

ABSTRACT
Staphylococcus aureus subverts innate defenses during infection in part by killing host immune cells to exacerbate disease. This human pathogen intercepts host cues and activates a transcriptional response via the S. aureus exoprotein expression (SaeR/SaeS [SaeR/S]) two-component system to secrete virulence factors critical for pathogenesis. We recently showed that the transcriptional repressor CodY adjusts nuclease (nuc) gene expression via SaeR/S, but the mechanism remained unknown. Here, we identified two CodY binding motifs upstream of the sae P1 promoter, which suggested direct regulation by this global regulator. We show that CodY shares a binding site with the positive activator SaeR and that alleviating direct CodY repression at this site is sufficient to abrogate stochastic expression, suggesting that CodY represses sae expression by blocking SaeR binding. Epistasis experiments support a model that CodY also controls sae indirectly through Agr and Rot-mediated repression of the sae P1 promoter. We also demonstrate that CodY repression of sae restrains production of secreted cytotoxins that kill human neutrophils. We conclude that CodY plays a previously unrecognized role in controlling virulence gene expression via SaeR/S and suggest a mechanism by which CodY acts as a master regulator of pathogenesis by tying nutrient availability to virulence gene expression.
IMPORTANCE Bacterial mechanisms that mediate the switch from a commensal to pathogenic lifestyle are among the biggest unanswered questions in infectious disease research. Since the expression of most virulence genes is often correlated with nutrient depletion, this implies that virulence is a response to the lack of nourishment in host tissues and that pathogens like S. aureus produce virulence factors in order to gain access to nutrients in the host. Here, we show that specific nutrient depletion signals appear to be funneled to the SaeR/S system through the global regulator CodY. Our findings reveal a strategy by which S. aureus delays the production of immune evasion and immune-cell-killing proteins until key nutrients are depleted.
KEYWORDS: CodY, Staphylococcus aureus, Sae TCS, virulence, branched-chain amino acids, Sae, two-component system, gene regulation
INTRODUCTION
Staphylococcus aureus is a Gram-positive bacterium that colonizes the nasal epithelium of up to ∼30% of the human population (1, 2). As an opportunistic pathogen, S. aureus is a leading cause of skin and soft tissue infections, health care-associated infections, endocarditis, osteomyelitis, and bacteremia (3). While health care-acquired infections have been around as long as health care workers, aggressive staphylococcal strains now circulate in the community; multidrug resistance has compounded this problem (4, 5). For example, community acquired, methicillin-resistant S. aureus (CA-MRSA) has killed tens of thousands of people (6). As a result, CA-MRSA infections burden the U.S. health care system with between $560 million and $2.7 billion in annual treatment-associated costs (7).
The success of S. aureus as a pathogen stems in part from its ability to produce a plethora of virulence factors aiding in adhesion, nutrient acquisition, and host immune escape. For example, agglutination in the bloodstream, a characteristic of S. aureus but not of related coagulase-negative staphylococci, provides a mechanism for survival and dissemination to organs throughout the body (8). However, the production of all virulence factors simultaneously is costly to the bacterium from a resource and energy conservation perspective (9). Thus, the precise location and timing of virulence factor production are just as important as the factors themselves, and interfering with this choreography represents an exciting approach for developing new treatments and therapies.
The SaeR/SaeS (SaeR/S) two-component system (TCS) contributes to the regulation of over 20 virulence factors, most notably α-hemolysin, bicomponent leukocidins, surface proteins, nuclease, superantigens and superantigen-like proteins, coagulase, and proteases (10–18). The organization of the sae locus and its regulation are complex and incompletely understood (Fig. 1A) (14). The saeRS operon is transcribed constitutively from the weak sae P3 promoter and encodes the response regulator SaeR and an intramembrane-sensing histidine kinase, SaeS. In the presence of neutrophils or factors produced by neutrophils, including human neutrophil peptides 1, 2, and 3 (HNP1–3), the metal chelator calprotectin, and hydrogen peroxide (19–22), SaeS is phosphorylated (SaeS∼P) on a conserved histidine residue; then the phosphoryl group is transferred to a conserved aspartate residue on SaeR. Upon activation, the fraction of activated SaeR (SaeR∼P) rises and binds to the inducible sae P1 promoter just upstream of saeP, activating transcription of saeRS as well as saeP and saeQ, whose products form a complex in the membrane to stimulate the phosphatase activity of SaeS and, ultimately, reduce the fraction of SaeR∼P in the cell (Fig. 1B) (23).
FIG 1.
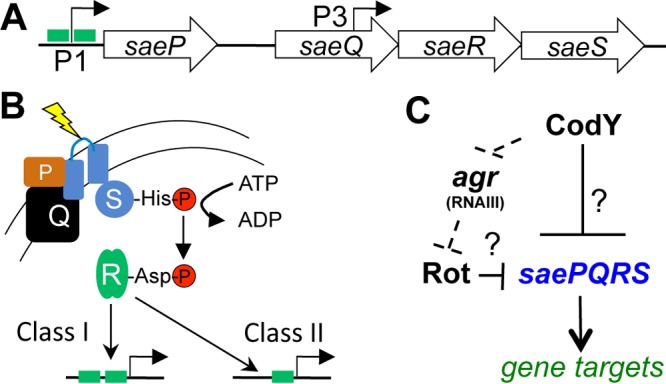
CodY and Sae control virulence gene expression. (A) Schematic of the saePQRS locus. The green boxes at promoters denote SaeR binding sites. The weak but constitutive P3 promoter drives expression of saeRS; the P1 promoter is inducible and drives expression of saePQRS. (B) SaeS responds to multiple stimuli (yellow), and this promotes autophosphorylation of SaeS and phosphotransfer to SaeR. SaeR∼P can then bind its various targets. (C) Regulatory relationships between Agr, Rot, CodY, and Sae. Major global regulators under CodY control and known to affect sae P1 promoter activity are shown. The dashed lines indicate a potential indirect mechanism of CodY-mediated repression; the solid line indicates potential direct regulation. Question marks indicate an unknown or postulated regulatory mechanism.
Direct targets of SaeR can be assigned to one of two classes (Fig. 1B) (14, 24). Class II targets have high affinity for SaeR∼P and contain a single SaeR binding site (e.g., hla, α-toxin; hlb, β-toxin). Under uninduced conditions, low levels of SaeR∼P are sufficient to activate transcription of these promoters. Class I targets are low-affinity promoters possessing two SaeR binding sites (e.g., coa, coagulase; eap, extracellular adherence protein; efb, extracellular fibrinogen binding protein; fnbA, fibronectin binding protein) and require high SaeR∼P levels to activate their expression. The sae P1 promoter is also a class I target. Thus, SaeR is a positive regulator of its own operon as well as of virulence genes.
In addition to SaeS-specific signals, the Sae system is subject to additional transcriptional and posttranscriptional regulation though regulation appears to vary depending on strain background or growth conditions. For example, the sae P1 promoter has been shown to be repressed by the alternative sigma factor B (σB) and positively regulated by the agr locus, an autoinduced global regulatory system sensitive to population density (14, 25–27). More recently, it was shown that RNAIII, the small regulatory RNA effector of the Agr system, acts as an indirect, positive regulator of the Sae TCS by antagonizing the production of the repressor of toxins (Rot) (28, 29). Finally, the saePQRS transcript is a target of the endonuclease RNase Y (30). RNase Y, a known driver of RNA decay and maturation, cleaves within an intergenic region between saeP and saeQ. The resulting mRNA processing has been proposed to increase the expression of saeRS (31).
CodY is a global transcriptional regulator that directly or indirectly controls the expression of dozens of genes in a variety of Gram-positive bacteria with low G+C content. In pathogens including S. aureus, CodY links nutrient availability, metabolism, and virulence gene expression by sensing branched-chain amino acids (isoleucine, leucine, valine [ILV]) and GTP (32, 33). Upon binding these key metabolites, CodY is activated as a DNA-binding protein and interacts with regions of chromosomal DNA containing a core sequence motif (i.e., AATTTTCWGAAAATT) (34–37). As the abundance of ILV and GTP drops in the cell, so too does the fraction of active CodY molecules, giving rise to prioritized gene expression as CodY loses affinity for its target sequences (38, 39). The picture that has emerged is that S. aureus uses CodY to first turn on nutrient uptake. As limitation intensifies, the bacterium turns on the expression of secreted digestive enzymes and cytolytic, pore-forming toxins, delaying the production of these factors until nutrients are critically low (39). In essence, induction of virulence gene expression when nutrients are depleted may be considered a normal response of the bacterium to starvation, and S. aureus potentially produces virulence factors to gain access to nutrients in the host in an effort to restore nutrient availability.
