Yoshimura M, Ijichi H, Okamura H, Murakami S, Ibata Y. Evidence for a digitalis-like substance in the hypothalamopituitary axis in rats: implications in the central cardiovascular regulation associated with an excess intake of sodium. Jpn Circ J 51: 1199–1207, 1987. doi: 10.1253/jcj.51.1199. [DOI] [PubMed] [Google Scholar]
- 276.Tamura M, Harris TM, Phillips D, Blair IA, Wang YF, Hellerqvist CG, Lam SK, Inagami T. Identification of two cardiac glycosides as Na+-pump inhibitors in rat urine and diet. J Biol Chem 269: 11972–11979, 1994. [PubMed] [Google Scholar]
- 277.Tamura M, Konishi F, Sakakibara M, Inagami T. Large scale purification of an endogenous Na+/K+-pump inhibitor from bovine adrenal glands. In: The Sodium Pump: Structure, Mechanism, Hormonal Control and its Role in Disease, edited by Bamberg E, Schoner W. Darmstadt: Steinkopf, 1994, p. 763–766. doi: 10.1007/978-3-642-72511-1_137. [DOI] [Google Scholar]
- 278.Tamura M, Lam TT, Inagami T. Specific endogenous Na, K-ATPase inhibitor purified from bovine adrenal. Biochem Biophys Res Commun 149: 468–474, 1987. doi: 10.1016/0006-291X(87)90391-3. [DOI] [PubMed] [Google Scholar]
- 279.Tentori S, Messaggio E, Brioni E, Casamassima N, Simonini M, Zagato L, Hamlyn JM, Manunta P, Lanzani C. Endogenous ouabain and aldosterone are coelevated in the circulation of patients with essential hypertension. J Hypertens 34: 2074–2080, 2016. doi: 10.1097/HJH.0000000000001042. [DOI] [PubMed] [Google Scholar]
- 280.ter Keurs HE. Heart failure and Starling’s Law of the heart. Can J Cardiol 12: 1047–1057, 1996. [PubMed] [Google Scholar]
- 281.Thabet R, Rouault JD, Ayadi H, Leignel V. Structural analysis of the α subunit of Na+/K+ ATPase genes in invertebrates. Comp Biochem Physiol B Biochem Mol Biol 196-197: 11–18, 2016. doi: 10.1016/j.cbpb.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 282.Tymiak AA, Norman JA, Bolgar M, DiDonato GC, Lee H, Parker WL, Lo LC, Berova N, Nakanishi K, Haber E. Physicochemical characterization of a ouabain isomer isolated from bovine hypothalamus. Proc Natl Acad Sci USA 90: 8189–8193, 1993. doi: 10.1073/pnas.90.17.8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Tyrer JH. A comparison of the direct peripheral actions upon systemic venous pressure of k-strophanthosid, digoxin and ouabain. Q J Exp Physiol Cogn Med Sci 38: 177–184, 1953. [DOI] [PubMed] [Google Scholar]
- 284.Valvassori SS, Resende WR, Lopes-Borges J, Mariot E, Dal-Pont GC, Vitto MF, Luz G, de Souza CT, Quevedo J. Effects of mood stabilizers on oxidative stress-induced cell death signaling pathways in the brains of rats subjected to the ouabain-induced animal model of mania: mood stabilizers exert protective effects against ouabain-induced activation of the cell death pathway. J Psychiatr Res 65: 63–70, 2015. doi: 10.1016/j.jpsychires.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 285.van Breemen C, Lukeman S, Leijten P, Yamamoto H, Loutzenhiser R. The role of superficial SR in modulating force development induced by Ca entry into arterial smooth muscle. J Cardiovasc Pharmacol 8, Suppl 8: S111–S116, 1986. doi: 10.1097/00005344-198600088-00023. [DOI] [PubMed] [Google Scholar]
