

Abstract
Inherited syndromic retinopathies are a highly heterogeneous group of diseases that involve retinal anomalies and systemic manifestations. They include retinal ciliopathies, other well-defined clinical syndromes presenting with retinal alterations and cases of non-specific multisystemic diseases. The heterogeneity of these conditions makes molecular and clinical characterization of patients challenging in daily clinical practice. We explored the capacity of targeted resequencing and copy-number variation analysis to improve diagnosis of a heterogeneous cohort of 47 patients mainly comprising atypical cases that did not clearly fit a specific clinical diagnosis. Thirty-three likely pathogenic variants were identified in 18 genes (ABCC6, ALMS1, BBS1, BBS2, BBS12, CEP41, CEP290, IFT172, IFT27, MKKS, MYO7A, OTX2, PDZD7, PEX1, RPGRIP1, USH2A, VPS13B, and WDPCP). Molecular findings and additional clinical reassessments made it possible to accurately characterize 14 probands (30% of the total). Notably, clinical refinement of complex phenotypes was achieved in 4 cases, including 2 de novo OTX2-related syndromes, a novel phenotypic association for the ciliary CEP41 gene, and the co-existence of biallelic USH2A variants and a Koolen-de-Vries syndrome–related 17q21.31 microdeletion. We demonstrate that combining next-generation sequencing and CNV analysis is a comprehensive and useful approach to unravel the extensive phenotypic and genotypic complexity of inherited syndromic retinopathies.
Introduction
Inherited syndromic retinopathies (ISR) are clinically and genetically heterogeneous diseases in which retinal alteration is accompanied by systemic anomalies that can involve one or more systems, such as auditory, neurological, musculoskeletal, renal, cardiac, and hepatic systems1,2. These disorders can be inherited in an autosomal recessive, autosomal dominant, X-linked, or mitochondrial fashion. Triallelism and oligogenic forms have also been described3. To date, ISRs have been related to at least 90 genes based on the Retinal Information Network database (RetNet, https://sph.uth.edu/retnet/).
ISRs include clinically well-defined syndromes such as ciliopathies4 (Alström5, Bardet-Biedl6, Joubert7, Senior-Løcken8, and Usher syndrome9), syndromes related to other cellular components (Cohen syndrome10 and peroxisome biogenesis disorders [PBDs]11), and even extracellular molecular defects (Stickler syndrome)12. In other cases, patients present with diverse non-specific systemic symptoms and signs in which a clear clinical diagnosis cannot be easily achieved13. As many of these entities share medical and molecular features that hinder their clinical management and follow-up13, ISR diagnosis is quite difficult and challenging, and patients often visit various specialists over a period of years without a conclusive diagnosis being made. Thus, molecular characterization is essential for an accurate and definitive diagnosis and helps to guide reproductive risk assessment, counseling, surveillance, management, and prognosis1,13. Our study may also provide clues for appropriate selection of patients to be enrolled in future clinical trials such as those examining gene-based therapies.
Traditional methods for mutation screening are costly and labor-intensive and entail a low molecular diagnostic rate, even for patients with a clear suspicion of ciliopathy14,15. Next-generation sequencing (NGS) is a cost-effective approach for the genetic diagnosis of retinal diseases16,17. Several studies have focused on specific retinal ciliopathies, such as Bardet-Biedl syndrome (BBS)18 and Joubert syndrome7,19–21, with a wide range of reported mutation detection rates depending on the number of targeted genes, NGS platform, and use of pre-screened or “naive” cases. In patients with complex atypical phenotypes, chromosomal microarray analysis may also be very useful for assessing contiguous gene deletion syndromes as a cause of disease co-occurrence22. To date, few authors have studied the role of copy number variations (CNVs) in retinal dystrophies (RD)23–27. We performed a molecular analysis using a custom targeted resequencing approach combined with CNV analysis in order to characterize a heterogeneous cohort of ISR patients whose specific disease had not yet been determined. Our results provide new insights into the genetic complexity of these diseases.
Results
A cohort of 47 genetically uncharacterized ISR cases underwent a comprehensive analysis of single-nucleotide variants (SNVs) and CNVs, based mainly on a custom targeted NGS for 121- gene panel of ISR-associated and candidate genes (Supplementary Table S1). The genes were selected based on previous associations with ISR reported in the literature, many of which showed a high proportion of associated phenotypes and appeared to be functionally related (Supplementary Fig. S1).
Patients were classified into 4 major phenotypic categories based on clinical features and suspected clinical diagnosis (Fig. 1): i) ciliopathies, including 10 cases with Alström, BBS, or Joubert syndrome; ii) ciliopathy-like, including 11 patients with RD and some ciliopathy-related systemic anomalies (polydactyly, obesity, diabetes, hearing, loss, and/or nephronophthisis); iii) other known syndromic retinopathies, a total of 5 patients with a clear diagnosis of either PBDs, Stickler syndrome, or pseudoxanthoma elasticum (PXE); iv) miscellanea, including 21 cases with heterogeneous phenotypes involving retinal disease with 1 or more non-specific systemic features, such as congenital malformations, ataxia, dwarfism, intellectual disability (ID), or neuroendocrine alterations.
Figure 1.
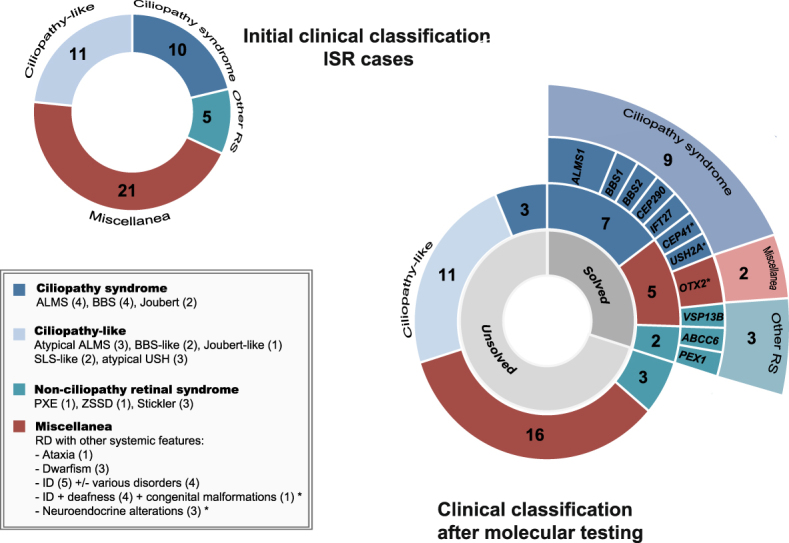
Clinical classification of patients with inherited syndromic retinopathies (ISR) and molecular findings obtained in this study. The two charts summarize initial and reassessed clinical classifications, before and after molecular testing, respectively. Four main phenotypic categories of cases were considered: i) clinically-defined ciliopathy syndromes, represented in dark blue color; ii) ciliopathy-like cases, in which retinal degeneration was found in association with multiple ciliopathy-like features (in light blue color); iii) patients with a clear diagnosis of a non-ciliopathy RD, represented in green, and iv) miscellaneous cases (in red color) with a variety of phenotypes that involved RD with one or more unspecific systemic symptoms, in which an obvious diagnosis could not be clearly established. The different levels of circles of the right chart (from inner to outside) reflect molecular yield depending on these 4 clinical categories and the mutated genes. Inner circle in grey shapes represents the fraction of solved (30%, 14/47) vs unsolved (70%, 33/47) cases after molecular testing. Second circle indicates the yield of molecular characterization based on the initial classification. Third level shows the causal genes found in this study and their respective color indicate the associated syndromic retinal disease. Outer level indicated the final classification of the solved cases. *CEP41, USH2A, and VSP13B cases, which were initially included in the miscellanea group, were re-classified after genetic testing. Dual diagnosis of USH2A biallelic pathogenic variants and de novo 17q21.31 mosonomy associated with Usher and Koolen de Vries syndromes, respectively. ALMS: Alström syndrome; BBS: Bardet-Biedl Syndrome; ID: Intellectual Disability; ISR: inherited syndromic retinopathies; RD: Retinal Dystrophy; SLS: Senior-Løcken syndrome; USH: Usher syndrome; ZSSD: Zellweger syndrome spectrum disorder.
