

ABSTRACT
Human embryonic stem cells (hESCs) are exquisitely sensitive to WNT ligands, which rapidly cause differentiation. Therefore, hESC self-renewal requires robust mechanisms to keep the cells in a WNT inactive but responsive state. How they achieve this is largely unknown. We explored the role of transcriptional regulators of WNT signaling, the TCF/LEFs. As in mouse ESCs, TCF7L1 is the predominant family member expressed in hESCs. Genome-wide, it binds a gene cohort involved in primitive streak formation at gastrulation, including NODAL, BMP4 and WNT3. Comparing TCF7L1-bound sites with those bound by the WNT signaling effector β-catenin indicates that TCF7L1 acts largely on the WNT signaling pathway. TCF7L1 overlaps less with the pluripotency regulators OCT4 and NANOG than in mouse ESCs. Gain- and loss-of-function studies indicate that TCF7L1 suppresses gene cohorts expressed in the primitive streak. Interestingly, we find that BMP4, another driver of hESC differentiation, downregulates TCF7L1, providing a mechanism of BMP and WNT pathway intersection. Together, our studies indicate that TCF7L1 plays a major role in maintaining hESC pluripotency, which has implications for human development during gastrulation.
KEY WORDS: BMP4, Gastrulation, Primitive streak, TCF7L1, WNT signaling, Human ES cells
Summary: TCF7L1 represses PS gene expression to maintain the primed, pluripotent state, with relief of this repression by BMP4 leading to upregulation of PS-promoting genes (including NODAL, WNT3 and BMP4) to drive hESC differentiation.
INTRODUCTION
Human embryonic stem cells (hESCs) represent an important tool in the study of human biology, especially early development, since they may be most closely related to cells of the early post-implantation epiblast (reviewed by Nichols and Smith, 2009, 2012). Information gleaned from these studies could improve our understanding of normal cellular differentiation, which, in turn, might be translated into developing better-directed differentiation methods for making specialized cells to develop new treatments for human diseases. However, there are large gaps in our knowledge of the mechanisms regulating the choice stem cells make between self-renewal and differentiation. A major issue is the role of WNT signaling in pluripotent stem cell growth. WNT ligands are not required for hESC self-renewal but the cells respond to these WNT ligands by rapid differentiation (Dravid et al., 2005; Davidson et al., 2012; Xu et al., 2016). This implies that hESCs have all the components to respond to WNT ligands but somehow, during self-renewal, are inhibited from doing so.
Vital effectors of WNT signaling are the T cell factor (TCF)/Lymphoid enhancer factor (LEF) family of transcription factors (Cadigan and Waterman, 2012; Mao and Byers, 2011). In mice and humans there are four TCF/LEF factors, each with distinct abilities to modulate the output of WNT signaling: TCF7 (formerly TCF1), TCF7L1 (formerly TCF3), TCF7L2 (formerly TCF4) and LEF1. TCF/LEFs are members of the high mobility group (HMG)-box DNA-binding proteins that function as binding partners for β-catenin, a key molecule in the transcriptional output of the WNT signaling pathway. WNT ligand binding to its receptors causes β-catenin, which itself does not directly bind DNA, to be stabilized and move into the nucleus where it binds to TCF/LEFs. TCF/LEFs are themselves bound to chromatin at recognition sequences termed WNT-response elements (WREs). Binding of β-catenin to TCF/LEFs at WREs can lead to WNT target gene transcription. An emerging concept of TCF/LEFs, however, is that each TCF/LEF has distinct potential in regulating WNT targets. TCF7 and LEF1 are strong activators, whereas TCF7L1 and TCF7L2 appear to act more as weak activators and better as repressors (Cadigan and Waterman, 2012; Chodaparambil et al., 2014; Nguyen et al., 2009). Thus, the TCF/LEFs, acting at a key nexus in the WNT pathway, can modulate, positively or negatively, the transcriptional output of this crucial developmental pathway.
One would therefore expect TCF/LEFs to play important roles in the overall response of cells to WNT signals. Indeed, previous studies demonstrated that Tcf7l1 plays a key role in gastrulation (Merrill et al., 2004). Mice lacking Tcf7l1 show embryonic axis defects associated with ectopic expression of Foxa2, a key endoderm regulator, indicating a non-redundant role for Tcf7l1 in embryonic axis formation at gastrulation. Studies in mouse ESCs (mESCs) propose that Tcf7l1 acts to limit pro-self-renewal mechanisms to ensure timely and effective response to differentiation cues, perhaps partly explaining the knockout phenotype (Cole et al., 2008; Marson et al., 2008; Pereira et al., 2006; Yi et al., 2008). Importantly, though, in mice Tcf7l1 is needed to mediate the transition from the ʻnaïve' to the ʻprimed' state of pluripotency (Guo et al., 2011; Hoffman et al., 2013; Pereira et al., 2006). Our current understanding of pluripotency suggests that mESCs, like the blastocyst inner cell mass (ICM) from which they are derived, represent a naïve state of pluripotency in which the cells are pluripotent and can give rise to all the germ layers and germ cells in chimeras (Kalkan and Smith, 2014; Morgani et al., 2017). Around the time of embryo implantation the ICM gives rise to the epiblast, which is also pluripotent but now primed for differentiation into cells of the three primary germ layers. In experiments using Tcf7l1 mutant mESCs and embryos, these mutant cells have difficulty proceeding to a primed state and retain aspects of the naïve state (Guo et al., 2011; Hoffman et al., 2013; Pereira et al., 2006). Thus, in these systems, the function of Tcf7l1 in primed pluripotency has not been examined because disruption of Tcf7l1 in the naïve state disrupts progression to the primed state. Moreover, no studies to date have comprehensively examined the functions of individual TCF/LEFs in hESCs despite the role they play in mESC self-renewal and differentiation and in development. Experiments using hESCs, representing the primed state of pluripotency, could also inform our knowledge of this stage of development.
Here, we examine the role of TCF/LEFs in undifferentiated primed hESCs. Of the four TCF/LEFs, TCF7L1 is the most highly expressed. TCF7L1 mRNA and protein are rapidly downregulated upon directed differentiation. Using chromatin immunoprecipitation and next-generation sequencing (ChIP-seq) and gene ontology (GO) analysis, as well as loss- and gain-of-function experiments, we find that TCF7L1 is bound at genes largely connected to vertebrate gastrulation and primitive streak (PS) formation, including NODAL, WNT3 and BMP4. These data suggest that in pluripotent hESCs TCF7L1 acts to suppress genes involved in PS differentiation, aiding maintenance of hESC pluripotency. We also find that TCF7L1 is less integrated with the core pluripotency transcriptional regulators [OCT4 (POU5F1) and NANOG] and moreover is downregulated by BMP4, a known inducer of mesendoderm differentiation at the PS (Winnier et al., 1995). Our studies provide new information on how pluripotency may be maintained, while also improving our understanding of WNT signaling in vertebrate development, especially at the time of human gastrulation.
RESULTS
TCF7L1 is the dominant TCF/LEF in hESCs
We investigated whether WNT signaling plays a role in the maintenance of pluripotency or in directing differentiation in hESCs. Examining β-catenin-dependent WNT signaling in hESCs using the TOPflash WNT reporter we found that undifferentiated hESCs were in a WNT-inactive state (Fig. 1A). Furthermore, we used immunocytochemistry to interrogate β-catenin localization in undifferentiated hESCs. Corroborating our TOPflash result, all detectable β-catenin was localized at the plasma membrane in normal cultures, indicating lack of β-catenin-dependent WNT transcriptional activity (Fig. 1B). When hESCs were stimulated with WNT3A we observed robust migration of β-catenin into the nucleus, concomitant upregulation of TOPflash activity, and PS gene expression (Fig. 1A-C). Moreover, β-catenin in the nucleus was the active, unphosphorylated form, consistent with the above data (Fig. S1A,B). These results indicate that undifferentiated hESCs have very low or no β-catenin-dependent WNT activity, but are also highly responsive to WNTs, consistent with the findings of other studies (Blauwkamp et al., 2012; Davidson et al., 2012; Dravid et al., 2005; Frank et al., 2012; Xu et al., 2016).
Fig. 1.

Inactive WNT signaling and TCF/LEF expression in hESCs. (A) TOPflash WNT signaling reporter analysis (n=3) of undifferentiated (Ut.) H9 hESCs or those treated with WNT3A (100 ng/ml) for 24 h. One-tailed t-test, *P≤0.05. Error bars indicate s.e.m. (B) Confocal analysis of OCT4 and β-catenin localization in untreated (mTeSR1) or 24 h WNT3A (100 ng/ml) stimulated conditions. (C) qPCR analysis (n=3) shows that WNT3A induces primitive streak (PS) gene expression after 48 h. T, brachyury. Two-tailed t-test, *P≤0.05, **P≤0.01, ***P≤0.001. (D) Averaged Ct values from LEF/TCF qPCR analysis in H9 hESCs (left) and normalized comparative analysis of LEF/TCF qPCR data showing LEF/TCF mRNA levels relative to TCF7L1 (n=3). Two-tailed t-test, **P≤0.01. (E) PCR analysis of TCF7L1 mRNA expression levels in H1, H9 and H14 hESCs. MEF-only sample illustrates species specificity of the TCF7L1 primers. β-actin was the template loading control. (F) Confocal immunofluorescence analysis of OCT4 and TCF7L1 in H9 hESCs under feeder-free conditions.
Not all WNTs signal through β-catenin. Therefore, we asked if any type of WNT-triggered signaling acts through an autocrine mechanism necessary for hESC pluripotency. We treated hESC cultures with IWP-2, which prevents secretion of all WNTs. After 7 days of continuous IWP-2 exposure, hESCs were morphologically undifferentiated and levels of OCT4, SOX2 and NANOG were unaffected (Fig. S2A,B), suggesting that no WNT ligand family members are required for hESC self-renewal and pluripotency, consistent with reports from other studies (Davidson et al., 2012; Kurek et al., 2015).
TCF/LEFs mediate the last step in the WNT signaling cascade by recruiting β-catenin to control WNT target genes. This final step thus constitutes one point at which WNT signaling could be held in an inactive, but highly responsive, state. To address TCF/LEF functions in hESCs, we first profiled their mRNA. qPCR and reverse transcription PCR (PCR) analyses indicated that TCF7L1 is the most highly expressed TCF/LEF in hESCs (Fig. 1D,E). Staining of TCF7L1 and pluripotency markers (OCT4, NANOG and SOX2) verified TCF7L1 protein expression and nuclear localization in hESCs (Fig. 1F, Fig. S1C).