- 286.Van Huysse JW, Dostanic I, Lingrel JB, Hou X, Wu H. Hypertension from chronic central sodium chloride in mice is mediated by the ouabain-binding site on the Na,K-ATPase α2-isoform. Am J Physiol Heart Circ Physiol 301: H2147–H2153, 2011. doi: 10.1152/ajpheart.01216.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Vemuri R, Longoni S, Philipson KD. Ouabain treatment of cardiac cells induces enhanced Na+-Ca2+ exchange activity. Am J Physiol Cell Physiol 256: C1273–C1276, 1989. [DOI] [PubMed] [Google Scholar]
- 288.Villa L, Buono R, Ferrandi M, Molinari I, Benigni F, Bettiga A, Colciago G, Ikehata M, Messaggio E, Rastaldi MP, Montorsi F, Salonia A, Manunta P. Ouabain contributes to kidney damage in a rat model of renal ischemia-reperfusion injury. Int J Mol Sci 17: 17, 2016. doi: 10.3390/ijms17101728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Vogeser M, Baecher S. Correspondence concerning the article “No endogenous ouabain is detectable in human plasma by ultra-sensitive UPLC-MS/MS.” (Clin Chim Acta 2014;431:87–92 by S. Baecher et al.). Clin Chim Acta 448: 250–251, 2015. doi: 10.1016/j.cca.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 290.Vogt M, Motz W, Strauer BE. Long-term treatment in arterial hypertension for protecting hypertrophic myocardium. Basic Res Cardiol 86, Suppl 3: 223–233, 1991. [DOI] [PubMed] [Google Scholar]
- 291.Walker JM, Lourie EM, Burn JH. The effects of digoxin and ouabain on the heart-lung preparation of the dog. Br J Pharmacol Chemother 5: 306–314, 1950. doi: 10.1111/j.1476-5381.1950.tb01015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Wang H, Haas M, Liang M, Cai T, Tian J, Li S, Xie Z. Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J Biol Chem 279: 17250–17259, 2004. doi: 10.1074/jbc.M313239200. [DOI] [PubMed] [Google Scholar]
- 293.Wang H, Huang BS, Leenen FH. Brain sodium channels and ouabainlike compounds mediate central aldosterone-induced hypertension. Am J Physiol Heart Circ Physiol 285: H2516–H2523, 2003. doi: 10.1152/ajpheart.00299.2003. [DOI] [PubMed] [Google Scholar]
- 294.Wang HW, Huang BS, Chen A, Ahmad M, White RA, Leenen FH. Role of brain aldosterone and mineralocorticoid receptors in aldosterone-salt hypertension in rats. Neuroscience 314: 90–105, 2016. doi: 10.1016/j.neuroscience.2015.11.055. [DOI] [PubMed] [Google Scholar]
- 295.Wansapura AN, Lasko V, Xie Z, Fedorova OV, Bagrov AY, Lingrel JB, Lorenz JN. Marinobufagenin enhances cardiac contractility in mice with ouabain-sensitive alpha1 Na+-K+-ATPase. Am J Physiol Heart Circ Physiol 296: H1833–H1839, 2009. doi: 10.1152/ajpheart.00285.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Wansapura AN, Lasko VM, Lingrel JB, Lorenz JN. Mice expressing ouabain-sensitive α1-Na,K-ATPase have increased susceptibility to pressure overload-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol 300: H347–H355, 2011. doi: 10.1152/ajpheart.00625.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Weber KT, Clark WA, Janicki JS, Shroff SG. Physiologic versus pathologic hypertrophy and the pressure-overloaded myocardium. J Cardiovasc Pharmacol 10, Suppl 6: S37–S50, 1987. doi: 10.1097/00005344-198700106-00006. [DOI] [PubMed] [Google Scholar]
- 298.Wendt-Gallitelli MF, Voigt T, Isenberg G. Microheterogeneity of subsarcolemmal sodium gradients. Electron probe microanalysis in guinea-pig ventricular myocytes. J Physiol 472: 33–44, 1993. doi: 10.1113/jphysiol.1993.sp019934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Werdan K, Wagenknecht B, Zwissler B, Brown L, Krawietz W, Erdmann E. Cardiac glycoside receptors in cultured heart cells—II. Characterization of a high affinity and a low affinity binding site in heart muscle cells from neonatal rats. Biochem Pharmacol 33: 1873–1886, 1984. doi: 10.1016/0006-2952(84)90542-2. [DOI] [PubMed] [Google Scholar]
