

ABSTRACT
Zebrafish regenerate cardiac tissue through proliferation of pre-existing cardiomyocytes and neovascularization. Secreted growth factors such as FGFs, IGF, PDGFs and Neuregulin play essential roles in stimulating cardiomyocyte proliferation. These factors activate the Ras/MAPK pathway, which is tightly controlled by the feedback attenuator Dual specificity phosphatase 6 (Dusp6), an ERK phosphatase. Here, we show that suppressing Dusp6 function enhances cardiac regeneration. Inactivation of Dusp6 by small molecules or by gene inactivation increased cardiomyocyte proliferation, coronary angiogenesis, and reduced fibrosis after ventricular resection. Inhibition of Erbb or PDGF receptor signaling suppressed cardiac regeneration in wild-type zebrafish, but had a milder effect on regeneration in dusp6 mutants. Moreover, in rat primary cardiomyocytes, NRG1-stimulated proliferation can be enhanced upon chemical inhibition of Dusp6 with BCI. Our results suggest that Dusp6 attenuates Ras/MAPK signaling during regeneration and that suppressing Dusp6 can enhance cardiac repair.
KEY WORDS: Zebrafish, Heart regeneration, Cardiac repair, Dual specificity phosphatase 6, Ras/MAPK signaling, Cardiomyocyte proliferation
Summary: Following heart injury, zebrafish dusp6 mutants show increased cardiomyocyte proliferation and angiogenesis and early downregulation of fibrosis genes, indicating accelerated cardiac repair associated with enhanced signaling via PDGFR and Nrg1/Erbb2.
INTRODUCTION
Myocardial infarction (MI) is a leading cause of morbidity and mortality in industrialized countries (Mozaffarian et al., 2016). The effects of MI include massive cardiomyocyte death, which leads to the formation of a non-contractile scar (Mill et al., 1990). Humans have a poor ability to replace damaged cardiac tissue after an MI event, mostly because adult cardiomyocytes are postmitotic in mammals (Ahuja et al., 2007). Therefore, a major goal of cardiac regenerative medicine is to induce cardiomyocyte proliferation or to transdifferentiate cardiac fibroblasts into beating cardiomyocytes (Bersell et al., 2009; Ebelt et al., 2008; Zhou et al., 2015). Although there have been intensive studies towards the direct reprogramming of fibroblasts, including the use of chemical agents, there is still much progress to be made before an effective treatment is achieved (Cao et al., 2016b; Qian et al., 2012; Zhang et al., 2016).
Unlike mammals, zebrafish (Danio rerio) can regenerate the heart throughout the lifespan (Itou et al., 2012a; Poss et al., 2002). After resection of the ventricular apex, a blood clot is formed to seal the wound that is subsequently replaced by fibrin and collagen (Poss et al., 2002). Within hours of the injury, the epicardium, a layer of cells surrounding the heart, is activated and these cells proliferate and migrate to the injury area after undergoing epithelial-to-mesenchymal transition (EMT). The function of activated epicardial cells is to promote the formation of new blood vessels that support the regenerating myocardium (González-Rosa et al., 2012; Lepilina et al., 2006). Between 4 and 14 days post amputation (dpa), spared cardiomyocytes dedifferentiate and proliferate (Jopling et al., 2010; Kikuchi et al., 2010). By 30-60 dpa, the heart is fully regenerated, with the fibrotic tissue resolved and replaced by new cardiac muscle and vasculature.
The molecular mechanisms activated after injury that drive cardiomyocyte proliferation are complex and involve ligands such as Fibroblast growth factors (FGFs) (Lepilina et al., 2006; Lien et al., 2006), Neuregulins (Nrg) (Gemberling et al., 2015), Transforming growth factor β (TGFβ) (Chablais and Jazwinska, 2012b), Platelet derived growth factor β (PDGFβ) (Lien et al., 2006), Insulin-like growth factor (IGF) (Huang et al., 2013) and Bone morphogenetic proteins (BMPs) (Wu et al., 2016). A number of these secreted factors bind to receptor tyrosine kinases (RTKs), resulting in activation of the mitogen-activated protein kinase (MAPK) pathway. For example, the epidermal growth factor Nrg1 has been shown to promote cardiomyocyte dedifferentiation and proliferation in zebrafish (Gemberling et al., 2015), mice (D'Uva et al., 2015) and infant human hearts (Polizzotti et al., 2015). Likewise, FGFs activate MAPK signaling to promote cellular proliferation and differentiation (Tsang and Dawid, 2004). During zebrafish heart regeneration, FGFs stimulate epicardial cell activation and EMT together with neovascularization (Choi et al., 2013; Lepilina et al., 2006).
To control proper signaling levels, a number of feedback inhibitors, including Dual specificity phosphatase 6 (Dusp6; also known as Mkp3) function to attenuate FGF-stimulated MAPK signaling during embryonic development (Kawakami et al., 2003; Li et al., 2007; Tsang and Dawid, 2004). Dusp6 is a cytoplasmic ERK1/2 phosphatase and its expression is regulated by active FGF signaling during development (Li et al., 2007). Two groups have independently generated Dusp6 mutant mice. Li et al. (2007) reported that Dusp6 mutant mice have higher pERK levels and phenotypes reminiscent of hyperactive FGFR signaling, such as corona craniosynotosis and dwarfism. By contrast, Maillet et al. (2008) noted that Dusp6 mutant mice were phenotypically normal in appearance, but had an enlarged heart owing to increased cardiomyocyte proliferation during development. Importantly, after permanent ligation of the left coronary artery, Dusp6 mutant mice have a smaller area of scar tissue and improved cardiac output, showing that deletion of Dusp6 can promote cardiac repair in mammals. Consistent with these findings, mice overexpressing Dusp6 in the heart have myocardial failure, increased fibrosis and apoptosis (Purcell et al., 2007). In agreement, regenerative capacity is suppressed in zebrafish overexpressing dusp6 (Han et al., 2014).
Here, we report that Dusp6 function limits the regenerative response in the heart. dusp6 mutant zebrafish were generated and showed increased cardiomyocyte proliferation and angiogenesis of the coronary vasculature, accompanied by faster resolution of fibrotic tissue after ventricular resection. Importantly, dusp6 mutant zebrafish can efficiently regenerate the heart even in the presence of Nrg or PDGF receptor (PDGFR) inhibitor, which block regeneration in wild-type (WT) zebrafish. These findings suggest that Dusp6 functions to attenuate Nrg1 and PDGFR signaling after cardiac injury. Moreover, chemical inhibition of Dusp6 using small molecules enhanced cardiac regeneration, offering potential new tools for therapeutic treatments.
RESULTS
dusp6 is expressed in endothelial cells and cardiomyocytes in the adult heart
To determine if dusp6 is expressed in the zebrafish heart and during regeneration, we performed Q-PCR before and after ventricle apex amputation. Within 1 dpa, dusp6 expression was induced after injury and continued to 7 dpa (Fig. 1A). To determine the cell-specific expression of dusp6 we used RNAscope in cryosectioned Tg(fli1a:EGFP)y1 to delineate endothelial cells from cardiomyocytes in adult hearts. In uninjured hearts, dusp6 expression was detected diffusely within the myocardium and compact myocardium (Fig. 1B). dusp6 transcripts were also localized to endothelial cells, as evidenced by colocalization with EGFP+ cells (Fig. 1B). At 3 dpa, dusp6 expression was detected in the myocardium and within endothelial cells (Fig. 1B, Fig. S1). Studies have shown that Dusp6 is expressed in the myocardium and epicardium, thus expression in vessel-like structures was not previously noted (Han et al., 2014).
Fig. 1.

dusp6 is induced upon cardiac injury and expressed in cardiomyocytes and endothelial cells. (A) Q-PCR analysis of dusp6 in adult zebrafish hearts at 1 and 7 days post amputation (dpa), compared with uninjured hearts. dusp6 is induced after ventricle apex amputation. Values are normalized to β-actin and rnap expression. Data represent three independent replicates. *P<0.05, one-way ANOVA. (B) dusp6 expression in uninjured hearts and at 3 dpa using RNAscope. In uninjured hearts, dusp6 (red) is weakly detected throughout the heart including endothelial cells [Tg(fli1a:EGFP)y1], myocardium (Myo) and compact myocardium (CM). At 3 dpa, dusp6 expression was detected in the myocardium and in endothelial cells. Dotted line demarcates the amputation plane. Sections were counterstained with DAPI (blue) to visualize nuclei. Boxed areas are magnified beneath. (C) Confocal image from Tg(dusp6:memGFP)pt21 heart immunostained with anti-GFP and anti-Mef2C, showing that cardiomyocytes express memGFP in uninjured hearts. (D) AFOG images (left) and confocal images (center and right) of Tg(dusp6:memGFP)pt21; Tg(kdrl:NLS:mCherry)is5 hearts at multiple time points after ventricle apex amputation (n=3 for each time point). memGFP is expressed in the vessels of the compact myocardium in uninjured hearts and in the nascent vessels inside the clot after injury. Sections were counterstained with DAPI (blue) to visualize nuclei. Scale bars: 100 µm.
