

Abstract
Proteostasis, or protein homeostasis, encompasses the maintenance of the conformational and functional integrity of the proteome and involves an integrated network of cellular pathways. Molecular chaperones, such as the small heat shock proteins (sHsps), are key elements of the proteostasis network that have crucial roles in inhibiting the aggregation of misfolded proteins. Failure of the proteostasis network can lead to the accumulation of misfolded proteins into intracellular and extracellular deposits. Deposits containing fibrillar forms of α-synuclein (α-syn) are characteristic of neurodegenerative disorders including Parkinson's disease and dementia with Lewy bodies. Here we show that the sHsp Hsp27 (HSPB1) binds to α-syn fibrils, inhibiting fibril growth by preventing elongation. Using total internal reflection fluorescence (TIRF)–based imaging methods, we show that Hsp27 binds along the surface of α-syn fibrils, decreasing their hydrophobicity. Binding of Hsp27 also inhibits cytotoxicity of α-syn fibrils. Our results demonstrate that the ability of sHsps, such as Hsp27, to bind fibrils represents an important mechanism through which they may mitigate cellular toxicity associated with aberrant protein aggregation. Fibril binding may represent a generic mechanism by which chaperone-active sHsps interact with aggregation-prone proteins, highlighting the potential to target sHsp activity to prevent or disrupt the onset and progression of α-syn aggregation associated with α-synucleinopathies.
Keywords: alpha-synuclein, α-synuclein, amyloid, molecular chaperone, neurodegeneration, neurodegenerative disease, protein aggregation, small heat shock protein, sHsp, Lewy body dementia, Parkinson's disease, alpha-synucleinopathy, α-synucleinopathy
Introduction
Cells have an intricate network of protein quality control machinery that maintains proteostasis and hence the functionality of the proteome. The small heat shock molecular chaperone proteins (sHsps)3 are a crucial component of the proteostasis network as they are a first line of defense against protein aggregation (1, 2). The sHsps are defined by the presence of a conserved α-crystallin domain and typically have a relatively low monomeric mass (12–43 kDa) (3, 4). However, some mammalian sHsps, including αB-crystallin (αB-c) (HSPB5) and Hsp27 (HSPB1), form large polydisperse oligomeric assemblies that undergo rapid subunit exchange and display chaperone activity (5–8). Indeed, αB-c and Hsp27 are capable of inhibiting the aggregation of a wide range of model and disease-relevant amyloidogenic targets (9–15).
The sHsps have traditionally been classified as ATP-independent “holdases,” because they can recognize and bind misfolded or aggregation-prone protein intermediates to form stable high–molecular mass complexes (16–18). More recently, it has been demonstrated that some sHsps can prevent protein aggregation without the need to form stable, high-molecular mass complexes containing the aggregation-prone protein (11, 15). Instead, interactions with aggregation-prone proteins can be weak and transient, and do not result in stable complexes. Thus, rather than solely acting as holdases, the molecular mechanisms underlying the interactions between sHsps and aggregating target proteins are multifaceted and are likely to depend on the stability of the target protein (10, 11, 19).
The failure of the proteostasis network to prevent protein aggregation can lead to the deposition of insoluble proteins, often in the form of amyloid fibrils, into intracellular inclusions and extracellular plaques. These deposits are hallmarks of a range of neurodegenerative disorders, including Alzheimer's disease and Parkinson's disease (20, 21). In particular, inclusions, known as Lewy bodies, are detected within the neuronal tissue of patients with Parkinson's disease and dementia with Lewy bodies. These inclusions are composed primarily of the intrinsically disordered protein, α-synuclein (α-syn) (22). The aggregation of soluble proteins to form amyloid fibrils is well-established to involve a nucleation-dependent polymerization reaction (21, 23). The initial step is the formation of oligomers in a primary nucleation process, followed by growth and elongation of these species by the addition of monomers. It is increasingly evident that other secondary processes exist, including fragmentation and secondary nucleation on the surface of aggregates as they are formed (24).
Understanding the pathogenesis of α-syn requires detailed knowledge of the fundamental nature of each of the steps within the aggregation pathway, including primary and secondary nucleation processes. The soluble oligomer model of amyloid cytotoxicity suggests that mature fibrils are relatively inert end products of aggregation (21, 25, 26); however, the fibrils themselves can be a significant source of these small cytotoxic species. Thus, secondary processes such as catalysis of nucleation at the fibril surface and fibril fragmentation not only increase fibrillar load but can also directly increase the abundance of potentially cytotoxic oligomers (27). Fibril fragmentation has been highlighted as an important mechanism by which both disease-associated and model amyloid-forming proteins are cytotoxic (24, 28). In addition, fibrillar material released into the extracellular space may interact with neighboring cells and exert toxic effects (25, 26, 29, 30). As such, the formation of amyloid-rich inclusions in cells has been postulated to be a protective mechanism (31, 32), because the deposition of mature amyloid fibrils could sequester harmful small aggregates that would otherwise be cytotoxic.
In addition to α-syn, close to 300 other proteins have also been identified in Lewy bodies, including proteins that assist folding, trafficking, and degradation of proteins, as well as those involved in synaptic transmission and vesicular transport (33, 34). There remains debate as to whether these proteins are sequestered as a consequence of aggregation or if there is a physiological role for their presence. The recently described ability of the Hsp70 chaperone complex to bind and facilitate disassembly of α-syn fibrils (35) provides support for an active role of these chaperones within aggregates. Previous studies have reported that αB-c and Hsp27 (as well as other sHsps) are present in Lewy bodies (36–40). The presence of sHsps in Lewy bodies has often been regarded to be a consequence of their inability to prevent α-syn aggregation, because their chaperone activity may be overwhelmed when the rate of aggregation exceeds the capacity of the sHsps to supply chaperone-active subunits (15). Moreover, αB-c can interact with mature amyloid fibrils (41–44), and thus may play a specific functional role within inclusions. For example, by binding to fibrils formed by apolipoprotein C-II (apoC-II), αB-c has been shown to induce dissociation of monomeric units from fibril ends, inhibit fibril elongation and fragmentation, and cause the fibrils to entangle into larger deposits (43). However, the mechanism of this interaction and whether or not it provides a protective effect by inhibiting the cytotoxicity of the fibrils themselves is not yet known. Furthermore, it remains to be established if other sHsps, such as Hsp27, can bind fibrils, and thus whether fibril binding is likely to be a generic property of chaperone-active sHsps. In this work, we specifically address these issues by examining the ability of Hsp27 to interact with fibrillar forms of α-syn. Using a variety of techniques, including single aggregate visualization by enhancement (SAVE) imaging (45), we show that Hsp27 binds to α-syn fibrils and, in doing so, decreases their overall hydrophobicity and the cellular toxicity of exogenous, fibrillar α-syn.
Results
sHsps inhibit elongation of fibril fragments
To enable direct measurement of the elongation rate of small amyloid fibril fragments (seeds) (Fig. 1A), seeded aggregation assays were performed. Under these conditions, aggregation occurs primarily through monomer addition at fibril ends, as primary nucleation processes do not contribute significantly to the observed aggregation kinetics (46). Indeed, incubation of monomeric α-syn in the presence of 5% (w/w) seeds did not result in an observable lag phase typical of non-seeded aggregation (in which nucleation processes also occur), and was best described by a one-phase association function (Fig. 1B). Elongation was inhibited by the addition of Hsp27 and this effect was specific to the chaperone as no inhibition was seen when a non-chaperone control protein (BSA) was used at the same concentration (Fig. 1B). When the concentration of monomeric α-syn was increased in the presence of a constant concentration of seed fibrils (i.e. 2.5 μm), the rate of elongation and total fibril formation increased (Fig. 1C). By considering the linear portion of the elongation phase during which the increase in thioflavin T (ThT) fluorescence is directly proportional to fibril growth (in this case at times between 1 and 3 h) (Fig. 1D), the relationship between the concentration of monomeric α-syn and elongation rate was best described by two-step Michaelis-Menten behavior, as found previously (46) (Fig. 1G). However, the increase in the rate of elongation and total fibril formation was inhibited by the presence of Hsp27 (Fig. 1, E and F). In the presence of Hsp27 (50 μm), there was no significant increase in the rate of elongation at low concentrations of monomeric α-syn (up to 50 μm) (Fig. 1G). However, as the concentration of monomeric α-syn was increased (to 75 and 100 μm), such that the molar ratio of monomeric α-syn:Hsp27 exceeded 1:1, a small but significant increase in the rate of α-syn aggregation was observed. Importantly, in the presence of Hsp27, the elongation rate of the α-syn fibrils was found to no longer follow Michaelis-Menten kinetics (Fig. 1G). Together, these data suggest that the interaction between Hsp27 and monomeric α-syn is dependent on the rate of fibril elongation. However, these findings may also reflect an interaction between Hsp27 and the fibrillar seeds of α-syn, whereby Hsp27 is in direct competition with monomeric α-syn for access to fibril ends, thus inhibiting elongation of the fibril seeds. As the concentration of monomeric α-syn increases, it may outcompete Hsp27 for binding to fibril ends leading to the observed increase in the rate of elongation.
