

Abstract
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) resistance mediated by T790M-independent mechanisms remains a major challenge in the treatment of non-small cell lung cancer (NSCLC). We identified a targetable mechanism of EGFR inhibitor resistance whereby stress hormones activate beta2-adrenergic receptors (β2-AR) on NSCLC cells, which cooperatively signal with mutant EGFR, resulting in the inactivation of the tumor suppressor, liver kinase B1 (LKB1), and subsequently induce IL-6 expression. We show that stress and β2-AR activation promote tumor growth and EGFR inhibitor resistance, which can be abrogated with beta blockers or IL-6 inhibition. IL-6 was associated with worse outcome in EGFR TKI-treated NSCLC patients, and beta blocker use was associated with lower IL-6 concentrations and improved benefit from EGFR inhibitors. These findings provide evidence that chronic stress hormones promote EGFR TKI resistance via β2-AR signaling by an LKB1/CREB/IL-6-dependent mechanism and suggest that combinations of beta blockers with EGFR TKIs merit further investigation as a strategy to abrogate resistance.
INTRODUCTION
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) are effective therapies for NSCLC patients with EGFR activating mutations. However, resistant disease inevitably emerges (1). The most common mechanism of resistance to EGFR TKIs is the acquisition of secondary EGFR T790M mutations (2, 3). Second and third-generation EGFR inhibitors have shown activity against T790M-driven resistance (4, 5). However, in nearly 50% of cases, resistance is T790M-independent. Interleukin-6 (IL-6) has been implicated as a mediator of T790M-independent EGFR TKI resistance (6). The identification of targetable mechanisms of T790M-independent resistance is critical for improving outcomes in these patients.
Chronic stress results in increased production of stress hormones such as norepinephrine (NE) and epinephrine (E) from the adrenal medulla and sympathetic neurons. The effects of stress hormones are mediated through binding to β1,2- or β 1,2,3-adrenergic receptors (AR) on target cells. The AR pathway contributes to tumor development and progression in multiple malignancies including NSCLC (7–9). In lung cancer patients, psychological distress is a known predictor of mortality(10), and in preclinical models, beta adrenergic receptor (β-AR) signaling is associated with lung adenocarcinoma development and initiation of proliferative and invasive cellular programs (7, 11, 12). AR signaling has been linked to increased expression of cytokines including IL-6(13). We previously reported that stress hormones including NE and E activate ARs present on ovarian cancer cells and increase tumor-derived IL-6(14). Given the reports implicating IL-6 as a mediator of T790M-independent EGFR TKI resistance, we investigated whether stress hormones promote IL-6 expression in NSCLC cells and whether activation of ARs facilitates resistance to EGFR TKIs.
Here, we report that stress hormone activation of β2-AR promotes EGFR TKI resistance in cell lines and in mouse models of EGFR mutant NSCLC and this can be blocked by anti-IL-6 antibodies or beta blockers. Using reverse phase protein array (RPPA) to identify signaling pathways activated by β2-AR in NSCLC cells, we found that β2-AR signaling inactivates the tumor suppressor, LKB1, the loss of which promotes therapeutic resistance and metastasis in NSCLC (15). Analysis of clinical datasets revealed that high circulating concentrations of IL-6 were associated with a worse PFS and OS in patients treated with EGFR inhibitors, and circulating IL-6 concentrations were lower in patients incidentally receiving beta blockers. Furthermore, our analysis of incidental beta blocker use in the LUX-Lung3 study comparing afatinib vs chemotherapy in EGFR-mutant NSCLC suggests that patients receiving beta blockers may receive greater benefit from EGFR-targeting agents. Collectively, these data suggest the need for clinical testing of EGFR TKIs in combination with beta blockers.
RESULTS
Acquired resistance to EGFR TKIs is associated with increased IL-6
IL-6 is implicated as a driver of EGFR TKI resistance (6). We cultured HCC827 and HCC4006 (both EGFR mutant) cells with increasing concentrations of erlotinib, an EGFR inhibitor, until resistant variants emerged. EGFR TKI resistant variants HCC827 ER-1, ER-3, and ER-6 and HCC4006 ER-3, ER-5, and ER-6 were T790M negative, as determined by quantitative PCR (fig S1). T790M-negative erlotinib-resistant cells were also resistant to gefitinib, osimertinib, afatinib, and CO-1686 (fig. S2A–D) and expressed significantly higher IL-6 compared to parental cells (HCC827 ER-1,3,6 vs HCC827, p = 0.005; HCC4006 ER-3,5,6 vs HCC4006, p = 0.0005; Fig. 1A). IL-6 expression was minimal in cells where EGFR TKI resistance was associated with MET amplification (HCC827 ER-2); or T790M (PC9 ER-8, ER-9, and ER-11, and H1975; Fig. 1A).
Fig. 1. IL-6 is associated with resistance to EGFR TKIs and is induced by stress hormones.

(A) IL-6 secretion in NSCLC cells with acquired resistance to EGFR TKIs. Data are graphed as mean ± s.d. (B) Median OS in NSCLC patients with high or low circulating concentrations of IL-6 treated with erlotinib. (C) mRNA expression of ADRB1,2,3 in 159 patient samples (left) and in 116 NSCLC cell lines (right). (D) NSCLC cells were stimulated with NE (24 hours). IL-6 secretion was determined by ELISA. *p ≤ 0.001. Bars are mean ± s.e.m. (E) IL-6 mRNA expression after NE stimulation (10 μM for 3 hours). *p ≤ 0.001. Data are graphed as means ± s.d. (F) NE-induced IL-6 secretion after treatment with the β-AR inhibitor propranolol (PPL; 1 μM) or the α-AR inhibitor phentolamine hydrochloride (1 μM). *p≤0.01; one-way ANOVA. Data are graphed as means ± s.d. (G) IL-6 production after treatment with a β1-AR agonist (dobutamine; 50 μM) or a β2-AR agonist (salbutamol; 50 μM) for 24 hours. *p≤0.02; one-way ANOVA. Data are graphed as mean ± s.d. (H) Effect of the adenylyl cyclase activator, forskolin (10 μM), on IL-6 secretion. *p≤0.0002. p-value calculated by two-tailed Student’s t-test. (I) Circulating concentrations of IL-6 in NSCLC patients with or without incidental pan beta blocker use (BB; *p=0.02). Data are graphed as means ± s.e.m.