We next generated transgenic lines using the 10 kb dusp6 promoter driving membrane-targeted green fluorescent protein (memGFP) [Tg(dusp6:memGFP)pt21] in order to reveal the activity of dusp6 regulatory sequences by activation of exogenous gene expression in the adult heart. In uninjured adult hearts, weak expression of memGFP in cardiomyocytes and stronger expression in the blood vessels were noted in the compact myocardium (Fig. 1C). To confirm memGFP expression in the vasculature, Tg(dusp6:memGFP)pt21; Tg(kdrl:NLS:mCherry)is5 hearts were analyzed, in which endothelial cell nuclei are labeled in red and dusp6 regulatory sequences drive memGFP expression in green. After injury, memGFP/mCherry double-positive cells were observed in the nascent vessels in the injured area (Fig. 1D). Moreover, memGFP+ cells were detected as early as 3 dpa, suggesting the presence of endothelial cells within the regenerate (Fig. 1D). These observations suggest that dusp6 might serve a crucial early function during cardiac regeneration and is expressed in cardiomyocytes and in the endothelial cells after injury.
dusp6 mutant hearts develop cardiomegaly and have a thicker compact myocardium
To examine the role of Dusp6 in development and heart regeneration, we generated mutant alleles of dusp6 using transcriptional activator-like effector nucleases (TALENs) (Fig. 2A-C) (Bedell et al., 2012; Cade et al., 2012). We generated TALEN constructs targeting 30 bp downstream of the dusp6 initiation codon (Fig. 2A). Four dusp6 mutant alleles (named pt30a-d) were identified (Fig. 2B,C). Western blot analysis confirmed the absence of Dusp6 protein in dusp6pt30a/pt30a embryos, implicating pt30a as a null allele (Fig. 2D).
Fig. 2.

dusp6 mutant hearts exhibit mild cardiomegaly. (A) The TALENs targeting dusp6 exon 1. (B) Recovery of dusp6 mutant alleles pt30a-d. Mismatches created by the 1 bp insertion in pt30d are indicated in red. (C) Predicted amino acid sequence of dusp6pt30 alleles. (D) Western blot showing absence of Dusp6 protein in dusp6pt30a/pt30a embryos. β-actin was used as a loading control. (E,F) Whole-mount images of uninjured WT (E) and dusp6pt30a/pt30a (F) hearts at 5 months of age. dusp6 mutant hearts show cardiomegaly. A, atrium; V, ventricle; BA, bulbus arteriosus. (G) Quantification of the ratio of ventricle area/body weight (VA/BW) in WT (n=5) and dusp6 mutant (n=5) fish. dusp6pt30a/pt30a fish have a larger VA/BW ratio than WT fish. **P<0.01, Student's t-test. (H,I) AFOG staining of uninjured heart sections at 5 months of age. dusp6 mutant hearts (I) have a thicker compact myocardium (brackets) than WT hearts (H). (J) Quantification of compact myocardium thickness in uninjured hearts at 5 months of age for WT (n=4) and dusp6 mutant (n=5). ****P<0.0001, Student's t-test. (K,L) Sections of uninjured WT (n=4) and dusp6 mutant (n=4) fish with the Tg(fli1a:EGFP)y1 background to visualize the endothelium. dusp6 mutant hearts (L) have a thicker compact myocardium (red arrows) containing more vessels than WT hearts (K). (M,N) pERK is detected in fli1a:EGFP+ vessels of the compact myocardium. dusp6 mutant hearts (n=4) (N) have a thicker compact myocardium with more vessels showing pERK staining than in WT uninjured hearts (n=4) (M). Scale bars: 100 µm.
In all cases, homozygous dusp6 mutants developed normally, survived to adulthood at the expected Mendelian frequency and were fertile. Although we did not observe defects during development, we did detect a mild increase in heart size by 5 months of age in dusp6 mutants (Fig. 2E-G). This was accompanied by a thicker compact myocardium (Fig. 2H-J, Fig. S2A) compared with WT zebrafish matched by age and length. This was only noted in adults older than 5 months, as younger and length-matched controls had hearts of normal size. To understand the mechanism underlying the increase in heart size in dusp6 mutants, we measured cardiomyocyte proliferation at 3 and 5 months of age. Cardiomyocyte proliferation index was quantified by immunostaining for both Proliferating cell nuclear antigen (Pcna) and Mef2c (cardiomyocyte marker). dusp6 mutant hearts have significantly more proliferating cardiomyocytes than WT hearts at 3 months (Fig. S2B,C). However, by 5 months there was no difference, suggesting that the dusp6 mutant hearts do not proliferate indefinitely. In addition, measuring cardiomyocyte cell size with wheat germ agglutinin (WGA) staining showed that there was no difference between WT and dusp6 mutant hearts, suggesting that the increase in heart mass was a result of increased total cardiomyocyte number (Fig. S2D,E).
Next, we crossed the dusp6 mutants into Tg(fli1a:EGFP)y1, a transgenic line that marks endothelial cells, and noted that mutant hearts had a thicker compact myocardium containing more vessels than WT hearts (Fig. 2K,L, Fig. S2F). Moreover, the vessels in the compact myocardium of dusp6pt30a/pt30a hearts show increased activated (phosphorylated) ERK (pERK) when compared with WT hearts (Fig. 2M,N, Fig. S3A,B). These findings reveal that dusp6 mutants are phenotypically normal during development but as adults they develop mild cardiomegaly with thickened compact myocardium. This phenotype has features similar to those described for ectopic expression of Nrg1 in the cardiomyocytes of adult zebrafish (Gemberling et al., 2015). nrg1 has been shown to be induced in perivascular cells that surround blood vessels in the compact myocardium after ventricular resection. Given the induction of dusp6 expression in endothelial cells after injury, we hypothesized that Dusp6 could attenuate Nrg1 signaling during heart regeneration.
dusp6 mutant zebrafish exhibit increased cardiomyocyte proliferation and enhanced cardiac regeneration
Since Dusp6 suppresses ERK activity and this pathway is activated by a number of growth factors during regeneration, we reasoned that loss of dusp6 could augment cardiac regeneration. Ventricular apex resection in dusp6pt30a/pt30a zebrafish was performed, and at 7 dpa a significant increase in proliferating cardiomyocytes was noted (Fig. 3A,B). This observation was confirmed in dusp6pt30d/pt30d mutants (Fig. S4A-D). We next determined whether the increased cardiomyocyte proliferation in dusp6 mutants was prevalent throughout heart regeneration. At 4 and 7 dpa, a significant increase in cardiomyocyte proliferation index was noted, but not at later stages (12 and 20 dpa) (Fig. S5A,B), indicating that increased proliferation in dusp6 mutants was limited to the first 10 days after injury.
Fig. 3.
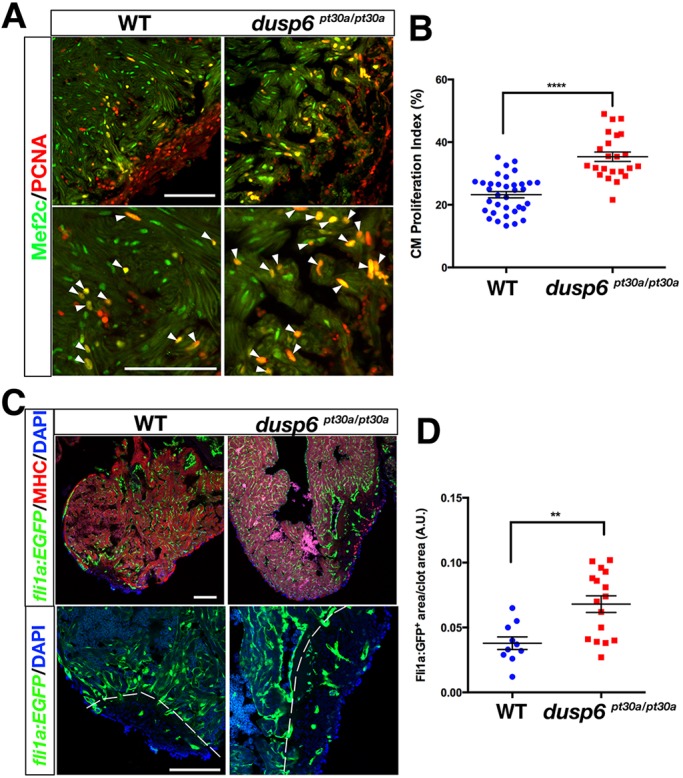
Increased cardiomyocyte proliferation and angiogenesis in dusp6 mutant hearts after cardiac injury. (A) Hearts at 7 dpa, immunostained for Mef2c (green; cardiomyocyte nuclei) and Pcna (red; proliferation marker). Cardiomyocyte proliferation is increased in dusp6 mutant hearts (n=23) compared with WT hearts (n=35). Arrowheads indicate proliferating cardiomyocytes. (B) Quantification of cardiomyocyte proliferation index in WT and dusp6 mutant hearts. ****P<0.0001, Student's t-test. (C) EGFP+ vessels were visualized in Tg(fli1a:EGFP)y1; dusp6pt30a/pt30a (n=16) and Tg(fli1a:EGFP)y1 (n=10) hearts at 8 dpa. Dashed line demarcates the resection plane. (D) Quantification of new vessels formed inside the clot area of hearts at 8 dpa. **P<0.01, Student's t-test. Scale bars: 100 µm.
In order for proper heart regeneration to occur, new blood vessels are required to form to support the regenerating myocardium (Lepilina et al., 2006). We therefore crossed the dusp6pt30a mutation into the Tg(fli1a:EGFP)y1 line. We observed an increased presence of GFP+ cells within the injury by 8 dpa in mutant hearts (Fig. 3C,D). The increased presence of fli1a:EGFP+ vessels in the regenerate at an earlier time point suggests that heart regeneration was accelerated in the dusp6 mutant.
At 20 dpa, dusp6 mutants consistently showed reduced scar tissue in the resected hearts (Fig. 4A-C). Moreover, cardiac function appeared normal in dusp6 mutant hearts based on decreased arrhythmic (irregular heart beat rhythm) events as measured by electrocardiography (ECG) at 20 dpa (Fig. 4D,E, Fig. S6). This was detected as variation in the R-R interval, which represents the time between heart beats. Uninjured zebrafish had normal heart rhythm with a regular R-R interval, and thus the standard deviation of the delta R-R values was low. After ventricular apex amputation, heart rhythm variation in the R-R interval was high in both WT and dusp6pt30a/pt30a zebrafish. By 20 dpa, WT hearts showed irregular R-R intervals, whereas dusp6 mutant heart presented regular R-R intervals, suggesting a restoration of cardiac function (Fig. 4D,E, Fig. S5).