CodY adjusts the expression of many of the known virulence genes in S. aureus, demonstrating the intimate linkage between nutrient availability and virulence gene expression (36, 37, 39, 40). However, the mechanistic underpinnings are incomplete. For example, in a codY null mutant, about half of the virulence genes are overexpressed because they are positively regulated by the Agr system. Here, CodY represses the agr locus, effectively blocking the quorum sensing regulatory signal even at low population density as long as nutrients are abundant (36, 41). The remaining genes are presumably under direct or indirect regulation by CodY. We previously demonstrated that CodY controls the expression of nuc (thermonuclease) via the Sae TCS (39). Since CodY and SaeR regulate an overlapping set of gene targets, we sought to define the regulatory circuit responsible for this apparent cascading regulation. The genetic and biochemical data presented in this study reveal that CodY binds to the sae P1 promoter region and appears to block the binding of the positive regulator SaeR. Such direct regulation collaborates with indirect regulation via Agr and Rot to tightly control the activity of the Sae TCS, the expression of virulence genes, and production of cytotoxic exoproteins.
RESULTS
CodY regulates the Sae TCS by both Rot-dependent and Rot-independent mechanisms.
CodY regulates the expression of many of the known virulence genes in S. aureus (36, 37, 39). However, the mechanistic details regarding how CodY accomplishes this feat are relatively obscure. For example, nuc, coding for a secreted nuclease that is important for in vivo survival (15), is overexpressed in the codY null mutant but lacks a definable CodY binding site. Moreover, nuc is directly controlled by the SaeR/S TCS, and SaeR is required for nuc overexpression in the codY null mutant, indicating that the CodY overexpression phenotype is at least partly routed through the Sae TCS (39). When nutrients are abundant, CodY represses the agr locus (36, 41). We reasoned that this would promote Rot-mediated repression of sae and surmised that this represents an indirect model of nutritional regulation by CodY (Fig. 1C). To test this hypothesis, we constructed a suite of single and double mutants in two independent S. aureus backgrounds, the methicillin-susceptible osteomyelitis isolate UAMS-1 (42) and CA-MRSA USA300 LAC (43), and measured saeP transcript abundance using quantitative, real-time reverse transcription-PCR (qRT-PCR). We chose to measure saeP transcript abundance because the gene lies immediately downstream of the sae P1 promoter, whose activity is known to be influenced by Agr and Rot (27, 44). During exponential growth, saeP transcript abundance in both UAMS-1 and LAC (wild type [WT]) cells is relatively low (0.11 and 0.09 copies relative to the rpoC transcript level, respectively) (Fig. 2A). We observed a 3- to 4-fold derepression of saeP when the rot gene was knocked out (Fig. 2A, compare rot mutant and the wild type). However, an analysis of variance (ANOVA) using Tukey's multiple-comparison posttest suggests that the transcript abundances in the rot single mutant are not significantly different from wild-type levels. In contrast, we observed a significant derepression of saeP (∼12-fold) in the codY knockout strain (Fig. 2A, compare codY mutant and the WT). We detected a small yet statistically different transcript abundance in the rot codY double mutant compared to that in the codY single mutant in the UAMS-1 background. Low levels of RNAIII in UAMS-1 relative to those in other clinical isolates might lead to increased residual Rot protein and may account for this discrepancy (45). Deleting the response regulator gene agrA prevented full derepression of saeP when codY was knocked out in UAMS-1 (see Fig. S1A in the supplemental material), consistent with the idea that CodY regulates the sae P1 promoter in part via Rot.
FIG 2.

CodY regulates saePQRS in a Rot-dependent and Rot-independent manner. Mean transcript abundances are shown for the indicated strains during exponential growth (OD600 of ∼0.5) in TSB medium using qRT-PCR. saeP (A) and nuc (B) transcripts were normalized to rpoC transcript in two independent lineages of S. aureus, UAMS-1 and USA300 LAC. Statistical significance for each condition was assessed using one-way ANOVA and Tukey's multiple comparison posttest (*, P < 0.05; **, P < 0.01; N.S., not significant). Dotted lines denote significance for the indicated background and strains. Error bars represent the standard errors of the means from at least two independent experiments. When not visible, error bars are too small to see and are obscured by the bars.
To determine whether a downstream target of the SaeR/S system exhibits the same pattern of regulation, we measured nuc transcript abundance in the same collection of strains. In agreement with our previous results, we observed a 17- to 32-fold increase in nuc transcript when codY was knocked out in the UAMS-1 and LAC backgrounds (Fig. 2B, compare the codY mutant to the wild type) (we note that nuc transcript abundance in LAC was generally higher than that in UAMS-1). Interestingly, deleting rot had no effect on nuc expression in either the UAMS-1 or USA300 LAC strain (Fig. 2B, compare the wild-type strain and the rot mutant), suggesting that SaeR∼P levels are insufficient to activate nuc transcription. While we detected a statistically significant difference in nuc transcript abundance between the LAC codY and the rot codY strains suggesting that Rot does have an effect on nuc in the absence of CodY in this background, the effect is less than 2-fold (Fig. 2B). While this could be a strain-specific difference, its biological significance is unclear at this time. Deleting agrA ensured Rot-mediated repression even when codY was disrupted (Fig. S1B). Taken together, these results suggest that CodY represses the sae P1 promoter and that CodY regulates the sae locus in both a Rot-dependent and Rot-independent manner.
CodY binds within the sae P1 promoter region.
Rot-mediated repression appears to account for only part of the CodY effect on saeP expression. Seeking the source of that missing regulation, we analyzed the upstream region of the sae P1 promoter and identified a putative CodY binding motif that resembles the 15-bp CodY binding consensus sequence AATTTTCWGAAAATT previously determined in Lactococcus lactis, validated in Bacillus subtilis, and refined for S. aureus (34–36). The motif, located between positions −98 and −83 relative to the saeP transcriptional start site (46), has four mismatches with respect to the consensus motif. An additional motif with six mismatches with respect to the consensus is located immediately downstream (Fig. 3A). To test whether CodY interacts with these motifs, we performed electrophoretic mobility shift assays (EMSAs) with a 6-carboxyfluorescein (6-FAM)-labeled PCR product and purified recombinant, histidine-tagged S. aureus CodY protein (SaCodY-His6). We synthesized a 235-bp DNA fragment (saeP235p+) containing the saeP regulatory region including the putative CodY motifs and the translation start codon and found that CodY bound to this fragment with moderate affinity, exhibiting an apparent equilibrium dissociation constant (KD) of ∼60 nM (here, KD reflects the concentration of CodY required to shift 50% of DNA fragments under conditions of vast CodY excess over DNA) (47) (Fig. 3B). CodY was efficiently competed off the saeP235p+ fragment when an excess of nonlabeled DNA fragment was included in the binding reaction mixture (Fig. 3C). Further, we determined this interaction to be specific, as equivalent amounts of CodY failed to bind and form stable DNA:CodY complexes when a DNA fragment lacking a definable binding site was tested (Fig. S2).
FIG 3.

S. aureus CodY interacts with the sae P1 upstream region. (A) The region surrounding the sae P1 regulatory region is displayed (coding strand; 5′ to 3′). The known translated sequence, transcriptional start site (+1), −10, and −35 sites are displayed and annotated in boldface. The identified SaeR∼P footprint is bracketed with the direct and indirect repeat motifs indicated above the sequence with a dotted line. A dashed line above the sequence identifies the putative CodY motifs. mm, mismatch. (B) Purified SaCodY-His6 protein was incubated with a 6-FAM-labeled 235-bp DNA fragment containing the upstream regulatory region of saeP in the presence of ILV and GTP. The unbound fragment is indicated with open arrows while CodY-DNA complexes are indicated by filled arrows. Increasing amounts of CodY monomer were incubated with the fragment. The molar concentration (of monomeric protein) used in each reaction mixture is indicated above each lane. (C) CodY monomer (120 nM) was incubated with the fragment in the presence of a 30× molar excess of unlabeled probe. The presence of each protein and unlabeled probe is indicated beneath each lane.
CodY and SaeR compete for binding to the SaeP regulatory region.
The proximity of the putative CodY motifs to the previously reported SaeR binding site (17) suggests that the binding sites for the two proteins at least partially overlap. In order to identify the CodY binding site, we performed DNase I footprinting experiments using automated capillary electrophoresis (48). We found that CodY protected a 31-bp region from −98 to −67 relative to the saeP transcriptional start site using 60 nM and 120 nM SaCodY-His6 protein. The protected sequence ATTTTCAAATTATTCTTTCTTCAATATTAGT encompasses the identified CodY motifs, and all but 4 nucleotides (nt) of the CodY protected region fall within the region containing the imperfect repeat sequence previously shown to be protected by SaeR∼P and required for activation of sae P1 (Fig. 4) (17).