A total of 33 pathogenic or likely pathogenic variants were identified in 49% of patients (23/47) (Tables 1 and 2 and Supplementary Table S2), including 30 SNVs and 3 CNVs in 18 different genes (ABCC6, ALMS1, BBS1, BBS2, BBS12, CEP41, CEP290, IFT172, IFT27, MKKS, MYO7A, OTX2, PDZD7, PEX1, RPGRIP1, USH2A, VPS13B, and WDPCP). Thirteen of the SNVs were novel (Supplementary Table S3), including 2 non-synonymous and 2 in-frame variants (ABCC6: p.(Leu495del), BBS12: p.(Ser440del), CEP41: p.(Ser2Phe), IFT27: p.(Tyr35Cys)), which were considered potentially pathogenic based on in silico prediction, evolutionary conservation of the wild-type residues, previous knowledge of ISR-associated genes, and disease co-segregation.
Table 1.
Clinical and genetic information of cases carrying likely disease-causal variants.
| Family | Initial Dx | Final Dx | Clinical Features | Gene | Allele 1 | Allele 2 | Other alterations | Methods | Segregation |
|---|---|---|---|---|---|---|---|---|---|
| Ciliopathies | |||||||||
| RP-1232 | Alström | Alström | RP, hearing loss, obesity, diabetes, NPHP, hypogonadism, hypothyroidism, hyperinsulinemia, and acantosis nigricans | ALMS1 |
NM_015120.4:c.4252del;
p.(Arg1418Glyfs*55) |
NM_015120.4:c.4252del;
p.(Arg1418Glyfs*55) |
NGS | Y (AR) | |
| RP-2186 | Alström | Alström | Early RD, overweight, dilated cardiomyopathy, and diabetes | ALMS1 |
NM_015120.4:c.4477G>T; p.(Glu1493*)61 |
NM_015120.4: c.7571_7572del; p.(His2524Argfs*11)61 |
NGS | Y (AR) | |
| RP-2177 | Alström | Alström | CRD, hearing loss, and dilated cardiomyopathy | ALMS1 |
NM_015120.4:c.808C>T; p.(Arg270*)31 |
NM_015120.4:c.11618_11619del; p.(Ser3873Tyrfs*19)61 |
NGS | na | |
| RP-2069 | BBS | BBS | CRD, polydactyly, maturation and learning delay, obesity, and chronic renal failure | IFT27 |
NM_006860.4:c.104A>G; p.(Tyr35Cys) |
NM_006860.4:c.350-2A>G | NGS | na | |
| RP-2167 | BBS | BBS | CRD, obesity, polydactyly and brachydactyly, psychomotor and learning delay, and behaviour disorder | BBS2 |
NM_031885.2:c.471G>A; Affecting 5′ splicing site31 |
NM_031885.2:c.1237C>T; p.(Arg413*)30 | NGS | na | |
| RP-2228 | BBS | BBS | RP, ID, overweight since infancy, brachydactyly, chronic renal failure, and renal transplant | BBS1 |
NM_024649.4:c.1645G>T; p.(Glu549*)33 |
NM_024649.4:c.118del; p.(Cys40Alafs*2)62 |
MKKS NM_018848.3:c.724G>T; p.(Ala242Ser)32 | chip + NGS | Y* |
| RP-1814 | Joubert | Joubert | RD, nystagmus, psychomotor delay, cerebellar atrophy, chronic renal failure, and macrocephaly | CEP290 |
NM_025114.3:c.4028del; p.(Lys1343Argfs*2)34 |
NM_025114.3:c.7341dup; p.(Leu2448Thrfs*8)35 |
chip + NGS | Y (AR) | |
| Other specific syndromes | |||||||||
| RP-1724 | ZSSD | ZSSD | Early RP, congenital deafness, neonatal jaundice, intrahepatic biliary dysgenesis, encephalopathy, ID, hiperprolactinemia, and gynecomastia | PEX1 |
NM_000466.2:c.2528G>A; p.(Gly843Asp)36 |
NM_000466.2:c.2528G>A; p.(Gly843Asp)36 |
NGS | NA | |
| V-0799 | Pseudoxanthoma elasticum | Pseudoxanthoma elasticum | Macular atrophy, angioid streaks, and neovascular membrane | ABCC6 |
NM_001171.5:c.1483_1485del;
p.(Leu495del) |
NM_001171.5:c.1483_1485del;
p.(Leu495del) |
NGS | Y (AR) | |
| Miscellanea | |||||||||
| RP-0485 | RP + ID + deafness + congenital malformations | Usher + Koolen de Vries | RP, mild ID, hearing loss, ASD, macrocephaly, and low-set ears | USH2A | NM_206933.2:c.1876C>T; p.(Arg626*)43 | NM_206933.2:c.13010C>T; p.(Thr4337Met)44 | 721Kb monosomy at 17q21.31† | NGS + aCGH | Y (USH2A: AR CNV: de novo) |
| RP-1430 | RP + ID | Cohen | RP and ID | VPS13B | NM_017890.4:c.1512del; p.(Glu505Lysfs*23) | NM_017890.4:c.1512del; p.(Glu505Lysfs*23) | NGS | Y* | |
| RP-1613 | RP + neuroendocrine alteration | OTX2-related | LCA, congenital nystagmus, psychomotor delay, compulsive eating, muscular hypotonia, obesity, and hypogonadism | OTX2 | NM_172337.2:c.255 G > A; p.(Trp85*) | NGS | Y (de novo) | ||
| RP-2140 | RP + neuroendocrine alteration | OTX2-related | Congenital nystagmus, early-onset CRD, developmental delay, and panhypopituitarism | OTX2 |
NM_172337.2:c.559
C
>
T;
p.(Gln187*) |
NGS | Y (de novo) | ||
| RP-2273 | RP + neuroendocrine alteration | BBS | Congenital nystagmus, LCA, developmental delay, congenital hypothalamic obesity, hypogonadism, acantosis nigricans, and clinodactyly | CEP41 |
NM_018718.2:c.5
C
>
T;
p.(Ser2Phe) |
NM_018718.2:c.5 C > T; p.(Ser2Phe) | NGS | na | |
Novel likely pathogenic variants found in this study are represented in bold. *Segregation of families RP-2228 and RP-1430 could be only done in an unaffected sister and father, respectively. †Monosomy at 17q21.3 (arr[GRCh37] 17q21.31(43417434_44138572)x1) encompassed the following genes: ARHGAP27, PLEKHM1, MIR4315-1, MIR4315-2, LRRC37A4, LOC101929001, DND1P1, LOC644172, RPS26P8, CRHR1, MGC57346, SPPL2C, MAPT, MAPT-IT1, STH, KANSL1. Abbreviations: aCGH: array–based comparative genomic hybridization; AR: autosomal recessive; ASD: atrial septal defect; BBS: Bardet-Biedl syndrome; CRD: cone-rod dystrophy; Dx: diagnosis; ID: intellectual disability; LCA: Leber congenital amaurosis; na: not available; NGS: next-generation sequencing; NPHP: nephronophthisis; RD: retinal dystrophy; RP: retinitis pigmentosa; Y: yes; ZSSD: Zellweger syndrome spectrum disorder.