Fig. 4.

dusp6 mutant hearts show accelerated regeneration. (A,B) Sections of hearts at 20 dpa stained with AFOG to visualize the deposition of fibrotic tissue (arrows). dusp6 mutant hearts (n=13) (B) have reduced fibrotic tissue area compared with WT hearts (n=17) (A). (C) Quantitation of fibrotic tissue area at 20 dpa. ***P<0.001, Student's t-test. (D) Representative ECG from WT (n=25) and dusp6 mutants (n=24) at 20 dpa. dusp6 mutant fish exhibit fewer arrhythmic events than WT. ECG are shown from four fish (#1, #10, #11, #13). (E) Quantitation of the arrhythmic events, measured as R-R intervals, in WT and dusp6pt30a/pt30a zebrafish at 20 dpa. ****P<0.0001, Student's t-test. (F) RNA-seq analysis of uninjured hearts and hearts at 20 dpa. At 20 dpa, WT hearts express fibrosis genes, but in dusp6pt30a hearts fibrosis genes are downregulated, suggesting faster regeneration. (G) Q-PCR analysis of selected fibrosis genes in hearts at 20 dpa. dusp6pt30a hearts show downregulation of fibrosis genes. Values are normalized to β-actin and rnap expression. Data are from one representative experiment from three independent biological replicates.
To confirm that dusp6pt30a/pt30a hearts regenerate faster than WT we performed transcriptome analysis of uninjured hearts at 20 dpa (Fig. 4F). In WT hearts, genes known to be associated with cardiac fibrosis were significantly upregulated at 20 dpa. This includes fibronectin (fn1a and fn1b) (Wang et al., 2013), collagen (col1a1b, col1a2, col5a1, col12a1a) (Chablais and Jazwinska, 2012a), TGFβ signaling (smad3b) (Chablais and Jazwinska, 2012b), lysyl oxidase (loxl2a), tenascin C (tnc), caveolin 1 (cav1) (Cao et al., 2016a), lipoprotein lipase (lpl) and periostin (postna and postnb) (Ito et al., 2014) (Fig. 4F, Table S1). By contrast, expression of these fibrosis genes in the dusp6 mutant hearts had already returned to the baseline levels of uninjured hearts, suggesting that regeneration was almost complete at 20 dpa. We performed Q-PCR experiments on several of these genes and confirmed the RNA-seq findings (Fig. 4G). Taken together, these data show that genetic disruption of dusp6 results in enhanced cardiac regeneration.
Chemical inhibition of Dusp6 increases cardiomyocyte proliferation and improves cardiac regeneration
Previously, we employed a transgenic FGF reporter chemical screen for small molecules that modulate FGF/Ras/MAPK activity (Molina et al., 2007; Saydmohammed et al., 2011). We identified 2-benzylidene-3-(cyclohexylamino)-3H-inden-1-one chloride (BCI), which enhanced Ras/MAPK signaling (Molina et al., 2009). BCI, and its analog BCI215, are allosteric inhibitors of Dusp6 (Korotchenko et al., 2014). Since genetic disruption of dusp6 resulted in accelerated heart regeneration, we examined whether chemical suppression of Dusp6 activity would have similar effects. Because we observed toxicity in embryos with long-term treatments of BCI, our first objective was to determine the maximum tolerated dose (MTD) in adult zebrafish of BCI and BCI215. Retro-orbital injections were performed daily for 4 days in adult zebrafish with 0.5, 2.5, 12.5 and 25 mg/kg BCI and BCI215. At 0.5 and 2.5 mg/kg, BCI and BCI215 were well tolerated (Fig. S7A,B), but higher BCI doses (12.5 and 25 mg/kg) resulted in more deaths than BCI215, confirming our previous observations in embryos (Korotchenko et al., 2014).
One day after ventricle apex amputation, BCI (0.5 mg/kg), BCI215 (0.5 mg/kg) or vehicle DMSO was injected for 6 consecutive days and hearts were extracted at 7 dpa to assess cardiomyocyte proliferation, at 8 dpa to assess angiogenesis, and at 25 dpa to measure fibrotic tissue area. Both molecules improved cardiac regeneration in adult zebrafish by increasing the cardiomyocyte proliferation index (Fig. 5A-D) and blood vessel formation (Fig. 5E-H), and reducing scar size (Fig. 5I-L). Importantly, BCI and BCI215 did not increase cell proliferation in uninjured hearts (Fig. S8A,B) and did not cause cardiomegaly (Fig. S8C).
Fig. 5.

Chemical inhibition of Dusp6 increases cardiomyocyte proliferation and angiogenesis and reduces fibrosis after cardiac injury. (A-C) Adult zebrafish hearts at 7 dpa, injected for 6 days with DMSO (n=16) (A), 0.5 mg/kg BCI (n=13) (B) or 0.5 mg/kg BCI215 (n=12) (C) and stained for Mef2c and Pcna. (D) Quantification of proliferating cardiomyocytes at 7 dpa. BCI and BCI215 increased cardiomyocyte proliferation compared with DMSO vehicle. (E-G) Tg(fli1a:EGFP)y1 hearts at 8 dpa injected for 6 days with BCI (n=16) (F), BCI215 (n=19) (G) or DMSO (n=14) (E) and stained for Mef2c. (H) Quantification of new vessels formed inside the clot area of hearts at 8 dpa. (I-K) Sections of hearts at 25 dpa stained with AFOG to visualize the scar. Intact cardiac muscle stains orange-brown, fibrin stains red and collagen blue. Fish injected with BCI (n=12) (J) and BCI215 (n=18) (K) resolved the injury faster than DMSO-injected fish (n=14) (I). (L) Quantification of clot areas at 25 dpa. (D,H,L) ****P<0.0001, ***P<0.001, **P<0.01, *P<0.05, one-way ANOVA. Scale bars: 100 µm.
Several studies have reported that BCI and BCI215 can inhibit other Dusp family members, including Dusp1 (Korotchenko et al., 2014; Molina et al., 2009). To confirm the specificity of BCI in suppressing Dusp6 function we tested whether lower BCI215 doses could enhance cardiac regeneration in heterozygous dusp6pt30a/+ zebrafish. As predicted, a suboptimal dose of BCI215 did not increase cardiomyocyte proliferation in WT hearts at 7 dpa (Fig. S9A,B). However, this suboptimal dose of BCI215 significantly increased cardiomyocyte proliferation in dusp6pt30a/+ hearts (Fig. S9A,B). To confirm BCI215 specificity, we treated dusp6pt30a/pt30a mutants with BCI215 and did not observe increased cardiomyocyte proliferation (Fig. S9C,D). Our data show that chemical inhibition of Dusp6 can enhance cardiac regeneration, offering a potential therapeutic target for enhancing cardiac repair.
PDGFR inhibition reduces the effects of dusp6 mutations on injury-induced cardiomyocyte proliferation
Studies have shown that during zebrafish heart regeneration, several growth factors activating the Ras/MAPK pathway are crucial for proper regeneration to occur. To determine the ligands that Dusp6 attenuates after ventricular resection, we tested the effects of suppressing specific RTKs with small molecules. PDGFβ is induced after cardiac injury and has been demonstrated to stimulate cardiomyocyte proliferation (Lien et al., 2006). WT and dusp6pt30a/pt30a mutant zebrafish were treated with a specific inhibitor of PDGFRs called PDGFR tyrosine kinase inhibitor III (PTKI III) (Matsuno et al., 2002). In WT hearts, we observed a significant decrease in the cardiomyocyte proliferation index after treatment with PTKI III (Fig. 6A,C,E). In the dusp6pt30a/pt30a mutant hearts, by contrast, cardiomyocyte proliferation was not affected by PTKI III treatment (Fig. 6B,D,E). Moreover, in WT hearts PTKI III treatment caused a failure to resolve fibrotic tissue at 20 dpa, as noted from fibrin deposition in the injury area (Fig. 6F,G,J). However, in dusp6pt30a/pt30a hearts, the injured area was much reduced and lacked fibrin, suggesting that even in the presence of a PDGFR inhibitor, normal cardiac regeneration can occur (Fig. 6H-J). These observations suggest that Dusp6 attenuates PDGFR signaling during cardiac regeneration.
Fig. 6.

dusp6 mutant hearts are mildly affected by PDGFR inhibition. (A-D) Hearts at 7 dpa were injected for 6 days with PTKI III (C,D) or vehicle DMSO (A,B) and stained for Mef2c and Pcna to determine cardiomyocyte proliferation. WT hearts (DMSO, n=22; PTKI III, n=19) (A,C) and dusp6pt30a/pt30a hearts (DMSO, n=21; PTKI III, n=15) (B,D). Arrowheads indicate proliferating cardiomyocytes. (E) Quantification of cardiomyocyte proliferation in WT and dusp6 mutant hearts after injection of PTKI III or DMSO. Each point represents the average proliferation index from one heart. (F-I) AFOG staining of WT or dusp6pt30a/pt30a hearts at 20 dpa injected for 6 days with PTKI III (WT, n=8; dusp6pt30a/pt30a, n=12) (H,I) or DMSO (WT, n=10; dusp6pt30a/pt30a, n=7) (F,G). Dashed line demarcates injury zone. (J) Quantification of fibrotic tissue area in hearts at 20 dpa. (E,J) ****P<0.0001, ***P<0.001, **P<0.01; ns, not significant; one-way ANOVA.