FIG 4.

DNase I footprinting reveals that the CodY and SaeR binding sites overlap. A 5′-FAM-labeled saeP235p+ fragment incubated in the presence of 0, 60, and 120 nM purified CodY or bovine serum albumin (BSA; control) was challenged with 0.075 to 0.1 U of DNase I. The resulting fragments were separated using capillary electrophoresis and aligned to the sequenced PCR product as a reference. Relative fluorescence (y axis) is displayed as a function of nucleotide position (x axis). Reaction mixtures containing CodY (gray trace) were compared to control reaction mixtures containing bovine serum albumin (black trace). A drop in peak intensity at a given nucleotide position indicates protection by CodY. CodY binds to a 31-bp region (colored nucleotides). Data are representative of at least two independent experiments.
The above in vitro EMSAs and footprinting experiments suggested that CodY regulates the sae P1 promoter directly. These results led us to determine the extent to which the CodY binding site contributes to regulation of the sae P1 promoter during in vitro growth. To investigate this, we first constructed a green fluorescent protein (GFP) reporter fusion containing the sae P1 regulatory region used in EMSAs and measured fluorescence in cells during exponential growth when CodY activity is expected to be maximal. We note that no potential CodY binding sites were inadvertently introduced as fusion constructs did not result in sequences containing fewer than six mismatches with respect to the CodY consensus sequence. Consistent with qRT-PCR results, a fusion (saeP235p+-gfp) containing the 235-bp fragment including the entire sae P1 regulatory region was regulated ∼5-fold (Table 1, compare fluorescence levels in the wild-type and codY strains). Knocking out rot led to partial derepression of saeP235p+-gfp; similar to the results of the qRT-PCR analysis, we observed a small difference in promoter activity when we compared GFP fluorescence levels in the codY null mutant and the rot codY double mutant (Table 1).
TABLE 1.
CodY mediated regulation of saeP-GFP fusions
| Strain | Genotype | RFUa | Fold regulationb |
|---|---|---|---|
| Strains expressing the saeP235p+-gfp fusion | |||
| SRB1008 | Wild type | 1,663 ± 335 | |
| SRB1009 | ΔcodY | 8,991 ± 1,172** | 5.4 |
| SRB1042 | Δrot | 3,448 ± 333 | |
| SRB1043 | Δrot ΔcodY | 16,530 ± 402** | 4.8 |
| Strains expressing the saeP235pscrambled-gfp fusion | |||
| SRB1162 | Wild type | 2,086 ± 212 | |
| SRB1163 | ΔcodY | 3,442 ± 170 | 1.6 |
| SRB1164 | Δrot | 2,367 ± 731 | |
| SRB1165 | Δrot ΔcodY | 5,108 ± 992* | 2.2 |
Relative fluorescence units (RFU) were determined by using the measured fluorescence values normalized to the OD600 value and subtracting the background value of cells lacking the fusion. Reported values (means ± standard errors of the means) are the results of three independent experiments. Values were compared to those of SRB1008, and significance was determined by one-way ANOVA with Tukey's multiple-comparison test (*, P < 0.01; **, P < 0.001).
Fold regulation is reported as RFUΔcodY mutant/RFUwild type or RFUΔcodY Δrot mutant/RFUΔrot mutant, where the subscript indicates the relevant strain.
To address the role of direct CodY repression of the saeP P1 promoter, we created a 235-bp fragment of the saeP P1 promoter region in which both CodY binding motif sequences were scrambled (saeP235pscrambled). Scrambling the newly identified CodY binding motifs prevented the formation of the stable DNA:CodY complexes observed in the wild-type fragment (Fig. S3A). We note that we observed a decrease in probe signal intensity and smearing as CodY concentration was increased in the presence of the scrambled fragment. At this time, we cannot rule out the possibility that at high concentrations of CodY, the protein binds nonspecifically to other regions within the probe in the absence of the CodY binding site or weakly to the scrambled site. Nevertheless, these complexes are unstable during electrophoresis, and we conclude that the affinity of CodY for the native saeP promoter fragment is likely higher. Next, given the significant overlap between the CodY and SaeR binding sites and the importance of SaeR in activating the saeP P1 promoter, we sought to ensure that this interaction was unaffected. Using purified cytosolic SaeS histidine kinase (SaeSc), we phosphorylated recombinant SaeR protein and found that SaeR∼P alone bound the saeP235p+ fragment with an apparent KD of ∼500 nM (Fig. S4). Importantly, there was no apparent decrease in affinity of SaeR∼P for the saeP235pscrambled fragment as determined by EMSAs (Fig. S3B).
The saeP235pscrambled fragment was then fused to gfp. This mutant fusion showed slightly elevated promoter activity in a wild-type background compared to that of the wild-type fusion and less than a 2-fold difference when we knocked out codY. However, the activities were not significantly different (Table 1). We note that overall promoter activity is decreased with this mutant fusion, complicating the interpretation of these data. Given the significant overlap between the CodY and SaeR binding sites and the loss of promoter activity, these data do suggest that CodY shares a binding site with the positive activator of the SaeR/S system. Taken all together, these results suggest that CodY directly represses sae P1.
CodY maintains tight repression of sae P1 across the cell population.
The expression of the Sae-dependent target nuc was shown to remain shut off in most S. aureus cells within in vitro biofilms; only a fraction of the cells within the population express nuc, giving rise to a stochastic gene expression phenotype (49). CodY and SaeR have opposing roles in regulating the sae P1 promoter (repression and activation, respectively) and appear to compete for the same site. This led us to question whether CodY/SaeR competition might contribute to the stochastic expression pattern for SaeR-dependent genes that require a relatively high cellular abundance of SaeR∼P, including saeP and nuc. To test this, we first examined cells carrying one of two reporter constructs, saeP235p+-gfp or saeP235pscrambled-gfp, and examined the level of reporter expression across the cell population in planktonic cells using fluorescence microscopy and flow cytometry. With respect to the wild-type (saeP235p+-gfp) fusion, relatively few wild-type UAMS-1 cells expressed the GFP reporter (Fig. S5). Reporter expression in rot mutant cells also appeared stochastic. However, when we knocked out codY, the fraction of cells expressing the fusions appeared to be greatly increased using fluorescence microscopy. To determine the fraction of GFP-positive (GFP+) cells in the population, we turned to flow cytometry analysis. We found that the fraction of cells expressing saeP235p+-gfp is very low (∼1%) in wild-type UAMS-1 cells (Fig. 5A). Knocking out rot increased the frequency of saeP235p+-gfp-expressing cells in the population (∼20%), while the frequencies were further increased in the codY mutant and rot codY double mutant (∼76 and 66%, respectively). We note not only that the frequency of GFP+ cells is increasing in these mutants but also that the observed fluorescence intensity is increasing. Notably, flow analysis revealed that scrambling the CodY binding motifs increased the population of GFP+ cells to ∼70% regardless of strain genotype (Fig. 5B), which is in agreement with fluorescence microscopy experiments (Fig. S5, compare saeP235pscrambled-gfp and saeP235p+-gfp). This supports the notion that in the absence of efficient binding of CodY to the promoter, SaeR∼P is free to bind and activate expression from sae P1.
FIG 5.

CodY contributes to the stochastic expression pattern of saeP. Flow cytometry analysis was performed of UAMS-1 cells harboring either the saeP235p+-gfp (A) or saeP235pscrambled-gfp (B) reporter fusions during exponential growth in TSB medium. Histograms indicate the relative number of cells (y axis) exhibiting a given fluorescence intensity (x axis) and are scaled to 30,000 cells to account for various sample sizes. Data are the means ± the standard errors of the means of at least two independent experiments.
Next, we looked at cells carrying the nuc-gfp reporter plasmid pMRSI-nuc. Congruent with fluorescence microscopy and qRT-PCR analyses, <1% of planktonic cells carrying the nuc-gfp reporter were GFP+, and knocking out codY turned on the fusion in nearly all cells (Fig. S5 and S6). Last, we examined expression of the nuc-gfp reporter fusion during the early stages of in vitro biofilm development under biologically relevant flow conditions. As with planktonic cells, we observed that the number of GFP+ cells was remarkably higher in the codY mutant than in the wild-type parent strain during the early stages of biofilm development (Fig. 6). Taken together, these data suggest that in UAMS-1, CodY is a more potent repressor of the sae P1 promoter than Rot and that direct repression by CodY blocks activation of SaeR/S and Sae-dependent genes when nutrients are abundant. These data also indicate that CodY contributes to the regulation of stochastic expression of at least two Sae-dependent genes during both planktonic and biofilm growth.
FIG 6.