Table 2.
Copy Number Variants found by coverage depth analysis or array–based comparative genomic hybridization.
| Family | Initial diagnosis | Cytogenetic band | Rearrangement type | Genomic coordinates (hg19) | Involved genes | Minimun Size (kb) | Method | Validation | Causality | Additional genetic findings |
|---|---|---|---|---|---|---|---|---|---|---|
| RP-0485 | RP + ID + deafness + congenital malformations | 17q21.31 | monosomy | chr17:43417434–44138572 | ARHGAP27, PLEKHM1, MIR4315-1, MIR4315-2, LRRC37A4, LOC101929001, DND1P1, LOC644172, RPS26P8, CRHR1, MGC57346, SPPL2C, MAPT, MAPT-IT1, STH, KANSL1 | 721 | aCGH | yes | Yes (Koolen de Vries) | Biallelic USH2A variants, responsible of Usher related symptoms |
| RP-1581 | RP + ID + various disorders | 11q13.2 | whole gene duplication | chr11: 66277760–66300760 | BBS1 | 23 | NGS + aCGH | yes | VUS | in cis with a VUS in BBS1(maternally inherited) |
| RP-2009 | RP + ID + various disorders | 14q11.2 | exon 17–19 deletion | chr14:21795786–21798554 | RPGRIP1 | 2,77 | NGS + MLPA | yes | Unlikely | Biallelic SCAPER variants55 |
aCGH: array–based comparative genomic hybridization; ID: intellectual disability; NGS: next-generation sequencing; RP: retinitis pigmentosa; VUS: variant of uncertain significance.
From the total identified variants, 23 were further considered likely causative in 14 cases based on phenotypic concordance, and also on co-occurrence with a second allele in recessive genes or the presence of de novo events in dominant genes (Table 1). Thus, we obtained a diagnosis rate of 30%, including known and likely disease-associated variants (Table 1 and Fig. 1). In terms of inheritance patterns, 86% of the cases had an autosomal recessive inheritance, 2 cases carried de novo autosomal dominant variants, and in one family, triallelism for BBS1 and MKKS was suspected. Additionally, in 9 cases (19%) which carried heterozygous likely pathogenic alleles in genes responsible for recessive RD (Supplementary Table S2), no second allele was found using NGS, multiplex ligation-dependent probe amplification (MLPA), and/or array–based comparative genomic hybridization (aCGH).
Revision of Cases with Concordant Molecular Findings and Initial Clinical Diagnosis
Our approach confirmed the initial clinical suspicion in 9 cases. Based on our first phenotypic classification, we characterized up to 70% of cases (7/10) with a clear phenotypic manifestation of ciliopathy and 40% of cases (2/5) presenting other well-defined ISRs (Fig. 1).
Three out of 4 cases with clinical suspicion of Alström syndrome carried biallelic causal ALMS1 variants (families RP-1232, RP-2177, and RP-2186, Table 1 and Fig. 2). Similarly, 3 out of 4 of the cases previously classified as BBS carried likely pathogenic variants in known BBS-associated genes (Table 1). In family RP-2069, which comprised 2 affected siblings with RP, neurodevelopment delay, obesity, and renal alterations (Table 1 and Fig. 2), the index case was found to have two novel heterozygous likely pathogenic variants in IFT27 (Supplementary Table S3), a splicing variant in exon 6 (c.350-2A>G), and a missense variant p.(Tyr35Cys), which affects a highly conserved residue located in a nucleotide-binding domain with GTPase activity28. In addition, the IFT27-associated phenotype is clearly consistent with the clinical presentation of our family29. In sporadic case RP-2167, which had characteristic BBS-associated features (Table 1), we found 2 known heterozygous BBS2 alleles, a nonsense variant30, and a synonymous change that had previously been reported in homozygosis as a cause of retinitis pigmentosa31. This silent variant, which was located at the last nucleotide of exon 3, likely abolishes the canonical donor splice site by interfering with U1snRNA recognition. In the sporadic BBS case RP-2228, which carried 2 previously identified heterozygous variants in MKKS32 and BBS133, a second loss-of-function (LOF) allele in BBS1 was found by NGS analysis (Table 1). In this case, triallelism could not be completely confirmed, owing to an uninformative pedigree structure with only 1 affected patient (Fig. 2). In addition, we characterized 1 of the 2 Joubert syndrome families in our cohort. Two affected siblings presenting with early-onset RD, cerebellar atrophy, late-stage renal failure, and ID (family RP-1814, Table 1) carried previously described biallelic LOF variants in CEP29034,35 (Fig. 2).
Figure 2.

Pedigrees and co-segregation analysis for cases carrying likely disease-causing variants. Genotype of each available family member is represented below the individual symbol being “+” normal allele, and “m, m1, m2, and m3”, mutated alleles.
We also confirmed the clinical diagnosis of 2 families presenting with a clear non-ciliopathy retinal syndrome (Fig. 1). The sporadic case RP-1724, who was born to an endogamous couple and presented infantile Refsum disease, homozygously carried a PEX1 allele, p.(Gly843Asp), which is the most common causal allele in Zellweger syndrome spectrum disorders (ZSSD)36,37. Similar to other cases with this variant36,37, the patient exhibited a mild form of ZSSD with a progressive disease course. Finally, we studied an apparently pseudo-dominant family in which PXE has been suspected in 2 affected siblings and their father (V-0799, Fig. 2). All affected individuals showed a similar phenotype of angioid streaks, vision loss around 55 years, atrophic macular degeneration and dyschromatopsia. After NGS and segregation analysis, both affected siblings (II:1 and II:2) carried a novel homozygous in-frame deletion in the ABCC6 gene, namely p.(Leu495del) (Table 1 and Fig. 2). This variant affects a phylogenetically conserved amino-acid position, where a missense change had previously been described for this disease38 and was predicted as deleterious by in silico analysis. CNV depth-read and MLPA analysis ruled out whole-gene or multi-exon deletions in the ABCC6 gene in the family (Supplementary Fig. S2). In addition, haplotype analysis of the ABCC6 locus and the surrounding genomic regions confirmed that an identity-by-descent (IBD) mechanism was involved in the homozygous transmission of the variant (Supplementary Fig. S2). This finding was clearly consistent with the highly endogamous ancestry of both parents. Thus, the suspected autosomal dominant transmission of this variant has been clearly ruled out in this family, accordingly to which PXE is a well recognized autosomal recessive disorder39,40. Therefore, the apparent pseudo-dominance in this family is most likely due to the presence of a second different pathogenic SNV in the father, that remains to be discovered after NGS sequencing. However, we cannot exclude the presence of additional non-coding variants that could explain the PXE phenotype.