Next, we analyzed pre-treatment plasma concentrations of IL-6 by ELISA in 209 patients in the erlotinib arm of the phase III ZEST (ZACTIMA Efficacy Study versus Tarceva; NCT00364351) (16) clinical study. All patients had platinum-refractory NSCLC, and were not genotype selected. In this population, high IL-6 (greater than median) was associated with a significantly worse overall survival (OS) compared to patients with lower than median IL-6 concentrations (p<0.0001; Fig. 1B; table S1). The median OS for erlotinib-treated patients with high IL-6 was 4.8 months, whereas the median OS was 11.5 months in patients with low IL-6. High IL-6 was also associated with a significantly worse progression-free survival (p=0.0092; table S1; fig. S2E). Smokers had higher concentrations of IL-6 than nonsmokers (table S2). The findings are consistent with earlier preclinical studies (6) and suggest that higher IL-6 may be associated with EGFR TKI resistance.
Stress hormones induce IL-6 expression in NSCLC cells through activation of β2-AR
NE and E are increased under stress conditions, and can be substantially higher in tissues compared to circulating concentrations (17, 18). Effects of stress hormones are mediated through binding to α-AR1,2 or β-AR1,2,3. We examined ADRB1, ADRB2, and ADRB3 gene expression in 159 NSCLC clinical samples and 116 lung cancer cell lines. Tumor samples and cell lines tested positive for ADRB1, ADRB2, and ADRB3 expression (Fig. 1C; fig. S3A). NSCLC cell lines harboring EGFR activating mutations expressed constitutively phosphorylated EGFR (fig. S3B). We treated NSCLC cells harboring EGFR activating mutations (HCC827, HCC4006, H3255) with 1 or 10 μM of NE for 24 h and measured IL-6 secretion by ELISA. NE induced a striking rise in IL-6 (Fig. 1D). IL-6 mRNA expression was significantly increased after NE (10 μM) stimulation for 3 hours, as determined by quantitative real-time RT-PCR (p< 0.001; Fig. 1E). NE concentrations of 0.1 μM and 0.01 μM also increased IL-6 (fig. S4A&B). Epinephrine (E) and the β-AR agonist, isoproterenol (ISO), similarly induced IL-6 in EGFR-mutant NSCLC cells, however, the effect on non-malignant human bronchial epithelial cells (HBEC) was minimal (fig. S4C&D).
Propranolol (PPL; β-AR inhibitor) but not phentolamine hydrochloride (α-AR inhibitor) blocked NE-induced IL-6 (Fig. 1F; fig. S4E). Treatment with a β2-AR agonist (salbutamol) but not a β1-AR agonist (dobutamine) increased IL-6 (Fig. 1G). β-ARs activate the cAMP signaling pathway though stimulation of adenylyl cyclase (19). We treated NSCLC cells with an adenylyl cyclase activator, forskolin, and observed an increase in IL-6 similar to that induced by NE (Fig. 1H). β-ARs can also activate Epac (20), however, treatment with 8-CPT-2Me-cAMP, a cAMP analog that specifically targets Epac, did not affect IL-6 secretion (fig. S4F).
To determine whether beta blocker use may be associated with a reduction of IL-6 in patients, we measured circulating concentrations of IL-6 by ELISA in patient samples from the BATTLE trial (21). IL-6 concentrations were significantly lower in patients using pan-beta blockers compared to patients not receiving beta blockers, supporting the contribution of β-ARs in regulating IL-6 concentrations (p = 0.02; Fig. 1I).
β-AR signals cooperatively with mutant EGFR
Although NE induced a robust increase in IL-6 in cells with EGFR activating mutations (HCC827, HCC4006, H3255, PC9, H1975, H1650), the effect was minimal in EGFR wild-type cells (A549, H441, Calu6, H460, H661, H23, HCC15, H1993) (Fig. 2A). Similarly, salbutamol (a β2 -AR agonist) and forskolin induced IL-6 expression in NSCLC cells with mutant but not wild-type EGFR (fig. S5A&B). Dobutamine (a β1 -AR agonist) treatment did not affect IL-6 secretion. β2-AR expression was similar across EGFR mutant and wild-type cell lines (fig. S3A). To determine whether EGFR interacts with β2 -AR in cells with EGFR activating mutations, HCC827 cells were treated with or without NE, lysates were immunoprecipitated with anti-EGFR antibodies, and blots were probed with anti-β2-AR antibodies. We observed interaction between endogenous β2 -AR and EGFR in HCC827 cells (EGFR mutant) (Fig. 2B). A similar experiment was performed using HCC827 cells transfected with a FLAG-tagged β2 -AR expression vector. Lysates were immunoprecipitated with anti-EGFR antibodies, and blots were probed with anti-FLAG antibodies. We observed interaction between β2 -AR and EGFR in HCC827 cells (EGFR mutant) (fig. S5C). To visualize in situ interaction between EGFR and β2 -AR, we performed a Duolink proximity ligation assay. HCC827 (EGFR mutant) and A549 (EGFR wild-type) cells were transfected with a control vector or a FLAG-tagged β2 -AR expression vector and treated with or without NE for 15 minutes. Confocal microscopy revealed NE-induced interactions between β2 -AR and mutant EGFR but not wild-type EGFR (Fig. 2C&D, red spots).
Fig. 2. β-ARs signal cooperatively with mutant EGFR and inactivate LKB1.