codY mutant cells express thermonuclease (nuc) at early stages of S. aureus biofilm development. S. aureus (UAMS-1) wild-type and codY mutant cells containing the nuc-gfp reporter plasmid pMRSI-nuc were grown in the BioFlux 1000 microfluidic system. Bright-field and epifluorescence microscopic images were acquired at a magnification of ×200 after 6 h of biofilm development. Images are representative of multiple experiments.
CodY restrains Sae-dependent production of leukocidins.
The SaeR/S system enables S. aureus to produce numerous cell-associated and secreted proteins including the bicomponent, pore-forming leukocidins, which contribute to polymorphonuclear leukocyte (PMN) lysis (50). Since CodY represses the expression of sae and appears to suppress the activity of the SaeR/S system, we next assessed the toxicity of supernatants prepared from USA300 LAC and its codY null mutant. To do this, we exposed human PMNs (i.e., neutrophils) to supernatants of cells grown to exponential phase (i.e., after 3 h of growth) and postexponential phase (i.e., after 5 h of growth) in tryptic soy broth (TSB) medium and determined the percent killing. At both time points, supernatants from cultures of the codY null mutant displayed enhanced PMN killing compared to that of its wild-type parent strain (Fig. 7, compare codY and WT strains). The toxic effect was more potent at the 5-h time point, as evidenced by the increased cell death at relatively low percentages of culture filtrates. Knocking out saeR in both the WT and the codY null mutant essentially eliminated toxicity, suggesting that the increased PMN killing by the codY mutant is Sae dependent. We conclude that nutrient sufficiency, a condition monitored by CodY, restrains the production of factors that kill human immune cells via the regulation of the Sae TCS.
FIG 7.

CodY-enhanced PMN killing is dependent on the Sae TCS. Primary human PMNs were intoxicated with supernatants obtained from codY null mutants at either 3 h (A) or 5 h (B) following subculture. Intoxication experiments were performed on two independent occasions, with a total of four blood donors. Statistical analysis was performed by ANOVA with Dunnett's multiple-comparison test. Asterisks indicate P values for comparisons between results for the wild type (LAC) and the codY null mutant (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001). Error bars represent the standard errors of the means.
DISCUSSION
CodY's role in controlling virulence during infection has been firmly established in multiple Gram-positive pathogens, including S. aureus, Listeria monocytogenes, and Bacillus anthracis (51–58). Surprisingly, a comparison of the set of genes differentially regulated by S. aureus CodY in vitro with the gene targets identified using an in vitro DNA:CodY pulldown experiment revealed that fewer than half of the virulence genes under CodY control are directly regulated (36). Thus, how nutrient sufficiency is linked mechanistically to virulence gene expression remains incompletely understood. In the present study, we demonstrate that CodY indirectly represses the sae P1 promoter via Agr and Rot and directly represses sae expression binding to the sae P1 regulatory region (Fig. 2 and 5; see also Fig. S1 in the supplemental material). We further show that loss of CodY regulation at sae results in an Sae-dependent increase in the production of cytotoxic factors that kill human neutrophils (Fig. 7). Mechanistically, CodY likely funnels the nutrient depletion signal (ILV/GTP depletion specifically) through the Sae TCS to upregulate virulence gene expression. We posit that CodY acts in part as a physical blockade and generally as a nutritional checkpoint protein to prevent SaeR binding and to keep saePQRS tightly shut off when ILV/GTP are plentiful.
Our flow cytometry data reveal that some cells fail to express the reporter fusions even in the absence of Rot- and CodY-mediated repression (Fig. 5A and S6). Moreover, some areas of the codY mutant biofilm show nuc expression, while other areas remain dark (Fig. 6). This may be due to SaeS phosphatase activity responding to an as yet unknown signal, reducing the fraction of SaeR∼P in the cell, since upregulating sae P1 by removing CodY repression would lead to an increase in saePQ expression. Performing the experiments in an saePQ mutant could test this. Experiments to reveal a mechanism for the observed phenotypic heterogeneity are currently in progress.
The logic of nutritional and phagocytosis-associated signals.
We note that our study focuses solely on the role of nutrient sufficiency (i.e., ILV and GTP levels, specifically) on the Sae TCS under noninduced conditions. However, neutrophils provide the primary response to S. aureus infection, ingesting the bacteria and exposing them to reactive oxygen species and antimicrobial peptides like human neutrophil peptide 1 (HNP-1). Interestingly, the sae P1 promoter is induced up to 6-fold in the presence of HNP-1 in vitro under conditions that promote high CodY activity (21). Further, during invasive disease (most intensely studied in renal tissues), phagocytes degrade surrounding tissues, and the resulting necrosis and deposition of fibrin result in the formation of distinctive and replicative niches for S. aureus (59, 60). These abscesses are bathed in calprotectin, a metal chelator secreted by neutrophils known to protect SaeS from inactivation by zinc and iron (19, 20). SaeS kinase activity is stimulated by these signals (14, 18). Finally, the nutritive properties of host tissues are largely unknown, and these environments are potentially nutritionally restricted or depleted of key nutrients, including ILV and purine nucleotides (61). Experiments to determine the contribution of nutritional regulation by CodY on virulence in vitro and in vivo, with particular focus on whether the nutrient depletion and neutrophil-associated signals are independent or exclusive, are in progress.
Working model.
A major question that arises from this work is why the cell would evolve to have CodY turn on and off the Sae system simultaneously. A straightforward strategy for activating the Sae system during nutrient depletion would be to simply repress the P3 promoter to control the expression of saeRS. However, the P3 promoter has been shown previously to be constitutive, and, more importantly, ectopic overexpression of saeRS from an inducible promoter does not increase the expression of the Sae regulon when multiple gene targets are interrogated (24). Rather, the fraction of SaeR∼P in the cell determines Sae-dependent gene activation; CodY control of saePQRS expression is likely only part of the story.
The sequence of events is potentially very interesting in vivo and informs our working model (Fig. 8). That is, under conditions where nutrients are in excess, CodY repression is strong, and expression from the sae P1 promoter and SaeR-dependent targets is relatively low. Given CodY's moderate affinity for its binding site, intermediate nutrient levels may foster competition between active CodY and SaeR∼P for binding at saeP P1. Relieving direct CodY repression at the P1 promoter would increase expression of not only saeR and saeS but also saeP and saeQ (39), whose products are known to help return the Sae system to its basal activity state by stimulating the phosphatase activity of SaeS (23). Thus, while SaeR and SaeS are produced, their activity may be blocked by SaeP/Q and would remain poised to receive an additional signal. Such a strategy would allow the cells to avoid the costly synthesis of new proteins when ILV (among the most abundant amino acids in proteins [62]) are scarce. Alternatively, the strategy could allow amino acids and GTP to be spared for the production of digestive enzymes and toxins used to replenish those needed nutrients.
FIG 8.

Working model of how nutritional regulation controls SaeR/S. CodY integrates the nutritional status of the cell with host cues at the sae P1 promoter. Under conditions of nutrient excess, sae P1 transcription is repressed. As nutrients become depleted, CodY repression is lifted, eventually resulting in an increase in SaeR/S activity and the expression of exoproteins to damage host tissue. This allows S. aureus to scavenge nutrients from the host environment.
During severe ILV depletion, CodY activity is expected to plummet, and repression of agr is likely lifted. As population density increases, so too does the abundance of RNAIII, antagonizing the cellular abundance of Rot. The mechanism by which Rot regulates the Sae system is currently unknown (Fig. 1C); perhaps the lack of Rot simply increases SaeR and SaeS levels. Although phosphorylation may be inefficient, it may be enough to induce class I genes. This could be through indirect or direct means; it is possible that Rot binds in close proximity to the CodY-SaeR site. Alternatively, lack of Rot may result in the overexpression of a gene product or the accumulation of a small metabolite signal that instigates a posttranscriptional or posttranslational change in one or more components of the Sae system, mitigating SaeP/Q activity and leading to SaeS activation. Indeed, SaeR/S was recently shown to be responsive to cellular respiratory status (63). Further, the Sae system has been described as having switch-like behavior (64); it is tempting to speculate that a critical drop in the ILV (or GTP) pool and the concomitant drop in CodY activity increase SaeR∼P levels and trigger SaeR/S. This would support the notion that CodY delays the production of potent Sae-dependent toxins until nutrients are fully exhausted and may be one important mechanism governing the switch of S. aureus between commensal and invasive lifestyles. Experiments to test these questions are ongoing in our laboratory.
MATERIALS AND METHODS
Ethics statement.
Buffy coats were obtained from anonymous blood donors with informed consent from the New York Blood Center. Because all of the samples were collected anonymously prior to their delivery, the New York University Langone Medical Center (NYULMC) Institutional Review Board determined that our study was exempt from further ethics approval requirements.
Bacterial strains and growth conditions.