Molecular Characterization of Clinically Unclassified Cases and Refinement of Clinical Diagnosis
Five cases (11%) were phenotypically reclassified based on our molecular findings and further clinical reassessment (Table 1). Of them, 3 sporadic cases had common features of neuroendocrine disorder, psychomotor impairment, and Leber congenital amaurosis (LCA). It is noteworthy that 2 of these LCA cases (RP-1613 and RP-2140, Table 1), had diverse symptoms of hypopituitarism, and both bore de novo nonsense OTX2 variants: p.(Trp85*) and p.(Gln187*), respectively (Fig. 2). LOF variants in this transcription factor were previously associated with a wide spectrum of pituitary dysfunction and congenital eye disorders ranging from anophthalmia to retinal dystrophies41. In a third sporadic LCA consanguineous patient (RP-2273, Table 1), an extremely rare missense variant, p.(Ser2Phe), was homozygously found in CEP41. This change affects a highly conserved residue in the N-terminal domain of the ciliary protein CEP41 and was predicted as damaging by in silico analysis (Supplementary Table S3). Although CEP41 has previously been associated with Joubert syndrome42, no cerebellar abnormality was observed in the patient here reported, and his clinical picture (early-onset RD, mild ID, brachydactyly, and neuroendocrine alteration) was in principle more consistent with a BBS-like diagnosis.
In the sporadic case RP-0485, who had RD, neurosensorial hypoacusia, mild ID, and several congenital malformations (Fig. 3 and Table 1), the co-occurrence of 2 rare genetic disorders was identified. First, NGS analysis identified 2 biallelic USH2A variants, both of which had previously been associated with Usher syndrome43,44, that were responsible for the retinal and hearing manifestations. Further high-resolution aCGH analysis enabled us to identify a 721-kb de novo microdeletion at 17q21.31 (Fig. 3), including the KANSL1 gene, which had been reported to cause Koolen-de Vries syndrome45. Finally, in a consanguineous Middle-Eastern patient (RP-1430) with RD and mental disability, a novel causative homozygous frameshift variant was found in the VPS13B gene, which is associated with Cohen syndrome. Unfortunately, a more accurate diagnosis could not be established, since few clinical data were available, and clinical re-evaluation of this patient could not be performed.
Figure 3.

Dual diagnosis of Usher and Koolen de Vries syndromes in case index of family RP-0485 by targeted NGS and array–based comparative genomic hybridization (aCGH) (A). Pedigree and segregation analysis for genetic findings showed a recessive inheritance for biallelic pathogenic USH2A variants and a de novo CNV. Genotype of each available family member is represented below the individual symbol being “ + ” normal allele, and “m1, m2, and m3”, mutated alleles. Electropherograms of heterozygous carrier and wild-type individual for USH2A variants in exons 11 and 63 were also shown (B). Genomic rearrangement on 17q21.31 region identified in by aCGH. Chromosome and gene views of the affected chromosome show an aberrant deletion of minimum size of 721Kb (genomic coordinates: chr17:43,417,434-44,138,572) and maximum ~1,4 Mb (chr17:43,398,423-44,841,644), which is highlighted in dark and light red colour, respectively. An abnormal pattern of one copy was observed for 80 probes covering 16 genes (ARHGAP27, PLEKHM1, MIR4315-1, MIR4315-2, LRRC37A4, LOC101929001, DND1P1, LOC644172, RPS26P8, CRHR1, MGC57346, SPPL2C, MAPT, MAPT-IT1, STH, KANSL1). The vertical axis shows the position along the genome (hg19) and the horizontal axis the log2 intensity ratio values (−2/−1: deletions, 0: normal pattern, + 1/ + 2: duplications).
Discussion
ISRs comprise a diverse spectrum of highly heterogeneous and overlapping clinical conditions. Clinical diagnosis is often challenging, as some of the characteristic systemic features may not develop until later in infancy and adulthood or may even evolve and change over time. Several multigene panel approaches have been used for molecular characterization of specific retinal ciliopathies, such as BBS, Joubert syndrome, and Usher syndrome, yielding variable diagnostic rates7,18,19,46,47. Here, we first studied a more heterogeneous and large cohort of “non-naive”, i.e. previously screened ISR patients, who were all recruited in a single center, and were similar to those found in daily clinical practice. We included not only cases with a clear suspicion of syndromic forms of retinal disease, but also atypical cases that did not exactly fit any known ISR.
Our findings confirm the clinical utility of targeted NGS strategies, demonstrating an overall diagnostic rate of 30%, which is comparable with those obtained from similar studies using prescreened RD patients25,48. As expected from the heterogeneity of the cases included, the diagnostic yield obtained in the present ISR cohort was not uniform. While we obtained a high mutation detection rate (70%) in patients with clinically suspected specific ciliopathies, similar to previous studies46,47, the diagnostic rate decreased in patients with unspecific symptoms of ISR. Notably, the responsable molecular mechanism was identified in at least 5 of these patients, thus our study highlighted the potential of NGS to improve diagnostic accuracy in complex phenotypes. In this sense, the panel led us to clinically characterize 2 sporadic cases in which LCA was accompanied by neuroendocrine symptoms as bearing de novo OTX2 variants. The clinical symptoms of both cases overlapped with the previously OTX2-associated phenotypes, including pituitary dysfunction and retinal dystrophy49. Consequently, both patients were also genetically reclassified as having a condition based on dominant inheritance. The identification of de novo events highlights the relevance of obtaining a correct genetic diagnosis in sporadic genetic cases as first step to enable a more accurate genetic counseling50.
We were able to reclassify a miscellaneous case within the ciliopathy group owing to the identification of a novel homozygous likely causal CEP41 variant. Although this ciliary gene was previously associated with Joubert syndrome42, this sporadic case had RP with neuroendocrine symptoms that seemed to be more compatible with a BBS-like form. In addition, the normal brain MRI scan in this patient excluded Joubert syndrome as clinical diagnosis. Furthermore, we found biallelic pathogenic variants in the IFT27 gene in a BBS patient. To our knowledge, this is the second case of BBS bearing IFT27 alterations in the literature29.
In 9 cases, we found only a monoallelic likely pathogenic variant in a well-known recessive gene. The presence of a second pathogenic variant was not fully excluded, as it might reside in deep intronic, regulatory, and even highly repetitive regions that were not targeted in our gene panel. The clinical presentation in most cases was reasonably consistent with the phenotypic features of the suspected gene. For instance, 3 of 11 ciliopathy-like patients carried heterozygous pathogenic variants in ciliopathy genes, such as ALSM1, USH2A, and WDPCP, thus it might support the initial clinical suspicion. Therefore, some of these inconclusive genetic findings could also be helpful to guide further management and follow-up of these patients. However, additional molecular analysis of the above-mentioned unexplored regions should be performed using custom targeted resequencing or even whole-genome sequencing (WGS).