Figure 1.

Hsp27 inhibits the elongation of α-syn fibrils. A, amyloid seeds (left) and mature fibrils (right) of α-syn were imaged via transmission electron microscopy. Scale bars represent 100 nm. B, recombinant monomeric α-syn (50 μm) was incubated in 50 mm phosphate buffer (pH 7.4) in the absence or presence of 50 μm Hsp27 or the control protein BSA. After equilibration at 37 °C, α-syn seeds (2.5 μm monomer equivalent) were added to each sample containing monomeric α-syn and the elongation of these seeds was monitored via the change in ThT fluorescence at 490 nm over time. C–F, monomeric α-syn was incubated in 50 mm phosphate buffer (pH 7.4) at concentrations ranging from 10 to 100 μm in the absence (C) or presence (E) of 50 μm Hsp27. After equilibration at 37 °C, α-syn seeds (2.5 μm) were added to each sample and the elongation monitored via the change in ThT fluorescence at 490 nm over time. D and F, the data from 1–3 h (outlined in the red box in C and E) of incubation were fitted to a linear regression function. Data shown are mean ± S.E. of triplicate samples. G, the rate of elongation of the α-syn seeds, in the absence and presence of Hsp27, was calculated using values from these fits and correlated with the monomeric α-syn concentration. Data for α-syn alone were fitted to a Michaelis-Menten kinetic model. The data in C–F are representative of four independent repeats. The data in G are presented as the mean ± S.E. of these four repeats.
To explore further the possible interaction of Hsp27 with α-syn seed fibrils, the ratio of fibril seeds to monomeric α-syn was varied and Hsp27 added to the samples at a range of concentrations (Fig. 2). As expected, incubation of monomeric α-syn in the presence of 2.5% (w/w) α-syn seeds resulted in a characteristic aggregation profile with no observable lag phase (Fig. 2A). Addition of Hsp27 inhibited this aggregation, such that there was a concentration-dependent decrease in the rate and total amount of α-syn aggregation as the concentration of Hsp27 was increased (Fig. 2, A and B). By repeating this experiment using different concentrations of α-syn fibril seeds (from 1 to 10% (w/w) of the monomeric α-syn concentration) the relationship between the concentration of Hsp27 and subsequent α-syn fibril elongation rate was established (Fig. 2C). This relationship is best described by the single exponential decay function (Fig. 2C) whereby the half-maximal inhibition from this fit provides a measure of the ability of Hsp27 to inhibit fibril elongation at each ratio of seed to monomeric α-syn (Fig. 2D). There was a strong exponential correlation between the concentration of seed fibrils and the ability of Hsp27 to inhibit elongation, i.e. as the concentration of α-syn seed increased, more Hsp27 was required to inhibit fibril elongation. Taken together, these data therefore provide evidence that Hsp27 interacts directly with fibrillar α-syn.
Figure 2.

Hsp27 interacts with α-syn fibrils. Recombinant monomeric α-syn was incubated in 50 mm phosphate buffer (pH 7.4) at 50 μm in the absence or presence of Hsp27 at concentrations ranging from 0.1 to 10 μm. After equilibration at 37 °C, α-syn seeds were added at concentrations ranging from 0.5 to 5 μm (i.e. 1–10% (w/w) when expressed as a percentage of the soluble protein concentration) and elongation monitored via the change in ThT fluorescence at 490 nm over time. A, a representative trace is shown in the presence of 5% seeds. B, the linear portion (0–2.5 h; outlined in the red box in A) was fitted to a linear regression curve. C, the rate of elongation was calculated using values from this fit (i.e. slope of the line in B), which was correlated with the Hsp27 concentration at each seed ratio. These data were then fitted to a single exponential decay function. Inset shows a magnified view of the 1% (w/w) seed sample. D, the parameters of this fit were used to compare the chaperone efficacy of Hsp27 at each seed ratio, and the data fitted to a single exponential growth function. The data in A–C are representative of at least two independent experiments, and the data in D are reported as the mean ± S.E. of these repeats.
Hsp27 forms a stable complex with mature α-syn fibrils, and the interaction is mediated by the N and/or C termini
Although the interaction between aggregated α-syn and Hsp27 significantly influences the rate of α-syn aggregation in vitro, we were yet to establish the mechanism by which Hsp27 interacts with fibrillar α-syn. Sucrose centrifugation assays were therefore employed to investigate the ability of Hsp27 to form stable complexes with α-syn fibrils (Fig. 3). When a sample containing aggregated α-syn was applied to the top of the sucrose gradient, monomeric and small oligomeric α-syn forms were retained predominantly in the upper fractions, whereas fibrillar α-syn sedimented through the sucrose gradient and was detected in the lowest fraction (fraction 10) (Fig. 3A). In addition, some insoluble material remained trapped in the wells, characteristic of SDS-insoluble fibrillar aggregates (Fig. S1). When Hsp27 was incubated alone, it did not sediment and was localized in fractions 1–6 from the sucrose gradient, consistent with the large, polydisperse and oligomeric nature of wildtype Hsp27 under these conditions (Fig. 3B) (47). However, pre-incubation of Hsp27 with α-syn fibrils resulted in Hsp27 co-sedimenting with the fibrils and therefore being detected in the lowest fraction from the sucrose gradient (Fig. 3C).
Figure 3.

Hsp27 binds to α-syn fibrils, and this interaction is mediated by the N and/or C termini. α-Syn fibrils (75 μm) were incubated in the absence (A) or the presence of wildtype Hsp27 (C), Hsp273D (E), or Hsp27core (G) isotypes (15 μm). Wildtype Hsp27 (B), Hsp273D (D), and Hsp27core (F) isoforms, incubated in the absence of α-syn fibrils, are also included for comparison. Samples were layered on top of a 20% (w/v) sucrose cushion and centrifuged at 200,000 × g for 20 min. Sequential fractions were collected from the top of the cushion (top, fraction 1; bottom, fraction 10) and analyzed via SDS-PAGE.
To gain further insight into the mechanism by which Hsp27 binds to mature α-syn fibrils, an isoform of Hsp27 incorporating only the α-crystallin core domain (Hsp27core, i.e. a form of Hsp27 lacking the flanking N- and C-terminal domains) and an isoform mimicking a phosphorylated form of Hsp27 (S15D, S78D, S82D Hsp27; Hsp273D) were used in this fibril-binding assay. These variants were used as they both form only monomers and dimers under the experimental conditions used in this work, rather than large polydisperse oligomers such as those formed by wildtype Hsp27 (47, 48). Thus, these Hsp27 variants enabled us to test whether only large oligomeric forms of Hsp27 are capable of binding to α-syn fibrils. Importantly, both these Hsp27 variants have previously been shown to retain the ability to inhibit monomeric α-syn aggregation and therefore are chaperone active (15). Consistent with their existence as monomers and dimers under these experimental conditions, Hsp273D (Fig. 3D) and Hsp27core (Fig. 3F) were found predominantly in fractions 1–4 from the sucrose gradient when incubated alone. Incubation of α-syn fibrils with Hsp273D resulted in the protein co-sedimenting with the fibrils (Fig. 3E), however, Hsp27core did not co-sediment with the fibrils (Fig. 3G). This demonstrates that the N- and/or C-terminal regions of Hsp27 are required for stable binding to α-syn fibrils.
Binding of Hsp27 to α-syn fibrils occurs along the surface, decreasing exposed hydrophobicity
In light of the observation of the stable association of Hsp27 with α-syn fibrils, we sought to visualize this interaction directly using total internal reflection fluorescence (TIRF) imaging. In these experiments, fibrils were detected using SAVE imaging, whereby the fluorescence from ThT dye emitted as it binds fibrils is used to image them (Fig. 4; α-syn). CF647-labeled Hsp27 was imaged via its conjugated fluorophore in the absence and presence of α-syn fibrils (Fig. 4, A and C). In the presence of α-syn, the merged image (Fig. 4C) showed the chaperone distributed along the fibril surface. Quantitatively, the increase in co-incidence between ThT-reactive and CF647-fluorescent pixels when fibrils were co-incubated with Hsp27 was 66 ± 6%, compared with 20 ± 7% in the presence of the control (non-chaperone) protein α-lactalbumin (α-lac), or 21 ± 5% when Hsp27 was incubated with ThT in the absence of fibrils.