All bacterial strains used in this study are listed in Table S1 in the supplemental material. Staphylococcus aureus strains were routinely cultured in tryptic soy broth (TSB) (containing 0.25% [wt/vol] dextrose; Becton, Dickinson), and Escherichia coli strains were grown in modified Lennox (L) medium consisting of 10 g liter−1 tryptone, 5 g liter−1 yeast extract, and 5 g liter−1 NaCl (65). When necessary, medium was solidified with agar to 1.5% (wt/vol), and antibiotics were included in the medium at the following concentrations to maintain selection: ampicillin (Ap), 50 μg ml−1; chloramphenicol (Cm), 10 μg ml−1; kanamycin (Km), 35 μg ml−1; tetracycline (Tc), 3 μg ml−1; and erythromycin (Em), 5 μg ml−1. In vitro shake flask experiments were performed in 125-ml DeLong shake flasks as previously described (39). Briefly, precultures were inoculated from overnight cultures to an optical density at 600 nm (OD600) of 0.05 at a 5:1 flask-to-medium ratio in TSB. The inoculated cultures were grown in a gyrotory shaking water bath to an OD600 of ∼1.0 and rediluted to 0.05 to generate experimental cultures to ensure that cells were safely in exponential phase. Growth was monitored as the increase in absorbance at 600 nm using a Beckman DU350 UV-visible light spectrophotometer (Beckman Coulter). All exponential-phase samples were collected within one generation of one another at an OD600 of ∼0.5 after back dilution unless otherwise noted. All broth cultures were grown at 37°C with vigorous shaking at 280 rpm unless otherwise noted.
Recombinant DNA and genetic techniques.
Oligonucleotides for this study were synthesized by Integrated DNA technologies (Coralville, IA) and are listed in Table S2. Plasmids used in this study (Table S3) were constructed using Escherichia coli cloning strain NEB 5α (New England BioLabs) and confirmed by nonradioactive Sanger sequencing (Genewiz). Plasmids were introduced into S. aureus strain RN4220 by electroporation as previously described (66). As needed, plasmids and marked chromosomal mutations were transferred into select strain backgrounds using ϕ11-mediated transduction (67).
Construction of plasmids. (i) Construction of GFP reporter fusions.
To create an saeP-gfp promoter fusion, a 235-nucleotide (nt) PCR fragment containing the sae upstream region along with the ribosome binding site (RBS) and the translation initiation codon (ATG) of saeP (open reading frame [ORF] QV15_03495) was amplified using oligonucleotides oKM026 and oSRB484. To scramble the CodY binding motifs, nucleotides were arbitrarily assigned values from 1 to 4, and a random number generator was used to create a 25-nt sequence (TCCTTACGCAGGGACTTAAGTAGCC). Oligonucleotides oKM062 and oKM063 were used with oKM026 and oSRB484, respectively, to generate 139-nt and 137-nt fragments. These purified PCR products were mixed at an equal molarity and used as the template DNA in an overlapping PCR with oKM026 and oSRB484, creating an sae P1 promoter fragment featuring the scrambled CodY binding motifs. In each case, PCR fragments were digested with SphI and ligated to the SphI/SmaI site of plasmid pMRSI (39).
(ii) Construction of a TEV-cleavable CodY-His6 overproduction plasmid.
An 827-nt fragment containing the codY (ORF QV15_05910) coding sequence lacking the translational stop codon (TAA) was amplified from UAMS-1 genomic DNA using oligonucleotides oKM1 and oSRB410 (36). The PCR fragment was purified and subjected to a second round of PCR using oKM1 and oSRB411 to append a tobacco etch virus (TEV) protease cleavage sequence followed by six histidine (CAT) codons and a TAA stop codon. The 847-nt fragment was digested with SacI/SphI and ligated to the same sites of pBAD30 (68).
RNA extraction.
Methods for RNA extraction and quantitative real-time reverse transcription-PCR were performed as previously described in detail (39). Briefly, experimental cultures of the strains indicated in the figures were grown exponentially to an OD600 of 0.5, at which point a 4-ml sample was mixed with an equal volume of 1:1 (vol/vol) ethanol-acetone prechilled to −20°C and immediately frozen on dry ice. Prior to processing, samples were stored at −80°C. Once thawed, cells were pelleted and washed twice with TE buffer (10 mM Tris-Cl [pH 8], 1 mM EDTA) and mechanically disrupted in TRIzol using a Precellys 24 homogenizer with three 30-s pulses at 6,800 rpm. Samples were incubated on wet ice for 1 min between pulses. Nucleic acids were extracted using a Direct-Zol kit (Zymo Research Corporation) according to the manufacturer's instructions. Contaminating genomic DNA was depleted in each sample using a Turbo DNA-free DNase removal kit (Ambion) according to the manufacturer's instructions.
qRT-PCR.
Quantitative, real-time RT-PCR (qRT-PCR) was performed essentially as described previously (39). Two hundred fifty nanograms of total RNA from each sample was used as the template and reverse transcribed using ProtoScript first-strand cDNA synthesis (NEB) and random primers as per the manufacturer's instructions. To control for remaining residual DNA contamination during qRT-PCR analysis, parallel reactions were performed with mixtures lacking reverse transcriptase. Transcript abundance was determined in each sample using a C1000 thermal cycler fitted with a CFX96 detection module and SsoAdvanced Universal SYBR green Supermix (Bio-Rad). Target-specific oligonucleotides were each used at a concentration of 400 nM. Reaction mixtures were incubated at 98°C for 2 min and then cycled between 98°C and 60°C. Standard controls (no template and no reverse transcriptase) were run on each plate. To calculate transcript abundance, standard curves were generated from serial dilutions of genomic DNA spanning at least five orders of magnitude. Target transcript abundance was normalized to level of the rpoC transcript, whose abundance remained constant across all strains analyzed.
Flow cytometry analysis.
Cells were collected and fixed in paraformaldehyde (PFA) as described above, stained with 4′,6-diamidino-2-phenylindole (DAPI) for 20 min, and resuspended in phosphate-buffered saline (PBS). Stained cells were analyzed using a computer-controlled FACStar Plus dual-laser system (Becton, Dickinson) and FCS Express software (version 4.0; DeNovo Software). Populations were selected by gating forward scatter in conjunction with DAPI stain. The limit of detection was first determined using wild-type cells lacking the reporter construct (SRB337) and defined as the signal intensity encompassing 99% of this population. Then, GFP+ cells were identified as any cell exceeding the limit of detection and enumerated for each genotype.
BioFlux 1000 biofilm assays.
S. aureus biofilm development was assessed using a BioFlux 1000 microfluidic system (Fluxion Biosciences, Inc., San Francisco, CA) as described previously (69). Briefly, to grow biofilms in the BioFlux system, the channels were first primed for 5 min with 200 μl of TSB supplemented with 0.25% glucose at 5.0 dynes/cm2. To seed the channels with bacteria, excess TSB was aspirated from the output wells and replaced with 200 μl of fresh exponentially growing cultures diluted to an OD600 of 0.8 and then pumped into the channels at 2.0 dynes/cm2 for 5 s. Cells were allowed to attach to the surface of the channels for 1 h at 37°C. Then, the remaining inoculum was aspirated from the output well, and 1.3 ml of 50% TSB supplemented with 0.125% glucose was added to the input wells and pumped at 0.6 dyne/cm2 for 18 h (flow rate, 64 μl h−1). Bright-field and epifluorescence (with fluorescein isothiocyanate [FITC] filter) images were taken at a magnification of ×200 at 5-min intervals at a total of 217 time points for each strain tested; the gain and exposure settings were kept constant for all images.
Confocal laser scanning microscopy.
A 1-ml sample of cells containing the fusion indicated in Fig. S5 was pelleted during exponential growth, washed with PBS, and fixed with 200 μl of 3% (wt/vol) PFA (in PBS) for 45 min. The PFA was then washed away, and a 50-μl suspension of cells in PBS was applied to poly-l-lysine-treated glass coverslips and allowed to adhere for 30 min. Nonadherent cells were removed from coverslips by rinsing with distilled water for ∼10 s. Remaining cells were mounted using Vectashield antifade mounting medium with DAPI (Vector Laboratories) and imaged using a Zeiss LSM 880 confocal laser scanning microscope, optimizing excitation and detection based on the fluorescence intensity detected in codY null mutant cells. Images were processed using ImageJ (70).
GFP reporter assays.
Strains containing the reporter fusion indicated in Table 1 were grown as described above and were collected at an OD600 of ∼0.4 during exponential growth. At these points a 2-ml sample of cells was pelleted, washed once with PBS to remove residual medium, and resuspended in 0.5 ml of PBS. Fluorescence was measured using a computer-controlled Tecan Infinite F200 Pro plate reader (equipped with a 485-nm filter for excitation and 535-nm filter for emission) and i-control software (version 1.11). GFP signal acquisition settings were kept constant: gain, 50%; flash number, 25; integration time, 20 μs; lag time, 0 μs; settle time, 0 ms. Relative fluorescence units (RFU) were calculated as previously described (39) by subtracting the fluorescence of the SRB337 strain (lacks GFP reporter) and dividing by the OD600 value to correct for cell density.