Erbb2 inhibition reduces the effects of dusp6 mutation on injury-induced cardiomyocyte proliferation
The mild cardiomegaly and thickened compact myocardium phenotypes in the dusp6 mutants resemble the effects of transgenic Nrg1 overexpression in the zebrafish heart (Gemberling et al., 2015). We hypothesized that Dusp6 attenuates Nrg/Erbb activity. To test this, we monitored expression of errfi1b (also known as mig6), a reported target and feedback attenuator of EGF signaling (Chung et al., 2004; Ferby et al., 2006). Q-PCR analysis showed no difference in errfi1b expression in WT hearts upon injury (Fig. 7A). However, in dusp6 mutant hearts, errfi1b expression was significantly increased, suggesting that Nrg1 signaling was elevated (Fig. 7A).
Fig. 7.

EGF receptor inhibition in dusp6 mutants dampens cardiac regeneration. (A) Q-PCR analysis of errfi1b at 0 and 3 dpa. After amputation, dusp6 mutant hearts show higher errfi1b expression compared with WT. Data represent six independent replicates. (B-E) Hearts at 7 dpa injected for 6 days with AG1478 (C,E), or DMSO vehicle (B,D) and stained for Mef2c and Pcna to determine cardiomyocyte proliferation. Arrowheads indicate proliferating cardiomyocytes. For WT: DMSO, n=36; 10 µM and 25 µM AG1478, n=17 and n=14, respectively. For dusp6pt30a/pt30a: DMSO, n=31; 10 µM and 25 µM AG1478, n=19 and n=13, respectively. (F) Quantification of cardiomyocyte proliferation in WT and dusp6 mutant hearts after injection of AG1478 or DMSO. (G-J) Tg(fli1a:EGFP)y1 hearts at 8 dpa injected with AG1478 (H,J) or DMSO (G,I). MHC (red) marks the resection plane. AG1478 treatment blocked angiogenesis in WT hearts, but not in dusp6 mutant hearts. Dashed line demarcates resection plane. n=17 for DMSO in WT and dusp6pt30a/pt30a; n=7 for 25 µM AG1478 in WT and dusp6pt30a/pt30a. (K) Quantification of new vessels formed at 8 dpa. (L) Quantification of cardiomyocyte proliferation in erbb2st61/+ hearts at 7 dpa, injected with BCI215 (n=13) or DMSO (n=14) as compared with sibling WT hearts (DMSO, n=17). erbb2st61/+ hearts have a diminished cardiomyocyte proliferation index compared with WT hearts, and this is rescued by BCI215. (A,F,K,L) ****P<0.0001, ***P<0.001, *P<0.05; ns, not significant; one-way ANOVA. Scale bars: 100 µm.
We next injected AG1478, a known inhibitor of EGF receptors including Erbb2 (Gemberling et al., 2015), into dusp6 mutant zebrafish after ventricular amputation to determine if cardiac regeneration can be suppressed. Retro-orbital injection of AG1478 efficiently decreased cardiomyocyte proliferation in WT hearts (Fig. 7B-F, Fig. S10A,B). Although injecting the same dose of AG1478 reduced cardiomyocyte proliferation in dusp6 mutant hearts (Fig. 7D-F), the proliferation index was similar to that of control DMSO-treated WT hearts. We injected AG1478 into dusp6pt30a/pt30a; Tg(fli1a:EGFP)y1 to monitor endothelial cells in the regenerate. EGF receptor inhibition blocked new vessel formation in WT hearts (Fig. 7G,H,K), but did not affect vessel formation in dusp6pt30a/pt30a hearts (Fig. 7I-K). Fibrotic tissue was still present in WT hearts after AG1478 treatment, suggesting that heart regeneration was inhibited by blocking Nrg signaling (Fig. S11). However, dusp6pt30a/pt30a hearts exhibited much reduced fibrotic tissue area (Fig. S11). These data suggest that Nrg signaling is increased in dusp6 mutant hearts and that suppressing Nrg receptor activity is not sufficient to block regeneration.
We next tested whether cardiomyocyte proliferation can be rescued in zebrafish harboring heterozygous mutations in erbb2 (erbb2st61/+). Homozygous erbb2 mutants do not survive to adults as they fail to trabeculate their hearts (Lyons et al., 2005). Because Nrg1 is induced during heart regeneration in zebrafish (Gemberling et al., 2015), we reasoned that heterozygous erbb2st61/+ carriers might show decreased cardiomyocyte proliferation. This was confirmed as there was a significant decrease in the presence of Pcna+ cardiomyocytes at 7 dpa (Fig. 7L). Treatment of heterozygous erbb2st61/+ zebrafish with BCI215 resulted in a mild rescue of cardiomyocyte proliferation (Fig. 7L).
To directly test the effects of BCI on Nrg1 activity, we employed the rat primary cardiomyocyte proliferation assay. In these experiments, primary neonatal rat ventricular cardiomyocytes (NRVM) are cultured in vitro in the presence or absence of NRG1, a known cardiomyocyte mitogen (Bersell et al., 2009). NRG1 treatment increased cardiomyocyte proliferation as marked by increased phosphorylated Histone H3 (H3P) (Fig. 8). The addition of BCI together with NRG1 resulted in a significant increase in cardiomyocyte proliferation (Fig. 8). Taken together, our data show that the effects of blocking Nrg and PDGF signaling during heart regeneration are significantly reduced in the absence of Dusp6.
Fig. 8.
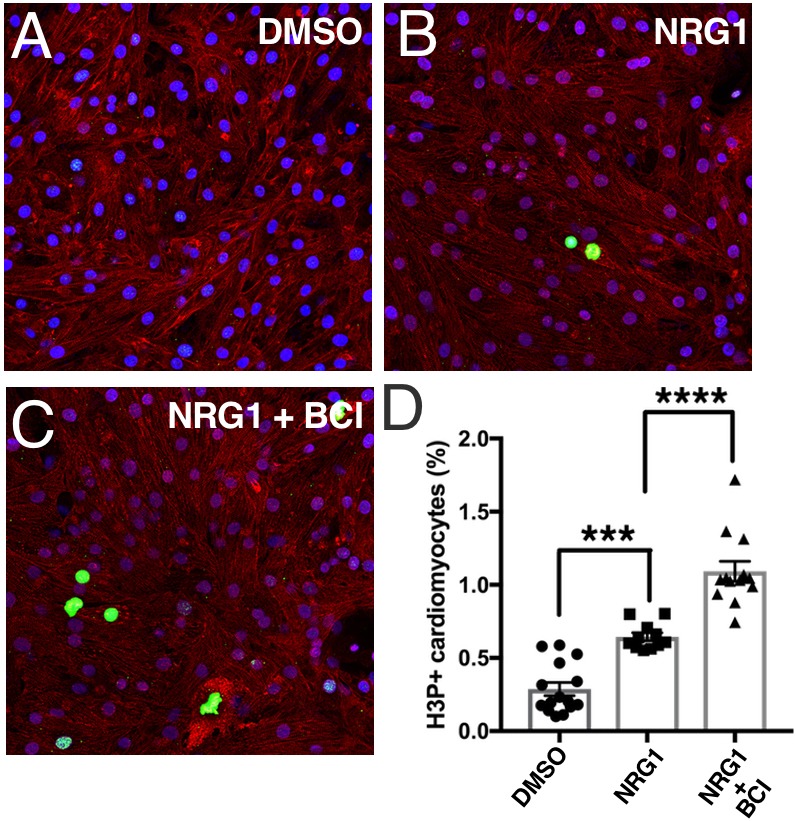
BCI augments NRG1 activity in primary rat cardiomyocytes. Postnatal day 1 rat neonatal ventricular myocytes were isolated and cultured in 10% FBS. Cells were exposed to DMSO vehicle (A), NRG1 (50 ng/ml) (B) or NRG1 plus BCI at 5 µM (C) for 3 days. All treatments were performed in the presence of 10% FBS. Cardiomyocyte proliferation was quantified by H3P staining (n=2). (D) BCI treatment augments the effect of NRG1 on neonatal cardiomyocyte proliferation. ****P<0.0001, ***P<0.001, ANOVA followed by Bonferroni post-hoc test.
DISCUSSION
Dusp6 is an important regulator of Ras/MAPK signaling, and this pathway is utilized to control epicardial cell activation, cardiomyocyte proliferation and angiogenesis. Previous reports based on in situ hybridization staining have revealed dusp6 expression within cardiomyocytes and in the epicardial layer (Han et al., 2014; Itou et al., 2012b). Here, using double-transgenic zebrafish, we found that the dusp6 promoter can drive memGFP expression in endothelial cells of the compact myocardium in uninjured hearts and, after amputation, that memGFP is induced in the nascent vessels in the injury area. This expression in endothelial cells is compelling given a recent study that showed nrg1 induction in perivascular cells and the rapid appearance of vessels soon after injury (Gemberling et al., 2015; Marin-Juez et al., 2016). Thus, dusp6 is expressed in the epicardium and cardiomyocytes and, after cardiac injury, is induced in endothelial cells at sites juxtaposed to cells expressing nrg1. Marin-Juez et al. (2016) demonstrated a requirement for rapid revascularization in zebrafish heart regeneration and that suppressing this process affected cardiomyocyte proliferation. These findings suggest that a positive interaction exists between endothelial cells and cardiomyocytes that supports heart regeneration.