(A) NSCLC cells harboring wild-type EGFR or EGFR activating mutations were stimulated with NE (10 μM), and IL-6 secretion was measured by ELISA. *p≤0.001. Data are graphed as means ± s.d. (B) Co-immunoprecipitation of endogenous β2-AR and EGFR in HCC827 cells (EGFR mutant) after NE stimulation. (C) HCC827 (EGFR mutant) and A549 (EGFR wild-type) cells were transfected with control vector or a FLAG-tagged β2-AR expression vector. After treatment with NE, cells were immunostained with antibodies directed against EGFR or FLAG-tag and assessed by Duolink proximity ligation assay. Data are graphed as means ± s.d. (D) Representative images from Duolink assay. Red foci indicate interactions between endogenous EGFR and FLAG-tagged β2-AR. Scale bar = 10 μm. (E) Heatmap depicting mean protein expression after treatment with NE (10 μM; 15 minutes). (F) Alterations in phosphorylation of LKB1 at the inhibitory site S428, AMPK activation, and mTOR activity (p70S6K) after NE stimulation as determined by RPPA. Data are graphed as means ± s.d. (G) RPPA results were confirmed by Western blotting. (H) Western blot demonstrating the effect of NE (10 μM; 15 minutes) on phosphorylation of LKB1 at the inhibitory site S428 and phosphorylation of mTOR and p70S6K in H1975 cells.
Stress hormone signaling inactivates the tumor suppressor LKB1 in a PKC-dependent manner
We stimulated NSCLC cells with NE and conducted a proteomic analysis by RPPA. NE induced LKB1S428 phosphorylation, a modification which inhibits LKB1 function (22) (Fig. 2E&F). LKB1 plays a pivotal role in energy sensing, suppressing the mTOR pathway via activation of AMPK and TSC2 (23). NE concurrently decreased AMPK activation and increased mTOR activity, as indicated by p70S6K phosphorylation (Fig. 2E–G). Total amounts of LKB1, AMPK, and p70S6K were unchanged (fig. S6A). RPPA data were confirmed by Western blotting (Fig. 2G). The effect of NE on LKB1 inactivation was also observed at lower concentrations of NE (fig. S6B). NE had a minimal effect on p-LKB1S428 in two EGFR wild-type cell lines (A549 and H661; fig. S6C). Forskolin induced a similar rise in p-LKB1S428 (fig. S6D). NE-induced inactivation of LKB1 was abrogated by propranolol (Fig. 2G). Similarly, in H1975 cells (EGFR-mutant), NE stimulation induced a rise in p-LKB1S428 and increased p-mTOR and p-p70S6K (Fig. 2H).
The LKB1 S428 residue can be phosphorylated by PKC(24) and p90RSK (22), and activation of β-AR triggers activation of PKC(25) and the MAPK/p-90RSK pathways (26) (fig. S6E). Ro31-8220, an inhibitor of PKC and p90RSK, reduced NE-induced phosphorylation of PKC and LKB1S428 (Fig. 3A; fig. S6F) and abrogated NE-induced IL-6 (Fig. 3B; fig. S6G). To distinguish between the potential contributions of the PKC and MAPK/p-90RSK pathways, we treated HCC827 cells with control medium, a negative control peptide, or a PKC inhibitor peptide (27) with or without NE. PKC inhibition blocked NE-induced p-LKB1S428 (Fig. 3C), whereas p90RSK knockdown failed to inhibit NE-induced increases in p-LKB1S428 or IL-6 (fig. S6H&I). β-AR signaling can also activate PKA (28, 29). The PKA inhibitor H89 did not block NE-induced p-LKB1S428 (fig. S6J). To investigate the contribution of LKB1 inactivation in β-AR-induced IL-6, we overexpressed LKB1 in HCC827 cells (fig. S6K). NE increased IL-6 in GFP control cells, but not in cells overexpressing LKB1 (Fig. 3D).
Fig. 3. β-AR signaling induces IL-6 in NSCLC cells via activation of PKC and CREB.

(A) HCC827 and HCC4006 cells were treated with NE with or without Ro31-8220 (5 μM). LKB1S428 and p-CREBS133 were quantified by Western blotting and (B) IL-6 was quantified by ELISA (*p≤0.01; one-way ANOVA). Data are graphed as means ± s.d. Representative data from two independent experiments performed in triplicate. (C) HCC827 cells were treated with a PKC inhibitor peptide and then stimulated with NE. LKB1S428 and p-CREBS133 were quantified by Western blotting. (D) LKB1 was stably overexpressed in HCC827 cells. After treatment with NE, IL-6 secretion was evaluated by ELISA assay. *p=0.009; by two-tailed Student’s t-test. Data are graphed as means ± s.d. (E) NSCLC cells harboring EGFR activating mutations (HCC827, HCC4006, H1650, PC9) or wild-type EGFR (H460, A549) were stimulated with NE, and p-CREBS133 was quantified by Western blotting. (F) HCC8227 and HCC4006 cells were treated with NE alone or in the presence of the CREBBP inhibitor, SGC-CBP30 (1 μM), and IL-6 production was evaluated by ELISA. *p≤0.01; one-way ANOVA. Data are graphed as means ± s.e.m.
Stress hormones induce IL-6 expression in a CREB-dependent manner
Transcription of the IL6 gene is complex, involving multiple transcription factors including NF-κB and CREB. In HCC827 cells, NE treatment increased p-CREBS133 (fig. S7A&B), and inhibition of β-AR but not α-ARs abrogated NE-induced p-CREBS133 (fig. S7C). The NE-induced rise in p-CREBS133 in EGFR mutant cells was not observed in EGFR wild-type cells (Fig. 3E). Activated CREB binds CREB-binding proteins (CREBBP), allowing transcription of target genes. SGC-CBP30 (CREBBP inhibitor) abrogated β2 -AR-induced IL-6 (Fig. 3F). EGFR activation was not necessary for β-AR-mediated IL-6 expression, because erlotinib (1 μM) failed to inhibit NE-induced p-CREB and pLKB1S428 (fig. S7D) and failed to block NE-induced IL-6 expression (fig. S7E). NE had minimal effects on NF-κB nuclear localization; NF-κB inhibition failed to block NE-induced IL-6 (fig. S8). Inhibition of PKC by RO31-8220 or a PKC inhibitor peptide diminished NE-induced rises in p-CREB (Fig. 3A&C). In contrast, p90RSK knockdown failed to inhibit NE-induced p-CREB (fig. S6H). NE increased p-CREBS133 in GFP control cells, but not in cells overexpressing LKB1 (fig. S6L), suggesting that CREB activation occurs downstream of LKB1 suppression.