Figure 4.
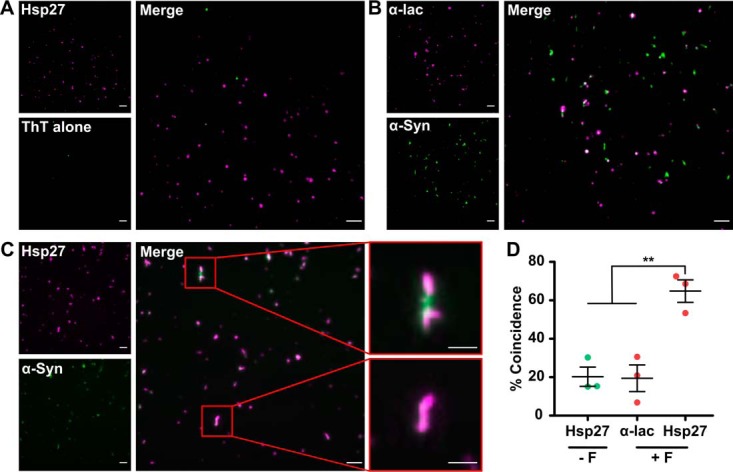
Hsp27 binds on the surface of α-syn fibrils. α-Syn fibrils (50 μm) were incubated in the presence of 1 μm CF647-labeled Hsp27, or the control protein α-lac. Samples were incubated in PBS containing 50 μm ThT, to allow α-syn fibrils to be visualized and imaged via TIRF microscopy. A–C, example TIRF images are shown for Hsp27 incubated with ThT (A), α-syn fibrils in the presence of α-lac (B), and α-syn fibrils in the presence of Hsp27 (C), along with the corresponding merge images. The scale bars represent 5 μm or 2 μm (zoom). D, the percentage of ThT-reactive pixels coincident with magenta pixels. The data are the mean ± S.E. (n = 3 repeats of nine images each) and were analyzed via a one-way analysis of variance (ANOVA) with a Bonferroni post hoc test (** denotes p < 0.01). The data are representative of two separate experiments.
In an attempt to further characterize the nature of the interaction between Hsp27 and α-syn fibrils, we used spectral PAINT (sPAINT), which allows regions of hydrophobicity along the fibril surface to be identified at the nanometer scale. This technique relies on the use of Nile Red in solution (in place of ThT) to image the fibrils. When Nile Red interacts with amyloid it undergoes a characteristic shift in its emission wavelength according to the hydrophobicity of its surrounding environment (49, 50). Thus, sPAINT enables regions of hydrophobicity along the face of fibrils to be mapped at the super-resolution level (Fig. 5A and Fig. S2). The addition of fluorescently labeled Hsp27, which was imaged below the diffraction limit using stochastic optical reconstruction microscopy (STORM) (51), allows the association of the chaperone with the fibrils to be mapped (Fig. 5B) and correlated with regions of higher or lower hydrophobicity (Fig. 5C and Fig. S2). The emission wavelengths recorded for each localization in the absence (Fig. 5D) or presence of Hsp27 were grouped to describe regions of high (550–600 nm), medium (600–650 nm), or low (650–700 nm) hydrophobicity (Fig. 5E). Notably, binding of Hsp27 to α-syn fibrils significantly affected the emission wavelengths associated with the fibrils (F(2, 27) = 3917, p < 0.0001). The presence of Hsp27 significantly decreased the proportion of localizations corresponding to high levels of hydrophobicity (p < 0.001) and significantly increased the proportion of localizations corresponding to low levels of hydrophobicity (p < 0.001) (Fig. 5E).
Figure 5.

Hsp27 decreases the relative hydrophobicity at the surface of α-syn fibrils. α-Syn fibrils (50 μm) were incubated in the presence of CF647-labeled Hsp27 in GLOX buffer containing 100 nm Nile Red to allow α-syn fibrils to be visualized and imaged via TIRF microscopy. A–C, an example super-resolution image is shown for α-syn (A) in the presence of labeled Hsp27 (B), along with the corresponding merge image (C). D, an example super-resolution image is shown for α-syn in the absence of labeled Hsp27. Localizations in (A–D) corresponding to α-syn are colored according to the wavelength of Nile Red emission (color scale given at the bottom of the image). Scale bars represent 2 μm, or 1 μm in inset. E, the percentage of localizations in each wavelength range in the absence and presence of Hsp27 were quantified and data are displayed as the mean ± S.E. (n = 6 images). Data were analyzed via a Student's t test, where *** denotes a significant (p < 0.001) difference between group means. Data are representative of two separate experiments.
Binding of sHsps protects against the cellular toxicity associated with exogenous α-syn fibrils
Finally, we investigated if Hsp27 decreases the cytotoxicity associated with exogenous α-syn fibrils. The dye dihydroethidium (DHE) has been used extensively to monitor the intracellular production of reactive oxygen species (ROS), specifically superoxide (52–54), as a measure of cellular toxicity. This method was therefore chosen to measure the toxicity of α-syn fibrils when added exogenously to cells. Traces of the fluorescence ratio (reduced DHE: oxidized ethidium) before and after treatment with monomeric or fibrillar α-syn (Fig. 6, A and B) demonstrate that there was an increase in cellular ROS production upon introduction of the α-syn fibrils. The difference in the rate of change in the fluorescence ratio (reported as a -fold change relative to the PBS control) is a measure of potential cytotoxicity, i.e. a faster rate of change in the fluorescence ratio is because of higher rates of ROS production associated with increased toxicity. We observed a significant effect of treatment on the generation of ROS within cells (F(7,247) = 33.96, p < 0.001). Post hoc tests revealed the α-syn fibrils induced significantly more ROS production than monomeric α-syn (p < 0.0001) (Fig. 6B). However, when the α-syn fibrils were pre-incubated with Hsp27 this dramatically reduced the rate of ROS production (by 65 ± 14% compared with fibrils alone) (Fig. 6B). Importantly, the production of ROS by cells associated with the addition of exogenous α-syn fibrils was not significantly decreased when the non-chaperone control proteins BSA and α-lac were pre-incubated with α-syn fibrils. Thus, the cytoprotective effect was specific to Hsp27. Also, incubation of the chaperone or control proteins in the absence of α-syn fibrils did not significantly alter basal levels of ROS production.
Figure 6.

Hsp27 reduces the generation of ROS by Neuro2a cells exposed to exogenous α-syn fibrils. Neuro-2a cells were incubated in PBS containing DHE (2 μm) before the addition of monomeric (M) or fibrillar (F) α-syn (20 μm) that had been pre-incubated in the absence or presence of Hsp27 (or the control proteins BSA and α-lac; each at 2 μm). The ratio of oxidized to reduced DHE before and after treatment was determined for 20 cells per treatment. A, example traces are shown for individual cells treated with monomeric α-syn, fibrillar α-syn, fibrillar α-syn preincubated with Hsp27 or Hsp27 alone. The fluorescence ratio before and after treatment was fitted by a linear regression function and the change in the rate of ROS production because of treatment was calculated as the difference in the gradient of the fitted lines. B, the -fold change in ROS production was then determined relative to a buffer only control (dashed line) and is reported as mean ± S.E. (n = 3 biological repeats, with each repeat an average of 20 cells). Data were analyzed via one-way ANOVA with a Bonferroni multiple comparison post hoc test, where *** denotes a significant (p < 0.001) difference compared with the fibril alone sample.
Discussion
The ability of Hsp27 (and other sHsps such as αB-c, Hsp20, HspB8, and HspB2/HspB3) to inhibit the aggregation of a range of proteins has been well-characterized (9, 10, 37, 44, 55). We have recently shown that αB-c and Hsp27 interact transiently with monomeric α-syn to inhibit its nucleation and subsequent aggregation, a process that is dependent on the aggregation rate (i.e. the efficiency of sHsps to inhibit α-syn aggregation decreases as the rate of aggregation increases) (15). Here, we demonstrate that Hsp27 is also capable of binding to α-syn fibrils, a process that requires the N- and/or C-terminal domains of the protein, and that by binding these fibrils Hsp27 inhibits elongation, and decreases their relative hydrophobicity and cytotoxicity.