To delineate Dusp6 function in zebrafish heart regeneration, we disrupted Dusp6 using TALENs. dusp6 mutants are overtly normal and do not show embryonic or adult lethality. Previous studies have shown that Dusp6 attenuates FGF signaling during early development (Li et al., 2007; Tsang et al., 2004). We anticipated developmental defects including dorsal/ventral patterning, heart, central nervous system, craniofacial and fin formation as FGFs are known to be crucial for the proper development of these tissues. However, analysis of dusp6 mutant embryos did not reveal any developmental defects normally associated with increased FGF activity. It might not be surprising that zebrafish dusp6 mutant embryos are normal, as a similar lack of developmental defects was described for mouse Dusp6 mutants (Li et al., 2007; Maillet et al., 2008). These findings suggest that either dusp6 is dispensable for embryonic development or that redundancy with other Dusp family members can compensate for the absence of dusp6. For example, Dusp2, Dusp4, Dusp5 and Dusp7 can dephosphorylate ERK1/2 and compensate for the loss of dusp6 (Kidger and Keyse, 2016; Kondoh and Nishida, 2007). Alternatively, since several FGF negative-feedback regulators such as sef (also known as il17rd) and sprouty family genes are expressed early in development, the loss of dusp6 may be compensated by their activity (Fürthauer et al., 2001, 2002; Tsang et al., 2002). Although early embryonic and larval development appeared normal, we did observe differences between dusp6 mutant and WT adult zebrafish. After ventricular resection, dusp6pt30a/pt30a mutants showed accelerated regeneration, in line with the observations from Dusp6 null mice subjected to MI (Li et al., 2007; Maillet et al., 2008). In addition, dusp6 mutant zebrafish show elevated cardiomyocyte proliferation and angiogenesis. The induction of GFP expression by the dusp6 enhancer in endothelial cells (Fig. 1) coupled with increased pERK levels (Fig. 2N) suggests that the main function of Dusp6 is in the coronary vasculature after injury.
To determine which growth factor signaling pathway Dusp6 attenuates during heart growth and regeneration, we blocked RTK signaling with specific small molecule inhibitors of PDGFR and Erbb2. In the absence of Dusp6, PTKI III, an inhibitor of PDGFR, was unable to suppress heart regeneration. A similar effect was noted with the Erbb2 inhibitor AG1478. Our findings point to a general role for Dusp6 as an attenuator of Ras signaling via multiple ligands during heart regeneration. The phenotypic similarity between dusp6 mutant hearts and Nrg1 overexpression in zebrafish is intriguing and suggests that Dusp6 might attenuate Nrg1 signaling during physiological growth of the heart. Both dusp6 mutant and Nrg1 transgenic adult hearts show cardiomegaly and an expanded compact myocardium, albeit much milder in the dusp6 mutants (Gemberling et al., 2015). For this reason, we hypothesized that dusp6 mutant zebrafish have higher levels of Nrg1/Erbb2 signaling. This was confirmed by monitoring the expression of a specific target of EGF signaling, errfi1b, which was increased in dusp6 mutant hearts. A consequence of Dusp6 deficiency is that the threshold concentration of ligand required to stimulate proliferation is reduced. Previous work has shown a role for FGFR, IGF and PDGFR in angiogenesis and cardiomyocyte proliferation after injury (Kim et al., 2010; Lepilina et al., 2006). It will be of interest to determine in the future whether Dusp6 can also impact these pathways for heart regeneration. Furthermore, the exact cell type in which Dusp6 functions is not clearly defined in this study. One model is that Dusp6 has multiple roles in attenuating growth factors and this is achieved by its activity in different cells. Another interpretation from this work is based on the observation of strong dusp6 enhancer activity in endothelial cells after injury. This is crucial in regulating cardiomyocyte proliferation, suggesting that any role of Dusp6 is indirect. Tissue-specific inactivation of Dusp6 is likely to uncover the exact role of this phosphatase during heart regeneration.
We confirmed that inhibiting Dusp6 with small molecules could enhance heart regeneration in zebrafish (Han et al., 2014). Increased cardiomyocyte proliferation, the earlier formation of coronary vessels within the regenerate, and reduced scar tissue deposition within the wound apex with BCI or BCI215 treatment was observed. The increased proliferation of cardiomyocytes in amputated hearts was observed within 4-7 dpa and not detected beyond 12 dpa. This is likely to reflect growth factor activity being at its highest within this time frame to support cardiomyocyte proliferation, and this is the period when Dusp6 functions to attenuate the Ras pathway. Han et al. (2014) reported that cardiac injury resulted in induction of Dusp6 expression through H2O2 generation, and that this induction is a limiting factor in heart regeneration. In their studies, the focus was on Dusp6 activity after 7 dpa and they noted that BCI could increase the cardiomyocyte proliferation index at 14 dpa in the absence of H2O2. In our study, we focused on the earlier events (from 1 to 7 dpa) and noted that blocking Dusp6 early was also sufficient to enhance cardiac regeneration. Importantly, this enhancement in proliferation was observed only after cardiac amputation and not in uninjured hearts, implying that the effects of these compounds are dependent upon injury. It will be interesting to determine whether BCI and BCI215 can augment heart regeneration in other injury models, such as cryoinjury (Chablais and Jazwinska, 2012a), cardiomyocyte ablation (He et al., 2016; Wang et al., 2011) or in the neonatal mouse (Polizzotti et al., 2016). Recent findings show that Nrg1 can stimulate heart repair in mammals (D'Uva et al., 2015; Polizzotti et al., 2015), so the possibility of using BCI to augment Nrg1 signaling could prove to be important in the treatment of heart disease. One complication with Nrg1 therapies is that they have been implicated to induce tumor formation: in NRG1 trials it was found that breast cancer can arise from activated mutation in the NRG1 receptor ERBB2 (Rimawi et al., 2015). The utility of recombinant human NRG1 in cardiac regenerative therapy could be limited owing to this potential side effect of increasing cancer risk. However, a recent study in mice points to an absence of neoplastic growth with Nrg1 administration (Ganapathy et al., 2016). Nevertheless, using lower concentrations of Nrg1 together with chemical inhibition of Dusp6 could stimulate cardiomyocyte proliferation for cardiac repair.
MATERIALS AND METHODS
Generation of dusp6 mutant zebrafish
Two pairs of TALENs were designed against the translational initiation site in exon 1 of the dusp6 gene. TALEN repeat variable diresidues (RVDs) were assembled in two steps by the Golden Gate method using pT3TS (Cermak et al., 2015) and subcloned into pCS2+ containing the Fok1 homodimer domain. mRNA was synthesized using the mMESSAGE mMACHINE Kit (Ambion). mRNAs encoding each TALEN were co-injected into one-cell stage zebrafish embryos. Heritable TALEN-induced mutations in the F1 and F2 generation were evaluated by genomic DNA PCR (primers 5′-GTGGCTCGCGCACTCACAGGCTA-3′ and 5′-GGAGCCGCCGTCGATGTTTTCA-3′) followed by restriction fragment length polymorphisms that detect disruption of the ClaI site in exon 1 of dusp6.
Zebrafish maintenance, ventricular amputation, and retro-orbital injections
The zebrafish experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Pittsburgh, which conforms to NIH guidelines. Adult (6- to 18-month-old) wild-type AB* and Tü, transgenic Tg(fli1a:EGFP)y1 (Lawson and Weinstein, 2002), Tg(kdrl:NLS:mCherry)is5 (Wang et al., 2010), and mutant dusp6pt30a-d and erbb2st61 (Lyons et al., 2005) zebrafish were maintained at 28°C. Tg(dusp6:memGFP)pt21 lines were generated using the 10 kb dusp6 promoter as previously described (Molina et al., 2007). Both homozygous and heterozygous dusp6pt30a and dusp6pt30d alleles were used in the experiments.
To measure compact myocardium thickness and cardiomyocyte proliferation rate in uninjured hearts, zebrafish of 3 and 5 months of age were used. Ventricle area normalized to body weight (VA/BW) was measured for each fish at the indicated time and expressed in mm2/g. Ventricle apex amputation was performed as described previously (Poss et al., 2002). Approximately 20% of the ventricle apex was resected and zebrafish returned to aquaria with standard feeding and husbandry. Retro-orbital injections were performed as previously described (Pugach et al., 2009). After surgery, fish were allowed to recover for 24 h, and compounds were retro-orbitally injected daily, for a maximum of 6 days. Control fish were injected with 3 µl 50% DMSO (Sigma, D8418) in filtered PBS. Fish were injected with 3 µl 0.5 mg/kg BCI (Sigma, 317496) (Molina et al., 2009) or its analog BCI215 (chemically synthesized by the University of Pittsburgh Chemistry Department) (Korotchenko et al., 2014) dissolved in DMSO. For the suboptimal dose experiment, fish were injected with 0.25 mg/kg BCI215. To inhibit PDGFR, 10 µM 4-(6,7-dimethoxyquinazolin-4-yl)-N-(4-phenoxyphenyl) piperazine-1-carboxamide (PTKI III; Santa Cruz, sc-204173) was injected daily for 6 days. To inhibit Erbb2, 10 µM or 25 µM AG1478 (Sigma, T4182) was injected daily for 6 days.
RNAscope
RNAscope [Advanced Cell Diagnostics (ACD)] was performed on uninjured and injured (3 or 7 dpa) hearts isolated from WT and Tg(fli1a:EGFP)y1 adult zebrafish. Hearts were fixed overnight at 4°C in 4% paraformaldehyde (PFA), transferred into a sucrose gradient (10-20-30% sucrose in PBS) the following day at 4°C before cryopreservation overnight. Tissue was embedded in Surgipath Cryo-Gel (Leica, 39475237) and sectioned at 14 µm. RNAscope probe hybridization, amplification and immunostaining were performed following the protocol provided in the RNAscope Multiplex Fluorescent Reagent Kit v2 user manual (ACD). ACD designed the dusp6 probe used in this study. Following the final wash step of the RNAscope probe hybridization protocol, immunofluorescent staining was performed to better visualize endogenous GFP or cardiomyocyte nuclei. Primary antibodies for immunostaining were chicken anti-GFP (1:1000; Aves Labs, GFP-1020) and rabbit polyclonal anti-Mef2c (1:500; Santa Cruz, sc-313). Secondary antibodies for immunostaining were fluorescein goat anti-chicken 488 (1:1000; Aves Labs, F-1005) and Alexa Fluor 488 goat anti-rabbit IgG peroxidase conjugate (1:1000; Invitrogen, A11008). Slides were sealed using ProLong Diamond Antifade Mountant with DAPI (Invitrogen, P36962). Images were taken on a Zeiss 700 confocal microscope at 20× and 40×. Image analysis was performed using ImageJ Fiji (NIH).