We next tested whether TCF7L1 was regulated by pro-pluripotency FGF and TGFβ signaling. We inhibited TGFβ signaling using SB431542, leading to a nearly 50% decrease in TCF7L1 mRNA expression (Fig. S2C). As expected, mRNA levels of NANOG, a known downstream target of the TGFβ pathway, were also downregulated (Fig. S2C) (Xu et al., 2008). Inhibiting FGF signaling with SU5402 for 24 and 48 h had no impact on TCF7L1 mRNA expression (Fig. S2D). OCT4 and NANOG were slightly downregulated under these conditions, whereas SOX2 (also involved in neuroectoderm differentiation) was modestly upregulated (Fig. S2D) (Eiselleova et al., 2009). These results suggest that TGFβ signaling, which is crucial for maintenance of hESC pluripotency, helps maintain TCF7L1 expression.
Genome-wide analysis of TCF7L1 localization
Next, we performed ChIP-seq to make inferences into TCF7L1 function in hESCs and identified 9760 high-confidence, reproducible TCF7L1 peaks from two biological replicates. We carried out an irreproducibility discovery rate (IDR) analysis and showed that there is a high degree of reproducibility in the two ChIP-seq biological replicates (Fig. S3). Scatter plot analysis illustrated the high degree of reproducibility of the two TCF7L1 ChIP-seq replicates; coverageBed (Quinlan and Hall, 2010) was used to determine the number of reads that map to each base-pair position ±5 kb of the transcription start site (TSS) of all human hg19 RefSeq genes (Fig. S3). Peak annotation indicated 41% (4021) of binding sites were within ±5 kb of a RefSeq TSS, 22% (2133) were in genic regions and 37% (3606) occurred in distal intergenic regions (Fig. 2A). Additionally, de novo peak motif analysis showed significant enrichment for the WRE sequence CTTTGA, indicating that we recovered specific TCF7L1-bound sites genome-wide (Fig. 2B).
Fig. 2.
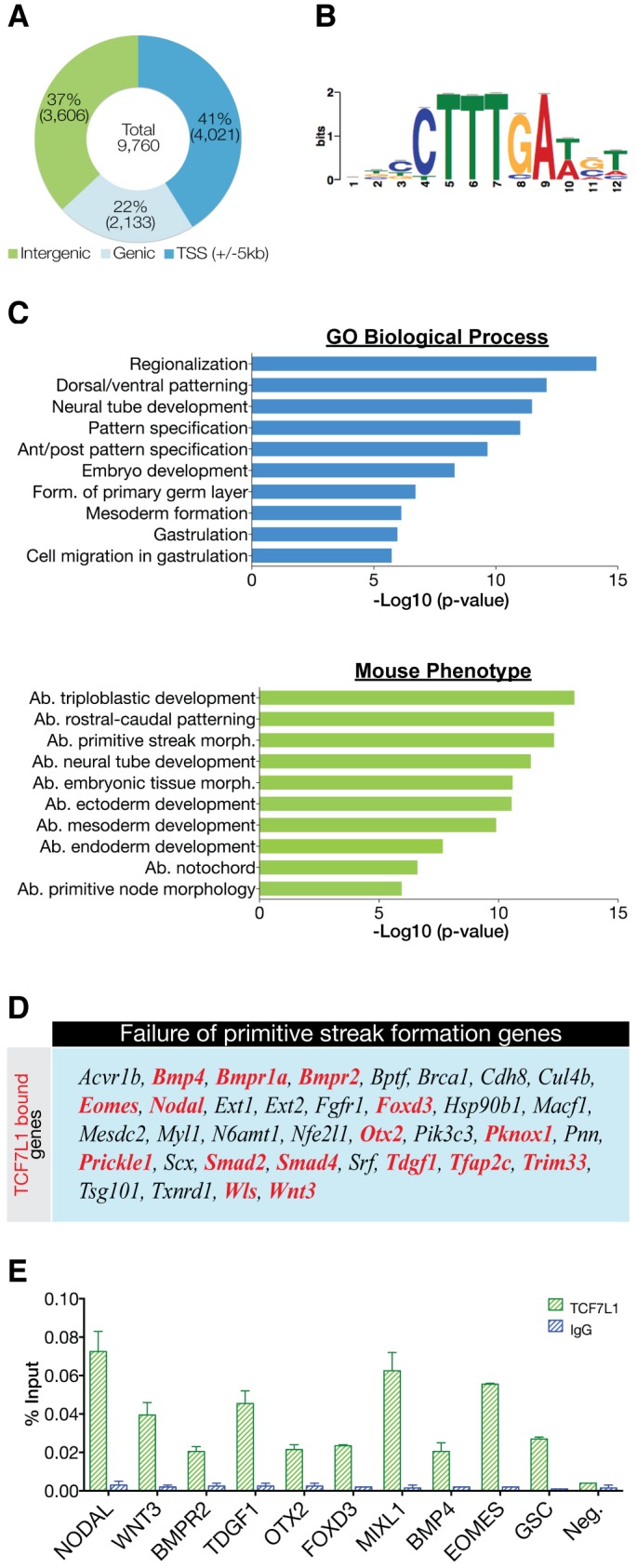
Characterization of TCF7L1 binding sites in hESCs. (A) Location of the top 9760 peaks (IDR<0.01325) relative to the nearest gene as determined by HOMER annotation. (B) De novo motif detected in TCF7L1/β-catenin-shared binding sites. The TCF7L1 site observed is a canonical WNT-response element (WRE). (C) GREAT analysis of underlying GO themes among TCF71 target genes. The top 1000 peaks were analyzed using ‘basal plus extension’ default settings. Data are presented as the –log10 of their respective P-values for convenience. Ab., abnormal; form., formation; morph., morphology. (D) TCF7L1-bound genes (red) represented in the mammalian phenotype category ‘failure of primitive streak formation’. Genes with TCF7L1 binding within ±5 kb of the TSS were used for this comparison. (E) ChIP-qPCR analysis (n=2) of TCF7L1 binding to crucial PS genes shown as percentage input recovery. Error bars indicate s.e.m.
Genomic regions enrichment of annotations tool (GREAT) analysis was used to examine functional GO categories of TCF7L1-occupied genes. Biological processes involved in gastrulation were highly represented among TCF7L1-bound genes, including ʻgastrulation', ʻanterior/posterior pattern specification', ʻformation of the primary germ layers' and ʻmesoderm formation' (Fig. 2C). Additionally, GREAT returned mouse phenotype analysis, categorizing mouse knockout phenotypes. Similar to the GO results, TCF7L1-bound genes were implicated in abnormal primitive streak morphology and in abnormal endoderm, ectoderm and mesoderm development (Fig. 2C).
Since the very first event of gastrulation is formation of the PS, the ChIP-seq data were analyzed to predict whether TCF7L1-bound genes were involved in this process. Mouse Genome Informatics provides a repository of mutant genotypes and phenotypes called the Mammalian Phenotype (MP) Browser (Eppig et al., 2015). This database has a category termed ʻfailure of primitive streak formation', which is defined as: ʻinability to form the epiblast ridge from which arises the germ layers of the embryo'. Interestingly, of the 36 genes listed, 16 were bound by TCF7L1 within ±5 kb of their TSS, including NODAL, WNT3, BMPR1 and TDGF1 and several DNA-binding proteins or transcriptional regulators including EOMES, FOXD3, OTX2, PKNOX1, SMAD2, SMAD4 and TFAP2C (Fig. 2D). We validated TCF7L1 binding by ChIP-qPCR on ten of these genes (Fig. 2E), implicating TCF7L1 as a potential transcriptional regulator of differentiation during PS formation.
Comparison of TCF7L1 with β-catenin, OCT4 and NANOG chromatin localization
It is known that TCF/LEFs act as binding partners for β-catenin, and so we examined potential interactions of TCF7L1 with β-catenin. Recent studies examined the expression of β-catenin in undifferentiated hESCs. β-catenin in hESCs is stimulated either with WNT3A or using the GSK3β inhibitor CHIR-99021. One study investigated β-catenin binding in undifferentiated cells and found only ∼1300 binding sites (Estarás et al., 2015), consistent with our β-catenin immunohistochemistry data (Fig. 1B) as well as other studies suggesting that there is little or no β-catenin in the nucleus of undifferentiated hESCs (Davidson et al., 2012; Dravid et al., 2005). When β-catenin genome binding was examined in hESCs stimulated for 4 h with WNT3A, the number of β-catenin binding sites increased to 12,684 (Estarás et al., 2015). Comparison of these peaks with TCF7L1 peaks demonstrated that approximately one-fifth (1998) of the 9760 TCF7L1 peaks detected in undifferentiated hESCs became bound by β-catenin within 4 h of WNT3A stimulation (Fig. 3A). Furthermore, the strongest TCF7L1 peaks were occupied by β-catenin following WNT pathway activation (Fig. 3B). As previously mentioned, the WRE was the highest-scoring motif enriched in TCF7L1 peaks, indicating TCF7L1 acts in part by suppressing activation of β-catenin-dependent WNT signaling and not through unique circuitry. GREAT analysis of these overlapping peaks indicates that the genes bound by TCF7L1 in undifferentiated hESCs and subsequently by β-catenin following WNT3A treatment are involved in embryogenesis (Fig. 3C). We also determined where TCF7L1 bound to genes relative to the TSS that would later become bound by β-catenin upon WNT3A stimulation (Fig. 3D). TCF7L1 bound to relatively few sites near the TSS, but those close to the TSS include key developmental genes such as NODAL and EOMES (Fig. 3E).
Fig. 3.

β-catenin is recruited to TCF7L1-occupied sites upon WNT3A stimulation of hESCs. (A) Overlap of TCF7L1 ChIP-seq binding sites and β-catenin sites upon WNT3A stimulation from Estarás et al. (2015). 1998 overlapping sites were identified. (B) Comparison of signal intensity maps for TCF7L1 and β-catenin ±2 kb from the observed binding sites. 1998 peaks are bound by both TCF7L1 and β-catenin with strong signal intensity. Peaks bound by TCF7L1 (7795 peaks) and β-catenin (10,686 peaks) independently are also shown. (C) GREAT enrichment GO categories for the 1998 overlapping TCF7L1 and β-catenin sites. (D) Relationship of the 1998 shared binding sites to the genome with respect to the nearest gene TSS. (E) Screenshots from the UCSC Genome Browser showing overlapping TCF7L1 and β-catenin binding sites within the human NODAL and EOMES loci [β-catenin data from Estarás et al. (2015)]. Vert., vertebrate.