Stress hormones promote EGFR TKI resistance and growth of NSCLC xenografts
We treated EGFR-mutant NSCLC cell lines with NE 24 hours before the addition of erlotinib. NE promoted erlotinib resistance in vitro (Fig. 4A). The addition of the β-AR inhibitor propranolol or IL-6 neutralizing antibodies blocked the effect on EGFR TKI resistance (Fig. 4B&C).
Fig. 4. Stress hormones promote EGFR TKI resistance in vitro and in vivo.

(A) EGFR mutant NSCLC cells were stimulated with NE (10 μM) for 24 hours and then treated with erlotinib. Cell viability was determined by MTS assay. *p<0.0001; by two-tailed Student’s t-test. Bars are means ± s.d. (B) HCC827 cells were treated with propranolol (PPL; *p≤0.03) or (C) IL-6 neutralizing antibodies (*p<0.001) before NE stimulation. After 24 hours, cells were treated with erlotinib for 5 days. Cell viability was evaluated by MTS assay. (D) Ten mice per cell line were injected subcutaneously (control n=5; stress n=5). Chronic stress was induced via restraint. For HCC4006, all mice developed tumors. For HCC827, 4 and 2 mice developed tumors in the stress and control groups, respectively. Data are graphed as means ± s.e.m. *p<0.04; two-tailed Student’s t-test. (E) HCC827 tumor-bearing mice were treated with isoproterenol (β-AR agonist) for three days. Tumors were collected, and IL-6 mRNA was measured by quantitative PCR (control group n=13; ISO group n=7, run as technical duplicates). *p=0.02 two-tailed Student’s t-test. Data are graphed as means ± s.e.m. (F) Erlotinib induced tumor regression in HCC827 xenografts. (G) After prolonged erlotinib treatment, resistant disease emerged in mice treated with erlotinib plus isoproterenol compared to mice receiving erlotinib alone (p=0.018). PPL (p=0.035) or IL-6 antibody, siltuximab (Silt, p=0.009), blocked the effect. *p=0.018; means ± s.e.m.
Next, we injected HCC827 and HCC4006 cells into the flanks of nude mice. Chronic stress was initiated using an established restraint model (30). Tumor volumes were more than doubled in stressed mice relative to control mice (Fig. 4D; p ≤ 0.04 for both). Immunohistochemistry revealed increased IL-6 expression in tumors from stressed mice compared to controls (p=0.002; fig. S9A&B). Tumor cell proliferation was not significantly different in stressed mice, but they had reduced microvessel density (fig. S9C). Treatment of HCC827 tumor-bearing mice with isoproterenol significantly increased IL-6 mRNA (p=0.02; Fig. 4E).
We established HCC827 xenografts in female nude mice. Once tumors reached 400 mm3, animals were randomized to receive erlotinib (100 mg/kg) alone or with daily isoproterenol. Erlotinib treatment resulted in near complete tumor regression (Fig. 4F). After prolonged erlotinib treatment, resistant disease emerged in mice treated with erlotinib plus isoproterenol compared to mice receiving erlotinib alone (p=0.018; Fig. 4G). The addition of propranolol (p=0.035), or the IL-6 antibody, siltuximab (p=0.009), inhibited the effect of isoproterenol on EGFR TKI resistance, indicating a role for IL-6 in β-AR-mediated resistance.
Beta blocker use is associated with improved benefit from afatinb in the LUX-Lung3 study
Finally, we evaluated the influence of incidental beta blocker use on treatment effect in the randomized phase III LUX-Lung3 study (31) comparing the EGFR TKI, afatinib, versus chemotherapy in EGFR mutation-positive NSCLC patients. In patients not receiving beta blockers, afatinib improved PFS time, with a median PFS of 11.1 and 6.9 months for afatinib and chemotherapy, respectively (Fig. 5, left; log-rank test p-value=0.001), and a hazard ratio (HR) of 0.60, corresponding to a 40% reduction in the likelihood of progression for afatinib. In patients receiving beta blockers, afatinib was associated with a greater relative PFS benefit, with a median PFS of 13.6 and 2.5 months for afatinib and chemotherapy, respectively (Fig. 5, right; HR=0.25, log-rank test p-value=0.0001), corresponding to a 75% reduction in the likelihood of progression. Although the analysis of these randomized trials was exploratory and limited by the modest number of patients receiving beta blockers, the findings are consistent with the preclinical data that beta blockade could delay the emergence of EGFR TKI resistance.
Fig. 5. Beta blocker use is associated with improved benefit from afatinib in LUX-Lung3.

Analysis of incidental beta blocker use in the LUX-Lung3 clinical study comparing the EGFR TKI afatinib versus chemotherapy in EGFR mutant-positive NSCLC. E = number of events; N = number of patients; pem = pemetrexed; cis = cisplatin.