Immunostaining, fibrotic tissue area, cell counting and size measurement
For histological examination, hearts were collected in cold PBS and fixed in 4% PFA in PBS for 2 h at room temperature. After two washes in PBS, a 30% sucrose solution in PBS was used to cryopreserve the tissues before immersion in embedding medium (Leica). 14 µm cryosections were collected and consecutive sections used for immunostaining, and for Acid Fuchsin Orange G (AFOG) staining as previously described (Poss et al., 2002). Images were taken with a Leica MZ16 microscope and Q Imaging Retiga 1300 camera. Fibrotic areas were measured using ImageJ. For each heart, the average of the sum of the scar area was calculated from the four largest central sections.
Primary antibodies used for immunostaining were chicken anti-GFP (Aves Labs, GFP-1020; 1:100), rabbit polyclonal anti-Mef2c (Santa Cruz, sc-313; 1:500), mouse monoclonal anti-PCNA (Sigma, P8825; 1:3000), mouse monoclonal anti-MAPK, activated (diphosphorylated ERK1/2) (Sigma, M8159; 1:100) and mouse anti-MHC (DSHB, F59; 1:50). Secondary antibodies were Alexa Fluor 488 goat anti-rabbit IgG peroxidase conjugate (Invitrogen, A11008; 1:1000), Alexa Fluor 594 goat anti-mouse IgG (H+L) (Invitrogen, A11005; 1:1000), Alexa Fluor 488 goat anti-mouse IgG (H+L) (Invitrogen, A11001; 1:1000) and Alexa Fluor 488 goat anti-chicken (Thermo Fisher, A11039; 1:300). Slides were mounted with Vectashield mounting medium with DAPI (Vector Laboratories, H-1200). Images were taken with a Zeiss LSM 700 confocal microscope. For each experiment, at least four sections were analyzed for each heart. Slides stained with only secondary antibody were used as negative control.
Cardiomyocyte proliferation index (%) was calculated from the number of Mef2c+ Pcna+ cells among Mef2c+ cells. Nascent vasculature was quantified by measuring (ImageJ) the areas of vessels formed in the regenerate in Tg(fli1a:EGFP)y1 hearts and dividing by the injury area. Vessel density in uninjured hearts was quantified by measuring (ImageJ) the areas of vessels within the compact myocardium in Tg(fli1a:EGFP)y1 hearts and dividing by the compact myocardium area, to give the percentage of vessels per compact myocardium. Four images were taken for each heart section and the average was calculated for each heart.
pERK1/2 staining (%) was measured with ImageJ as pERK1/2+ immunostained area divided by compact myocardium area. Two to ten sections were used for each heart and the average was calculated for each heart. The thickness of the compact myocardium was measured from the average of 16 measurements for each heart section.
To measure cardiomyocyte size, sections were stained with mouse anti-MHC (DSHB, F59; 1:50), Rhodamine wheat germ agglutinin (WGA) (Vector Laboratories, RL-1022; 1:500) for the detection of cell borders, and DAPI (1:1000) to label nuclei. We followed the methods described by Nguyen et al. (2014) to measure cardiomyocyte size. Only round cells triple positive for MHC, WGA and DAPI staining were measured using ImageJ. For each ventricle, four confocal images were taken using a 60× objective and the cardiomyocyte size was averaged. For each heart, at least four sections were analyzed and 120-150 cells were measured. The ventricular wall thickness was measured in transverse section after AFOG staining using ImageJ. Four measurements were made for each heart section, and for each heart at least four sections were analyzed.
RNA extraction, cDNA synthesis and quantitative PCR (Q-PCR)
Total RNA was extracted from uninjured hearts and hearts at 6 h post amputation or 1, 3 and 7 dpa using TRIzol (Invitrogen) and the RNeasy Micro Kit (Qiagen). Eight hearts were pooled together for each condition. RNA (1 µg) was reverse transcribed to cDNA with SuperScript reverse transcriptase (Invitrogen) using random hexamers. Q-PCR was performed as described previously (Missinato et al., 2015). β-actin (actb2) and RNA polymerase (rnap; also known as polr2d) were used to normalize gene expression in the Q-PCR experiments. Primers for Q-PCR are listed in Table S2. At least three independent biological replicates were performed.
RNA-seq sample preparation and data analysis
For the RNA-seq experiment, only ventricles were collected, dissecting out atria and outflow tract with the use of a micro-scalpel. Total RNA was extracted from uninjured and 20 dpa hearts using TRIzol (Invitrogen) and the RNeasy Micro Kit (Qiagen). Two ventricles were pooled for each condition. Tufts Genomic Core (http://tucf-genomics.tufts.edu) prepared the libraries and ran RNA-seq using an Illumina HiSeq 2500 with 150 bp single-end reads. RNA-seq reads were analyzed using the CLC Genomics Workbench software package (Qiagen).
Western blot
WT and dusp6 mutant zebrafish embryos (24 h post fertilization) were dechorionated and deyolked by gently pipetting in ice-cold PBS with proteinase/phosphatase inhibitor. Deyolked embryos were centrifuged at 5000 rpm (2300 g) for 5 min. After removing PBS, embryos were lysed in Laemmli buffer. Equal amounts of protein (50 µg) were heat denatured and separated by 10% SDS-PAGE and transferred to nitrocellulose membrane using a semi-dry blot system (Bio-Rad). Blots were scanned using the Li-COR Odyssey CLx infrared imaging system, and band intensities were quantified and normalized using Image Studio software (Li-COR). Antibodies were diluted in Odyssey Blocking Buffer containing 0.2% Tween 20 (National Diagnostics). Primary antibodies were mouse monoclonal Dusp6 (Sigma, WH0001848M1; 1:50) and goat polyclonal β-actin (Santa Cruz, sc-1615; 1:100). Secondary antibodies (1:15,000) were IRDye 680 donkey anti-goat (Li-COR, 926-68024) and IRDye 800 donkey anti-mouse (Rockland, 610-731-002).
Electrocardiogram (ECG)
ECG was performed in adult zebrafish 1 day prior to ventricle amputation, and zebrafish were kept in single tanks to allow tracking. At day 0, surgery was performed and ECG was recorded at 20 dpa using the iWorx ECG system, and selecting the zebrafish ECG settings. Each adult zebrafish was anesthetized for 2 min in Tricaine (MS-222, Sigma) and positioned on its back on the pedestal located on top of the grounded aluminium base. Gently, the Ag/AgCl surface electrodes were placed on the fish axially along the center line of the ventral surface. One electrode was placed near the gills and the other next to the heart. ECG was recorded for 3 min. Data were analyzed with Labscribe3 software (iWorx) and the R-R interval was measured as previously described (Zhang et al., 2014). Data were expressed as standard deviation of delta R-R values.
Neonatal ventricular cardiomyocyte (NRVM) isolation and treatments
NRVM from 1-day-old Sprague Dawley rats (Charles River) were isolated using a commercially available kit (Cellutron, nc-6031). Cells were digested to yield single-cell suspensions and pre-plated for 1 h to purify cardiomyocytes from other cell populations. Cells were cultured for 3 days in NS medium containing 5% FBS (Cellutron, m-8031) on glass coverslips coated with 10 µg/ml fibronectin (BD Biosciences). Isolated cardiomyocytes were treated with vehicle control (DMSO), recombinant Neuregulin 1 (NRG1, 50 ng/ml), or a combination of NRG1 and BCI (5 µM) for 72 h.
Immunofluorescence of cultured cardiomyocytes
The cultured cells were fixed with 4% PFA for 12 min at room temperature. Samples were blocked with 5% goat serum containing Triton X-100 (0.5 μl/ml) for 30 min at room temperature. Following blocking they were incubated in primary antibody solutions overnight at 4°C: anti-phospho-Histone H3 (rabbit IgG, Millipore, 06-570; 1:200), anti-α-actinin (mouse IgG1, Sigma, A7811; 1:200). Cells were then washed with PBS and incubated with secondary antibodies in PBS for 1 h at room temperature: Alexa Fluor 488 goat anti-rabbit IgG (Thermo Fisher, A11008; 1:500), Alexa Fluor 594 goat anti-mouse IgG (Thermo Fisher, A11032; 1:500); followed by nuclear counterstaining in Hoechst solution (Invitrogen; 1:1000) for 5 min at room temperature. The cells were mounted in 10 μl mounting medium containing 1% n-propyl gallate (NPG) dissolved in glycerol and sealed with nail polish. All imaging was performed on a Nikon A1-R confocal using a 40× oil immersion objective followed by analysis using Nikon Elements software. Scoring of H3P+ cells was performed in a blinded fashion for quantitative analysis. Rat NRVM were isolated from two separate litters for the experiment. In each litter, the animals were divided into two groups and NRVM were isolated independently from each group. Isolated cells were plated and treated on duplicate coverslip wells for staining and quantification. Imaging of 20× fields was performed on eight random fields per coverslip (∼1000 cardiomyocytes per field) and then averaged between the duplicate coverslips. Final data from isolations from the two different donor litters were statistically analyzed.
Statistical analysis
Statistical analyses were performed with GraphPad Prism version 7.0. Statistical significance was analyzed by unpaired Student's t-test and one-way ANOVA. Data are shown as mean±s.e.m. P<0.05 was considered significant. Statistical significance between the multiple treatments on rat cardiomyocytes was assessed by one-way ANOVA followed by post-hoc Bonferroni correction.