The seeded aggregation assays employed in this work allow us to measure the rate of elongation without the confounding effects of primary nucleation events (46). The results presented here support our earlier work (15) by showing that the ability of Hsp27 to inhibit the aggregation of monomeric α-syn is dependent on the rate of aggregation. Here, we also demonstrate that the elongation of α-syn seeds is strongly inhibited by Hsp27 when the relative concentration of monomeric α-syn to Hsp27 is low (i.e. below a 1:1 molar ratio). As the concentration of monomeric α-syn increases, such that the molar ratio of monomeric α-syn:Hsp27 exceeds 1:1, aggregation occurs. This finding is significant given the association of early onset Parkinson's disease with duplication or triplication of the SNCA gene (56–60). In these cases, increases in the concentration of α-syn may allow aggregation-prone forms of α-syn to escape the protective capacity of the sHsps.
The ability of Hsp27 to inhibit elongation of α-syn fibril seeds (as measured by the rate constant) was inversely correlated to the seed concentration. This suggests that the ability of Hsp27 to inhibit α-syn fibril elongation results from both transient interactions with monomeric α-syn (to prevent association and addition to fibril ends) as well as interactions with fibrillar α-syn (essentially outcompeting monomeric α-syn for access to the fibril ends and surface). We demonstrate that Hsp27 forms a stable complex with α-syn fibrils, as has been reported previously for αB-c (41). Moreover, we show, using Hsp273D (an isoform that mimics a phosphorylated state of the chaperone) and the isolated core domain of Hsp27 (Hsp27core), that dimeric forms of Hsp27 can bind to α-syn fibrils and binding is mediated by the N- and/or C-terminal regions. Given that Hsp27core is still chaperone active (15, 48), these data suggest that domains within sHsps play distinct functional roles with regard to the interaction of these chaperones with aggregation-prone proteins. The central α-crystallin domain can interact with monomeric proteins to prevent aggregation, whereas the flexible N- and/or C-terminal regions mediate stable interactions with aggregated (fibrillar) protein.
Using TIRF microscopy, we were able to visualize directly the interaction between Hsp27 and α-syn fibrils, observing that Hsp27 binds along the surface and ends of fibrils. By coating the fibril surface in this manner, Hsp27 may act to inhibit surface-dependent secondary nucleation or fibril fragmentation, both of which lead to increases in the abundance of small oligomeric species and total fibril load (28, 61, 62). Our previous work using apolipoprotein C-II fibrils and analytical ultracentrifugation demonstrated that the binding of αB-c to fibrils inhibits their fragmentation (43). With regard to α-syn fibril formation, at neutral pH and under quiescent conditions, surface-assisted nucleation and fragmentation processes are not detectable in vitro (46). However, these processes dominate the aggregation of α-syn under mildly acidic pH conditions, such as would be present in some intracellular locations including endosomes and lysosomes (46). Given this, the ability of sHsps to bind α-syn aggregates may have physiological roles in disrupting these secondary processes under conditions whereby surface-assisted aggregation drives amyloid formation. Similar functions have recently been described for the isolated BRICHOS domain as a result of its interaction with amyloid fibrils formed from the amyloid-β peptide (27).
In this work, we also noted that the distribution of Hsp27 bound to the fibril was not uniform, suggesting that it may bind preferentially to specific regions on the surface of the fibril. We therefore sought to characterize the hydrophobic nature of the fibril surface using a novel imaging technique, sPAINT. This TIRF microscopy-based technique utilizes Nile Red (a fluorescent dye exhibiting a characteristic emission wavelength shift according to the hydrophobic nature of the surrounding environment) (49) to provide a quantitative measure of the relative hydrophobicity at any given region along the fibril face. Importantly, we demonstrate that, by binding to α-syn fibrils, Hsp27 significantly decreases hydrophobicity at the fibril surface. This is noteworthy, given the toxicity of aggregates formed from both pathogenic and nonpathogenic proteins correlates with the level of exposed hydrophobicity at the aggregate surface (63). The observed reduction in the relative hydrophobicity of the Hsp27-bound α-syn fibrils suggests that Hsp27 binds to regions of high hydrophobicity and this may decrease the cytotoxicity of the aggregates.
In addition to hydrophobicity, the production of ROS associated with protein aggregates is thought to be a significant mediator of neuronal toxicity in neurodegenerative disorders, including Parkinson's disease (64, 65). As anticipated, exposure of cells to exogenous α-syn fibrils resulted in a specific, rapid, and significant increase in cellular ROS production. However, pre-incubation of the α-syn fibrils with Hsp27 reduced this toxicity. Thus, the binding of Hsp27 to fibrillar α-syn represents a previously uncharacterized protective function of these chaperones, i.e. by binding to fibrils, Hsp27 (and most likely other sHsps) inhibits the toxicity associated with amyloid fibrils. This parallels the recently described novel chaperone activity of the chaperonin CCT, which by binding to α-syn oligomers significantly decreases the toxicity of α-syn aggregates to human neuroblastoma cells (66). It remains to be definitively established how Hsp27 can prevent cytotoxicity of exogenous fibrils. By binding to fibrils and decreasing their relative hydrophobicity, Hsp27 may prevent the interaction of the fibrils with cell membranes.
In summary, our work demonstrates that by binding to fibrils Hsp27 (and by inference other sHsps that bind to fibrils, e.g. αB-c) prevents the toxicity associated with protein aggregation. Thus, apart from its role in inhibiting protein aggregation, Hsp27 can protect cells from protein aggregates if they have already formed. Our work rationalizes the co-localization of sHsps in protein deposits associated with disease. Rather than being deposited in inclusions as a by-product of their failed attempt to prevent aggregation, our work suggests that sHsps are found in these deposits because of their specific association with the aggregated protein. Given that αB-c also binds fibrils (41–43), fibril-binding may represent a generic mechanism by which chaperone-active sHsps interact with aggregation-prone proteins. This aspect of their chaperone activity represents a secondary line of defense through which they may mitigate cellular toxicity even in the face of aberrant protein aggregation. These results highlight the potential to therapeutically target sHsp levels and/or activity in cells as a means of halting both the onset and progression of α-syn aggregation associated with the α-synucleinopathies.
Experimental procedures
Materials
The pET24d and pET24a bacterial expression vectors, containing the human HSPB1 (Hsp27) and SCNA (α-syn) genes, respectively, were used for expression of recombinant wildtype proteins. Disease-related mutants of α-syn and sHsp variants used in this work were produced via site-directed mutagenesis of the wildtype gene (GenScript, Piscataway, NJ). In particular, a variant of Hsp27 designed to mimic phosphorylation of the protein was generated via mutation of serine residues to aspartic acid at sites that are known to be modified in vivo (i.e. S15D, S78D, and S82D) to produce Hsp273D. The construct for the expression of the core domain of Hsp27, i.e. residues 84–176 (Hsp27core), was a kind gift from Professor A. Laganowsky (Texas A&M Health Science Center, Bryan, TX). All recombinant proteins were expressed in Escherichia coli BL21(DE3), previously transformed with each plasmid, and purified as described previously (23, 67, 68). Protein molecular mass standards used in gel electrophoresis were Protein Precision Plus Dual Color obtained from Bio-Rad, and dithiothreitol (DTT) was purchased from Amresco LLC (Solon, OH). All other chemicals, including ThT, were obtained from Sigma-Aldrich, unless otherwise stated. All buffers contained 0.02% (w/v) NaN3 to prevent bacterial contamination, and were filtered using 0.2 μm sterile filters before use. Protein concentrations were determined using a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific), based upon A280 nm0.1% values of 0.44 for α-syn calculated using the ExPASy Prot-Param tool (69), and 1.65 for Hsp27 (70).
Seeded aggregation assays
In an attempt to differentiate between interactions of Hsp27 with monomeric and fibrillar α-syn, an adaption of a previously described seeded aggregation assay was employed (46). Briefly, to produce seed fibrils, 1-ml aliquots of monomeric α-syn were prepared at concentrations ranging from 100 to 200 μm in 50 mm sodium phosphate (pH 7.4), in the absence of added salt. Samples were incubated at 45 °C with maximal stirring with a Teflon flea on a WiseStir heat plate (Witeg, Wertheim, Germany) for 24 h, then sonicated using three cycles of 10 s at 30% power (Branson digital Sonifier, model 250, Branson Ultrasonics, Danbury, CT). Samples were then incubated for a further 24 h under the conditions described above, before being sonicated again. Finally, samples were distributed into aliquots, flash frozen in liquid N2 and stored at −20 °C. The concentration of fibril seeds is reported as the monomer-equivalent concentration.