ChIP-seq data sets also indicated that there were 7795 TCF7L1 peaks not bound by β-catenin following 4 h WNT3A exposure (Fig. 3A,B, Fig. S4). Analysis of these peaks indicated GO categories including ʻDNA-dependent transcription', ʻRNA biosynthetic processes' and ʻnucleobase-containing compound biosynthetic process' (Fig. S4). Whether β-catenin eventually binds at all the TCF7L1 peaks over a longer time frame is an interesting, and now testable, question. Additionally, we examined β-catenin binding at peaks not bound by TCF7L1. The intensity of the β-catenin ChIP peaks indicates that these are strong sites of occupancy and, therefore, are likely to be real β-catenin occupancies not associated with TCF7L1 in hESCs prior to WNT treatment (Fig. 3B). When we examined individual TCF7L1 and β-catenin binding sites in the 1998 genes co-occupied by TCF7L1 and β-catenin, we found strong overlapping binding at highly conserved sequences. Shown is analysis for NODAL and EOMES, two key gastrulation genes (Fig. 3E). These data therefore indicate that in undifferentiated hESCs TCF7L1 is bound partly to genes that can be bound by β-catenin and activated upon WNT3A-mediated differentiation. Together, these findings suggest that prior to WNT stimulation TCF7L1 is either actively repressing differentiation genes or occupying/marking (ʻpriming') genes for rapid activation upon WNT stimulation.
In mESCs, Tcf7l1 is considered one of the core transcriptional regulators of pluripotency alongside Oct4, Sox2 and Nanog (Cole et al., 2008; Marson et al., 2008). Of genes bound by Tcf7l1 in mESCs, 74% overlap with Oct4 and Nanog (Marson et al., 2008). We also compared our TCF7L1 ChIP-seq data with two separate published ChIP-seq data sets for OCT4 and NANOG, one from H1 (The ENCODE Project Consortium, 2012) and the other from H9 (Kushwaha et al., 2015) hESCs. The H1 and H9 data were compared and 2713 genes were found to overlap (data not shown). These overlapping genes were then compared with genes identified in our TCF7L1 data. Only 24% of TCF7L1 genes were co-bound by OCT4 and NANOG (Fig. S5A). In analyzing the annotated overlapping peak data – as opposed to the overlapping genes – we observed the same trend, namely a large reduction in overlap between TCF7L1 and OCT4/NANOG from 67% in mESCs to 14% in hESCs (Fig. S5B). Thus, in primed hESCs there is a substantial reduction in the overlap of core regulator binding profiles with TCF7L1 when compared with naïve mESCs.
TCF7L1 knockdown highlights an effect on key PS regulators
Next, we asked how downregulation of TCF7L1 might influence hESC gene expression and pluripotency. TCF7L1 mRNA was downregulated by transient siRNA knockdown (KD). After two rounds of siRNA treatment over 3 days, TCF7L1 mRNA and protein levels were reduced by ∼90% (Fig. 4A). Loss of TCF7L1 did not have dramatic effects on the morphology of hESC colonies, although some colonies lost their sharp, well-defined edges and a few cells at the colony periphery appeared migratory (Fig. 4B). We also analyzed whether loss of TCF7L1 triggered compensatory changes in expression of other TCF/LEFs. Using qPCR, we found that TCF7 and LEF1 were slightly upregulated by 1.2-fold and 1.8-fold, respectively, 3 days post-siRNA-mediated KD, whereas TCF7L2 mRNA levels were unaffected (Fig. S6A).
Fig. 4.

Loss of TCF7L1 upregulates PS gene expression. (A) (Left) siRNA reduced TCF7L1 mRNA expression by a factor of 10 after 3 days, as analyzed by qPCR (n=3). Two-tailed t-test, ***P≤0.001. (Right) Western blot confirmed concomitant downregulation of TCF7L1 protein. Non-S., non-silencing siRNA; Ut., untreated. siRNAs were used at 50 nM. (B) Phase contrast images (original magnification 4×) showing morphological changes in TCF7L1 knockdown colonies as compared with controls. (C) Scatter plot of microarray mean gene expression values from the TCF7L1 siRNA condition plotted against non-silencing siRNA condition. Uniform linear distribution signifies highly similar expression values in both siRNA conditions. (D) Results of 3 day TCF7L1 siRNA knockdown microarray analysis. Top upregulated and downregulated gene candidates (red and blue boxes, respectively) were selected based on the following criteria: P≤0.05; fold change (FC) ≥1.5 and ≤−1.5. Genes highlighted in bold (and others) were validated by qPCR (see E and F). (E) qPCR validation of selected candidate upregulated genes identified by microarray analysis (n=3). Two-tailed t-test, **P≤0.01, ***P≤0.001. (F) qPCR validation of selected candidate downregulated genes identified by microarray analysis (n=3). Two-tailed t-test, *P≤0.05, **P≤0.01, ***P≤0.001. Error bars indicate s.e.m. (G) DAVID GO analysis (P≤0.05) of genes upregulated by TCF7L1 siRNA knockdown. x-axis shows –log10 conversion of the P-values. Reg., regulation; dev., development.
We next ascertained the impact of TCF7L1 on the hESC transcriptome by comparing microarray analyses of mRNA from TCF7L1 KD cells with mRNA from cells treated with non-silencing siRNAs. Three days post-TCF7L1 KD only a few genes (DACT1, FST, NODAL and FAT4) – besides TCF7L1 itself – passed the statistical post-test analysis for significant mRNA level changes (Fig. 4C). Even with relaxed parameters (P≤0.05 cutoff and fold-change cutoff of 1.5 and −1.5), only a small number of additional genes were identified as upregulated or downregulated (49 or 37, respectively; Fig. 4D). qPCR validation of a panel of upregulated and downregulated genes corroborated the minimal gene expression changes observed by microarray (Fig. 4E,F). These results were confirmed using a second siRNA targeting TCF7L1 (Fig. S6C,D). Despite limited gene expression changes, the 49 upregulated genes provided clues as to a potential function of TCF7L1. GO analysis of genes upregulated following TCF7L1 KD indicated that they belonged in categories such as: ʻregulation of cell proliferation', ʻregulation of gene-specific transcription', ʻpositive regulation of development', ʻtissue morphogenesis', ʻpattern specification' and ʻBMP signaling pathway' (Fig. 4G). Specifically, genes upregulated in response to TCF7L1 KD, such as NODAL, BMP4 and WNT3, have known crucial regulatory functions in PS formation (reviewed by Tam and Loebel, 2007). The limited effect of TCF7L1 KD on the hESC transcriptome could be attributed to the strong pluripotency-maintaining growth factors FGF2 and TGFβ1 present in mTeSR1 medium. These, we reasoned, were likely to oppose the impact of TCF7L1 KD, an idea borne out by subsequent studies (see below).
TCF7L1 represses PS genes during hESC differentiation
To test if TCF7L1 functions as a transcriptional repressor opposing differentiation, we examined the effect of induced differentiation on TCF7L1 levels. We adopted a differentiation protocol for inducing a PS gene program [e.g. brachyury (TBXT), EOMES, MIXL1, GSC] in hESCs. To focus on immediate gene expression changes that occur as primed epiblast cells differentiate within the PS, we used an abridged version (48 h) of a definitive endoderm (DE) differentiation procedure (Teo et al., 2012). Importantly, this PS-like differentiation procedure was performed under feeder-free conditions using fully defined medium that contains BMP4 and activin A, allowing control over signals driving differentiation. We observed a downregulation of TCF7L1 protein levels after 48 h (Fig. 5A). Next, we created clonal hESC lines to maintain TCF7L1 expression during differentiation. These lines expressed an inducible full-length TCF7L1 with an N-terminal FLAG tag and a C-terminal HTBH tag (H9-TCF7L1) (Tagwerker et al., 2006). As a control, we created a cell line for induced expression of GFP (H9-GFP). Transgene expression was controlled using a doxycycline-inducible TET-ON system to maintain levels of TCF7L1 expression during differentiation (Fig. 5B, Fig. S7).
Fig. 5.

TCF7L1 overexpression impedes PS differentiation. (A) Western blot analysis of TCF7L1 protein after 48 h of PS differentiation as compared with untreated control (UT). (B) FLAG-TCF7L1-HTBH is robustly overexpressed in H9-TCF7L1 cells after 48 h of doxycycline (1 μg/ml) induction. The higher molecular weight band is FLAG-TCF7L1-HTBH (blue arrow). The lower band is endogenous TCF7L1 (red arrow). WT, H9 cells. (C) Scheme of the TCF7L1 overexpression microarray experiment. (D) Comparison of PS differentiated H9-TCF7L1 cells (without doxycycline induction) with mTeSR1-cultured H9-TCF7L1 cells (untreated). GO analysis of significantly differentially expressed genes is shown as –log10 of their respective P-values. (E) Comparison of PS differentiated H9-TCF7L1 cells with and without 48 h of doxycycline induction. GO analysis of significantly differentially expressed genes is presented as –log10 of their respective P-values. (F) GO analysis of shared PS+TCF7L1 downregulated versus PS upregulated genes, indicating enrichment of genes involved in gastrulation, embryonic patterning and embryonic morphogenesis. (G) qPCR validation of candidate TCF7L1-repressed genes identified by microarray analysis. Analysis was performed with H9-TCF7L1 and control H9-GFP cell lines (n=3). Two-tailed t-test, *P≤0.05, **P≤0.01, ***P≤0.001. Error bars indicate s.e.m. Ant/post, anterior/posterior; dev., development; diff., differentiation; form., formation; reg., regulation; neuro., neurological; neg., negative; surf., surface; sig., signaling.
First, we validated the PS-like differentiation gene signature by comparing global gene expression patterns in uninduced H9-TCF7L1 cells versus the same cells stimulated to undergo PS differentiation (Fig. 5C). PS differentiation upregulated 1409 genes and downregulated 1596 genes (Fig. 5D). There was a strong induction of PS differentiation-associated genes (EOMES, MIXL1, LHX1, TBXT, GSC, GATA4, GATA6, SOX17, FOXA2). GO analysis confirmed that genes involved in development and differentiation were upregulated (Fig. 5D).
Next, we addressed TCF7L1 function by comparing the gene signature of PS-directed differentiating cells with the signature of cells differentiating while TCF7L1 levels were maintained (PS medium+TCF7L1). Overall, 917 genes were increased and 970 genes were decreased in expression relative to the PS differentiation condition (Fig. 5E). Interestingly, genes associated with PS formation (TBXT, EOMES, MIXL1, GSC, WNT3) were significantly repressed when TCF7L1 expression was maintained during differentiation. Importantly, there was a notable overlap between the top genes normally upregulated during PS differentiation and the top genes that were repressed when TCF7L1 expression was maintained during the same differentiation. The data reveal a striking and strong inverse correlation between these two data sets (Fig. 5F). GO analysis indicated overlapping biological processes between genes suppressed by TCF7L1 in PS conditions and genes normally upregulated during differentiation, including regulators of ‘gastrulation’, ‘embryonic morphogenesis’ and ‘primary germ layer formation’ (Fig. 5F). Using mRNA from the H9-GFP control and TCF7L1-induced cell lines, we validated TCF7L1-specific repression of candidate PS markers TBXT, GSC, MIXL1 and WNT3 during PS differentiation (Fig. 5G). As hypothesized, TCF7L1 repressed genes that are normally activated during PS-induced differentiation and, in doing so, is likely to have contributed to pluripotency.