DISCUSSION
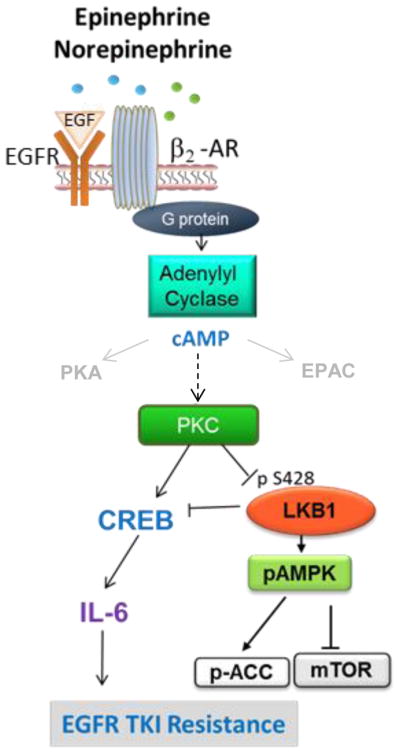
Acquired resistance to EGFR targeted therapies is a major challenge in the treatment of NSCLC patients. The development of second and third-generation EGFR inhibitors with activity against T790M-positive EGFR mutants is a clear advancement for targeting resistance. However, effective strategies are needed to delay and target resistant disease that emerges through T790M-independent mechanisms. This study revealed that stress hormones act on β2-AR on NSCLC tumor cells and promote EGFR inhibitor resistance through upregulation of IL-6 (fig. S10). By using a high-throughput proteomic analysis, we identified a mechanism of LKB1- and CREB-dependent induction of IL-6 that could be inhibited by beta blockers. Our preclinical findings were supported by clinical data indicating that high plasma concentrations of IL-6 are associated with a worse response to EGFR inhibitors, and beta blocker use is associated with lower circulating concentrations of IL-6. Finally, our analysis of the phase III LUX-Lung3 study suggests that beta blocker use may improve response to EGFR inhibitors in NSCLC patients with EGFR activating mutations.
The β-AR pathway contributes to tumor progression in a variety of malignancies, enhancing tumor cell proliferation and invasion (7, 32–36), inducing vascular and lymphatic remodeling (37), and enhancing lymph node metastasis (37). In lung cancer patients, psychological distress is a predictor of mortality (10). Retrospective clinical analyses have indicated that beta blocker use is associated with a reduced risk of cancer development (38) and decreased breast(39) and prostate cancer-specific mortality (40), but whether beta blockers improve clinical outcome remains controversial. Whereas first-generation beta blockers (propranolol, nadolol, timolol) are non-selective and thus block β1 and β2-ARs, second-generation cardioselective beta blockers (metoprolol, acebutolol, busoprolol) have activity against β1- but not β2-ARs. In the treatment of patients with cardiovascular conditions, cardioselective betablockers are more frequently used in effort to minimize adverse effects. Here, we show that β2-ARs are highly expressed on NSCLC tumor cells and that stress hormones activate signaling pathways that contribute to therapeutic resistance in NSCLC cell lines though β2-AR but not β1-AR. These findings indicate that pan beta blockers but not cardioselective beta blockers may improve the clinical outcome of this patient population. Moreover, there were differential effects between cells with or without EGFR activating mutations. It is feasible that the conflicting clinical data regarding the effect of beta blocker use on clinical outcome are related to the specificity of the beta blocker used and the mutational context of the tumors. It is worth noting that the tobacco-specific nitrosamine, NKK, is a high affinity agonist for β1 and β2 -ARs (12, 41). It is therefore feasible that the established association between smoking status and IL-6 expression occurs in part through activation of the adrenergic pathway as described here.
Findings from this study and others (6) indicate that IL-6 is a key driver of T790M-independent EGFR TKI resistance. The present report highlights the regulation of IL-6 by stress hormones, but smoking status and comorbidities including obesity and diabetes are also associated with chronic inflammation and enhanced expression of IL-6 (42–44) and thus may impact the emergence of therapeutic resistance. Moreover, the deleterious effects of chronic stress on the clinical outcome of EGFR-mutant NSCLC patients may extend beyond the induction of IL-6. Stress hormone-mediated activation of β2-ARs can further activate the β-arrestin pathway, triggering degradation of p53 and the accumulation of DNA damage (45). In addition, β-arrestins are thought to facilitate GPCR-mediated transactivation of EGFR in cancer cells(46).
Earlier studies in other settings have suggested that interactions between β-AR and mutant EGFR could occur. Many G-protein coupled receptors use EGFR as an intermediate signaling protein (47). β-AR transactivates EGFR, resulting in incomplete downstream signaling. However, when EGFR and β-AR are simultaneously activated, broader signal transduction pathways are initiated (48). In animal models of carcinogen-induced lung adenocarcinoma, activation of both β-AR and EGFR is observed, and data suggest receptor crosstalk (41). Our data indicate that the effects of stress hormones on lung cancer cells may be heightened in cells with EGFR activating mutations.
Here, we uncovered a mechanism of LKB1 inactivation in NSCLC cells, where intact LKB1 is inactivated by stress hormone signaling. Although the LKB1/STK11 tumor suppressor is frequently lost at the genomic level, or mutated, in NSCLC, the mechanism reported here may be particularly important in tumors with EGFR mutations because they nearly always have intact LKB1 (49). Consistent with our proposed model, in cardiac myocytes, β-AR stimulation inactivates the LKB1 target, AMPK (50), as part of metabolic changes induced by catecholamines. Our data provide a detailed mechanistic understanding of how this could occur in non-malignant cells and indicate that NSCLC cells may usurp this physiological process. LKB loss or inactivation has been linked to an enhanced CREB gene expression signature(51) as well as an immunosuppressive tumor microenvironment characterized by increased infiltration of myeloid-derived tumor suppressor cells and increased IL-6(52). Studies have found an association between reduced LKB1/AMPK pathway signaling and increased distant metastasis (15, 53). In an analysis of 722 NSCLC patients receiving definitive radiotherapy, beta blocker use was strongly associated with reduced distant metastasis(8). Studies are warranted to determine if stress hormones promote lung cancer metastasis through inactivation of LKB1.
Our analysis also demonstrated that β-AR activation stimulates the MAPK pathway in EGFR-mutant NSCLC cells. The effects of β-AR signaling on MAPK are notable because reactivation of the MAPK pathway has been established as a mechanism of EGFR TKI resistance (54, 55).