Seeded aggregation of α-syn was monitored using a microplate assay using clear 384 micro plates (Greiner Bio One, Frickenhausen, Germany) with each well initially containing a total of 40 μl of sample (all samples were run in duplicate). To probe the interaction of Hsp27 with monomeric α-syn, α-syn was incubated at concentrations from 10 to 100 μm in the absence or presence of 50 μm Hsp27 (or the negative control protein BSA) in 50 mm phosphate buffer (pH 7.4). Samples were equilibrated at 37 °C, then seed fibrils were added to each sample to a final concentration of 2.5 μm, resulting in a final volume of 50 μl for each sample. Alternatively, to probe the interaction of Hsp27 with fibrillar α-syn, 50 μm α-syn was incubated in the absence or presence of Hsp27 (or BSA as a control) at concentrations ranging from 0.1 to 10 μm in 50 mm phosphate buffer (pH 7.4). Samples were equilibrated at 37 °C, then seed fibrils were added into each sample to a final concentration of 0.5–5 μm, resulting again in a final volume of 50 μl for each sample. All wells contained a final concentration of 50 μm ThT, and the plates were sealed to prevent evaporation. The plates were then incubated in a FLUOstar Optima plate reader (BMG LABTECH, Ortenberg, Germany) at 37 °C without shaking, and ThT fluorescence measured using excitation and emission filters of 440 nm and 490 nm, respectively. Readings were taken every 300 s for periods of up to 20 h.
The change in ThT fluorescence intensity was calculated by subtracting the value at t = 0 h from subsequent measurements. The elongation rate for each sample was then determined by fitting data from the linear elongation phase of the assay (consisting of the initial 1–3 h or 0–2.5 h where appropriate) with linear regression using GraphPad Prism v 5 (GraphPad Software Inc., San Diego, CA). The relationship between rate and concentration was then determined by fitting using nonlinear regression.
Fibril formation
Monomeric α-syn, at concentrations ranging from 50 to 300 μm, was incubated in 50 mm phosphate buffer or PBS (pH 7.4) in the presence or absence of 1% (w/w) pre-formed α-syn seeds (prepared as described above). To monitor fibril growth, samples were incubated in the presence of 50 μm ThT using a black-walled, clear bottom 96-well microplate (Greiner Bio One), with each well containing 100 μl of sample. Plates were incubated with or without shaking (in the case of unseeded or seeded reactions, respectively) at 37 °C in a FLUOStar Optima plate reader (BMG LABTECH), with the plates sealed to prevent evaporation. The ThT fluorescence was monitored via excitation and emission at 440 nm and 490 nm, respectively, and readings were taken every 300 s for periods of up to 48 h. Samples were considered to contain mature amyloid fibrils once the ThT fluorescence had reached a plateau (typically after 48 h of incubation). The fibril samples were collected from the plates and stored at 4 °C for use.
Sucrose cushion centrifugation assays
Mature α-syn fibrils (formed with 1% (w/w) seeds) were prepared at a final concentration of 75 μm in 50 mm phosphate buffer (pH 7.4). Fibrils were incubated at 37 °C for 1 h in a VorTemp Shaking Incubator (Labnet International, Edison, NJ) with shaking at 60 rpm, in the absence or presence of Hsp27 (15 μm). Aliquots were taken (load samples) prior to samples (100 μl) being layered on top of a 20% (w/v) sucrose solution (900 μl) (Amresco LLC) prepared in 50 mm phosphate buffer containing 100 mm NaCl (pH 7.4). Following centrifugation at 200,000 × g for 20 min at 4 °C using an MTX150 ultracentrifuge (Thermo Fisher Scientific), 100 μl fractions were collected and any pelleted material resuspended in the final 100 μl (top, fraction 1; bottom, fraction 10). SDS-PAGE analysis of the load samples and fractions was performed using 12% (v/v) polyacrylamide gels (Bio-Rad), except in the case of samples containing Hsp27core which required 20% (v/v) acrylamide for appropriate separation. For analysis, gels were stained with Coomassie Brilliant Blue R250.
Fluorescent labeling of proteins
For imaging purposes, recombinant proteins of interest were fluorescently labeled with either succinimidyl ester (in the case of α-lac) or maleimide (in the case of Hsp27) derivatives of CF647 (Biotium, Hayward, CA) as described previously (15). The concentration and degree of labeling was calculated according to the manufacturer's instructions and found to be greater than 60%. The labeled proteins were then stored at −20 °C.
Total internal reflection fluorescence microscopy methods
TIRF microscopy was employed to visualize directly the interaction of Hsp27 with mature α-syn amyloid fibrils grown in the absence of seeds using a custom-built inverted optical microscope as described previously (45). To prepare slides for microscopy, glass coverslips were cleaned using an argon plasma cleaner (Diener Electronic, Ebhausen, Germany) for 1 h and 9 × 9 mm frame-seal slide chambers (Bio-Rad) were fixed to the cleaned surface. The glass surface within the chamber was then coated with poly-l-lysine (0.01% w/v), and incubated for 30 min at room temperature. The chamber was then washed three times with 0.02 μm filtered PBS, before the slide was transferred to the microscope stage and coupled to the lens using immersion oil (n = 1.518; Olympus, Tokyo, Japan). Samples for SAVE imaging were prepared in PBS containing 70 μm α-syn and 70 nm fluorescently labeled wildtype Hsp27 or the negative control protein, α-lac. Samples were incubated for 30 min at room temperature before being centrifuged at 20,000 × g for 10 min. The supernatant was removed and replaced with fresh PBS, and the wash repeated three times before the fibril pellet was resuspended to a final concentration of 100 pm in PBS containing 5 μm ThT immediately before imaging.
Images were collected (nine fields of view each at three random sites for each sample) using a custom script to prevent user bias (Beanshell script, Micro-Manager) and the resultant images were analyzed using custom scripts written in Igor Pro v6.3.4.1. Background and weak signals were removed by adaptive signal thresholding via functions available directly in Igor Pro. Fluorescently labeled proteins (spots) above the threshold were enumerated, and the position of a spot above the threshold in one channel was examined for the presence of a spot in the other channel. Chance coincidence was determined by rotating one of the images 90°, translating it horizontally by 10 pixels (to offset any central spots not affected by the rotation), and then testing for coincidence in an identical manner.
sPAINT imaging of mature α-syn fibrils was also performed using a custom-built inverted optical microscope. Briefly, an inverted microscope (Nikon Eclipse TI, Tokyo, Japan) was configured to operate in objective-type TIRF mode with three light sources, a 405-nm CW diode 200 milliwatt OBIS laser (Coherent, Santa Clara, CA), a 514-nm solid-state 200 milliwatt Sapphire laser (Coherent), and a 647-nm CW diode 200 milliwatt OBIS laser (Coherent). The lasers were directed via dichroic mirrors through a high numerical aperture, 60× oil immersion CFI Apochromate objective lens (Nikon Eclipse TI) to the sample coverslip. The emitted fluorescence was collected and directed through long-pass filters specific for each fluorophore (see below) and finally projected onto an electron-multiplying charged coupled device (EMCCD) camera (Evolve II 512 Delta, Photometrics, Tucson, AZ). A physical aperture (VA100/M, Thorlabs), and a transmission diffraction grating (300 Grooves/mm 8.6° Blaze Angle; GT13–03, Thorlabs) were mounted on the camera port path prior to the detector.
Slides were prepared and coupled to the microscope as described above. Samples were prepared in 50 mm phosphate buffer (pH 7.4) or PBS containing 50 μm α-syn in the absence or presence of 1 μm fluorescently labeled wildtype Hsp27. Samples were incubated for 5 min at room temperature, before being diluted 100-fold in the GLOX-mercaptoethylamine imaging buffer (0.5 mg/ml glucose oxidase, 40 μg/ml catalase and 10% (v/v) glucose in 50 mm Tris-HCl (pH 8.0) with 10 mm NaCl and 10 mm β-mercaptoethylamine) supplemented with 100 nm Nile Red. Fibrillar α-syn was detected via Nile Red fluorescence, which was excited at 514 nm and collected using an ET590LP long-pass filter (Semrock Inc., Rochester, NY). The CF647-labeled Hsp27 was imaged via excitation at 647 nm and the emitted fluorescence was detected using an ET655LP long-pass filter.
Data were analyzed to generate sPAINT images as described previously (50) using a typical signal strength threshold of ∼30. Quantitative co-localization and wavelength analyses were completed using custom scripts written for Python 2.7, and the localizations were binned into low, medium, and high wavelengths using GraphPad Prism v 5 (GraphPad Software).