Regulation of TCF7L1 by BMP4 signaling
An important question is how TCF7L1 itself is regulated, since conditions driving PS differentiation of hESCs cause downregulation of TCF7L1 mRNA (Fig. 5A). To understand what signals might control TCF7L1 expression, we used the PS-like differentiation conditions in which we could manipulate individual components. We found that BMP4 (but not activin A) strongly influenced TCF7L1 expression (Fig. 6A). This was confirmed by BMP4 addition to hESC culture and the observed decline in TCF7L1 protein levels over a 48 h timecourse (Fig. 6B).
Fig. 6.

TCF7L1 antagonizes BMP4-induced differentiation. (A) qPCR analysis (n=3) of TCF7L1 mRNA levels after 24 h in mTeSR1, complete PS differentiation medium, PS differentiation medium without BMP4 and activin A (−GFs), or PS differentiation medium with either BMP4 or activin A alone. Two-tailed t-test, *P≤0.05, **P≤0.01. (B) Western blot analysis of TCF7L1 levels at 12, 24 and 48 h of BMP4-induced differentiation. (C) H9-GFP and H9-TCF7L1 cells were grown under feeder-free conditions and treated with BMP4 (24 h) while inducing GFP or TCF7L1 with doxycycline (1 μg/ml). Phase contrast images, original magnification 10×. (D) Scheme of experiment in which hESCs were treated with BMP4 (10 ng/ml) while performing TCF7L1 siRNA (50 nM) knockdown under feeder-free conditions. Cells were harvested after 48 h of siRNA knockdown and 24 h of BMP4 treatment. Control experiments were performed without BMP4 treatment for each condition (not shown). Red numbers indicate days of procedure. (E) Simultaneous loss of TCF7L1 and treatment with BMP4 causes pronounced morphological changes in colonies. Phase contrast images (original magnification 10×) show TCF7L1 siRNA+BMP4-treated hESC colonies, which appear more flattened and differentiated than controls. (F) qPCR analysis showing synergistic upregulation of PS markers when BMP4 treatment is combined with TCF7L1 knockdown (n=3). Two-tailed t-test, *P≤0.05, **P≤0.01. Error bars indicate s.e.m.
To test if TCF7L1 is a downstream effector of the BMP4 pathway, we once again took advantage of cells in which we could conditionally induce TCF7L1. We induced TCF7L1 expression (or GFP control) during BMP4-induced differentiation. As expected, BMP4-induced differentiation of hESCs caused cell flattening and loss of packed colony morphology (Fig. 6C). This phenotype was also noted in both transgenic cell lines (H9-TCF7L1 and H9-GFP) in the absence of doxycycline. When the GFP transgene was induced in hESCs, BMP4 also caused differentiation of the cells, as expected. By contrast, when TCF7L1 was induced in the presence of BMP4, hESCs retained the tight-knit colony morphology and failed to display hallmarks of differentiation (Fig. 6C). Thus, one potential role of BMP4 in promoting differentiation is to downregulate TCF7L1 expression.
To test whether BMP4 and TCF7L1 act in the same pathway, we carried out TCF7L1 KD with or without BMP4 (Fig. 6D). BMP4 caused morphological changes consistent with induction of differentiation in untreated hESCs and in non-silencing siRNA-treated cells (Fig. 6E). In cells with TCF7L1-silencing siRNAs, BMP4 caused a more exaggerated differentiated morphology, suggesting more robust differentiation (Fig. 6E). To confirm that these changes were reflected at the molecular level we assayed expression of TBXT, MIXL1 and GSC. Each gene was dramatically upregulated when TCF7L1 levels were reduced during BMP4 exposure (Fig. 6F). Together, these data suggest that the PS-inducing function of BMP4 acts, in part, by downregulating the expression of TCF7L1.
We also noted that in these same conditions LEF1 expression is upregulated, perhaps to replace TCF7L1 to activate gene expression in concert with β-catenin (Fig. S8A). We tested this by carrying out ChIP-PCR for TCF7L1 and LEF1 during differentiation of hESCs into mesendoderm. We examined TCF7L1 and LEF1 binding at the EOMES and NODAL loci following induced differentiation. At both loci we found evidence for decreased binding of TCF7L1 and concomitant increased binding of LEF1 following stimulation with BMP4 and activin A (Fig. S8B). Therefore, one way that BMP4 could drive hESC differentiation is by decreasing TCF7L1-mediated gene repression and subsequent activation of WNT target genes. Although we found that WNT3A had little effect by itself on the expression of TBXT, MIXL1 and GSC, siRNA-mediated KD of TCF7L1 sensitized hESCs to WNT3A addition and caused upregulation of TBXT and MIXL1 (Fig. S9). These data are most consistent with our hypothesis that BMP4 relieves TCF7L1-mediated repression of target genes leading to upregulation of WNT3A and LEF1 (and possibly other TCF/LEFs). WNT-mediated stabilization of β-catenin could then knock TCF7L1 off target WREs and/or also stabilize LEF1 at those elements.
DISCUSSION
Groundbreaking studies have identified some of the key molecular players in the regulation of pluripotency including OCT4, SOX2, NANOG and TCF7L1. A major question about these molecules is how they act to maintain pluripotency in a primed state ESC – stem cells on the verge of gastrulation and differentiation. Here we report that TCF7L1, the most highly expressed TCF/LEF WNT transcription factor in primed hESCs, plays a crucial role in maintaining pluripotency by preventing differentiation. Early studies in mice that disrupted Tcf7l1 revealed a role at the naïve stage of pluripotency, but cells lacking Tcf7l1 have difficulty reaching the primed state and, therefore, the function of this factor at this stage was unknown. Our studies of TCF7L1 in hESCs have revealed new information about how this factor functions in primed cells, providing new information that is also likely to be relevant to human embryogenesis since hESCs are now thought of as in vitro equivalents of post-implantation epiblast cells (Nichols and Smith, 2009). Key to informing our studies was the generation of TCF7L1 ChIP-seq data revealing TCF7L1 genome-wide binding.
We found a substantial reduction in the overlaps between TCF7L1 and OCT4 and NANOG chromatin binding in hESCs compared with mESCs, suggesting that TCF7L1 becomes partially disengaged from the core transcriptional regulatory circuitry in hESCs (Cole et al., 2008; Marson et al., 2008). This could be because of an inherent difference between mouse and human cells or because they represent different pluripotent states – naïve and primed. Further experiments utilizing naïve and primed cells of the same species with extensive ChIP-seq analyses will be needed to address this question comprehensively. De novo motif analysis of TCF7L1 binding sites revealed an enrichment of consensus WREs, indicating that in many cases it is bound to genes typically controlled through WNT signaling. This suggests that TCF7L1 is acting in large part through its ability to regulate β-catenin-dependent WNT signaling rather than through a unique regulatory circuit.
Our data also indicate that TCF7L1 is bound in hESCs at genes involved in aspects of embryonic development, including axis formation, morphogenesis, PS formation and gastrulation. These data imply that TCF7L1 acts to control the activity of these genes in hESCs. Because, hypothetically, hESCs are an in vitro equivalent of epiblast cells, genes regulated by TCF7L1 might also represent a gene clade involved in human gastrulation, perhaps providing a new window into this period of the human lifecycle. Since TCF7L1 differs from other members of the TCF/LEF family in certain regards, we cannot necessarily infer its function from their action. In fact, TCF7L1 and TCF7L2 act as weak activators of WNT target genes, whereas TCF7 and LEF1 are strong activators of those genes (Cadigan and Waterman, 2012; Chodaparambil et al., 2014; Nguyen et al., 2009). Based on lack of expression of WNT targets in hESCs, our microarray studies demonstrating the effect of gain- and loss-of-function of TCF7L1 on gene expression, and the general absence of β-catenin in the hESC nucleus, we conclude that TCF7L1 is acting as a gene repressor in these cells. This finding agrees with its proposed role as a gene repressor in mESCs (Chambers et al., 2007; Cole et al., 2008; Pereira et al., 2006; Shy et al., 2013; Wu et al., 2012; Yi and Merrill, 2007). Because TCF7L1 and TCF7L2 act in a similar manner (Nguyen et al., 2009) and TCF7L2 is the second most abundant TCF/LEF in hESCs, it is possible that the TCF7L1 phenotype might be rescued to some extent by TCF7L2. Although in our hands there was no change in TCF7L2 mRNA expression levels following TCF7L1 KD, compensation for loss of TCF7L1 by TCF7L2 is still a formal possibility. Nevertheless, and in spite of this caveat, our conclusions about the role of TCF7L1 in humans seem most consistent with the notion that TCF7L1, irrespective of the state of potency, opposes the action of WNT signaling. In naïve cells, it opposes the action of WNT in driving self-renewal whereas in human cells it opposes the action of WNT in driving differentiation.
We also compared our TCF7L1 ChIP-seq data with β-catenin ChIP-seq data in hESCs (Estarás et al., 2015; Funa et al., 2015). In undifferentiated hESCs, very few β-catenin binding sites were observed (only ∼1300), consistent with the notion that WNT signaling is absent, very low or present in only a subset of cells in certain conditions (Blauwkamp et al., 2012; Davidson et al., 2012; Dravid et al., 2005; Kurek et al., 2015; Xu et al., 2016). Following 4 h of WNT3A treatment, β-catenin was bound to ∼10,000 sites genome-wide (Estarás et al., 2015). Notably, at this time β-catenin was bound to ∼20% of sites that were bound by TCF7L1 in undifferentiated cells. It will be interesting to determine whether longer WNT signaling results in β-catenin binding to more sites occupied by TCF7L1 in undifferentiated hESCs. Nevertheless, these data demonstrate that WNT signaling is likely to cause rapid transcriptional activation of many genes that are repressed by TCF7L1 in hESCs. Because of the known ability of β-catenin to remove TCF7L1 from chromatin (Wu et al., 2012), it is likely that β-catenin-induced transcription of target genes occurs in concert with other activating TCF/LEFs including TCF7 and LEF1. Indeed, our preliminary studies (Fig. S8) suggest that TCF7L1 is replaced at some key developmental genes (EOMES and NODAL) by LEF1. But this finding does not preclude the idea that other mechanisms might also act in concert to activate developmental genes.
hESCs are now regarded as in vitro equivalents of pluripotent epiblast cells, and therefore we might speculate that TCF7L1 could play a similar role in guarding those cells from premature differentiation during development. This role for TCF7L1 seems consistent with its role in mouse embryos where loss of function of Tcf7l1 leads to defective PS formation (Merrill et al., 2004). Key questions concerning transcriptional regulators are: which are the important regulated genes, and are these genes activated in a specific temporal order? Recent studies support our proposed role for TCF7L1 as a suppressor of PS differentiation genes (Hoffman et al., 2013; Morrison et al., 2016). Hoffman and colleagues surveyed Tcf7l1 protein expression within the epiblast during PS formation (Hoffman et al., 2013) and found that, as gastrulation proceeds, Tcf7l1 is downregulated in posterior epiblast cells during mouse PS formation. Additionally, others suggest that a direct target of Tcf7l1 repression in mESCs is Foxa2 (Morrison et al., 2016), consistent with previous studies indicating that Foxa2 is ectopically expressed in Tcf7l1−/− embryos (Merrill et al., 2004). Foxa2 is upregulated at the very beginning of gastrulation in mice and is considered a key endodermal regulator (Burtscher and Lickert, 2009; Levinson-Dushnik and Benvenisty, 1997). Together, these studies offer in vivo evidence that Tcf7l1 is involved in the regulation of epiblast differentiation within the forming PS.