Overproduction and purification of recombinant proteins. (i) CodY.
The gene coding for SaCodY-His6 was expressed in E. coli strain DH5α under the control of the arabinose-inducible ParaBAD promoter essentially as described previously (36). SRB628 was grown in L broth with 100 μg ml−1 ampicillin at 37°C to an OD600 of 0.3, at which point SaCodY-His6 overproduction was induced using 0.2% (wt/vol) l-(+) arabinose. After 4 h of induction, cells were harvested by centrifugation at 8,610 × g at 4°C for 10 min and frozen at −80°C. Cells were thawed and resuspended in buffer A (20 mM Tris-HCl [pH 7.9], 500 mM NaCl, 5% [vol/vol] glycerol, 5 mM imidazole) supplemented with 1 mM phenylmethylsulfonyl fluoride and 0.1% (vol/vol) Nonidet P-40, and lysed by sonication. The supernatant was clarified by centrifugation at 40,000 × g for 30 min at 4°C. SaCodY-His6 was purified to near homogeneity using a computer-controlled ÄKTAprime plus fast protein liquid chromatography (FPLC) system fitted with a HisTrap column (GE Healthcare Life Sciences) according to the manufacturer's recommendations for gradient elution using buffer B (20 mM Tris-HCl [pH 7.9], 500 mM NaCl, 5% [vol/vol] glycerol, 685 mM imidazole). Fractions containing purified protein were pooled, supplemented with glycerol to 50% (vol/vol), and stored at −20°C. Protein concentration was determined using a Pierce Coomassie (Bradford) protein assay kit according to the manufacturer's instructions.
(ii) SaeSc.
Overproduction of the His-tagged, cytosolic portion of SaeS containing the autokinase and phosphotransferase domains was carried out as previously described (17).
(iii) SaeR.
His6-SaeR was purified as previously described (17) except that we induced expression overnight at 18°C.
Electrophoretic mobility shift assays (EMSAs). (i) CodY.
The wild-type saeP235p+ DNA fragment was synthesized by PCR using oKM026 and oKM025 to create a 5′-6-FAM-labeled fragment. The saeP235pscrambled DNA fragment was synthesized similarly using pKM15 as the template DNA. The rpoC 5′-6-FAM-labeled fragment used as a control was synthesized by PCR using oKM027 and oSRB239. Incubation of labeled fragments with SaCodY-His6 was performed in binding buffer (20 mM Tris-Cl [pH 8.0], 50 mM KCl, 2 mM MgCl2, 5% [vol/vol] glycerol, 0.5 mM EDTA, 0.05% [vol/vol] Nonidet P-40, 1 mM dithiothreitol [DTT], and 25 μg ml−1 sheared salmon sperm DNA). Samples (11 μl) containing various amounts of CodY, 200 fmol of FAM-labeled DNA fragment, 10 mM (each) ILV, and 2 mM GTP were incubated for 20 min at 25°C in a ThermoMixer (Eppendorf). Samples were separated on 8% nondenaturing 35 mM HEPES–43 mM imidazole–10 mM ILV polyacrylamide gels in the same run buffer for 60 min at 200 V. Gels were imaged; fluorescent bands were detected and quantified using a computer-controlled ImageQuant LAS 4000 biomolecular imager (GE Healthcare Life Sciences) using a SYBR filter set with a 1-min exposure or a ChemiDoc MP imaging system (Bio-Rad) with a SYBR filter set following a 20-s exposure. When indicated in the figures, a 30-fold molar excess of unlabeled PCR product (synthesized using oKM026 and oSRB477) was included in the reaction mixture.
(ii) SaeR.
SaeR was first phosphorylated as described previously (17). Briefly, 20 μM His-tagged SaeR protein was incubated in the presence of 5 μM His-tagged SaeSc with 1 mM ATP in phosphorylation buffer (10 mM Tris-HCl [pH 7.4], 50 mM KCl, 5 mM MgCl2, 10% [vol/vol] glycerol) for 5 min. Phosphorylated SaeR was then used in gel shift assays with the fragments described above under the same conditions as CodY EMSAs.
DNase I footprinting of the sae P1 promoter.
Samples containing femtomole amounts of 5′-6-FAM-labeled saeP235p+ were incubated with 30, 60, and 120 nM CodY as described above in a 60-μl reaction mixture. Two microliters of 0.075 to 0.1 U of RQ1 DNase I (Promega) was then added to each reaction mixture. After 1 min, reactions were inactivated with 20 mM EGTA and purified with a QiaQuick PCR cleanup kit (Qiagen). DNA footprint analysis by capillary electrophoresis (DFACE) was performed as previously described at the Plant-Microbe Genomics Facility at The Ohio State University (48). Analysis of sequence results was performed using GeneMapper and Xplorer software to identify the protected regions.
Cytotoxicity assays.
To assess the cytotoxicity of secreted factors produced from each respective strain, primary human PMNs were intoxicated with culture filtrates obtained from S. aureus cultures. Experimental cultures were generated by diluting overnight-grown S. aureus cells 100-fold and then allowing the strains indicated in Fig. 7 to grow for either 3 or 5 h in TSB for exponential or stationary phase, respectively. Bacterial concentrations were confirmed to be equal at each time point by the quantification of CFU. Prior to intoxication, culture filtrates were diluted 2-fold in a 96-well plate (20 to 0.31%). PMNs were isolated as described previously (71), and amounts were normalized to 200,000 cells per 80 μl of RPMI medium (10 mM HEPES, 0.1% human serum albumin [HSA]). Eighty microliters of PMNs was then pipetted into each well, and the supernatant-PMN mixture was incubated at 37°C in 5% CO2 for 1 h. To assess toxicity, 10 μl of CellTiter 96 Aqueous One solution (CellTiter; Promega) was added to the 96-well plate, and the mixture was incubated at 37°C in 5% CO2 for 1.5 h. PMN viability was assessed with a PerkinElmer EnVision 2103 multilabel reader at an absorbance of 492 nm.
Statistics and reproducibility.
Data shown are the results of at least two independent experiments. Statistical significance was determined using ANOVA with Tukey's multiple comparison posttest using Prism software (version 4.0; GraphPad Software) unless otherwise noted.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anthony McCoy of the Plant-Microbe Genomics Facility at The Ohio State University for his help with DFACE analysis. We thank Taeok Bae for the gift of SaeR protein and plasmids and Richard Procter for the gift of the rot mutant. We also thank Boris Belitsky for helpful conversations and technical advice on EMSAs and DNase I footprinting as well as Steven Singer and Karen Craswell for helpful discussions, advice, and expertise with flow cytometry. Finally, we thank Susan Gottesman and Nadim Majdalani for the use of their ChemiDoc MP Imaging System.
This work was supported in part by a Pathway to Independence Award from the National Institute of General Medical Sciences (GM099893), a research grant from the National Institute of Allergy and Infectious Diseases (R21-AI123708), and Georgetown University Startup funds to S.R.B and by research grants from the National Institute of Allergy and Infectious Diseases (AI103268, AI099394, and AI105129) to V.J.T. and (P01-AI83211 and R01-AI125589) to K.W.B. W.E.S. is supported by a Ruth L. Kirschstein Institutional National Research Service Award (T32 AI007180).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00012-18.