Dihydroethidium assay to monitor cellular ROS production
A DHE cell-based assay, similar to that described previously (52, 71), was used to investigate the toxicity associated α-syn aggregates when added exogenously to cells. Neuro-2a cells were routinely cultured in Dulbecco's modified Eagle's medium: Ham's Nutrient Mixture F-12 (DMEM/F12) (Thermo Fisher Scientific) supplemented with 10% (v/v) fetal bovine serum (Bovagen Biologicals, East Keilor, Australia) and 2.5 mm l-glutamine (Thermo Fisher Scientific), and incubated in a Heracell 150i CO2 incubator (Thermo Fisher Scientific) under 5% CO2/95% air at 37 °C. The cells were seeded into 8-well chamber μSlides (Ibidi, Martinsried, Germany) and cultured to 60% confluency. Solutions containing monomeric or fibrillar α-syn (formed in the presence of pre-formed seeds) were prepared in PBS in the absence or presence of Hsp27 (or a control protein, BSA or α-lac) at a 1:100 molar ratio (Hsp27:α-syn) and incubated at 37 °C for 30 min. Immediately prior to use, DHE (to a final concentration of 2 μm) was also added to these solutions to eliminate the effects of dilution of the DHE.
Cells were washed with PBS immediately before DHE (2 μm in PBS) was added. Images were taken every 30 s for 15 min using an epifluorescence microscope (Leica DMi8 fluorescence microscope, Wetzlar, Germany) to quantify the amount of DHE (excitation 325–375 nm and emission 435–485 nm) and its oxidized product (i.e. ethidium, excitation 540–580 nm and emission 592–668 nm) as a measure of ROS production in cells (52). The solutions were then added to cells, and images were taken every 30 s for a further 15 min. Data analysis was completed using custom programs written for Python 2.7, which allows 20 cells to be selected and the mean fluorescence intensity for each of the cells to be determined from every image in both the red and blue channels. The ratio of DHE fluorescence to ethidium fluorescence was then calculated both before and after treatment and fitted with linear regression where appropriate using GraphPad v5 (GraphPad Software). The difference in the rate of change of the DHE/ethidium fluorescence ratio before and after treatment was then determined and normalized to the PBS control.
Author contributions
H. E. and D. C. conceived the experiments and coordinated the study. H. E., D. C., D. R. W., J. B., and M. H. H. designed the experiments. D. C., D. R. W., M. H. H., and R. S. G. performed the experiments. C. M. D., D. K., and A. O. helped in the design of experiments and acquisition and interpretation of data. All authors analyzed and reviewed the data. D. C. and H. E. wrote the paper. All authors read and approved the final manuscript.
Supplementary Material
Acknowledgments
We thank the Illawarra Health and Medical Research Institute for technical support.
This work was supported by grants from the Faculty of Science, Medicine and Health of the University of Wollongong and the Cambridge Centre for Misfolding Diseases (to J. B. and C. M. D.). This work was also supported by an Australian Postgraduate Award (to D. C.), Australian Research Council Future Fellowship FT110100586 (to H. E.), and Australian Research Council Laureate Fellowship FL1401000027 (to. A. O.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1 and S2.
- sHsp
- small heat shock protein
- αB-c
- αB-crystallin
- α-syn
- α-synuclein
- SAVE
- single aggregate visualization by enhancement
- ThT
- thioflavin T
- TIRF
- total internal reflection fluorescence
- α-lac
- α-lactalbumin
- DHE
- dihydroethidium
- ROS
- reactive oxygen species
- sPAINT
- spectrally resolved points accumulation for imaging in nanoscale topography.
References
- 1. Treweek T. M., Meehan S., Ecroyd H., and Carver J. A. (2015) Small heat-shock proteins: Important players in regulating cellular proteostasis. Cell Mol. Life Sci. 72, 429–451 10.1007/s00018-014-1754-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hartl F. U., Bracher A., and Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 10.1038/nature10317 [DOI] [PubMed] [Google Scholar]
- 3. van Montfort R. L. M., Basha E., Friedrich K. L., Slingsby C., and Vierling E. (2001) Crystal structure and assembly of a eukaryotic small heat shock protein. Nat. Struct. Biol. 8, 1025–1030 10.1038/nsb722 [DOI] [PubMed] [Google Scholar]
- 4. Stamler R., Kappé G., Boelens W., and Slingsby C. (2005) Wrapping the alpha-crystallin domain fold in a chaperone assembly. J. Mol. Biol. 353, 68–79 10.1016/j.jmb.2005.08.025 [DOI] [PubMed] [Google Scholar]
- 5. Benesch J. L. P., Ayoub M., Robinson C. V., and Aquilina J. A. (2008) Small heat shock protein activity is regulated by variable oligomeric substructure. J. Biol. Chem. 283, 28513–28517 10.1074/jbc.M804729200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jehle S., Rajagopal P., Bardiaux B., Markovic S., Kühne R., Stout J. R., Higman V. A., Klevit R. E., van Rossum B. J., and Oschkinat H. (2010) Solid-state NMR and SAXS studies provide a structural basis for the activation of alphaB-crystallin oligomers. Nat. Struct. Mol. Biol. 17, 1037–1042 10.1038/nsmb.1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jehle S., Vollmar B. S., Bardiaux B., Dove K. K., Rajagopal P., Gonen T., Oschkinat H., and Klevit R. E. (2011) N-terminal domain of αB-crystallin provides a conformational switch for multimerization and structural heterogeneity. Proc. Natl. Acad. Sci. U.S.A. 108, 6409–6414 10.1073/pnas.1014656108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lelj-Garolla B., and Mauk A. G. (2006) Self-association and chaperone activity of Hsp27 are thermally activated. J. Biol. Chem. 281, 8169–8174 10.1074/jbc.M512553200 [DOI] [PubMed] [Google Scholar]
- 9. Bruinsma I. B., Bruggink K. A., Kinast K., Versleijen A. A. M., Segers-Nolten I. M. J., Subramaniam V., Bea Kuiperij H., Boelens W., de Waal R. M. W., and Verbeek M. M. (2011) Inhibition of alpha-synuclein aggregation by small heat shock proteins. Proteins Struct. Funct. Bioinformat. 79, 2956–2967 10.1002/prot.23152 [DOI] [PubMed] [Google Scholar]
- 10. Ecroyd H., Meehan S., Horwitz J., Aquilina J. A., Benesch J. L. P., Robinson C. V., Macphee C. E., and Carver J. A. (2007) Mimicking phosphorylation of αB-crystallin affects its chaperone activity. Biochem. J. 401, 129–141 10.1042/BJ20060981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kulig M., and Ecroyd H. (2012) The small heat-shock protein alphaB-crystallin uses different mechanisms of chaperone action to prevent the amorphous versus fibrillar aggregation of alpha-lactalbumin. Biochem. J. 448, 343–352 10.1042/BJ20121187 [DOI] [PubMed] [Google Scholar]
- 12. Robertson A. L., Headey S. J., Saunders H. M., Ecroyd H., Scanlon M. J., Carver J. A., and Bottomley S. P. (2010) Small heat-shock proteins interact with a flanking domain to suppress polyglutamine aggregation. Proc. Natl. Acad. Sci. U.S.A. 107, 10424–10429 10.1073/pnas.0914773107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rekas A., Jankova L., Thorn D. C., Cappai R., and Carver J. A. (2007) Monitoring the prevention of amyloid fibril formation by α-crystallin. FEBS J. 274, 6290–6304 10.1111/j.1742-4658.2007.06144.x [DOI] [PubMed] [Google Scholar]
- 14. Hatters D. M., Lindner R. A., Carver J. A., and Howlett G. J. (2001) The molecular chaperone, α-crystallin, inhibits amyloid formation by apolipoprotein C-II. J. Biol. Chem. 276, 33755–33761 10.1074/jbc.M105285200 [DOI] [PubMed] [Google Scholar]