Our ChIP-seq data support these observations but go further to indicate a much broader role for TCF7L1 in repression of PS and gastrulation genes. Genes we found regulated by TCF7L1 are WNT3 and NODAL. Thus, one potential role of TCF7L1 in primed pluripotent cells might be inhibition of key factors that can induce their differentiation. Since WNT3 and NODAL are known to be involved in gastrulation in many vertebrates it seems logical that there would be mechanisms to prevent their premature expression. Recent studies have demonstrated that exposure of hESCs to BMP4 in turn causes upregulation of WNT3 (Kurek et al., 2015). Our results showing that BMP4 causes downregulation of TCF7L1 complement those studies and indicate that upregulation of WNT3 by BMP4 might be achieved in part by BMP4 relieving TCF7L1 repression of WNT3. There are multiple mechanisms by which BMP4 might regulate the repressive activity of TCF7L1. ChIP-seq analyses suggest that one mechanism might be through SMAD binding to TCF7L1 gene regulatory elements (Tsankov et al., 2015). Nevertheless, our findings build further on a model for control of hESC (and perhaps epiblast) differentiation in which BMP4 inhibits TCF7L1 repressive activities, leading to upregulation of WNT3 (and other PS genes) (Fig. 7). Of course this mechanism of downregulating TCF7L1 could occur in concert with other mechanisms of TCF7L1 regulation, including regulation by β-catenin (see above), C-MYC and miR-211 (Atlasi et al., 2013; Morrison et al., 2016; Shy et al., 2013). TCF7L1 displaced from chromatin might be replaced with newly transcribed activating TCF/LEFs (LEF1 and TCF7), while WNT signaling could mediate stabilization of β-catenin in the nucleus. There, β-catenin could bind to LEF1 and/or TCF7 to activate WNT target genes. These studies are also consistent with recent findings showing that activation of the WNT pathway in hESCs causes β-catenin to associate with numerous PS genes (Funa et al., 2015). It is tempting to speculate whether TCF7L1 might also modulate other BMP-directed processes during embryogenesis, organogenesis and tissue homeostasis. Interestingly, a similar node of control also exists in neural precursor cells (NPCs) in which TCF7L1 represses WNT signaling and maintains NPCs in an undifferentiated state (Kuwahara et al., 2014). This model has clear parallels with our findings since, in response to differentiation, we find that LEF1 and to a lesser extent TCF7 are upregulated in hESCs.
Fig. 7.

Model of TCF7L1 role in hESC and epiblast pluripotency and differentiation. Epiblast cells (blue) in vivo – which can be thought of as synonymous with hESCs in vitro – remain in an undifferentiated state until necessary inductive signals are received. In the undifferentiated state, TCF7L1 represses genes involved in PS differentiation (blue-orange gradient). At the onset of gastrulation, BMP4 signaling triggers downregulation of TCF7L1, thereby relieving repression of PS gene targets. Whether the effect of BMP4 on TCF7L1 is direct or indirect remains to be determined.
Altogether, our data place TCF7L1 at a critical juncture in hESC differentiation and, by inference, potentially also in primed epiblast cells in human embryos (Fig. 7). A greater understanding of how TCF7L1 regulates gene expression during this period could fill vital gaps in our knowledge of this crucial time of development of our own species.
MATERIALS AND METHODS
hESC culture
hESC lines H1, H9 and H14 were maintained on mitotically inactivated mouse embryonic fibroblasts (MEFs; Millipore). hESCs were grown in medium containing DMEM/F12 (Thermo Fisher), 20% KnockOut Serum (Thermo Fisher), 4 ng/ml recombinant human basic fibroblast growth factor [bFGF (FGF2), Thermo Fisher], 5 mM GlutaMax (Thermo Fisher), 0.1 mM non-essential amino acids (Thermo Fisher) and 0.1 mM β-mercaptoethanol. For general hESC propagation and maintenance, cells were mechanically passaged every 5-7 days using the StemPro EZPassage tool (Thermo Fisher). For feeder-free culture, hESCs were grown on Geltrex (Thermo Fisher) and maintained with mTeSR1 or, in some experiments (Fig. S1C), TeSR E8 medium (STEMCELL Technologies) on hESC-qualified Matrigel (Corning). Cells stocks had a normal karyotype and were tested for mycoplasma using the MycoAlert PLUS Kit (Lonza). hESC work was carried out under protocols approved by the UC Irvine Human Stem Cell Research Oversight Committee.
Reverse transcription PCR
Total RNA was isolated from hESCs using the RNeasy Mini Kit (Qiagen) according to manufacturer's protocol. In addition, total RNA samples were treated with RNase-free DNase I (Qiagen) to remove genomic DNA. Total RNA was quantitated using a NanoDrop 2000 (Thermo Scientific) and 0.25 μg total RNA was used for PCR analysis. PCR conditions followed the SuperScript One-Step (Thermo Fisher) protocol with an annealing temperature of 55°C for 35 cycles of amplification. The loading control β-actin (ACTB) was amplified for 25 cycles with an annealing temperature of 55°C. PCR primer sets used in this study are listed in Table S1.
Quantitative PCR (qPCR)
Total RNA was isolated from hESCs and genomic DNA removed as described above. Total RNA was quantitated using the NanoDrop 2000 and 1.0 μg total RNA was used for cDNA synthesis with the High-Capacity cDNA Reverse Transcriptase Kit (Thermo Fisher). TaqMan and/or SYBR Green assays (Thermo Fisher) were used to measure RNA expression levels using a ViiA 7 RT-PCR System (Thermo Fisher). Data were analyzed by the ΔΔCT method and normalized to either 18S or GAPDH (Livak and Schmittgen, 2001). GraphPad Prism versions 5 and 6 were used to create graphs and perform two-tailed Student's t-test statistical analyses. SYBR Green primer sets and TaqMan probes used in this study are listed in Table S1.
Immunocytochemistry
For immunofluorescence staining analysis, hESCs were grown on 35 mm glass-bottom imaging dishes (Matek) or, in some experiments (Fig. S1C), on hESC-qualified Matrigel-coated dishes/chamber slides. Cells were fixed with 4% paraformaldehyde in PBS(−) (without calcium and magnesium) for 10-20 min at room temperature (RT). After fixation, cells were gently washed with PBS(+) (with calcium and magnesium), permeabilized with 0.5% Triton X-100 in PBS(+) for 10 min at RT, rinsed and blocked with 10% serum (this was dependent upon the species of the secondary antibody) solution for 1 h at RT. Immunostaining was carried out using antibodies against non-phosphorylated β-catenin (Cell Signaling) (Fig. S1B) and antibodies to TCF7L1, OCT4, SOX2 and NANOG (all from Santa Cruz) (Fig. S1B,C). Cells were incubated in 0.125 M glycine for 10 min at RT and blocked for 30 min at RT with 10% goat and donkey serums and 0.2% Triton X-100 in PBS(−). Primary antibodies were diluted in blocking buffer containing 1% of the appropriate serum and incubated with the cells overnight at 4°C. Cells were then rinsed two or three times with PBS(−) and incubated with secondary antibodies conjugated to Alexa Fluor dyes (Thermo Fisher) for 1-2 h in the dark at RT. Cells were then rinsed with PBS(−), and incubated with Hoechst for 7 min or as part of the secondary incubation process. Cells were then rinsed again and placed in 1.5-2.0 ml PBS(−). Stained cells were imaged using a Zeiss LSM 510 confocal, Olympus FSX100 or Nikon Ti microscope. Antibodies and dilutions are detailed in Table S2.
TOPflash assays
hESCs were co-transfected with TOFflash+β-galactosidase or, for the negative control, FOPflash+β-galactosidase reporter plasmids (the generous gift of Hans Clevers, Hubrecht Institute, Utrecht, The Netherlands; 1 µg per plasmid) using FuGENE HD (Promega) as described (Wu et al., 2013). Whole-cell lysates were collected and luciferase activity measured as relative light units after normalization to β-galactosidase activity. Statistical evaluation was performed by Student's unpaired t-test. P<0.05 was considered significant.
SDS-PAGE
Whole-cell protein extracts were either trypsinized (0.05% trypsin-EDTA) into single-cell suspensions, counted (0.5-1×106 cells, depending on the number required for a given experiment), lysed in 95°C SDS sample buffer, boiled for 5 min at 95-100°C and briefly sonicated to shear DNA or, in other experiments (Fig. S8A), washed with PBS, lysed with RIPA buffer (15 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40, 0.7% deoxycholate) plus Protease Inhibitor Cocktail (P8340, Sigma-Aldrich; 1:10) and Phosphatase Inhibitor Cocktail 2 and 3 (P5726 and P0044, Sigma-Aldrich; 1:200), scraped, and centrifuged at high speed (14,000-18,000 g) for 5-10 min at 4°C. Bradford assay (Bio-Rad) was used to determine protein concentration for equal loading across samples. Acrylamide gels (10-12%) were used for SDS-PAGE. Proteins were transferred to a PVDF membrane (GE Healthcare) for immunoblotting and membranes were imaged using a VersaDoc MP 5000 (Bio-Rad). Antibodies and their dilutions are detailed in Table S2.