- 300.Westcott KV, Huang BS, Leenen FH. Brain renin-angiotensin-aldosterone system and ventricular remodeling after myocardial infarct: a review. Can J Physiol Pharmacol 87: 979–988, 2009. doi: 10.1139/Y09-067. [DOI] [PubMed] [Google Scholar]
- 301.Withering W. An Account of the Foxglove and Some of Its Medical Uses. Birmingham, UK: G.G.J. and J. Robinson, 1785, p. 207. [Google Scholar]
- 302.Wood AJ, Phelan EL, Simpson FO. Cardiovascular effects of prazosin in normotensive and genetically hypertensive rats. Clin Exp Pharmacol Physiol 2: 297–304, 1975. doi: 10.1111/j.1440-1681.1975.tb01836.x. [DOI] [PubMed] [Google Scholar]
- 303.Wu J, Akkuratov EE, Bai Y, Gaskill CM, Askari A, Liu L. Cell signaling associated with Na+/K+-ATPase: activation of phosphatidylinositide 3-kinase IA/Akt by ouabain is independent of Src. Biochemistry 52: 9059–9067, 2013. doi: 10.1021/bi4011804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Xie J, Ye Q, Cui X, Madan N, Yi Q, Pierre SV, Xie Z. Expression of rat Na-K-ATPase α2 enables ion pumping but not ouabain-induced signaling in α1-deficient porcine renal epithelial cells. Am J Physiol Cell Physiol 309: C373–C382, 2015. doi: 10.1152/ajpcell.00103.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Xie Z, Askari A. Na+/K+-ATPase as a signal transducer. Eur J Biochem 269: 2434–2439, 2002. doi: 10.1046/j.1432-1033.2002.02910.x. [DOI] [PubMed] [Google Scholar]
- 306.Yan X, Liang F, Li D, Zheng J. Ouabain elicits human glioblastoma cells apoptosis by generating reactive oxygen species in ERK-p66SHC-dependent pathway. Mol Cell Biochem 398: 95–104, 2015. doi: 10.1007/s11010-014-2208-y. [DOI] [PubMed] [Google Scholar]
- 307.Yan Y, Shapiro AP, Haller S, Katragadda V, Liu L, Tian J, Basrur V, Malhotra D, Xie ZJ, Abraham NG, Shapiro JI, Liu J. Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. J Biol Chem 288: 34249–34258, 2013. doi: 10.1074/jbc.M113.461020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Yatime L, Laursen M, Morth JP, Esmann M, Nissen P, Fedosova NU. Structural insights into the high affinity binding of cardiotonic steroids to the Na+,K+-ATPase. J Struct Biol 174: 296–306, 2011. doi: 10.1016/j.jsb.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 309.Yazaki Y, Tsuchimochi H, Kurabayashi M, Komuro I. Molecular adaptation to pressure overload in human and rat hearts. J Mol Cell Cardiol 21, Suppl 5: 91–101, 1989. doi: 10.1016/0022-2828(89)90775-X. [DOI] [PubMed] [Google Scholar]
- 310.Yoda A. Association and dissociation rate constants of the complexes between various cardiac monoglycosides and Na, K-ATPase. Ann NY Acad Sci 242: 598–616, 1974. doi: 10.1111/j.1749-6632.1974.tb19120.x. [DOI] [PubMed] [Google Scholar]
- 311.Yoda A. Structure-activity relationships of cardiotonic steroids for the inhibition of sodium- and potassium-dependent adenosine triphosphatase. I. Dissociation rate constants of various enzyme-cardiac glycoside complexes formed in the presence of magnesium and phosphate. Mol Pharmacol 9: 51–60, 1973. [PubMed] [Google Scholar]
- 312.Yoda A, Yoda S, Sarrif AM. Structure-activity relationships of cardiotonic steroids for the inhibition of sodium- and potassium-dependent adenosine triphosphatase. II. Association rate constants of various enzyme-cardiac glycoside complexes. Mol Pharmacol 9: 766–773, 1973. [PubMed] [Google Scholar]