Structural variants also represent an underestimated mutational burden that contributes to missing heritability and oligogenic mechanisms, not only in non-syndromic RD, but also in BBS and Joubert syndrome20,24–26,51,52. Here, we report novel heterozygous multi-exon structural variants for BBS1 and RPGRIP1 that were identified by targeted comparative read-depth analysis. However, their possible causality is unclear, since no second pathogenic allele was found in trans in these genes. Therefore, additional studies would be necessary to determine whether the variants represent random background variation or contribute to the disease. Nonetheless, our results also suggest that CNV and re-sequencing studies are both required to fully delineate the complexity of disease architecture in ISR phenotypes.
The low diagnostic rate found in cases with suspected atypical ciliopathies or complex presentations also suggests the participation of additional genes not present in the panel, whether novel candidate genes or genes recently associated with ISRs (e.g. CEP120 or GRID2)53,54. Further whole-exome sequencing (WES) or WGS could prove useful for characterization of additional patients in our cohort. In this sense, biallelic pathogenic variants were identified in SCAPER, a novel RD gene explaining RP with ID, in 2 uncharacterized cases of the miscellaneous group, as we recently published55. An additional plausible mechanism that could explain some negative cases with a complex phenotypic spectrum is the possibility of multiple clinical diagnoses due to the co-existence of molecular defects in several disease-causing genes. Dual molecular diagnosis was recently reported in at least 4% of cases with informative WES22,56. Here, we observed co-occurrence of Usher syndrome and Koolen de Vries syndrome, each segregating independently as recessive and de novo events, respectively. Biallelic USH2A variants, first identified by NGS, were responsible for RP and neurosensorial hypoacusia, but they do not completely explain the phenotypic complexity of this case. Other features, such as mild ID, atrial septal defect, macrocephaly and low-set ears, were caused by a coexistent de novo 17q21.31 microdeletion, including the KANSL1 gene, which is associated with Koolen-de Vries syndrome45. Thus, further NGS testing of non-syndromic associated RD genes in combination with aCGH analysis may prove very useful for ruling out the co-existence of several genetic events in uncharacterized atypical cases22.
Our study highlights the huge potential of genetic testing to obtain prompt molecular diagnoses in complex ISR cases using relatively low-cost, robust, accurate, and clinically useful multigene NGS panels and CNV analysis. Thus, this combined strategy increased our understanding of well-defined phenotypes of ISR, such as retinal ciliopathies. Implementation in routine clinical practice of similar approaches based on NGS panels with a wider gene spectrum including not only reported RD and ciliary genes, but also genes associated with other retinal conditions, would reduce the time and cost of diagnosing ISRs and provide an earlier diagnosis of systemic features, even before they appear. This is especially important for clinical follow-up in medically actionable diseases and also for enrollment of patients in future gene therapy–based treatments. Alternatively, clinical or WES seems to be a very useful choice for cases presenting with more unspecific or widely complex ISR phenotypes and could potentially improve molecular diagnostic yields.
Materials/Subjects and Methods
Clinical diagnosis and sample collection
We selected genetically non-characterized patients with syndromic retinopathies from our database of RD patients who attended Fundación Jiménez Díaz University Hospital (FJD, Madrid, Spain) in the last 25 years. We excluded typical Usher syndrome patients, as they were already assessed using a specific NGS Usher gene panel47. The clinical diagnosis was made according to previously established criteria and included ophthalmic, physical, and additional examinations10,12,14,57,58. The cohort included cases with various suspected patterns of inheritance, including autosomal recessive, autosomal dominant, and sporadic cases. This study was reviewed and approved by the FJD Ethics Committee and was performed according to the tenets of the Declaration of Helsinki and its reviews. The participants, or their legal guardians, signed a written informed consent form before the study. DNA samples were collected from FJD Biobank.
Clinical Genetic Testing
Following routine clinical procedures, all patients had previously been screened using commercial genotyping microarrays (AsperBiotech, Tartu, Estonia) for previously known pathogenic variants in several genes associated with BBS, Alström syndrome, LCA, or autosomal recessive retinitis pigmentosa. In cases compatible with BBS or Alström syndrome, Sanger-sequencing was performed to screen for the most prevalent mutated exons and/or genes, as previously reported57,58. Homozygosity mapping studies were also performed in some families57,59.
Target Enrichment and Bioinformatic Analysis
Gene Panel Design, Library Construction, and Sequencing
A custom targeted NGS approach was designed to specifically study ISR patients using the Nextera Rapid Capture kit (Illumina, Cambridge, UK) for target enrichment. The DesignStudio software tool (Illumina) was used to design the gene panel. We selected genes based on previous associations with ISRs, including 86 genes reported in the Retnet database (data accession in 2015) and 17 genes after a thorough revision of literature. In addition, 18 very likely candidate genes were also included due to their previously involvement in other non-syndromic ciliopathies.
Many of them show a high degree of phenotypic overlap and appear to be functionally related (Supplementary Fig. S1). The complete list of genes targeted is detailed in Supplementary Table S1 and includes the following: i) 71 ciliopathy-related genes accounting for RD and multisystemic disease; ii) 18 ciliary genes previously implicated in non-syndromic RD or associated with non-retinal ciliopathies, such as isolated nephronophthisis, were also added as likely candidate genes; and iii) 32 non-ciliopathy genes including PBDs and lysosomal disorders (5 genes), syndromic vitreoretinopathies (7 genes), and a miscellanea of syndromic retinal alterations with overlapping systemic features (20 genes). The gene panel included coding and non-coding exons and targeted flanking upstream and downstream 50-bp and 5′ and 3′ UTR regions and also known deep intronic variants previously identified in the BBS1, CEP290, OFD1, and USH2A genes (Supplementary Table S4). In total, the library contained 2,459 target regions covered by 3,813 probes spanning 577 kb.
Libraries for 47 samples were prepared following the manufacturer’s protocol. Briefly, DNA samples were quantified using fluorometric assays (PicoGreen, Invitrogen, Thermo Fisher Scientific, MA, USA) and normalized to prepare 50 ng of starting material at 5 ng/µl. Samples were tagmented, PCR amplified, and cleaned up. Libraries were pooled with 24 samples, multiplexed, hybridized for 90 min, and captured. A second round of hybridization, capture, PCR amplification, and clean-up was performed. Final libraries were quantified using qPCR with the KAPA Library Quantification Kit (Kapa Biosystems, London, UK) and validated by capillary electrophoresis using a 2100 Bionalyzer (Agilent Technologies, Santa Clara, CA, USA). Finally, 1.8 pm of 24 equimolecularly pooled libraries were loaded into a NextSeq500 flow cell and sequenced using the NextSeq500 Mid Output reagents Kit v1 (Illumina, Cambridge, UK) to obtain 150-bp paired-end reads. The sequencing had an average coverage of 435X with approximately 85% reads on target, and 93% of target regions of these 121 genes were completely covered (Supplementary Fig. S3).
The affected father (I:1) from family V-0799 has also been analyzed using targeted clinical exome. Libraries were prepared using TruSightOne (Illumina) following the manufacturer’s protocol. The captured libraries were sequenced with Illumina NextSeq500 with 150-bp paired-end reads.