Nuclear/cytoplasmic fractionation
hESCs were dissociated with 0.05% trypsin, counted, and 3×106 cells were lysed with cytoplasmic lysis buffer (50 mM Tris-HCl pH 7.5, 0.5% Triton X-100, 137.5 mM NaCl, 10% glycerol, 1 mM sodium vanadate, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, 5 mM EDTA, and protease inhibitors 1 mM PMSF, 4 µg/ml Aprotinin, 2 µg/ml Pepstatin A) on ice for 15 min. Insoluble nuclei were isolated by centrifugation at 13,000 rpm (16,000 g) for 5 min at 4°C. Supernatant (cytoplasmic fraction) was collected for analysis. The remaining nuclear pellet was washed three times with 500 μl cytoplasmic lysis buffer then lysed with nuclear lysis buffer (10 mM HEPES pH 7.9, 500 mM NaCl, 0.1 mM EDTA, 0.1 mM EGTA, 0.1% NP-40, and freshly added protease inhibitors 1 mM DTT, 1 mM PMSF, 4 µg/ml Aprotinin, 2 µg/ml Pepstatin A). Genomic DNA was sheared by sonication for 5 s with a Brandon Sonifier 450 at output two. The nuclear fraction was precleared by centrifugation at 13,000 rpm (16,000 g) for 15 min at 4°C and the supernatant transferred to a new tube. Nuclear or cytoplasmic fraction lysate was combined with an equal volume of 2× SDS lysis buffer for western blot analysis.
Signaling pathway inhibition
Cells were seeded at 50,000 per well on MEFs in a 12-well plate and allowed to grow for 4 days before treatment. Inhibitors of various signaling pathways were used: 10 μM SU5402 (EMD Millipore), 10 μM SB431542 (Tocris), 3 μM CHIR-99021 (Selleck), 2 μM IWP-2 (Stemgent) Cells treated with IWP-2 were grown on Geltrex (Thermo Fisher) under feeder-free conditions, with 0.1% DMSO as vehicle control.
siRNA knockdown
siRNA-mediated knockdowns in hESCs were performed under feeder-free conditions on Geltrex in mTeSR1 (STEMCELL Technologies) defined medium. hESCs were dissociated into single cells using 0.05% trypsin-EDTA (Thermo Fisher), filtered through a 40 μm filter to remove clumps and then 1.8-2×105 cells were plated per well of a 6-well plate in the presence of 10 μM ROCK inhibitor (Calbiochem) to increase single-cell survival (Watanabe et al., 2007). After 18-20 h, siRNAs were transfected into the hESCs using the RNAiMAX (Thermo Fisher) transfection reagent according to the manufacturer's protocol. Briefly, transfection of a single well of a 6-well plate with TCF7L1 or non-targeting siRNA (Dharmacon) at 50 nM was performed as follows: an Eppendorf tube containing 6 μl RNAiMAX and 94 μl Opti-MEM (Thermo Fisher) and a second tube containing 5 μl siRNA and 95 μl Opti-MEM were mixed together, incubated at RT for 15 min and then added to the well of hESCs. Twenty-four hours later this transfection procedure was repeated. RNA was isolated at appropriate time points and analyzed by qPCR as previously described. siRNAs are listed in Table S3.
Microarray
Microarray analyses were performed using GeneChip Human Gene 1.0 ST Arrays (Affymetrix). Probe cell intensity (.CEL) files were analyzed in Affymetrix Expression Console software v1.1.1 using the PLIER algorithm. Differential gene expression analysis was performed using Cyber-T (http://cybert.microarray.ics.uci.edu/). Genes were considered significant with a Benjamini and Hochberg value ≤0.05 and fold-change cutoff ±1.5. GO analysis was performed using DAVID 6.8 (david.ncifcrf.gov).
H9-TCF7L1 cell line generation
All cloning was performed using the GeneArt Seamless Cloning Kit (Thermo Fisher). 3×FLAG-TCF7L1-HTBH was inserted into pLenti-CMVtight-Puro (pLenti-CMVtight-TCF7L1-Puro).
The tetracycline repressor (rtTA3)-expressing lentiviral construct pLenti-CMV-rtTA3-Blast (a gift from Eric Campeau, Addgene plasmid #26429) was modified to generate pLenti-EF1alpha-rtTA3-Blast, so that the EF1alpha promoter would drive rtTA3 expression in hESCs. Transductions were performed using a Lentivector Packaging Kit (LV500A-1, System Biosciences) and virus concentrated using PEG-it Virus Precipitation Solution (LV810A-1, System Biosciences). Clonal lines were generated by serial single-cell dilutions and puromycin (1 μM) and blasticidin (2 μg/ml) selection.
Primitive streak-like differentiation assay
hESCs were trypsinized and 2×105 single cells were plated per well of a 6- or 12-well plate as described above under feeder-free conditions. Single cells were treated with 5-10 µM Rho-associated protein kinase inhibitor (ROCKi; Y-27632, Calbiochem) to improve survival. The following day, the cells were provided mTeSR1 medium to allow recuperation from single-cell passaging and to form small colonies. Twenty-four hours later, the cells were given primitive streak-like (PS) differentiation medium, which contains TeSR E5/E6 basal medium (Fig. S1B, Fig. S8) or RPMI 1640 (Thermo Fisher) supplemented with 2% B-27 supplement (Thermo Fisher), 1× non-essential amino acids (Thermo Fisher) and with 50 μg BMP4 (R&D Systems) and 50 μg activin A (R&D Systems or PeproTech). After 24 h in PS differentiation medium, hESCs were given fresh PS differentiation medium for another 24 h then harvested for analysis.
ChIP-seq
Each ChIP experiment was performed using ∼6×107 cells grown under feeder-free conditions in mTeSR1 defined medium as described above. Twenty million cells were used for each immunoprecipitation (IP) condition. Once the hESCs reached confluence, they were cross-linked in fixing buffer (PBS plus 1% methanol-free formaldehyde) for 10 min at RT. Cross-linking was inhibited by adding 2.5 M glycine to a final concentration of 0.125 M and incubated for 10 min at RT with gentle rocking. The wells were then aspirated and rinsed with cold (4°C) PBS containing protease inhibitors (Thermo Fisher; PIs) twice. Cells were scraped from the wells with a cell scraper (Corning) and added to a 50 ml conical tube on ice. The hESC suspension was centrifuged at 1350 g for 10 min at 4°C. The supernatant was carefully removed and the cell pellet resuspended with 10 ml cold lysis buffer (50 mM HEPES pH 7.9, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, fresh PIs) to release nuclei. The cell suspension was then transferred to a new 15 ml conical tube and incubated at 4°C on a nutator for 10 min. Cells were pelleted at 1350 g for 5 min at 4°C. The supernatant was removed and the pellet resuspended in 5 ml wash buffer (10 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, fresh PIs) and incubated again at 4°C for 10 min on a nutator. Finally, the nuclei were pelleted at 1350 g for 5 min at 4°C. and then resuspended in 1 ml buffer NUC (15 mM HEPES pH 7.5, 60 mM KCl, 15 mM NaCl, 0.32 mM sucrose, fresh 0.5 mM PMSF) and transferred to a new microfuge tube, bringing the total volume to 1.2 ml. Next, 12 μl 100× protease inhibitor (Thermo Scientific), 3.3 μl 1 M CaCl2 and 10 μl micrococcal nuclease (2000 units/μl; New England BioLabs) were added to the chromatin suspension and incubated at 37°C for 25 min with shaking. Immediately after 25 min, 1.2 ml 2× sonication buffer (90 mM HEPES pH 7.9, 220 mM NaCl, 10 mM EDTA pH 8.0, 1% NP-40, 0.2% Na deoxycholate, 0.2% SDS) was added to the sample in a 15 ml conical tube and placed on ice. The volume was then equally split between two 1.5 ml microfuge tubes for gentle sonication to release chromatin from the nuclei. The QSonica Q700 with micro-tip was set: amplitude 1; time on 5 s; time off 60 s. This cycle was repeated four times for each microfuge tube. Sheared and sonicated samples were centrifuged for 15 min at 18,000 rpm (22,000 g) at 4°C. Chromatin from both microfuge tubes was pooled in a 15 ml conical tube and the volume brought to 3 ml by adding 1× sonication buffer [a 50/50 mixture of 2× sonication buffer, buffer NUC plus Halt protease inhibitor cocktail (Thermo Fisher, 78430; 1:100)]. Lastly, 30 μl was removed for 1% input and DNA digestion analysis.
For a single ChIP, 5 μg antibody (for details see Table S2), or IgG for the control, was added to 1 ml of the chromatin suspension (roughly equivalent to the chromatin from 2×107 cells) and incubated overnight at 4°C on a nutator. Then, 50 μl Dynabeads (Thermo Fisher) were washed once with 1 ml 1× sonication buffer and transferred to the chromatin and antibody suspension, with incubation for 2 h at 4°C on a rotator. Beads were collected with a magnet and washed as follows: five times with 1 ml cold modified RIPA buffer (50 mM HEPES-KOH pH 7.9, 500 mM LiCl, 1 mM EDTA, 1% NP-40, 0.7% Na deoxycholate) and twice with 1 ml TE (10 mM Tris-HCl pH 7.5, 1 mM EDTA).
After the final wash, the beads were centrifuged at RT for 1 min at 1000 rpm (1200 g) and the supernatant discarded. Beads were resuspended in 210 μl direct elution buffer (10 mM Tris-HCl pH 8.0, 300 mM NaCl, 5 mM EDTA, 1% SDS, 0.2 μg/ml RNase A) and incubated at 65°C overnight with shaking. Then, beads were centrifuged for 1 min at 18,000 rpm (22,000 g) and 200 μl supernatant transferred to a new microfuge tube. Proteinase K (2 μl of 20 mg/ml stock; Thermo Scientific) was added, incubated at 55°C for 2 h, then purified using a Mini Elute PCR Purification Kit (Qiagen) according to the manufacturer's protocol. DNA was eluted with 30 μl EB buffer (50°C) and DNA concentration measured with a Qubit HS Kit (Thermo Fisher) following the manufacturer's instructions.
ChIP-seq peak calling and irreproducibility discovery rate (IDR)
TCF7L1 ChIP-seq and input samples from H9 hESCs were sequenced at the UCI High-Throughput Facility on an Illumina HiSeq 2500 platform. Reads were mapped to the human genome (hg19) with Bowtie version 0.12.8 using −v 2, −S, −k 1, −m 1 parameters to report only uniquely mapped reads with at most two mismatches. The resulting sam files were converted to bam files using SAMtools version 0.1.19. To ensure greater or equal read depth in input samples versus ChIP samples (as suggested by ENCODE), TCF7L1 ChIP bam files were randomly downsampled (using SAMtools −view, −b, −s parameters) so that they contained fewer reads than input. SAMtools was also used to remove PCR duplicates (using the rmdup parameter). The resulting number of mapped reads for each sample was: input, 55,127,188; TCF7L1 replicate #1, 53,797,735; TCF7L1 replicate #2, 54,273,005.
IDR analysis was performed as suggested by ENCODE to determine the consistency of the ChIP-seq biological replicates. IDR was performed using the Macs2 peak caller as in https://sites.google.com/site/anshulkundaje/projects/idr. Briefly, peaks were called on individual TCF7L1 biological replicates as well as on the pooled replicates with Macs2 using a P-value threshold of 0.01. The top 50,000 peaks from TCF7L1 ChIP-seq replicate #1 and replicate #2 were entered into IDR analysis. To achieve the final ChIP-seq peaks, an IDR of 0.05 was used, which is within the range suggested (https://sites.google.com/site/anshulkundaje/projects/idr).