Our preclinical findings indicate that stress hormones act on β2-AR on NSCLC cells, inducing increases in IL-6 and, in turn, resistance to EGFR TKIs. Our in vitro observations were supported by an animal model of NSCLC, where β-AR activation promoted the emergence of erlotinib-resistant disease, which was blocked by IL-6 neutralizing antibodies or beta blockers. To provide clinical validation, we analyzed the effect of incidental beta blocker use on benefit from afatinib versus chemotherapy in NSCLC patients with EGFR mutations in the LUX-Lung3 study. In patients receiving beta blockers, afatinib appeared to produce a larger improvement in PFS compared to patients not receiving beta blockers. However, interpreting this effect is difficult because patients receiving beta blockers also had a worse clinical response to chemotherapy (Fig. 5). Although our analysis of the LUX-Lung3 study supports our hypothesis that beta blocker use may improve clinical response to EGFR inhibitors, definitive clinical proof of this hypothesis would require direct clinical testing of EGFR TKIs alone and in combination with beta blockers. Given the potential benefits highlighted here, as well as the favorable tolerability profile and low cost of these drugs, we feel that such clinical testing merits consideration.
In summary, this study reveals a mechanism of EGFR inhibitor resistance and advances the understanding of how chronic stress may influence disease progression. These findings have immediate clinical implications because many well-tolerated and inexpensive beta blockers are available. These data support the future clinical testing of beta blockers in combination with EGFR-targeting agents.
MATERIALS AND METHODS
Study design
The objective of this study was to (i) determine whether IL-6 expression was associated with T790M-negative resistance to EGFR TKIs and (ii) determine whether stress hormones could signal on NSCLC tumor cells and promote EGFR TKI resistance through upregulation of IL-6. To investigate the effects of β-AR signaling on NSCLC cells and the effect of β-AR activation on EGFR TKI resistance, we used quantitative real-time PCR, in vitro assays, Western blotting, RPPA, ELISA assays, immunohistochemistry, and xenograft models of EGFR mutant NSCLC. All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Texas MD Anderson Cancer Center in accordance with NIH guidelines. Animals were randomly assigned to control or treatment groups. No statistical method was used to predetermine the sample size. Investigators were not blinded for the preclinical experiments.
Cell lines and reagents
Cell lines (table S3) were maintained in 10% fetal bovine serum (FBS; Sigma) RPMI medium. Norepinephrine, epinephrine, isoproterenol, phentolamine hydrochloride, salbutamol, dobutamine, and forskolin were obtained from Sigma-Aldrich. 8-pCPT-2-Me-cAMP was obtained from ThermoFisher Scientific. QNZ (EVP4593), SGC-CBP30, and RO31-8220 were obtained from Selleck Chemicals. H89 was obtained from Tocris Chemicals. IL-6 neutralizing antibodies were obtained from R&D Systems. Erlotinib and siltuximab were obtained from the institutional pharmacy at The University of Texas MD Anderson Cancer Center. LKB1 expression vectors were obtained from Addgene. The PKC inhibitor peptide 19–36 and control peptides (EMD Millipore) were used at a concentration of 100 μg/ml in ice-cold permeabilization buffer for 5 min. Cells recovered in serum-free medium for 2 hours before NE treatment. The following antibodies were used for Western blotting: IKB-α (4814; Cell Signaling), p-CREBS133 (9198; Cell Signaling), pEGFR (3777; Cell Signaling), EGFR (1005; Santa Cruz Biotechnology, Inc.), p-LKB1S428 (3482 and 3051; Cell Signaling), p-p90RSK (9344; Cell Signaling), p-AMPK (2535; Cell Signaling), p-p70S6K (9205; Cell Signaling), p-ERK (9101; Cell Signaling), vinculin (v9131, 1:10,000 Sigma-Aldrich), β-actin (A5441; 1:10,000; Sigma-Aldrich). For immunohistochemical studies, the following antibodies were used: anti-mouse CD31 antibody (1:400, PharMingen), Ki67 (Dako), Alexa 594–conjugated secondary antibody (Molecular Probes).
Detection of IL-6
NSCLC cells (200,000 cells/well in 6-well plates) were treated with 1 ml of serum-free medium with or without NE, E, or ISO for 24 hours, and medium was collected. IL-6 ELISA (R&D Systems) was performed according to manufacturer’s instructions. For IL-6 staining, antibodies (1:50; Millipore) were used on frozen tumor sections. For inhibitor studies, cells were pretreated with inhibitors for 1 hour before NE stimulation. The doses of catecholamines used for our studies were selected to reflect physiologic conditions of these hormones at the level of the tumor. Whereas circulating plasma concentrations of norepinephrine range from 10 to 1000 pM in a normal individual and may reach as high as 100 nM in conditions of stress (56), catecholamine concentrations in some tissues are at least 100 times higher, and may reach concentrations as high as 10 μM (17, 18).
ADRB gene expression profiling
The MD Anderson cohort was obtained from the Profiling of Resistance patterns and Oncogenic Signaling Pathways in Evaluation of Cancers of the Thorax (PROSPECT) study. Surgically resected tumors, collected between 2006 and 2010, from 189 patients were included in PROSPECT, and of these, 152 were lung adenocarcinomas. Gene expression analysis and sequencing of select genes were conducted as reported elsewhere (57–59).
Immunoprecipitation
Cells were stably transfected with FLAG-B2ADR or control vector using lentivirus. Cells were serum-starved for 1 hour and then treated with NE (10 μM) for 10 minutes. Cells were washed with PBS and incubated in dithiobis succinimidyl propionate (DSP; Thermo Scientific; 1 mM in PBS) crosslinker solution and incubated on ice. After two hours, Tris-HCl stop solution was added at a final concentration of 10 mM and incubated for 15 min. Cells were lysed in protein lysis buffer. Co-immunoprecipitation was performed using Pierce crosslink immunoprecipitation kit according to manufacturer’s instructions (Cat#26147). After elution, DSP-crosslinking was quenched using 20 mM DTT loading buffer in 1% SDS, 60 mM Tris-HCl, 10% glycerol, boiling at 100°C for 10 min.