Bioinformatic Analyses
A specific custom pipeline for Illumina Nextera technology implemented on the commercial DNAnexus platform (https://www.dnanexus.com/) was used for the bioinformatics analysis as previously described25,48. Briefly, Prinseq-lite v0.20.3 was used to calculate read statistics, and BWA-MEM v0.7.5a with default parameters was used to map the reads. Samtools v0.19 was then used to sort and index the BAM files, and Picard MarkDuplicates v1.119 was used to mark duplicates. Freebayes v9.9.13 was used for variant calling, Picard CalculateHSMetrics v1.119 to calculate metrics, and BedTools and custom code to determine minimum coverage. Identified variants were annotated using GATK v2.4. Gene coverage was calculated using BedTools to extract the number of reads overlapping any genome region from the BAM file of each sample. From this, coverage was calculated per gene and sample by counting the reads per nucleotide inside the gene coordinates. Regions with a coverage of fewer than 10 reads were excluded.
To verify the reliability of our targeted NGS approach, we used a validation cohort consisting of 5 out of the total 47 patients analyzed, which carried 6 different heterozygous variants previously detected by genotyping microarray (Supplementary Table S5). All variants used as controls were correctly identified.
A CNV analysis was performed with NGS data following the previously reported CoNVaDING method, which had been specifically developed for gene panel data at high-coverage and enables accurately single-exon detection60. Briefly, it uses the average coverage of each specified target to perform calculations starting from BAM files. Then, it selects a group of control samples with the coverage pattern that is most similar to that of the sample to be analyzed, which are used in the normalization step. Finally, it performs a filtering step that renders the list with deletions and duplications. The method includes QC metrics that make it possible to distinguish high-quality from low-quality samples60.
Variant Prioritization and Classification
Potentially pathogenic variants were sequentially assessed as previously described25,48, based on the following criteria: i) variants previously reported as pathogenic in mutation databases, including HGMD Professional (http://www.biobase-international.com/product/hgmd) and the Leiden Open Variation Database (http://www.lovd.nl/3.0/home); ii) LOF variants such as nonsense, frameshift, and those located at the canonical splice site; iii) variants filtered out, focusing on rare variants with a minor allele frequency (MAF) ≤0.005 in 1000 Genomes (http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/), Exome Variant Server (EVS) (http://evs.gs.washington.edu/EVS/), ExAC Browser (http://exac.broadinstitute.org/), and CIBERER Spanish Variant Server (http://csvs.babelomics.org/); iv) missense SNV and in-frame deletions that induced an alteration in the protein predicted as pathogenic in at least 2 of the in silico tools used to predict pathogenicity (SIFT, Polyphen2, Mutation Taster, Align GVGD, PROVEAN); v) non-canonical splicing variants in silico predicted to cause a deleterious effect by several splicing tools (Human Splicing Finder, MaxEntScan, Splice Site Finder-like, NNSPLICE, GeneSplicer, ESEFinder); vi) genes associated with the most likely clinical phenotype in each case. Alamut software (Interactive Biosoftware, Rouen, France) was used to infer in silico predictions of variant pathogenicity.
Validation Studies
Sequence variants of interest identified by NGS were verified and segregated in the respective families by Sanger sequencing following PCR amplification of the respective coding exons and adjacent intronic sequences by standard protocols (primers are available on request).
Multiplex Ligation-dependent Probe Amplification (MLPA) analysis
MLPA was performed according to manufacturer instructions using commercially available kits (MRC-Holland, Amsterdam, Netherlands). The RPGRIP1 deletion on exons 17–19 was validated using MLPA SALSA p222 covering exons 18 and 19. The MLPA SALSA P092 was used to rule out CNVs in the ABCC6 gene. The amplified fragments were separated by capillary electrophoresis through an ABI 3130xl automatic analyzer (Applied Biosystems) and analyzed using GeneMapper (Applied Biosystems) and Coffalyser (MRC-Holland) software programs.
Array–based comparative genomic hybridization
Contiguous gene deletion syndromes was assessed by means of aCGH in 14 patients with atypical complex phenotypes in which RD was accompanied by ID and/or congenital malformations. The aCGH analysis was performed using the Agilent SurePrint G3 CGH + SNP Microarray Kit 2 × 400 K (Agilent Technologies, Santa Clara, CA, USA), which contains 292 097 CGH probes and 119 091 SNP probes with median spacing of 7.2 kb. The CGH probes are gene- and exon-biased, focusing coverage on regions of clinical interest in the genome. Briefly, genomic DNA (500 ng) from the patient and from a sex-matched control were labeled with Cy3-dUTP and Cy5-dUTP fluorochromes using the Sure Tag DNA Labeling Kit (Agilent Technologies), and further hybridized on microarray according to Agilent protocols. The slide was scanned on a SureScan G4 900DA scanner (Agilent Technologies). TIFF image was analyzed using Agilent CytoGenomics v.2.7 based.
Haplotype Analysis
Five polymorphic microsatellite markers on 16p13.13-p12.1 with high heterozygosity (D16S3075, D16S3103, D16S3046, D16S420 and D16S3068) flanking around 13 Mb (chr16:12,115,341-25,549,627) of the ABCC6 gene were used to investigate the IBD mechanism in the family V-0779. For the STR genotyping, PCR products were analyzed using the automated ABI 3130xl Genetic Analyzer (Applied Biosystems) and further analyzed with the GeneMapper v3.5 software (Applied Biosystems). Haplotype reconstruction was performed using the software Cyrillic ver. 2.1 (Cyrillic Software, Wallingford, UK).
Electronic supplementary material
Acknowledgements
We would like to thank all patients who participated in the study and also everyone at the Clinical Genetics and Ophthalmology Services of Fundación Jiménez Díaz. Also, we would like to acknowledge Jacobo Herrera, Maria Luisa Martinez Frías and Francisco Galán for helping in patient recruiting, Berta Almoguera, Xiao Chang and Diego Mazzoti from Center for Applied Genomics, Children’s Hospital of Philadelphia for their technical and bioinformatic assistance, and Thomas O’Boyle for writing assistance. This study was funded by several grants from the Instituto de Salud Carlos III (ISCIII) from the Spanish Ministry of Health, including CIBERER (06/07/0036), FIS - FEDER (European Regional Development Fund) (PI016/00425), IIS-FJD Biobank PT13/0010/0012. In addition, sponsored chair HU-FJD-UAM “Cátedra de Patrocinio Medicina Genómica”, the Spanish National Organization of the Blind (ONCE) and the Spanish Fighting Blindness Foundation (FUNDALUCE) also supported our work. ISN and RSA are sponsored by Sara Borrell Postdoctoral Program (CD13-00085 and CD12-00676) from ISCIII/FEDER. MC is supported by the Miguel Servet Program (CP12/03256) from ISCIII/FEDER. RPC is supported by Fundación Conchita Rábago and LRJS is supported by CAPES Foundation, Ministry of Education of Brazil.