Genomic regions enrichment of annotations tool (GREAT) analysis
GREAT analysis (http://bejerano.stanford.edu/great/public/html/index.php) was performed on the top 1000 TCF7L1 peaks ±5 kb from annotated TSSs. The human GRCn37 hg19 build was selected as the ‘species assembly’ and the whole genome was chosen for the ‘background regions’ field. The ‘basal plus extension’ analysis method was used along with its default settings: proximal 5.0 kb upstream, 1.0 kb downstream, plus distal up to 1000.0 kb.
Comparison of TCF7L1 overlap with OCT4, NANOG and β-catenin in mESCs and hESCs
To determine overlapping peaks in different data sets and to annotate neighboring genes within ±5-8 kb away from TSSs we followed Zhu et al. (2010). Venny was used to compare common genes across data sets (Oliveros, 2016; R Development Core Team, 2013). The following public ChIP-seq data sets were used in the analyses: supplementary tables S2 and S3 from Marson et al. (2008), which contain gene lists of ChIP-seq data for Oct4, Nanog, Sox2 and Tcf7l1 in V6.5 mESCs; OCT4 and NANOG ChIP-seq data sets from H1 [WgEncodeEH001635 (GEO accession GSM803437) and WgEncodeEH001636 (GEO accession GSM803438); The ENCODE Project Consortium (2012)] and H9 [GEO accession GSE62983; Kushwaha et al. (2015)] hESCs. ChIP-seq data for β-catenin (CTNNB1) in hESCs (GEO accession GSE64758) were from Estarás et al. (2015).
TCF7L1 and LEF1 ChIP-PCR
ChIP was carried out as described above but with modifications. Cells were fixed with 1% methanol-free formaldehyde in PBS for 10 min at RT, and cross-linking was stopped with 0.125 M glycine in PBS for 10 min at RT. Fixed materials were proceed as previously described with modifications (Vuong et al., 2015). Cells were scraped into cold PBS and centrifuged at 1000 g for 5-10 min in 4°C. The pellet was resuspended in 1 ml hypotonic buffer (10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2) plus Protease Inhibitor Cocktail (1:20) and Phosphatase Inhibitor Cocktail 2 and 3 (1:200) and incubated for 10 min at 4°C. Then, MgCl2 and NP-40 were added to a final concentration of 5 mM and 0.1%, respectively, with additional incubation and centrifugation for 10 min at 4°C. The pellet was resuspended in 0.6 ml MNase buffer (20 mM Tris pH 7.5, 15 mM NaCl, 1 mM CaCl2) plus 3 μl (1.894 unit/μl) micrococcal nuclease (Worthington) and incubated at 37°C for 25 min with shaking; another 0.6 ml 2× sonication buffer plus Protease Inhibitor Cocktail (1:100) and Phosphatase Inhibitor Cocktail 2 and 3 (1:200). Cells were sonicated once using the Q700 sonicator (QSonica) with the following settings: amplitude 1%; time on 5 s; time off 60 s; processing time 30 s; power 10 W. After sonication, samples were centrifuged at maximum speed (14,000-18,000 g) for 5-10 min at 4°C. The supernatant was collected and pre-cleared with 10-20 μl prewashed Protein A/G magnetic beads (Pierce) for 30 min at 4°C. The lysate was then divided in half and anti-LEF1 (76010, Cell Signaling; 1:50), 10 μl anti-TCF7L1 (TCF3; 61125, Active Motif) or 2.5 μg rabbit IgG (12-370, Upstate) was added to each tube and incubated for ∼2 h at 4°C. IP samples were centrifuged at maximum speed (14,000-18,000 g) for 3 min at 4°C and 40-50 μg Protein A/G magnetic beads were added and incubated overnight at 4°C. Then, IP samples were incubated in the following buffers for 5 min at RT each in sequential order: TSE I (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100), II (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100) and III (10 mM Tris-HCl pH 8.0, 0.25 mM LiCl, 1 mM EDTA, 1% NP-40, 1% deoxycholate). Finally, IP samples were washed two or three times with 1× TE. The precipitated sample was eluted twice, each time with 150 μl IP elution buffer (0.1 M NaHCO3, 1% SDS) for 20 min at RT with shaking; prior to the second elution, samples were heated for 1 min at 100°C. To the eluted samples, 1 μl of 20 mg/ml proteinase K (E195, VWR) and 1/10 volume of proteinase K buffer (100 mM Tris-HCl pH 8.0, 500 mM NaCl, 50 mM EDTA) plus NaCl to a final concentration of 0.3 M were added and incubated at 65°C for at least 5 h or overnight to reverse the crosslinks and digest the protein. The QIAquick PCR Purification Kit (28104, Qiagen) was used to purify the DNA. PCR was performed on the purified DNA samples using the following primers (5′-3′, forward and reverse): EOMES, AAGCAGGAGGGAGTCAGTCA and TTGCTCTGCACTTGCTCTGT (product 499 bp); NODAL, AGGTACCAAACGCCTTGATG and GTGGGCAACAAGGGTAACAC (product 467 bp). UCSC In-Silico PCR (https://genome.ucsc.edu/cgi-bin/hgPcr) and Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/) programs were used to design the primers to promoters of EOMES and NODAL. An iCyder PCR machine (Bio-Rad) was used with the following setting: 94°C 3 min; 25-30 cycles of 94°C 45 s, 52°C 30 s, 72°C 1 min; then 72°C 10 min and 16°C hold. PCR samples were run in a 2% agarose gel. ImageJ (https://imagej.nih.gov/ij/index.html) was used to quantify intensity of DNA and protein bands. All qPCR primers used are listed in Table S4.
Supplementary Material
Acknowledgements
We extend our sincerest appreciation and gratitude to members of the P.J.D. and M.L.W. Laboratories, especially Dr Jenny Mastroianni, Norman Hsu, Kristyl Felix and George Todorov. We are especially grateful to Terry Nguyen for generating the data for Fig. S1C. We also thank Drs Xing Dai and Peter Kaiser for helpful and insightful suggestions and comments, and Drs Malcolm Casale, Harry Mangalam, Jenny Wu and G. Wesley Hatfield for advice on microarray analysis.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: R.A.S., N.P.H., R.N.R., L.M.V., A.M., B.J.M., M.L.W., P.J.D.; Methodology: R.A.S., N.P.H., R.N.R., L.M.V., A.M., B.J.M., M.L.W., P.J.D.; Software: N.P.H., R.N.R., L.M.V., A.M.; Validation: R.A.S., N.P.H., R.N.R., L.M.V., M.L.W., P.J.D.; Formal analysis: R.A.S., N.P.H., R.N.R., L.M.V., A.M., M.L.W., P.J.D.; Investigation: R.A.S., N.P.H., L.M.V., M.L.W., P.J.D.; Resources: M.L.W., P.J.D.; Data curation: R.A.S., N.P.H., R.N.R., L.M.V., M.L.W., P.J.D.; Writing - original draft: R.A.S., M.L.W., P.J.D.; Writing - review & editing: R.A.S., N.P.H., R.N.R., L.M.V., A.M., B.J.M., M.L.W., P.J.D.; Visualization: R.A.S., L.M.V., P.J.D.; Supervision: M.L.W., P.J.D.; Project administration: M.L.W., P.J.D.; Funding acquisition: M.L.W., P.J.D.
Funding
This work was supported by the California Institute of Regenerative Medicine (CIRM; RB2-01629 to M.L.W.), the National Institutes of Health (R01 GM096017 to P.J.D. and M.L.W.), a CIRM Training Fellowship (TG2-01152 to R.A.S.), National Cancer Institute (RO1-CA128571 to B.J.M.) and institutional funds from the University of California, Irvine (to P.J.D.). We also acknowledge the support of a National Cancer Institute award to the Chao Family Comprehensive Cancer Center (P30CA062203) and a CIRM Shared Research Laboratory Award (CL1-00520-1.2) to the Sue and Bill Gross Stem Cell Research Center. Deposited in PMC for release after 12 months.