Supplementary Material
Acknowledgements
We thank Takis Benos for discussion on RNA-seq data analysis; Donghun Shin and Neil Hukriede for critical reading of the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: M.A.M., M.S., D.A.Z., K.S.R., B.K., M.T.; Methodology: M.A.M., M.S., D.A.Z., K.S.R., G.W.O., B.K., M.T.; Validation: M.A.M., M.T.; Formal analysis: M.A.M., M.S., D.A.Z., K.S.R., G.W.O., B.K., M.T.; Investigation: M.A.M., M.S., D.A.Z., K.S.R., M.T.; Data curation: M.T.; Writing - original draft: M.A.M.; Writing - review & editing: M.A.M., M.S., D.A.Z., K.S.R., G.W.O., B.K., M.T.; Supervision: M.T.; Project administration: M.T.; Funding acquisition: B.K., M.T.
Funding
This work was supported by funding from the American Heart Association (14GRNT20480183) and the National Institutes of Health (NIH) (R01HD053287). D.A.Z. is supported by a T32 training grant from the NIH (T32 EB001026). This research was supported by the Richard King Mellon Foundation Institute for Pediatric Research (Children's Hospital of Pittsburgh of UPMC, to B.K.) and by a Transatlantic Network of Excellence grant from Fondation Leducq (15CVD03 to B.K.). Deposited in PMC for release after 12 months.
Data availability
RNA-seq data are deposited at Gene Expression Omnibus with accession number GSE90595.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.157206.supplemental
References
- Ahuja P., Sdek P. and MacLellan W. R. (2007). Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol. Rev. 87, 521-544. 10.1152/physrev.00032.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedell V. M., Wang Y., Campbell J. M., Poshusta T. L., Starker C. G., Krug R. G. II, Tan W., Penheiter S. G., Ma A. C., Leung A. Y. et al. (2012). In vivo genome editing using a high-efficiency TALEN system. Nature 491, 114-118. 10.1038/nature11537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersell K., Arab S., Haring B. and Kühn B. (2009). Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138, 257-270. 10.1016/j.cell.2009.04.060 [DOI] [PubMed] [Google Scholar]
- Cade L., Reyon D., Hwang W. Y., Tsai S. Q., Patel S., Khayter C., Joung J. K., Sander J. D., Peterson R. T. and Yeh J.-R. J. (2012). Highly efficient generation of heritable zebrafish gene mutations using homo- and heterodimeric TALENs. Nucleic Acids Res. 40, 8001-8010. 10.1093/nar/gks518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J., Navis A., Cox B. D., Dickson A. L., Gemberling M., Karra R., Bagnat M. and Poss K. D. (2016a). Single epicardial cell transcriptome sequencing identifies Caveolin 1 as an essential factor in zebrafish heart regeneration. Development 143, 232-243. 10.1242/dev.130534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao N., Huang Y., Zheng J., Spencer C. I., Zhang Y., Fu J.-D., Nie B., Xie M., Zhang M., Wang H. et al. (2016b). Conversion of human fibroblasts into functional cardiomyocytes by small molecules. Science 352, 1216-1220. 10.1126/science.aaf1502 [DOI] [PubMed] [Google Scholar]
- Cermak T., Starker C. G. and Voytas D. F. (2015). Efficient design and assembly of custom TALENs using the Golden Gate platform. Methods Mol. Biol. 1239, 133-159. 10.1007/978-1-4939-1862-1_7 [DOI] [PubMed] [Google Scholar]
- Chablais F. and Jazwinska A. (2012a). Induction of myocardial infarction in adult zebrafish using cryoinjury. J. Vis. Exp. 62, 3666 10.3791/3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chablais F. and Jazwinska A. (2012b). The regenerative capacity of the zebrafish heart is dependent on TGFbeta signaling. Development 139, 1921-1930. 10.1242/dev.078543 [DOI] [PubMed] [Google Scholar]
- Choi W.-Y., Gemberling M., Wang J., Holdway J. E., Shen M.-C., Karlstrom R. O. and Poss K. D. (2013). In vivo monitoring of cardiomyocyte proliferation to identify chemical modifiers of heart regeneration. Development 140, 660-666. 10.1242/dev.088526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H. A., Hyodo-Miura J., Kitayama A., Terasaka C., Nagamune T. and Ueno N. (2004). Screening of FGF target genes in Xenopus by microarray: temporal dissection of the signalling pathway using a chemical inhibitor. Genes Cells 9, 749-761. 10.1111/j.1356-9597.2004.00761.x [DOI] [PubMed] [Google Scholar]
- D'Uva G., Aharonov A., Lauriola M., Kain D., Yahalom-Ronen Y., Carvalho S., Weisinger K., Bassat E., Rajchman D., Yifa O. et al. (2015). ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat. Cell Biol. 17, 627-638. 10.1038/ncb3149 [DOI] [PubMed] [Google Scholar]
- Ebelt H., Zhang Y., Kampke A., Xu J., Schlitt A., Buerke M., Muller-Werdan U., Werdan K. and Braun T. (2008). E2F2 expression induces proliferation of terminally differentiated cardiomyocytes in vivo. Cardiovasc. Res. 80, 219-226. 10.1093/cvr/cvn194 [DOI] [PubMed] [Google Scholar]
- Ferby I., Reschke M., Kudlacek O., Knyazev P., Pantè G., Amann K., Sommergruber W., Kraut N., Ullrich A., Fassler R. et al. (2006). Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation. Nat. Med. 12, 568-573. 10.1038/nm1401 [DOI] [PubMed] [Google Scholar]
- Fürthauer M., Reifers F., Brand M., Thisse B. and Thisse C. (2001). sprouty4 acts in vivo as a feedback-induced antagonist of FGF signaling in zebrafish. Development 128, 2175-2186. [DOI] [PubMed] [Google Scholar]
- Fürthauer M., Lin W., Ang S. L., Thisse B. and Thisse C. (2002). Sef is a feedback-induced antagonist of Ras/MAPK-mediated FGF signalling. Nat. Cell Biol. 4, 170-174. 10.1038/ncb750 [DOI] [PubMed] [Google Scholar]
- Ganapathy B., Nandhagopal N., Polizzotti B. D., Bennett D., Asan A., Wu Y. and Kühn B. (2016). Neuregulin-1 administration protocols sufficient for stimulating cardiac regeneration in young mice do not induce somatic, organ, or neoplastic growth. PLoS ONE 11, e0155456 10.1371/journal.pone.0155456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemberling M., Karra R., Dickson A. L. and Poss K. D. (2015). Nrg1 is an injury-induced cardiomyocyte mitogen for the endogenous heart regeneration program in zebrafish. Elife 4, e05871 10.7554/eLife.05871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Rosa J. M., Peralta M. and Mercader N. (2012). Pan-epicardial lineage tracing reveals that epicardium derived cells give rise to myofibroblasts and perivascular cells during zebrafish heart regeneration. Dev. Biol. 370, 173-186. 10.1016/j.ydbio.2012.07.007 [DOI] [PubMed] [Google Scholar]
- Han P., Zhou X.-H., Chang N., Xiao C.-L., Yan S., Ren H., Yang X.-Z., Zhang M.-L., Wu Q., Tang B. et al. (2014). Hydrogen peroxide primes heart regeneration with a derepression mechanism. Cell Res. 24, 1091-1107. 10.1038/cr.2014.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J., Wang Y., Missinato M. A., Onuoha E., Perkins L. A., Watkins S. C., St Croix C. M., Tsang M. and Bruchez M. P. (2016). A genetically targetable near-infrared photosensitizer. Nat. Methods 13, 263-268. 10.1038/nmeth.3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Harrison M. R., Osorio A., Kim J., Baugh A., Duan C., Sucov H. M. and Lien C.-L. (2013). Igf signaling is required for cardiomyocyte proliferation during zebrafish heart development and regeneration. PLoS ONE 8, e67266 10.1371/journal.pone.0067266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Morioka M., Kimura S., Tasaki M., Inohaya K. and Kudo A. (2014). Differential reparative phenotypes between zebrafish and medaka after cardiac injury. Dev. Dyn. 243, 1106-1115. 10.1002/dvdy.24154 [DOI] [PubMed] [Google Scholar]
- Itou J., Kawakami H., Burgoyne T. and Kawakami Y. (2012a). Life-long preservation of the regenerative capacity in the fin and heart in zebrafish. Biol. Open 1, 739-746. 10.1242/bio.20121057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itou J., Oishi I., Kawakami H., Glass T. J., Richter J., Johnson A., Lund T. C. and Kawakami Y. (2012b). Migration of cardiomyocytes is essential for heart regeneration in zebrafish. Development 139, 4133-4142. 10.1242/dev.079756 [DOI] [PubMed] [Google Scholar]
- Jopling C., Sleep E., Raya M., Martí M., Raya A. and Izpisua Belmonte J. C. I. (2010). Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 464, 606-609. 10.1038/nature08899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Y., Rodríguez-León J., Koth C. M., Büscher D., Itoh T., Raya A., Ng J. K., Esteban C. R., Takahashi S., Henrique D. et al. (2003). MKP3 mediates the cellular response to FGF8 signalling in the vertebrate limb. Nat. Cell Biol. 5, 513-519. 10.1038/ncb989 [DOI] [PubMed] [Google Scholar]
- Kidger A. M. and Keyse S. M. (2016). The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin. Cell Dev. Biol. 50, 125-132. 10.1016/j.semcdb.2016.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K., Holdway J. E., Werdich A. A., Anderson R. M., Fang Y., Egnaczyk G. F., Evans T., Macrae C. A., Stainier D. Y. and Poss K. D. (2010). Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature 464, 601-605. 10.1038/nature08804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Wu Q., Zhang Y., Wiens K. M., Huang Y., Rubin N., Shimada H., Handin R. I., Chao M. Y., Tuan T.-L. et al. (2010). PDGF signaling is required for epicardial function and blood vessel formation in regenerating zebrafish hearts. Proc. Natl. Acad. Sci. USA 107, 17206-17210. 10.1073/pnas.0915016107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondoh K. and Nishida E. (2007). Regulation of MAP kinases by MAP kinase phosphatases. Biochim. Biophys. Acta 1773, 1227-1237. 10.1016/j.bbamcr.2006.12.002 [DOI] [PubMed] [Google Scholar]