Author Contributions
All authors revised and approved the final manuscript version. I.S.N. participated in study design, designed the customized gene panel, carried out NGS analysis and drafted the manuscript. L.R.J.S. carried out aCGH experiments, participated in data interpretation and drafted the manuscript. F.B.K. participated in the clinical evaluation, phenotypic assessment and classification of patients, case selection, previous molecular screening and manuscript revision. O.Z. and N.S.B. participated in validation of NGS data and segregation analysis by Sanger sequencing. C.V. performed validation of CNV findings by MLPA and ddPCR. M.I.L.M. and B.G.S. carried out ophthalmologic evaluations. S.T.S. participated in the clinical evaluation and classification of patients. P.M. carried out bioinformatic analysis and drafted the manuscript. R.R.A. participated in previous molecular screening of families and case selection. I.L. participated in the clinical evaluation and previous genetic screening, classification of patients. R.S.A. and R.P.C. participated in bioinformatic analysis, variant classification and manuscript revision. D.V. participated in previous molecular screening and variant interpretation. A.A.F. participated in previous molecular screening of families and case selection. H.H., L.T. and Y.L. participated in the CNV analyses using gene panel data and drafted the manuscript. M.C. participated in study design, NGS design, previous molecular screening of families, case selection, variant interpretation and drafted the manuscript. C.A.obtained funding, participated in study design, clinical evaluation and classification of patients, previous genetic screening, case selection and drafted the manuscript. All authors reviewed the manuscript. C.A. and M.C. are guarantors of this work and, as such, had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Competing Interests
The authors declare no competing interests.
Footnotes
Iker Sanchez-Navarro and Luciana R. J. da Silva contributed equally to this work.
Marta Corton and Carmen Ayuso jointly supervised this work.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-23520-1.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Marta Corton, Email: mcorton@fjd.es.
Carmen Ayuso, Email: cayuso@fjd.es.
References
- 1.Mysore, N. et al. A Review of Secondary Photoreceptor Degenerations in Systemic Disease. Cold Spring Harb Perspect Med5 (11) (2014). [DOI] [PMC free article] [PubMed]
- 2.Werdich XQ, Place EM, Pierce EA. Systemic diseases associated with retinal dystrophies. Semin Ophthalmol. 2014;29(5-6):319. doi: 10.3109/08820538.2014.959202. [DOI] [PubMed] [Google Scholar]
- 3.Gazzo AM, et al. DIDA: A curated and annotated digenic diseases database. Nucleic Acids Res. 2015;44(D1):D900. doi: 10.1093/nar/gkv1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.May-Simera, H., Nagel-Wolfrum, K. & Wolfrum, U. Cilia - The sensory antennae in the eye. Prog Retin Eye Res (2017). [DOI] [PubMed]
- 5.Marshall JD, Maffei P, Collin GB, Naggert JK. Alstrom syndrome: genetics and clinical overview. Curr Genomics. 2011;12(3):225. doi: 10.2174/138920211795677912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet. 2013;21(1):8. doi: 10.1038/ejhg.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bachmann-Gagescu R, et al. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet. 2015;52(8):514. doi: 10.1136/jmedgenet-2015-103087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ronquillo CC, Bernstein PS, Baehr W. Senior-Loken syndrome: a syndromic form of retinal dystrophy associated with nephronophthisis. Vision Res. 2012;75:88. doi: 10.1016/j.visres.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mathur P, Yang J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim Biophys Acta. 2014;1852(3):406. doi: 10.1016/j.bbadis.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandler KE, et al. Diagnostic criteria, clinical characteristics, and natural history of Cohen syndrome. J Med Genet. 2003;40(4):233. doi: 10.1136/jmg.40.4.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Folz SJ, Trobe JD. The peroxisome and the eye. Surv Ophthalmol. 1991;35(5):353. doi: 10.1016/0039-6257(91)90185-I. [DOI] [PubMed] [Google Scholar]
- 12.Nathaniel H Robin, MD., Rocio T Moran, MD. & Leena Ala-Kokko, MD, PhD. ed., Stickler Syndrome Synonym: Arthro-Ophthalmopathy. (1993–2016).
- 13.Sadagopan KA. Practical approach to syndromic pediatric retinal dystrophies. Curr Opin Ophthalmol. 2017;28(5):416. doi: 10.1097/ICU.0000000000000404. [DOI] [PubMed] [Google Scholar]
- 14.Pereiro I, et al. Arrayed primer extension technology simplifies mutation detection in Bardet-Biedl and Alstrom syndrome. Eur J Hum Genet. 2011;19(4):485. doi: 10.1038/ejhg.2010.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cremers FP, et al. Development of a genotyping microarray for Usher syndrome. J Med Genet. 2007;44(2):153. doi: 10.1136/jmg.2006.044784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee K, Garg S. Navigating the current landscape of clinical genetic testing for inherited retinal dystrophies. Genet Med. 2015;17(4):245. doi: 10.1038/gim.2015.15. [DOI] [PubMed] [Google Scholar]
- 17.Broadgate S, Yu J, Downes SM, Halford S. Unravelling the genetics of inherited retinal dystrophies: Past, present and future. Prog Retin Eye Res. 2017;59:53. doi: 10.1016/j.preteyeres.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 18.Janssen S, et al. Mutation analysis in Bardet-Biedl syndrome by DNA pooling and massively parallel resequencing in 105 individuals. Hum Genet. 2011;129(1):79. doi: 10.1007/s00439-010-0902-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kroes HY, et al. Joubert syndrome: genotyping a Northern European patient cohort. Eur J Hum Genet. 2015;24(2):214. doi: 10.1038/ejhg.2015.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watson CM, et al. Enhanced diagnostic yield in Meckel-Gruber and Joubert syndrome through exome sequencing supplemented with split-read mapping. BMC Med Genet. 2016;17:1. doi: 10.1186/s12881-015-0265-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vilboux, T. et al. Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center. Genet Med (2017). [DOI] [PMC free article] [PubMed]
- 22.Sawyer SL, et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet. 2016;89(3):275. doi: 10.1111/cge.12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eisenberger T, et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PLoS One. 2013;8(11):e78496. doi: 10.1371/journal.pone.0078496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindstrand A, et al. Copy-Number Variation Contributes to the Mutational Load of Bardet-Biedl Syndrome. Am J Hum Genet. 2016;99(2):318. doi: 10.1016/j.ajhg.2015.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perez-Carro R, et al. Panel-based NGS Reveals Novel Pathogenic Mutations in Autosomal Recessive Retinitis Pigmentosa. Sci Rep. 2016;6:19531. doi: 10.1038/srep19531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Cauwenbergh C, et al. arrEYE: a customized platform for high-resolution copy number analysis of coding and noncoding regions of known and candidate retinal dystrophy genes and retinal noncoding RNAs. Genet Med. 2016;19(4):457. doi: 10.1038/gim.2016.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Schil, K. et al. Mapping the genomic landscape of inherited retinal disease genes prioritizes genes prone to coding and noncoding copy-number variations. Genet Med (2017). [DOI] [PMC free article] [PubMed]