- 313.Yu Y, Wei SG, Zhang ZH, Gomez-Sanchez E, Weiss RM, Felder RB. Does aldosterone upregulate the brain renin-angiotensin system in rats with heart failure? Hypertension 51: 727–733, 2008. doi: 10.1161/HYPERTENSIONAHA.107.099796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Yuan CM, Manunta P, Hamlyn JM, Chen S, Bohen E, Yeun J, Haddy FJ, Pamnani MB. Long-term ouabain administration produces hypertension in rats. Hypertension 22: 178–187, 1993. doi: 10.1161/01.HYP.22.2.178. [DOI] [PubMed] [Google Scholar]
- 315.Zacharia J, Mauban JR, Raina H, Fisher SA, Wier WG. High vascular tone of mouse femoral arteries in vivo is determined by sympathetic nerve activity via α1A- and α1D-adrenoceptor subtypes. PLoS One 8: e65969, 2013. doi: 10.1371/journal.pone.0065969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Zanchetti A. Antihypertensive therapy: how to evaluate the benefits. Am J Cardiol 79: 3–8, 1997. doi: 10.1016/S0002-9149(97)00264-6. [DOI] [PubMed] [Google Scholar]
- 317.Zhang J, Hamlyn JM, Karashima E, Raina H, Mauban JR, Izuka M, Berra-Romani R, Zulian A, Wier WG, Blaustein MP. Low-dose ouabain constricts small arteries from ouabain-hypertensive rats: implications for sustained elevation of vascular resistance. Am J Physiol Heart Circ Physiol 297: H1140–H1150, 2009. doi: 10.1152/ajpheart.00436.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Zhang J, Lee MY, Cavalli M, Chen L, Berra-Romani R, Balke CW, Bianchi G, Ferrari P, Hamlyn JM, Iwamoto T, Lingrel JB, Matteson DR, Wier WG, Blaustein MP. Sodium pump α2 subunits control myogenic tone and blood pressure in mice. J Physiol 569: 243–256, 2005. doi: 10.1113/jphysiol.2005.091801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Zhang J, Ren C, Chen L, Navedo MF, Antos LK, Kinsey SP, Iwamoto T, Philipson KD, Kotlikoff MI, Santana LF, Wier WG, Matteson DR, Blaustein MP. Knockout of Na+/Ca2+ exchanger in smooth muscle attenuates vasoconstriction and L-type Ca2+ channel current and lowers blood pressure. Am J Physiol Heart Circ Physiol 298: H1472–H1483, 2010. doi: 10.1152/ajpheart.00964.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Zhang S, Malmersjö S, Li J, Ando H, Aizman O, Uhlén P, Mikoshiba K, Aperia A. Distinct role of the N-terminal tail of the Na,K-ATPase catalytic subunit as a signal transducer. J Biol Chem 281: 21954–21962, 2006. doi: 10.1074/jbc.M601578200. [DOI] [PubMed] [Google Scholar]
- 321.Zucker IH, Xiao L, Haack KK. The central renin-angiotensin system and sympathetic nerve activity in chronic heart failure. Clin Sci (Lond) 126: 695–706, 2014. doi: 10.1042/CS20130294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Zulian A, Baryshnikov SG, Linde CI, Hamlyn JM, Ferrari P, Golovina VA. Upregulation of Na+/Ca2+ exchanger and TRPC6 contributes to abnormal Ca2+ homeostasis in arterial smooth muscle cells from Milan hypertensive rats. Am J Physiol Heart Circ Physiol 299: H624–H633, 2010. doi: 10.1152/ajpheart.00356.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Zulian A, Linde CI, Pulina MV, Baryshnikov SG, Papparella I, Hamlyn JM, Golovina VA. Activation of c-SRC underlies the differential effects of ouabain and digoxin on Ca2+ signaling in arterial smooth muscle cells. Am J Physiol Cell Physiol 304: C324–C333, 2013. doi: 10.1152/ajpcell.00337.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]