- Korotchenko V. N., Saydmohammed M., Vollmer L. L., Bakan A., Sheetz K., Debiec K. T., Greene K. A., Agliori C. S., Bahar I., Day B. W. et al. (2014). In vivo structure-activity relationship studies support allosteric targeting of a dual specificity phosphatase. Chembiochem 15, 1436-1445. 10.1002/cbic.201402000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson N. D. and Weinstein B. M. (2002). In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 248, 307-318. 10.1006/dbio.2002.0711 [DOI] [PubMed] [Google Scholar]
- Lepilina A., Coon A. N., Kikuchi K., Holdway J. E., Roberts R. W., Burns C. G. and Poss K. D. (2006). A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 127, 607-619. 10.1016/j.cell.2006.08.052 [DOI] [PubMed] [Google Scholar]
- Li C., Scott D. A., Hatch E., Tian X. and Mansour S. L. (2007). Dusp6 (Mkp3) is a negative feedback regulator of FGF-stimulated ERK signaling during mouse development. Development 134, 167-176. 10.1242/dev.02701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien C.-L., Schebesta M., Makino S., Weber G. J. and Keating M. T. (2006). Gene expression analysis of zebrafish heart regeneration. PLoS Biol. 4, e260 10.1371/journal.pbio.0040260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons D. A., Pogoda H.-M., Voas M. G., Woods I. G., Diamond B., Nix R., Arana N., Jacobs J. and Talbot W. S. (2005). erbb3 and erbb2 are essential for schwann cell migration and myelination in zebrafish. Curr. Biol. 15, 513-524. 10.1016/j.cub.2005.02.030 [DOI] [PubMed] [Google Scholar]
- Maillet M., Purcell N. H., Sargent M. A., York A. J., Bueno O. F. and Molkentin J. D. (2008). DUSP6 (MKP3) null mice show enhanced ERK1/2 phosphorylation at baseline and increased myocyte proliferation in the heart affecting disease susceptibility. J. Biol. Chem. 283, 31246-31255. 10.1074/jbc.M806085200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín-Juez R., Marass M., Gauvrit S., Rossi A., Lai S.-L., Materna S. C., Black B. L. and Stainier D. Y. (2016). Fast revascularization of the injured area is essential to support zebrafish heart regeneration. Proc. Natl. Acad. Sci. USA 113, 11237-11242. 10.1073/pnas.1605431113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuno K., Ichimura M., Nakajima T., Tahara K., Fujiwara S., Kase H., Ushiki J., Giese N. A., Pandey A., Scarborough R. M. et al. (2002). Potent and selective inhibitors of platelet-derived growth factor receptor phosphorylation. 1. Synthesis, structure-activity relationship, and biological effects of a new class of quinazoline derivatives. J. Med. Chem. 45, 3057-3066. [DOI] [PubMed] [Google Scholar]
- Mill J. G., Stefanon I., Leite C. M. and Vassallo D. V. (1990). Changes in performance of the surviving myocardium after left ventricular infarction in rats. Cardiovasc. Res. 24, 748-753. 10.1093/cvr/24.9.748 [DOI] [PubMed] [Google Scholar]
- Missinato M. A., Tobita K., Romano N., Carroll J. A. and Tsang M. (2015). Extracellular component hyaluronic acid and its receptor Hmmr are required for epicardial EMT during heart regeneration. Cardiovasc. Res. 107, 487-498. 10.1093/cvr/cvv190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina G. A., Watkins S. C. and Tsang M. (2007). Generation of FGF reporter transgenic zebrafish and their utility in chemical screens. BMC Dev. Biol. 7, 62 10.1186/1471-213X-7-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina G., Vogt A., Bakan A., Dai W.,de Oliveira P. Q., Znosko W., Smithgall T. E., Bahar I., Lazo J. S., Day B. W. et al. (2009). Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat. Chem. Biol. 5, 680-687. 10.1038/nchembio.190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffarian D., Benjamin E. J., Go A. S., Arnett D. K., Blaha M. J., Cushman M., Das S. R., de Ferranti S., Després J.-P., Fullerton H. J. et al. (2016). Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation 133, e38-e60. 10.1161/CIR.0000000000000350 [DOI] [PubMed] [Google Scholar]
- Nguyen V. B., Probyn M. E., Campbell F., Yin K. V., Samuel C. S., Zimanyi M. A., Bertram J. F., Black M. J. and Moritz K. M. (2014). Low-dose maternal alcohol consumption: effects in the hearts of offspring in early life and adulthood. Physiol. Rep. 2, e12087 10.14814/phy2.12087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polizzotti B. D., Ganapathy B., Walsh S., Choudhury S., Ammanamanchi N., Bennett D. G., dos Remedios C. G., Haubner B. J., Penninger J. M. and Kuhn B. (2015). Neuregulin stimulation of cardiomyocyte regeneration in mice and human myocardium reveals a therapeutic window. Sci. Transl. Med. 7, 281ra245 10.1126/scitranslmed.aaa5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polizzotti B. D., Ganapathy B., Haubner B. J., Penninger J. M. and Kühn B. (2016). A cryoinjury model in neonatal mice for cardiac translational and regeneration research. Nat. Protoc. 11, 542-552. 10.1038/nprot.2016.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss K. D., Wilson L. G. and Keating M. T. (2002). Heart regeneration in zebrafish. Science 298, 2188-2190. 10.1126/science.1077857 [DOI] [PubMed] [Google Scholar]
- Pugach E. K., Li P., White R. and Zon L. (2009). Retro-orbital injection in adult zebrafish. J. Vis. Exp. 34, 1645 10.3791/1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell N. H., Wilkins B. J., York A., Saba-El-Leil M. K., Meloche S., Robbins J. and Molkentin J. D. (2007). Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc. Natl. Acad. Sci. USA 104, 14074-14079. 10.1073/pnas.0610906104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L., Huang Y., Spencer C. I., Foley A., Vedantham V., Liu L., Conway S. J., Fu J. D. and Srivastava D. (2012). In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485, 593-598. 10.1038/nature11044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimawi M. F., Schiff R. and Osborne C. K. (2015). Targeting HER2 for the treatment of breast cancer. Annu. Rev. Med. 66, 111-128. 10.1146/annurev-med-042513-015127 [DOI] [PubMed] [Google Scholar]
- Saydmohammed M., Vollmer L. L., Onuoha E. O., Vogt A. and Tsang M. (2011). A high-content screening assay in transgenic zebrafish identifies two novel activators of fgf signaling. Birth Defects Res. C Embryo Today 93, 281-287. 10.1002/bdrc.20216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang M. and Dawid I. B. (2004). Promotion and attenuation of FGF signaling through the Ras-MAPK pathway. Sci. STKE 2004, pe17. [DOI] [PubMed] [Google Scholar]
- Tsang M., Friesel R., Kudoh T. and Dawid I. B. (2002). Identification of Sef, a novel modulator of FGF signalling. Nat. Cell Biol. 4, 165-169. 10.1038/ncb749 [DOI] [PubMed] [Google Scholar]
- Tsang M., Maegawa S., Kiang A., Habas R., Weinberg E. and Dawid I. B. (2004). A role for MKP3 in axial patterning of the zebrafish embryo. Development 131, 2769-2779. 10.1242/dev.01157 [DOI] [PubMed] [Google Scholar]
- Wang Y., Kaiser M. S., Larson J. D., Nasevicius A., Clark K. J., Wadman S. A., Roberg-Perez S. E., Ekker S. C., Hackett P. B., McGrail M. et al. (2010). Moesin1 and Ve-cadherin are required in endothelial cells during in vivo tubulogenesis. Development 137, 3119-3128. 10.1242/dev.048785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Panáková D., Kikuchi K., Holdway J. E., Gemberling M., Burris J. S., Singh S. P., Dickson A. L., Lin Y.-F., Sabeh M. K. et al. (2011). The regenerative capacity of zebrafish reverses cardiac failure caused by genetic cardiomyocyte depletion. Development 138, 3421-3430. 10.1242/dev.068601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Karra R., Dickson A. L. and Poss K. D. (2013). Fibronectin is deposited by injury-activated epicardial cells and is necessary for zebrafish heart regeneration. Dev. Biol. 382, 427-435. 10.1016/j.ydbio.2013.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. C., Kruse F., Vasudevarao M. D., Junker J. P., Zebrowski D. C., Fischer K., Noel E. S., Grun D., Berezikov E., Engel F. B. et al. (2016). Spatially resolved genome-wide transcriptional profiling identifies BMP signaling as essential regulator of zebrafish cardiomyocyte regeneration. Dev. Cell 36, 36-49. 10.1016/j.devcel.2015.12.010 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Shimizu H., Siu K. L., Mahajan A., Chen J.-N. and Cai H. (2014). NADPH oxidase 4 induces cardiac arrhythmic phenotype in zebrafish. J. Biol. Chem. 289, 23200-23208. 10.1074/jbc.M114.587196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Cao N., Huang Y., Spencer C. I., Fu J. D., Yu C., Liu K., Nie B., Xu T., Li K. et al. (2016). Expandable cardiovascular progenitor cells reprogrammed from fibroblasts. Cell Stem Cell 18, 368-381. 10.1016/j.stem.2016.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Dickson M. E., Kim M. S., Bassel-Duby R. and Olson E. N. (2015). Akt1/protein kinase B enhances transcriptional reprogramming of fibroblasts to functional cardiomyocytes. Proc. Natl. Acad. Sci. USA 112, 11864-11869. 10.1073/pnas.1516237112 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.