REFERENCES
- 1.Gorwitz RJ, Kruszon-Moran D, McAllister SK, McQuillan G, McDougal LK, Fosheim GE, Jensen BJ, Killgore G, Tenover FC, Kuehnert MJ. 2008. Changes in the prevalence of nasal colonization with Staphylococcus aureus in the United States, 2001–2004. J Infect Dis 197:1226–1234. doi: 10.1086/533494. [DOI] [PubMed] [Google Scholar]
- 2.Graham PL III, Lin SX, Larson EL. 2006. A U.S. population-based survey of Staphylococcus aureus colonization. Ann Intern Med 144:318–325. doi: 10.7326/0003-4819-144-5-200603070-00006. [DOI] [PubMed] [Google Scholar]
- 3.Lowy F. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 4.Thurlow LR, Joshi GS, Richardson AR. 2012. Virulence strategies of the dominant USA300 lineage of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA). FEMS Immunol Med Microbiol 65:5–22. doi: 10.1111/j.1574-695X.2012.00937.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogers KL, Fey PD, Rupp ME. 2009. Coagulase-negative staphylococcal infections. Infect Dis Clin North Am 23:73–98. doi: 10.1016/j.idc.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 7.Lee B, Singh A, David M, Bartsch S, Slayton R, Huang S, Zimmer S, Potter M, Macal C, Lauderdale D, Miller L, Daum R. 2013. The economic burden of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA). Clin Microbiol Infect 19:528–536. doi: 10.1111/j.1469-0691.2012.03914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu W, Kim HK, Rauch S, Schneewind O, Missiakas D. 2017. Pathogenic conversion of coagulase-negative staphylococci. Microbes Infect 19:101–109. doi: 10.1016/j.micinf.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richardson AR, Somerville GA, Sonenshein AL. 2015. Regulating the intersection of metabolism and pathogenesis in Gram-positive bacteria. Microbiol Spectr 3:3. doi: 10.1128/microbiolspec.MDNA3-0061-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baroja ML, Herfst CA, Kasper KJ, Xu SX, Gillett DA, Li J, Reid G, McCormick JK. 2016. The SaeRS two-component system is a direct and dominant transcriptional activator of toxic shock syndrome toxin 1 in Staphylococcus aureus. J Bacteriol 198:2732–2742. doi: 10.1128/JB.00425-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benson MA, Lilo S, Nygaard T, Voyich JM, Torres VJ. 2012. Rot and SaeRS cooperate to activate expression of the staphylococcal superantigen-like exoproteins. J Bacteriol 194:4355–4365. doi: 10.1128/JB.00706-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giraudo AT, Cheung AL, Nagel R. 1997. The sae locus of Staphylococcus aureus controls exoprotein synthesis at the transcriptional level. Arch Microbiol 168:53–58. doi: 10.1007/s002030050469. [DOI] [PubMed] [Google Scholar]
- 13.Giraudo AT, Raspanti CG, Calzolari A, Nagel R. 1994. Characterization of a Tn551-mutant of Staphylococcus aureus defective in the production of several exoproteins. Can J Microbiol 40:677–681. doi: 10.1139/m94-107. [DOI] [PubMed] [Google Scholar]
- 14.Liu Q, Yeo WS, Bae T. 2016. The SaeRS two-component system of Staphylococcus aureus. Genes (Basel) 7:E81. doi: 10.3390/genes7100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olson M, Nygaard T, Ackermann L, Watkins R, Zurek O, Pallister K, Griffith S, Kiedrowski M, Flack C, Kavanaugh J, Kreiswirth B, Horswill A, Voyich J. 2013. Staphylococcus aureus nuclease is an SaeRS-dependent virulence factor. Infect Immun 81:1316–1324. doi: 10.1128/IAI.01242-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pantrangi M, Singh VK, Wolz C, Shukla SK. 2010. Staphylococcal superantigen-like genes, ssl5 and ssl8, are positively regulated by Sae and negatively by Agr in the Newman strain. FEMS Microbiol Lett 308:175–184. doi: 10.1111/j.1574-6968.2010.02012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun F, Li C, Jeong D, Sohn C, He C, Bae T. 2010. In the Staphylococcus aureus two-component system sae, the response regulator SaeR binds to a direct repeat sequence and DNA binding requires phosphorylation by the sensor kinase SaeS. J Bacteriol 192:2111–2127. doi: 10.1128/JB.01524-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zurek OW, Nygaard TK, Watkins RL, Pallister KB, Torres VJ, Horswill AR, Voyich JM. 2014. The role of innate immunity in promoting SaeR/S-mediated virulence in Staphylococcus aureus. J Innate Immun 6:21–30. doi: 10.1159/000351200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho H, Jeong DW, Liu Q, Yeo WS, Vogl T, Skaar EP, Chazin WJ, Bae T. 2015. Calprotectin increases the activity of the SaeRS two component system and murine mortality during Staphylococcus aureus infections. PLoS Pathog 11:e1005026. doi: 10.1371/journal.ppat.1005026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, Anderson KL, Dattilo BM, Dunman PM, Gerads R, Caprioli RM, Nacken W, Chazin WJ, Skaar EP. 2008. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 319:962–965. doi: 10.1126/science.1152449. [DOI] [PubMed] [Google Scholar]
- 21.Geiger T, Goerke C, Mainiero M, Kraus D, Wolz C. 2008. The virulence regulator Sae of Staphylococcus aureus: promoter activities and response to phagocytosis-related signals. J Bacteriol 190:3419–3428. doi: 10.1128/JB.01927-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joiner KA, Ganz T, Albert J, Rotrosen D. 1989. The opsonizing ligand on Salmonella typhimurium influences incorporation of specific, but not azurophil, granule constituents into neutrophil phagosomes. J Cell Biol 109:2771–2782. doi: 10.1083/jcb.109.6.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeong DW, Cho H, Jones MB, Shatzkes K, Sun F, Ji Q, Liu Q, Peterson SN, He C, Bae T. 2012. The auxiliary protein complex SaePQ activates the phosphatase activity of sensor kinase SaeS in the SaeRS two-component system of Staphylococcus aureus. Mol Microbiol 86:331–348. doi: 10.1111/j.1365-2958.2012.08198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mainiero M, Goerke C, Geiger T, Gonser C, Herbert S, Wolz C. 2010. Differential target gene activation by the Staphylococcus aureus two-component system saeRS. J Bacteriol 192:613–623. doi: 10.1128/JB.01242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balaban N, Novick RP. 1995. Autocrine regulation of toxin synthesis by Staphylococcus aureus. Proc Natl Acad Sci U S A 92:1619–1623. doi: 10.1073/pnas.92.5.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bischoff M, Dunman P, Kormanec J, Macapagal D, Murphy E, Mounts W, Berger-Bächi B, Projan S. 2004. Microarray-based analysis of the Staphylococcus aureus σB regulon. J Bacteriol 186:4085–4099. doi: 10.1128/JB.186.13.4085-4099.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Novick R, Jiang D. 2003. The staphylococcal saeRS system coordinates environmental signals with agr quorum sensing. Microbiology 149:2709–2717. doi: 10.1099/mic.0.26575-0. [DOI] [PubMed] [Google Scholar]
- 28.Geisinger E, Adhikari RP, Jin R, Ross HF, Novick RP. 2006. Inhibition of rot translation by RNAIII, a key feature of agr function. Mol Microbiol 61:1038–1048. doi: 10.1111/j.1365-2958.2006.05292.x. [DOI] [PubMed] [Google Scholar]
- 29.Novick RP, Ross HF, Projan SJ, Kornblum J, Kreiswirth B, Moghazeh S. 1993. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J 12:3967–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marincola G, Schäfer T, Behler J, Bernhardt J, Ohlsen K, Goerke C, Wolz C. 2012. RNase Y of Staphylococcus aureus and its role in the activation of virulence genes. Mol Microbiol 85:817–832. doi: 10.1111/j.1365-2958.2012.08144.x. [DOI] [PubMed] [Google Scholar]
- 31.Marincola G, Wolz C. 2017. Downstream element determines RNase Y cleavage of the saePQRS operon in Staphylococcus aureus. Nucleic Acids Res 45:5980–5994. doi: 10.1093/nar/gkx296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brinsmade SR. 2017. CodY, a master integrator of metabolism and virulence in Gram-positive bacteria. Curr Genet 63:417–425. doi: 10.1007/s00294-016-0656-5. [DOI] [PubMed] [Google Scholar]
- 33.Stenz L, Francois P, Whiteson K, Wolz C, Linder P, Schrenzel J. 2011. The CodY pleiotropic repressor controls virulence in Gram-positive pathogens. FEMS Immunol Med Microbiol 62:123–139. doi: 10.1111/j.1574-695X.2011.00812.x. [DOI] [PubMed] [Google Scholar]
- 34.Belitsky BR, Sonenshein AL. 2008. Genetic and biochemical analysis of CodY-binding sites in Bacillus subtilis. J Bacteriol 190:1224–1236. doi: 10.1128/JB.01780-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.den Hengst CD, van Hijum SA, Geurts JM, Nauta A, Kok J, Kuipers OP. 2005. The Lactococcus lactis CodY regulon: identification of a conserved cis-regulatory element. J Biol Chem 280:34332–34342. doi: 10.1074/jbc.M502349200. [DOI] [PubMed] [Google Scholar]