- 28.Huet D, Blisnick T, Perrot S, Bastin P. The GTPase IFT27 is involved in both anterograde and retrograde intraflagellar transport. Elife. 2014;3:e02419. doi: 10.7554/eLife.02419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aldahmesh MA, et al. IFT27, encoding a small GTPase component of IFT particles, is mutated in a consanguineous family with Bardet-Biedl syndrome. Hum Mol Genet. 2014;23(12):3307. doi: 10.1093/hmg/ddu044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fauser S, Munz M, Besch D. Further support for digenic inheritance in Bardet-Biedl syndrome. J Med Genet. 2003;40(8):e104. doi: 10.1136/jmg.40.8.e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang F, et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet. 2014;133(3):331. doi: 10.1007/s00439-013-1381-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stone DL, et al. Mutation of a gene encoding a putative chaperonin causes McKusick-Kaufman syndrome. Nat Genet. 2000;25(1):79. doi: 10.1038/75637. [DOI] [PubMed] [Google Scholar]
- 33.Mykytyn K, et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat Genet. 2002;31(4):435. doi: 10.1038/ng935. [DOI] [PubMed] [Google Scholar]
- 34.Perrault I, et al. Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum Mutat. 2007;28(4):416. doi: 10.1002/humu.9485. [DOI] [PubMed] [Google Scholar]
- 35.Sayer JA, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38(6):674. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 36.Reuber BE, et al. Mutations in PEX1 are the most common cause of peroxisome biogenesis disorders. Nat Genet. 1997;17(4):445. doi: 10.1038/ng1297-445. [DOI] [PubMed] [Google Scholar]
- 37.Ebberink MS, et al. Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder. Hum Mutat. 2011;32(1):59. doi: 10.1002/humu.21388. [DOI] [PubMed] [Google Scholar]
- 38.Miksch S, et al. Molecular genetics of pseudoxanthoma elasticum: type and frequency of mutations in ABCC6. Hum Mutat. 2005;26(3):235. doi: 10.1002/humu.20206. [DOI] [PubMed] [Google Scholar]
- 39.Plomp AS, Hu X, de Jong PT, Bergen AA. Does autosomal dominant pseudoxanthoma elasticum exist? Am J Med Genet A. 2004;126A(4):403. doi: 10.1002/ajmg.a.20632. [DOI] [PubMed] [Google Scholar]
- 40.Ringpfeil F, et al. Pseudoxanthoma elasticum is a recessive disease characterized by compound heterozygosity. J Invest Dermatol. 2006;126(4):782. doi: 10.1038/sj.jid.5700115. [DOI] [PubMed] [Google Scholar]
- 41.Ragge NK, et al. Heterozygous mutations of OTX2 cause severe ocular malformations. Am J Hum Genet. 2005;76(6):1008. doi: 10.1086/430721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee JE, et al. CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat Genet. 2012;44(2):193. doi: 10.1038/ng.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weston MD, et al. Genomic structure and identification of novel mutations in usherin, the gene responsible for Usher syndrome type IIa. Am J Hum Genet. 2000;66(4):1199. doi: 10.1086/302855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aller E, et al. Identification of 14 novel mutations in the long isoform of USH2A in Spanish patients with Usher syndrome type II. J Med Genet. 2006;43(11):e55. doi: 10.1136/jmg.2006.041764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koolen, D. A. et al. The Koolen-de Vries syndrome: a phenotypic comparison of patients with a 17q21.31 microdeletion versus a KANSL1 sequence variant. Eur J Hum Genet (2015). [DOI] [PMC free article] [PubMed]
- 46.Redin C, et al. Targeted high-throughput sequencing for diagnosis of genetically heterogeneous diseases: efficient mutation detection in Bardet-Biedl and Alstrom syndromes. J Med Genet. 2012;49(8):502. doi: 10.1136/jmedgenet-2012-100875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aparisi MJ, et al. Targeted next generation sequencing for molecular diagnosis of Usher syndrome. Orphanet J Rare Dis. 2014;9:168. doi: 10.1186/s13023-014-0168-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernandez-San Jose P, et al. Targeted Next-Generation Sequencing Improves the Diagnosis of Autosomal Dominant Retinitis Pigmentosa in Spanish Patients. Invest Ophthalmol Vis Sci. 2015;56(4):2173. doi: 10.1167/iovs.14-16178. [DOI] [PubMed] [Google Scholar]
- 49.Henderson RH, et al. A rare de novo nonsense mutation in OTX2 causes early onset retinal dystrophy and pituitary dysfunction. Mol Vis. 2009;15:2442. [PMC free article] [PubMed] [Google Scholar]
- 50.de Ligt J, Veltman JA, Vissers LE. Point mutations as a source of de novo genetic disease. Curr Opin Genet Dev. 2013;23(3):257. doi: 10.1016/j.gde.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 51.Khateb S, et al. Identification of genomic deletions causing inherited retinal degenerations by coverage analysis of whole exome sequencing data. J Med Genet. 2016;53(9):600. doi: 10.1136/jmedgenet-2016-103825. [DOI] [PubMed] [Google Scholar]
- 52.Martin-Merida I, et al. Analysis of the PRPF31 Gene in Spanish Autosomal Dominant Retinitis Pigmentosa Patients: A Novel Genomic Rearrangement. Invest Ophthalmol Vis Sci. 2017;58(2):1045. doi: 10.1167/iovs.16-20515. [DOI] [PubMed] [Google Scholar]
- 53.Roosing S, et al. Mutations in CEP120 cause Joubert syndrome as well as complex ciliopathy phenotypes. J Med Genet. 2016;53(9):608. doi: 10.1136/jmedgenet-2016-103832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Schil K, et al. Early-onset autosomal recessive cerebellar ataxia associated with retinal dystrophy: new human hotfoot phenotype caused by homozygous GRID2 deletion. Genet Med. 2014;17(4):291. doi: 10.1038/gim.2014.95. [DOI] [PubMed] [Google Scholar]
- 55.Tatour, Y. et al. Mutations in SCAPER cause autosomal recessive retinitis pigmentosa with intellectual disability. J Med Genet (2017). [DOI] [PubMed]
- 56.Posey JE, et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N Engl J Med. 2017;376(1):21. doi: 10.1056/NEJMoa1516767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pineiro-Gallego T, et al. Molecular approach in the study of Alstrom syndrome: analysis of ten Spanish families. Mol Vis. 2012;18:1794. [PMC free article] [PubMed] [Google Scholar]
- 58.Alvarez-Satta M, et al. Overview of Bardet-Biedl syndrome in Spain: identification of novel mutations in BBS1, BBS10 and BBS12 genes. Clin Genet. 2014;86(6):601. doi: 10.1111/cge.12334. [DOI] [PubMed] [Google Scholar]
- 59.Pereiro I, et al. New mutations in BBS genes in small consanguineous families with Bardet-Biedl syndrome: detection of candidate regions by homozygosity mapping. Mol Vis. 2010;16:137. [PMC free article] [PubMed] [Google Scholar]
- 60.Johansson LF, et al. CoNVaDING: Single Exon Variation Detection in Targeted NGS Data. Hum Mutat. 2016;37(5):457. doi: 10.1002/humu.22969. [DOI] [PubMed] [Google Scholar]
- 61.Marshall JD, et al. Alstrom Syndrome: Mutation Spectrum of ALMS1. Hum Mutat. 2015;36(7):660. doi: 10.1002/humu.22796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bravo-Gil N, et al. Improving the management of Inherited Retinal Dystrophies by targeted sequencing of a population-specific gene panel. Sci Rep. 2016;6:23910. doi: 10.1038/srep23910. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.