Duolink II fluorescence assay
Duolink II fluorescence assay was performed as previously described (60). Cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100, then blocked in 5% FBS in PBS and incubated with mouse anti-EGFR (1:2,000 dilution; Thermo Scientific) and rabbit anti-Flag (1:400 dilution; Cell Signaling) antibodies for 1 hour. The Duolink II PLA probe (Sigma) was used to detect the signals.
Reverse phase protein array
NSCLC cells were treated with 10 μM NE for 15 min, and protein lysates were collected. RPPA slides were printed from lysates, and RPPA studies were performed and analyzed as previously described (53, 61–63).
Cell viability assays
NSCLC cells (2,000 cells/well in 96-well plates) were treated with increasing concentrations of EGFR TKIs. After 5 days, MTS assays were performed. To evaluate the effect of NE on EGFR TKI resistance, cells were seeded in 24-well plates (40,000 cells/well) and treated with NE for 24 hours, and then erlotinib was added to the culture medium. After 5 days, cell viability was measured by MTS assay. Propranolol (1 μM) or IL-6 antibodies (1 μg/ml) were added one hour before the addition of NE.
Mice
Cancer cells (1 × 106) were injected subcutaneously into female nude mice. Chronic stress was induced using a restraint-stress procedure (64). To test the effect of ISO on IL-6, once tumors reached a volume of 400 mm3, animals received daily isoproterenol treatment (10 mg/kg, intraperitoneal (ip)) for three days. Tumors were processed for RNA collection and quantitative real-time PCR. For erlotinib resistance studies, 1 × 106 HCC827 cells were injected subcutaneously into nude mice. Once tumors reached 400 mm3, animals (n=5 mice/group) received vehicle, isoproterenol (10 mg/kg daily, ip), propranolol (10 mg/kg daily, ip), or siltuximab (20 mg/kg twice weekly, ip). Erlotinib was given at a dose of 100 mg/kg (oral gavage) on a 4 week on / 2 week off schedule.
Clinical analysis
Biospecimens were obtained after patients gave informed consent, under protocols approved by Institutional Review Boards at all participating institutions. Studies were conducted in accordance with the Declaration of Helsinki. The PROSPECT dataset has been detailed previously (65) and includes patients who underwent surgical resection of NSCLC with curative intent between 1996 and 2008 at MD Anderson Cancer Center. In the PROSPECT study, the majority of cases were lung adenocarcinomas. Clinical outcomes from BATTLE (NCT00409968, NCT00411671, NCT00411632, NCT00410059, NCT00410189), LUX-Lung3 study (NCT00949650) (31), and ZEST (NCT00364351)(16) have been reported. We analyzed baseline plasma samples collected during the BATTLE clinical trial(21) and the ZEST phase III clinical study and were blinded to clinical outcome. Each sample was analyzed in duplicate. For the analysis of circulating concentrations of IL-6 in the BATTLE trial, 185 patients received no beta blockers and 3 patients received pan beta blockers. Groups were compared using a two-tailed Student’s t-test.
Statistics
For in vitro studies, statistical analysis was performed using Student’s t-test (two-tailed) or one-way ANOVA. A p value ≤ 0.05 was considered statistically significant. For RPPA data, analysis of variance (ANOVA) was used on protein-by-protein basis. The resulting P values, computed from F-statistic, were modeled using the beta-uniform mixture (BUM) model and used to determine a false discovery rate (FDR) cutoff to identify significantly differentially expressed proteins. For in vivo studies, we fitted a linear model (with treatment and time effects) using generalized least squares (GLS) method to account for repeated measurements correlation. Dunnett’s test was used to adjust for multiple comparisons in comparing many treatments with a single control.
Supplementary Material
Fig. S1. Frequency of T790M EGFR secondary mutations in erlotinib resistant cells.
Fig. S2. Resistance of HCC827 ER and HCC4006 ER cells to EGFR TKIs.
Fig. S3. ADRB expression and EGFR status in NSCLC cell lines.
Fig. S4. Induction of IL-6 following β-AR activation.
Fig. S5. Differential effects of β2-AR signaling on EGFR mutant and wild-type cells.
Fig. S6. Stress hormone-induced effects on LKB1, PKC and CREB.
Fig. S7. Effect of EGFR inhibition on β-AR-induced p-LKB1, p-CREB, and IL-6.
Fig. S8. Effect of β-AR signaling on NF-κB activity.
Fig. S9. Effect of chronic stress on IL-6 expression and MVD in NSCLC xenografts.
Fig. S10. Stress hormones promote EGFR inhibitor resistance in NSCLC.
Table S1. PFS and OS of NSCLC patients with high or low IL-6 treated with erlotinib.
Table S2. Smoking status of patients with high or low circulating IL-6 in the ZEST trial.
Table S3. Lung cancer cell lines used in ADRB gene expression analysis.
Fig 6.
Acknowledgments
We thank Emily Roarty, Ph.D. for helpful scientific advice and editorial assistance. HA-Tagged β2-AR expression vector was a kind gift from Michael Tuvim (The University of Texas MD Anderson Cancer Center). Dr. Minna at The University of Texas Southwestern provided NSCLC cell lines.
Funding
This work was supported by LUNGevity Foundation; Lung SPORE grant 5 P50 CA070907; Lung Cancer Moon Shot Program, NIH CCSG(CA016672); 1R01 CA190628, Hallman Fund, Bruton Endowed Chair in Tumor Biology, Stading Fund for EGFR inhibitor resistance, the Fox Lung EGFR Inhibitor, the Rexanna Foundation for Fighting Lung Cancer. Additional support was provided through CA109298 and the American Cancer Society Research Professor award (Dr. Anil Sood).