- 15. Cox D., Selig E., Griffin M. D., Carver J. A., and Ecroyd H. (2016) Small heat shock proteins prevent α-synuclein aggregation via transient interactions and their efficacy is affected by the rate of aggregation. J. Biol. Chem. 291, 22618–22629 10.1074/jbc.M116.739250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ehrnsperger M., Gräber S., Gaestel M., and Buchner J. (1997) Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J. 16, 221–229 10.1093/emboj/16.2.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Feder M. E., and Hofmann G. E. (1999) Heat-shock proteins, molecular chaperones, and the stress response: Evolutionary and ecological physiology. Annu. Rev. Physiol. 61, 243–282 10.1146/annurev.physiol.61.1.243 [DOI] [PubMed] [Google Scholar]
- 18. Carver J. A., Rekas A., Thorn D. C., and Wilson M. R. (2003) Small heat-shock proteins and clusterin: intra- and extracellular molecular chaperones with a common mechanism of action and function? IUBMB Life 55, 661–668 10.1080/15216540310001640498 [DOI] [PubMed] [Google Scholar]
- 19. Devlin G. L., Carver J. A., and Bottomley S. P. (2003) The selective inhibition of serpin aggregation by the molecular chaperone, α-crystallin, indicates a nucleation-dependent specificity. J. Biol. Chem. 278, 48644–48650 10.1074/jbc.M308376200 [DOI] [PubMed] [Google Scholar]
- 20. Irvine G. B., El-Agnaf O. M., Shankar G. M., and Walsh D. M. (2008) Protein aggregation in the brain: The molecular basis for Alzheimer's and Parkinson's diseases. Mol. Med. 14, 451–464 10.1007/s00894-008-0296-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chiti F and Dobson C. M. (2017) Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 86, 27–68 10.1146/annurev-biochem-061516-045115 [DOI] [PubMed] [Google Scholar]
- 22. Martí M. J., Tolosa E., and Campdelacreu J. (2003) Clinical overview of the synucleinopathies. Mov. Disord. 18, 21–27 10.1002/mds.10559 [DOI] [PubMed] [Google Scholar]
- 23. Narhi L., Wood S. J., Steavenson S., Jiang Y., Wu G. M., Anafi D., Kaufman S. A., Martin F., Sitney K., Denis P., Louis J.-C., Wypych J., Biere A. L., and Citron M. (1999) Both familial Parkinson's disease mutations accelerate α-synuclein aggregation. J. Biol. Chem. 274, 9843–9846 10.1074/jbc.274.14.9843 [DOI] [PubMed] [Google Scholar]
- 24. Knowles T. P. J., Waudby C. A., Devlin G. L., Cohen S. I. A., Aguzzi A., Vendruscolo M., Terentjev E. M., Welland M. E., and Dobson C. M. (2009) An analytical solution to the kinetics of breakable filament assembly. Science 326, 1533–1537 10.1126/science.1178250 [DOI] [PubMed] [Google Scholar]
- 25. Glabe C. G. (2009) Amyloid oligomer structures and toxicity. Open Biol. J. 2, 222–227 10.2174/1874196700902010222 [DOI] [Google Scholar]
- 26. Glabe C. G. (2008) Structural classification of toxic amyloid oligomers. J. Biol. Chem. 283, 29639–29643 10.1074/jbc.R800016200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cohen S. I. A., Arosio P., Presto J., Kurudenkandy F. R., Biverstål H., Dolfe L., Dunning C., Yang X., Frohm B., Vendruscolo M., Johansson J., Dobson C. M., Fisahn A., Knowles T. P. J., and Linse S. (2015) A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nat. Struct. Mol. Biol. 22, 207–213 10.1038/nsmb.2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xue W.-F., Hellewell A. L., Gosal W. S., Homans S. W., Hewitt E. W., and Radford S. E. (2009) Fibril fragmentation enhances amyloid cytotoxicity. J. Biol. Chem. 284, 34272–34282 10.1074/jbc.M109.049809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee H. J., Suk J. E., Bae E. J., Lee J. H., Paik S. R., and Lee S. J. (2008) Assembly dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 40, 1835–1849 10.1016/j.biocel.2008.01.017 [DOI] [PubMed] [Google Scholar]
- 30. Lee H. J., Suk J. E., Bae E. J., and Lee S. J. (2008) Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 372, 423–428 10.1016/j.bbrc.2008.05.045 [DOI] [PubMed] [Google Scholar]
- 31. Tyedmers J., Mogk A., and Bukau B. (2010) Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 11, 777–788 10.1038/nrm2993 [DOI] [PubMed] [Google Scholar]
- 32. Yerbury J. J., Ooi L., Dillin A., Saunders D. N., Hatters D. M., Beart P. M., Cashman N. R., Wilson M. R., and Ecroyd H. (2016) Walking the tightrope: Proteostasis and neurodegenerative disease. J. Neurochem. 137, 489–505 10.1111/jnc.13575 [DOI] [PubMed] [Google Scholar]
- 33. Wakabayashi K., Tanji K., Mori F., and Takahashi H. (2007) The Lewy body in Parkinson's disease: Molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 27, 494–506 10.1111/j.1440-1789.2007.00803.x [DOI] [PubMed] [Google Scholar]
- 34. Leverenz J. B., Umar I., Wang Q., Montine T. J., McMillan P. J., Tsuang D. W., Jin J., Pan C., Shin J., Zhu D., and Zhang J. (2007) Proteomic identification of novel proteins in cortical Lewy bodies. Brain Pathol. 17, 139–145 10.1111/j.1750-3639.2007.00048.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gao X., Carroni M., Nussbaum-Krammer C., Mogk A., Nillegoda N. B., Szlachcic A., Guilbride D. L., Saibil H. R., Mayer M. P., and Bukau B. (2015) Human Hsp70 disaggregase reverses Parkinson's-linked α-synuclein amyloid fibrils. Mol. Cell 59, 781–793 10.1016/j.molcel.2015.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McLean P. J., Kawamata H., Shariff S., Hewett J., Sharma N., Ueda K., Breakefield X. O., and Hyman B. T. (2002) TorsinA and heat shock proteins act as molecular chaperones: Suppression of α-synuclein aggregation. J. Neurochem. 83, 846–854 10.1046/j.1471-4159.2002.01190.x [DOI] [PubMed] [Google Scholar]
- 37. Outeiro T. F., Klucken J., Strathearn K. E., Liu F., Nguyen P., Rochet J.-C., Hyman B. T., and McLean P. J. (2006) Small heat shock proteins protect against α-synuclein-induced toxicity and aggregation. Biochem. Biophys. Res. Commun. 351, 631–638 10.1016/j.bbrc.2006.10.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lowe J., Landon M., Pike I., Spendlove I., McDermott H., and Mayer R. J. (1990) Dementia with β-amyloid deposition: Involvement of alpha B-crystallin supports two main diseases. Lancet 336, 515–516 [DOI] [PubMed] [Google Scholar]
- 39. Iwaki T., Wisniewski T., Iwaki A., Corbin E., Tomokane N., Tateishi J., and Goldman J. E. (1992) Accumulation of alpha B-crystallin in central nervous system glia and neurons in pathologic conditions. Am. J. Pathol. 140, 345–356 [PMC free article] [PubMed] [Google Scholar]
- 40. Mizutani T., Inose T., Nakajima S., Kakimi S., Uchigata M., Ikeda K., Gambetti P., and Takasu T. (1998) Familial parkinsonism and dementia with ballooned neurons, argyrophilic neuronal inclusions, atypical neurofibrillary tangles, tau-negative astrocytic fibrillary tangles, and Lewy bodies. Acta Neuropathol. 95, 15–27 [DOI] [PubMed] [Google Scholar]
- 41. Waudby C. A., Knowles T. P. J., Devlin G. L., Skepper J. N., Ecroyd H., Carver J. A., Welland M. E., Christodoulou J., Dobson C. M., and Meehan S. (2010) The interaction of αB-crystallin with mature α-synuclein amyloid fibrils inhibits their elongation. Biophys. J. 98, 843–851 10.1016/j.bpj.2009.10.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shammas S. L., Waudby C. A., Wang S., Buell A. K., Knowles T. P. J., Ecroyd H., Welland M. E., Carver J. A., Dobson C. M., and Meehan S. (2011) Binding of the molecular chaperone αB-crystallin to Aβ amyloid fibrils inhibits fibril elongation. Biophys. J. 101, 1681–1689 10.1016/j.bpj.2011.07.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Binger K. J., Ecroyd H., Yang S., Carver J. A., Howlett G. J., and Griffin M. D. W. (2013) Avoiding the oligomeric state: αB-crystallin inhibits fragmentation and induces dissociation of apolipoprotein C-II amyloid fibrils. FASEB J. 27, 1214–1222 10.1096/fj.12-220657 [DOI] [PubMed] [Google Scholar]