- 36.Majerczyk CD, Dunman PM, Luong TT, Lee CY, Sadykov MR, Somerville GA, Bodi K, Sonenshein AL. 2010. Direct targets of CodY in Staphylococcus aureus. J Bacteriol 192:2861–2877. doi: 10.1128/JB.00220-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pohl K, Francois P, Stenz L, Schlink F, Geiger T, Herbert S, Goerke C, Schrenzel J, Wolz C. 2009. CodY in Staphylococcus aureus: a regulatory link between metabolism and virulence gene expression. J Bacteriol 191:2953–2963. doi: 10.1128/JB.01492-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brinsmade S, Alexander E, Livny J, Stettner A, Segrè D, Rhee K, Sonenshein A. 2014. Hierarchical expression of genes controlled by the Bacillus subtilis global regulatory protein CodY. Proc Natl Acad Sci U S A 111:8227–8232. doi: 10.1073/pnas.1321308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waters NR, Samuels DJ, Behera RK, Livny J, Rhee KY, Sadykov MR, Brinsmade SR. 2016. A spectrum of CodY activities drives metabolic reorganization and virulence gene expression in Staphylococcus aureus. Mol Microbiol 101:495–514. doi: 10.1111/mmi.13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Majerczyk CD, Sadykov MR, Luong TT, Lee C, Somerville GA, Sonenshein AL. 2008. Staphylococcus aureus CodY negatively regulates virulence gene expression. J Bacteriol 190:2257–2265. doi: 10.1128/JB.01545-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roux A, Todd D, Velázquez J, Cech N, Sonenshein A. 2014. CodY-mediated regulation of the Staphylococcus aureus Agr system integrates nutritional and population density signals. J Bacteriol 196:1184–1196. doi: 10.1128/JB.00128-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gillaspy AF, Hickmon SG, Skinner RA, Thomas JR, Nelson CL, Smeltzer MS. 1995. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect Immun 63:3373–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boles BR, Thoendel M, Roth AJ, Horswill AR. 2010. Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One 5:e10146. doi: 10.1371/journal.pone.0010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li D, Cheung A. 2008. Repression of hla by rot is dependent on sae in Staphylococcus aureus. Infect Immun 76:1068–1075. doi: 10.1128/IAI.01069-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blevins JS, Beenken KE, Elasri MO, Hurlburt BK, Smeltzer MS. 2002. Strain-dependent differences in the regulatory roles of sarA and agr in Staphylococcus aureus. Infect Immun 70:470–480. doi: 10.1128/IAI.70.2.470-480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steinhuber A, Goerke C, Bayer MG, Döring G, Wolz C. 2003. Molecular architecture of the regulatory locus sae of Staphylococcus aureus and its impact on expression of virulence factors. J Bacteriol 185:6278–6286. doi: 10.1128/JB.185.21.6278-6286.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Belitsky BR, Brinsmade SR, Sonenshein AL. 2015. Intermediate levels of Bacillus subtilis CodY activity are required for derepression of the branched-chain amino acid permease, BraB. PLoS Genet 11:e1005600. doi: 10.1371/journal.pgen.1005600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zianni M, Tessanne K, Merighi M, Laguna R, Tabita FR. 2006. Identification of the DNA bases of a DNase I footprint by the use of dye primer sequencing on an automated capillary DNA analysis instrument. J Biomol Tech 17:103–113. [PMC free article] [PubMed] [Google Scholar]
- 49.Moormeier DE, Bose JL, Horswill AR, Bayles KW. 2014. Temporal and stochastic control of Staphylococcus aureus biofilm development. mBio 5:e01341-. doi: 10.1128/mBio.01341-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Flack CE, Zurek OW, Meishery DD, Pallister KB, Malone CL, Horswill AR, Voyich JM. 2014. Differential regulation of staphylococcal virulence by the sensor kinase SaeS in response to neutrophil-derived stimuli. Proc Natl Acad Sci U S A 111:E2037–E2045. doi: 10.1073/pnas.1322125111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bennett HJ, Pearce DM, Glenn S, Taylor CM, Kuhn M, Sonenshein AL, Andrew PW, Roberts IS. 2007. Characterization of relA and codY mutants of Listeria monocytogenes: identification of the CodY regulon and its role in virulence. Mol Microbiol 63:1453–1467. doi: 10.1111/j.1365-2958.2007.05597.x. [DOI] [PubMed] [Google Scholar]
- 52.Château A, van Schaik W, Six A, Aucher W, Fouet A. 2011. CodY regulation is required for full virulence and heme iron acquisition in Bacillus anthracis. FASEB J 25:4445–4456. doi: 10.1096/fj.11-188912. [DOI] [PubMed] [Google Scholar]
- 53.Lobel L, Sigal N, Borovok I, Belitsky BR, Sonenshein AL, Herskovits AA. 2015. The metabolic regulator CodY links Listeria monocytogenes metabolism to virulence by directly activating the virulence regulatory gene prfA. Mol Microbiol 95:624–644. doi: 10.1111/mmi.12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lobel L, Sigal N, Borovok I, Ruppin E, Herskovits AA. 2012. Integrative genomic analysis identifies isoleucine and CodY as regulators of Listeria monocytogenes virulence. PLoS Genet 8:e1002887. doi: 10.1371/journal.pgen.1002887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mahdi LK, Deihimi T, Zamansani F, Fruzangohar M, Adelson DL, Paton JC, Ogunniyi AD, Ebrahimie E. 2014. A functional genomics catalogue of activated transcription factors during pathogenesis of pneumococcal disease. BMC Genomics 15:769. doi: 10.1186/1471-2164-15-769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Montgomery CP, Boyle-Vavra S, Roux A, Ebine K, Sonenshein AL, Daum RS. 2012. CodY deletion enhances in vivo virulence of community-associated methicillin-resistant Staphylococcus aureus clone USA300. Infect Immun 80:2382–2389. doi: 10.1128/IAI.06172-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sadaka A, Palmer K, Suzuki T, Gilmore MS. 2014. In vitro and in vivo models of Staphylococcus aureus endophthalmitis implicate specific nutrients in ocular Infection. PLoS One 9:e110872. doi: 10.1371/journal.pone.0110872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Schaik W, Château A, Dillies MA, Coppée JY, Sonenshein AL, Fouet A. 2009. The global regulator CodY regulates toxin gene expression in Bacillus anthracis and is required for full virulence. Infect Immun 77:4437–4445. doi: 10.1128/IAI.00716-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheng A, Kim H, Burts M, Krausz T, Schneewind O, Missiakas D. 2009. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J 23:3393–3404. doi: 10.1096/fj.09-135467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rigby KM, DeLeo FR. 2012. Neutrophils in innate host defense against Staphylococcus aureus infections. Semin Immunopathol 34:237–259. doi: 10.1007/s00281-011-0295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Valentino M, Foulston L, Sadaka A, Kos V, Villet R, Santa Maria JJ, Lazinski D, Camilli A, Walker S, Hooper D, Gilmore M. 2014. Genes contributing to Staphylococcus aureus fitness in abscess- and infection-related ecologies. mBio 5:e01729-. doi: 10.1128/mBio.01729-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sonenshein AL. 2007. Control of key metabolic intersections in Bacillus subtilis. Nat Rev Microbiol 5:917–927. doi: 10.1038/nrmicro1772. [DOI] [PubMed] [Google Scholar]
- 63.Mashruwala AA, Gries CM, Scherr TD, Kielian T, Boyd JM. 2017. SaeRS Is Responsive to Cellular Respiratory Status and Regulates Fermentative Biofilm Formation in Staphylococcus aureus. Infect Immun 85:e00157-. doi: 10.1128/IAI.00157-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu Q, Cho H, Yeo WS, Bae T. 2015. The extracytoplasmic linker peptide of the sensor protein SaeS tunes the kinase activity required for staphylococcal virulence in response to host signals. PLoS Pathog 11:e1004799. doi: 10.1371/journal.ppat.1004799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lennox ES. 1955. Transduction of linked genetic characters of the host by bacteriophage P1. Virology 1:190–206. doi: 10.1016/0042-6822(55)90016-7. [DOI] [PubMed] [Google Scholar]
- 66.Schenk S, Laddaga RA. 1992. Improved method for electroporation of Staphylococcus aureus. FEMS Microbiol Lett 73:133–138. doi: 10.1111/j.1574-6968.1992.tb05302.x. [DOI] [PubMed] [Google Scholar]
- 67.Novick RP. 1991. Genetic systems in staphylococci. Methods Enzymol 204:587–636. doi: 10.1016/0076-6879(91)04029-N. [DOI] [PubMed] [Google Scholar]
- 68.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moormeier D, Endres J, Mann E, Sadykov M, Horswill A, Rice K, Fey P, Bayles K. 2013. Use of microfluidic technology to analyze gene expression during Staphylococcus aureus biofilm formation reveals distinct physiological niches. Appl Environ Microbiol 79:3413–3424. doi: 10.1128/AEM.00395-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reyes-Robles T, Alonzo F III, Kozhaya L, Lacy DB, Unutmaz D, Torres VJ. 2013. Staphylococcus aureus leukotoxin ED targets the chemokine receptors CXCR1 and CXCR2 to kill leukocytes and promote infection. Cell Host Microbe 14:453–459. doi: 10.1016/j.chom.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.