Data availability
Data generated in the course of the present studies and submitted to the NCBI Gene Expression Omnibus comprise: GSE69911, expression data from TCF7L1 siRNA knockdown in H9 hESCs; GSE71668, expression data from TCF7L1 overexpression during PS-like differentiation in H9 hESCs; and GSE80331, TCF7L1 ChIP-seq in H9 hESCs.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.161075.supplemental
References
- Atlasi Y., Noori R., Gaspar C., Franken P., Sacchetti A., Rafati H., Mahmoudi T., Decraene C., Calin G. A., Merrill B. J. et al. (2013). Wnt signaling regulates the lineage differentiation potential of mouse embryonic stem cells through Tcf3 down-regulation. PLoS Genet. 9, e1003424 10.1371/journal.pgen.1003424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauwkamp T. A., Nigam S., Ardehali R., Weissman I. L. and Nusse R. (2012). Endogenous Wnt signalling in human embryonic stem cells generates an equilibrium of distinct lineage-specified progenitors. Nat. Commun. 3, 1070 10.1038/ncomms2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burtscher I. and Lickert H. (2009). Foxa2 regulates polarity and epithelialization in the endoderm germ layer of the mouse embryo. Development 136, 1029-1038. 10.1242/dev.028415 [DOI] [PubMed] [Google Scholar]
- Cadigan K. M. and Waterman M. L. (2012). TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol. 4, a007906 10.1101/cshperspect.a007906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers I., Silva J., Colby D., Nichols J., Nijmeijer B., Robertson M., Vrana J., Jones K., Grotewold L. and Smith A. (2007). Nanog safeguards pluripotency and mediates germline development. Nature 450, 1230-1234. 10.1038/nature06403 [DOI] [PubMed] [Google Scholar]
- Chodaparambil J. V., Pate K. T., Hepler M. R. D., Tsai B. P., Muthurajan U. M., Luger K., Waterman M. L. and Weis W. I. (2014). Molecular functions of the TLE tetramerization domain in Wnt target gene repression. EMBO J. 33, 719-731. 10.1002/embj.201387188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole M. F., Johnstone S. E., Newman J. J., Kagey M. H. and Young R. A. (2008). Tcf3 is an integral component of the core regulatory circuitry of embryonic stem cells. Genes Dev. 22, 746-755. 10.1101/gad.1642408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson K. C., Adams A. M., Goodson J. M., McDonald C. E., Potter J. C., Berndt J. D., Biechele T. L., Taylor R. J. and Moon R. T. (2012). Wnt/beta-catenin signaling promotes differentiation, not self-renewal, of human embryonic stem cells and is repressed by Oct4. Proc. Natl. Acad. Sci. USA 109, 4485-4490. 10.1073/pnas.1118777109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dravid G., Ye Z., Hammond H., Chen G., Pyle A., Donovan P., Yu X. and Cheng L. (2005). Defining the role of Wnt/beta-catenin signaling in the survival, proliferation, and self-renewal of human embryonic stem cells. Stem Cells 23, 1489-1501. 10.1634/stemcells.2005-0034 [DOI] [PubMed] [Google Scholar]
- Eiselleova L., Matulka K., Kriz V., Kunova M., Schmidtova Z., Neradil J., Tichy B., Dvorakova D., Pospisilova S., Hampl A. et al. (2009). A complex role for FGF-2 in self-renewal, survival, and adhesion of human embryonic stem cells. Stem Cells 27, 1847-1857. 10.1002/stem.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppig J. T., Blake J. A., Bult C. J., Kadin J. A., Richardson J. E. and The Mouse Genome Database Group. (2015). The Mouse Genome Database (MGD): facilitating mouse as a model for human biology and disease. Nucleic Acids Res. 43, D726-D736. 10.1093/nar/gku967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estarás C., Benner C. and Jones K. A. (2015). SMADs and YAP compete to control elongation of beta-catenin:LEF-1-recruited RNAPII during hESC differentiation. Mol. Cell 58, 780-793. 10.1016/j.molcel.2015.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S., Zhang M., Schöler H. R. and Greber B. (2012). Small molecule-assisted, line-independent maintenance of human pluripotent stem cells in defined conditions. PLoS ONE 7, e41958 10.1371/journal.pone.0041958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funa N. S., Schachter K. A., Lerdrup M., Ekberg J., Hess K., Dietrich N., Honoré C., Hansen K. and Semb H. (2015). beta-Catenin regulates primitive streak induction through collaborative interactions with SMAD2/SMAD3 and OCT4. Cell Stem Cell 16, 639-652. 10.1016/j.stem.2015.03.008 [DOI] [PubMed] [Google Scholar]
- Guo G., Huang Y., Humphreys P., Wang X. and Smith A. (2011). A piggybac-based recessive screening method to identify pluripotency regulators. PLoS ONE 6, e18189 10.1371/journal.pone.0018189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman J. A., Wu C.-I. and Merrill B. J. (2013). Tcf7l1 prepares epiblast cells in the gastrulating mouse embryo for lineage specification. Development 140, 1665-1675. 10.1242/dev.087387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkan T. and Smith A. (2014). Mapping the route from naive pluripotency to lineage specification. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130540 10.1098/rstb.2013.0540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurek D., Neagu A., Tastemel M., Tüysüz N., Lehmann J., van de Werken H. J. G., Philipsen S., van der Linden R., Maas A., van IJcken W. F. J. et al. (2015). Endogenous WNT signals mediate BMP-induced and spontaneous differentiation of epiblast stem cells and human embryonic stem cells. Stem Cell Rep. 4, 114-128. 10.1016/j.stemcr.2014.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwaha R., Jagadish N., Kustagi M., Tomishima M. J., Mendiratta G., Bansal M., Kim H. R., Sumazin P., Alvarez M. J., Lefebvre C. et al. (2015). Interrogation of a context-specific transcription factor network identifies novel regulators of pluripotency. Stem Cells 33, 367-377. 10.1002/stem.1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara A., Sakai H., Xu Y., Itoh Y., Hirabayashi Y. and Gotoh Y. (2014). Tcf3 represses Wnt-β-catenin signaling and maintains neural stem cell population during neocortical development. PLoS ONE 9, e94408 10.1371/journal.pone.0094408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson-Dushnik M. and Benvenisty N. (1997). Involvement of hepatocyte nuclear factor 3 in endoderm differentiation of embryonic stem cells. Mol. Cell. Biol. 17, 3817-3822. 10.1128/MCB.17.7.3817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K. J. and Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 25, 402-408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Mao C. D. and Byers S. W. (2011). Cell-context dependent TCF/LEF expression and function: alternative tales of repression, de-repression and activation potentials. Crit. Rev. Eukaryot. Gene Expr. 21, 207-236. 10.1615/CritRevEukarGeneExpr.v21.i3.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson A., Levine S. S., Cole M. F., Frampton G. M., Brambrink T., Johnstone S., Guenther M. G., Johnston W. K., Wernig M., Newman J. et al. (2008). Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 134, 521-533. 10.1016/j.cell.2008.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill B. J., Pasolli H. A., Polak L., Rendl M., García-García M. J., Anderson K. V. and Fuchs E. (2004). Tcf3: a transcriptional regulator of axis induction in the early embryo. Development 131, 263-274. 10.1242/dev.00935 [DOI] [PubMed] [Google Scholar]
- Morgani S., Nichols J. and Hadjantonakis A.-K. (2017). The many faces of Pluripotency: in vitro adaptations of a continuum of in vivo states. BMC Dev. Biol. 17, 7 10.1186/s12861-017-0150-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison G., Scognamiglio R., Trumpp A. and Smith A. (2016). Convergence of cMyc and beta-catenin on Tcf7l1 enables endoderm specification. EMBO J. 35, 356-368. 10.15252/embj.201592116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H., Merrill B. J., Polak L., Nikolova M., Rendl M., Shaver T. M., Pasolli H. A. and Fuchs E. (2009). Tcf3 and Tcf4 are essential for long-term homeostasis of skin epithelia. Nat. Genet. 41, 1068-1075. 10.1038/ng.431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J. and Smith A. (2009). Naive and primed pluripotent states. Cell Stem Cell 4, 487-492. 10.1016/j.stem.2009.05.015 [DOI] [PubMed] [Google Scholar]
- Nichols J. and Smith A. (2012). Pluripotency in the embryo and in culture. Cold Spring Harbor Perspect. Biol. 4, a008128 10.1101/cshperspect.a008128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveros J. C. (2016). Venny. An interactive tool for comparing lists with Venn's diagrams. 2007–2015. http://bioinfogp.cnb.csic.es/tools/venny/index.html. [Google Scholar]
- Pereira L., Yi F. and Merrill B. J. (2006). Repression of Nanog gene transcription by Tcf3 limits embryonic stem cell self-renewal. Mol. Cell. Biol. 26, 7479-7491. 10.1128/MCB.00368-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A. R. and Hall I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features Bioinformatics, 26, 841-842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Shy B. R., Wu C.-I., Khramtsova G. F., Zhang J. Y., Olopade O. I., Goss K. H. and Merrill B. J. (2013). Regulation of Tcf7l1 DNA binding and protein stability as principal mechanisms of Wnt/β-catenin signaling. Cell Rep. 4, 1-9. 10.1016/j.celrep.2013.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagwerker C., Flick K., Cui M., Guerrero C., Dou Y., Auer B., Baldi P., Huang L. and Kaiser P. (2006). A tandem affinity tag for two-step purification under fully denaturing conditions: application in ubiquitin profiling and protein complex identification combined with in vivocross-linking. Mol. Cell. Proteomics 5, 737-748. 10.1074/mcp.M500368-MCP200 [DOI] [PubMed] [Google Scholar]
- Tam P. P. L. and Loebel D. A. F. (2007). Gene function in mouse embryogenesis: get set for gastrulation. Nat. Rev. Genet. 8, 368-381. 10.1038/nrg2084 [DOI] [PubMed] [Google Scholar]
- Teo A. K. K., Ali Y., Wong K. Y., Chipperfield H., Sadasivam A., Poobalan Y., Tan E. K., Wang S. T., Abraham S., Tsuneyoshi N. et al. (2012). Activin and BMP4 synergistically promote formation of definitive endoderm in human embryonic stem cells. Stem Cells 30, 631-642. 10.1002/stem.1022 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57-74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankov A. M., Gu H., Akopian V., Ziller M. J., Donaghey J., Amit I., Gnirke A. and Meissner A. (2015). Transcription factor binding dynamics during human ES cell differentiation. Nature 518, 344-349. 10.1038/nature14233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong L. M., Chellappa K., Dhahbi J. M., Deans J. R., Fang B., Bolotin E., Titova N. V., Hoverter N. P., Spindler S. R., Waterman M. L. et al. (2015). Differential effects of hepatocyte nuclear factor 4α isoforms on tumor growth and T-cell factor 4/AP-1 interactions in human colorectal cancer cells. Mol. Cell. Biol. 35, 3471-3490. 10.1128/MCB.00030-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K., Ueno M., Kamiya D., Nishiyama A., Matsumura M., Wataya T., Takahashi J. B., Nishikawa S., Nishikawa S.-I., Muguruma K. et al. (2007). A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 25, 681-686. 10.1038/nbt1310 [DOI] [PubMed] [Google Scholar]
- Winnier G., Blessing M., Labosky P. A. and Hogan B. L. (1995). Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 9, 2105-2116. 10.1101/gad.9.17.2105 [DOI] [PubMed] [Google Scholar]
- Wu C.-I., Hoffman J. A., Shy B. R., Ford E. M., Fuchs E., Nguyen H. and Merrill B. J. (2012). Function of Wnt/β-catenin in counteracting Tcf3 repression through the Tcf3-β-catenin interaction. Development 139, 2118-2129. 10.1242/dev.076067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B., Piloto S., Zeng W., Hoverter N. P., Schilling T. F. and Waterman M. L. (2013). Ring Finger Protein 14 is a new regulator of TCF/β-catenin-mediated transcription and colon cancer cell survival. EMBO Rep. 14, 347-355. 10.1038/embor.2013.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R.-H., Sampsell-Barron T. L., Gu F., Root S., Peck R. M., Pan G., Yu J., Antosiewicz-Bourget J., Tian S., Stewart R. et al. (2008). NANOG is a direct target of TGFβ/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell 3, 196-206. 10.1016/j.stem.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Robitaille A. M., Berndt J. D., Davidson K. C., Fischer K. A., Mathieu J., Potter J. C., Ruohola-Baker H. and Moon R. T. (2016). Wnt/beta-catenin signaling promotes self-renewal and inhibits the primed state transition in naïve human embryonic stem cells. Proc. Natl. Acad. Sci. USA 113, E6382-E6390. 10.1073/pnas.1613849113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F. and Merrill B. J. (2007). Stem cells and TCF proteins: a role for beta-catenin--independent functions. Stem Cell Rev. 3, 39-48. 10.1007/s12015-007-0003-9 [DOI] [PubMed] [Google Scholar]
- Yi F., Pereira L. and Merrill B. J. (2008). Tcf3 functions as a steady-state limiter of transcriptional programs of mouse embryonic stem cell self-renewal. Stem Cells 26, 1951-1960. 10.1634/stemcells.2008-0229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L. J., Gazin C., Lawson N. D., Pagès H., Lin S. M., Lapointe D. S. and Green M. R. (2010). ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics 11, 237 10.1186/1471-2105-11-237 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.