- 44. Yerbury J. J., Gower D., Vanags L., Roberts K., Lee J. A., and Ecroyd H. (2013) The small heat shock proteins αB-crystallin and Hsp27 suppress SOD1 aggregation in vitro. Cell Stress Chaperones 18, 251–257 10.1007/s12192-012-0371-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Horrocks M. H., Lee S. F., Gandhi S., Magdalinou N. K., Chen S. W., Devine M. J., Tosatto L., Kjaergaard M., Beckwith J. S., Zetterberg H., Iljina M., Cremades N., Dobson C. M., Wood N. W., and Klenerman D. (2016) Single-molecule imaging of individual amyloid protein aggregates in human biofluids. ACS Chem. Neurosci. 7, 399–406 10.1021/acschemneuro.5b00324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Buell A. K., Galvagnion C., Gaspar R., Sparr E., Vendruscolo M., Knowles T. P., Linse S., and Dobson C. M. (2014) Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. U.S.A. 111, 7671–7676 10.1073/pnas.1315346111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jovcevski B., Kelly M. A., Rote A. P., Berg T., Gastall H. Y., Benesch J. L., Aquilina J. A., and Ecroyd H. (2015) Phosphomimics destabilize Hsp27 oligomeric assemblies and enhance chaperone activity. Chem. Biol. 22, 186–195 10.1016/j.chembiol.2015.01.001 [DOI] [PubMed] [Google Scholar]
- 48. Hochberg G. K., Ecroyd H., Liu C., Cox D., Cascio D., Sawaya M. R., Collier M. P., Stroud J., Carver J. A., Baldwin A. J., Robinson C. V., Eisenberg D. S., Benesch J. L., and Laganowsky A. (2014) The structured core domain of alpha B-crystallin can prevent amyloid fibrillation and associated toxicity. Proc. Natl. Acad. Sci. U.S.A. 111, 1562–1570 10.1073/pnas.1322673111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Greenspan P., and Fowler S. D. (1985) Spectrofluorometric studies of the lipid probe, nile red. J. Lipid Res. 26, 781–789 [PubMed] [Google Scholar]
- 50. Bongiovanni M. N., Godet J., Horrocks M. H., Tosatto L., Carr A. R., Wirthensohn D. C., Ranasinghe R. T., Lee J. E., Ponjavic A., Fritz J. V., Dobson C. M., Klenerman D., and Lee S. F. (2016) Multi-dimensional super-resolution imaging enables surface hydrophobicity mapping. Nat. Commun. 7, 13544 10.1038/ncomms13544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rust M. J., Bates M., and Zhuang X. (2006) Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–795 10.1038/nmeth929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang X., and Soldati T. (2013) Detecting, visualizing and quantitating the generation of reactive oxygen species in an amoeba model system. J. Vis. Exp. 81, e50717 10.3791/50717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang X., Fang H., Huang Z., Shang W., Hou T., Cheng A., and Cheng H. (2013) Imaging ROS signaling in cells and animals. J. Mol. Med. 91, 917–927 10.1007/s00109-013-1067-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao H., Kalivendi S., Zhang H., Joseph J., Nithipatikom K., Vásquez-Vivar J., and Kalyanaraman B. (2003) Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: Potential implications in intracellular fluorescence detection of superoxide. Free Radic. Biol. Med. 34, 1359–1368 10.1016/S0891-5849(03)00142-4 [DOI] [PubMed] [Google Scholar]
- 55. Wilhelmus M. M., Boelens W. C., Otte-Höller I., Kamps B., de Waal R. M., and Verbeek M. M. (2006) Small heat shock proteins inhibit amyloid-beta protein aggregation and cerebrovascular amyloid-beta protein toxicity. Brain Res. 1089, 67–78 10.1016/j.brainres.2006.03.058 [DOI] [PubMed] [Google Scholar]
- 56. Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W. G., Lazzarini A. M., Duvoisin R. C., Di Iorio G., Golbe L. I., and Nussbaum R. L. (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047 10.1126/science.276.5321.2045 [DOI] [PubMed] [Google Scholar]
- 57. Baba M., Nakajo S., Tu P. H., Tomita T., Nakaya K., Lee V. M., Trojanowski J. Q., and Iwatsubo T. (1998) Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884 [PMC free article] [PubMed] [Google Scholar]
- 58. Krüger R., Kuhn W., Müller T., Woitalla D., Graeber M., Kösel S., Przuntek H., Epplen J. T., Schöls L., and Riess O. (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat. Genet. 18, 106–108 10.1038/ng0298-106 [DOI] [PubMed] [Google Scholar]
- 59. Campbell B. C. V., McLean C. A., Culvenor J. G., Gai W. P., Blumbergs P. C., Jäkälä P., Beyreuther K., Masters C. L., and Li Q.-X. (2001) The solubility of α-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson's disease. J. Neurochem. 76, 87–96 [DOI] [PubMed] [Google Scholar]
- 60. Zarranz J. J., Alegre J., Gómez-Esteban J. C., Lezcano E., Ros R., Ampuero I., Vidal L., Hoenicka J., Rodriguez O., Atarés B., Llorens V., Gomez Tortosa E., del Ser T., Muñoz D. G., and de Yebenes J. G. (2004) The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173 10.1002/ana.10795 [DOI] [PubMed] [Google Scholar]
- 61. Jakhria T., Hellewell A. L., Porter M. Y., Jackson M. P., Tipping K. W., Xue W.-F., Radford S. E., and Hewitt E. W. (2014) β2-Microglobulin amyloid fibrils are nanoparticles that disrupt lysosomal membrane protein trafficking and inhibit protein degradation by lysosomes. J. Biol. Chem. 289, 35781–35794 10.1074/jbc.M114.586222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shvadchak V. V., Claessens M. M., and Subramaniam V. (2015) Fibril breaking accelerates alpha-synuclein fibrillization. J. Phys. Chem. B 119, 1912–1918 10.1021/jp5111604 [DOI] [PubMed] [Google Scholar]
- 63. Bolognesi B., Kumita J. R., Barros T. P., Esbjorner E. K., Luheshi L. M., Crowther D. C., Wilson M. R., Dobson C. M., Favrin G., and Yerbury J. J. (2010) ANS binding reveals common features of cytotoxic amyloid species. ACS Chem. Biol. 5, 735–740 10.1021/cb1001203 [DOI] [PubMed] [Google Scholar]
- 64. Tabner B. J., Turnbull S., El-Agnaf O., and Allsop D. (2001) Production of reactive oxygen species from aggregating proteins implicated in Alzheimer's disease, Parkinson's disease and other neurodegenerative diseases. Curr. Top. Med. Chem. 1, 507–517 10.2174/1568026013394822 [DOI] [PubMed] [Google Scholar]
- 65. Jomova K., Vondrakova D., Lawson M., and Valko M. (2010) Metals, oxidative stress and neurodegenerative disorders. Mol. Cell Biochem. 345, 91–104 10.1007/s11010-010-0563-x [DOI] [PubMed] [Google Scholar]
- 66. Sot B., Rubio-Muñoz A., Leal-Quintero A., Martínez-Sabando J., Marcilla M., Roodveldt C., and Valpuesta J. M. (2017) The chaperonin CCT inhibits assembly of α-synuclein amyloid fibrils by a specific, conformation-dependent interaction. Sci. Rep. 7, 40859–40871 10.1038/srep40859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Horwitz J., Huang Q. L., Ding L., and Bova M. P. (1998) Lens alpha-crystallin: chaperone-like properties. Methods Enzymol. 290, 365–383 10.1016/S0076-6879(98)90032-5 [DOI] [PubMed] [Google Scholar]
- 68. Laganowsky A., Benesch J. L., Landau M., Ding L., Sawaya M. R., Cascio D., Huang Q., Robinson C. V., Horwitz J., and Eisenberg D. (2010) Crystal structures of truncated alphaA and alphaB crystallins reveal structural mechanisms of polydispersity important for eye lens function. Protein Sci. 19, 1031–1043 10.1002/pro.380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Walker J. M. (2005) The Proteomics Protocols Handbook, Springer, New York, NY [Google Scholar]
- 70. Hayes D., Napoli V., Mazurkie A., Stafford W. F., and Graceffa P. (2009) Phosphorylation dependence of Hsp27 multimeric size and molecular chaperone function. J. Biol. Chem. 284, 18801–18807 10.1074/jbc.M109.011353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cremades N., Cohen S. I. A., Deas E., Abramov A. Y., Chen A. Y., Orte A., Sandal M., Clarke R. W., Dunne P., Aprile F. A., Bertoncini C. W., Wood N. W., Knowles T. P. J., Dobson C. M., and Klenerman D. (2012) Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 149, 1048–1059 10.1016/j.cell.2012.03.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.