Footnotes
Author Contributions
M.N. designed the research, performed experiments, interpreted data and wrote the manuscript. H.Y. performed in vitro and in vivo experiments. A.P. assisted with animal experiments. G.A P. performed animal experiments using restraint which was supervised by A.K.S. Y.F. performed RPPA analysis. L.L., L.D., P.T. and D.L. performed statistical and computational analysis, and J.W. and J.J.L. supervised and designed the analysis. S.L. performed duo-link assays, and M.C.H supervised the work and provided direction and insight. K.H. and V.H. conducted statistical analyses related to the ZEST clinical study. D.G assisted with data interpretation. H.T. analyzed and supervised the evaluation of circulating levels of IL-6 in patient samples from ZEST and BATTLE clinical studies. L.S. and J.Y. conducted the LUX-Lung3 clinical study. R.H., E.K., W.K.H., and I.W. conducted and designed the BATTLE study and assisted with data interpretation. J.V.H. supervised the work, designed the research, interpreted data, and wrote the manuscript.
Data and materials availability
Affymetrix microarray results were previously published and archived at the Gene Expression Omnibus repository. Cell line data are available at GEO accession GSE4824. PROSPECT gene expression data are available at GEO accession GSE42127.
Competing Interests
K.H. and V.H. are employees of Astrazeneca. L.S. is a non-compensated consultant for Boehringer Ingelheim, Clovis, Merrimack, Novartis, Taiho, a compensated consulting for AZ and Ariad and receives institutional support from Boehringer Ingelheim for the LL3 clinical trial. J.Y. received honorarium for speech or participated in compensated advisory board of Boehringer Ingelheim, Eli Lilly, Bayer, Roche/Genentech/Chugai, Astellas, MSD, Merck Serono, Pfizer, Novartis, Clovis Oncology, Celgene, innopharma, Merrimack, BMS, Ono pharmaceutical, and was part of an uncompensated advisory board of AstraZeneca. J.V.H has received honoraria and paid consulting from AstraZeneca and Boehringer Ingelheim and has received research funding from AstraZeneca.
REFERENCES AND NOTES
- 1.Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14:2895–2899. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 2.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. The New England journal of medicine. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 3.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, Red Brewer M, Ichihara E, Sun J, Jin H, Ballard P, Al-Kadhimi K, Rowlinson R, Klinowska T, Richmond GH, Cantarini M, Kim DW, Ranson MR, Pao W. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, Frewer P, Cantarini M, Brown KH, Dickinson PA, Ghiorghiu S, Ranson M. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. The New England journal of medicine. 2015;372:1689–1699. doi: 10.1056/NEJMoa1411817. [DOI] [PubMed] [Google Scholar]
- 6.Yao Z, Fenoglio S, Gao DC, Camiolo M, Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V, Kenner L, Sordella R. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci U S A. 2010;107:15535–15540. doi: 10.1073/pnas.1009472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Wadei HA, Plummer HK, 3rd, Ullah MF, Unger B, Brody JR, Schuller HM. Social stress promotes and gamma-aminobutyric acid inhibits tumor growth in mouse models of non-small cell lung cancer. Cancer Prev Res (Phila) 2012;5:189–196. doi: 10.1158/1940-6207.CAPR-11-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang HM, Liao ZX, Komaki R, Welsh JW, O’Reilly MS, Chang JY, Zhuang Y, Levy LB, Lu C, Gomez DR. Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Ann Oncol. 2013 doi: 10.1093/annonc/mds616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banerjee J, Papu John AM, Schuller HM. Regulation of nonsmall-cell lung cancer stem cell like cells by neurotransmitters and opioid peptides. Int J Cancer. 2015;137:2815–2824. doi: 10.1002/ijc.29646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamer M, Chida Y, Molloy GJ. Psychological distress and cancer mortality. J Psychosom Res. 2009;66:255–258. doi: 10.1016/j.jpsychores.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Park PG, Merryman J, Orloff M, Schuller HM. Beta-adrenergic mitogenic signal transduction in peripheral lung adenocarcinoma: implications for individuals with preexisting chronic lung disease. Cancer Res. 1995;55:3504–3508. [PubMed] [Google Scholar]
- 12.Schuller HM, Tithof PK, Williams M, Plummer H., 3rd The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer Res. 1999;59:4510–4515. [PubMed] [Google Scholar]
- 13.Cole SW, Arevalo JM, Takahashi R, Sloan EK, Lutgendorf SK, Sood AK, Sheridan JF, Seeman TE. Computational identification of gene-social environment interaction at the human IL6 locus. Proc Natl Acad Sci U S A. 2010;107:5681–5686. doi: 10.1073/pnas.0911515107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nilsson MB, Armaiz-Pena G, Takahashi R, Lin YG, Trevino J, Li Y, Jennings N, Arevalo J, Lutgendorf SK, Gallick GE, Sanguino AM, Lopez-Berestein G, Cole SW, Sood AK. Stress hormones regulate interleukin-6 expression by human ovarian carcinoma cells through a Src-dependent mechanism. J Biol Chem. 2007;282:29919–29926. doi: 10.1074/jbc.M611539200. [DOI] [PubMed] [Google Scholar]
- 15.Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, Liang MC, Cai D, Naumov GN, Bao L, Contreras CM, Li D, Chen L, Krishnamurthy J, Koivunen J, Chirieac LR, Padera RF, Bronson RT, Lindeman NI, Christiani DC, Lin X, Shapiro GI, Janne PA, Johnson BE, Meyerson M, Kwiatkowski DJ, Castrillon DH, Bardeesy N, Sharpless NE, Wong KK. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–810. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- 16.Natale RB, Thongprasert S, Greco FA, Thomas M, Tsai CM, Sunpaweravong P, Ferry D, Mulatero C, Whorf R, Thompson J, Barlesi F, Langmuir P, Gogov S, Rowbottom JA, Goss GD. Phase III trial of vandetanib compared with erlotinib in patients with previously treated advanced non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:1059–1066. doi: 10.1200/JCO.2010.28.5981. [DOI] [PubMed] [Google Scholar]
- 17.Lara HE, Porcile A, Espinoza J, Romero C, Luza SM, Fuhrer J, Miranda C, Roblero L. Release of norepinephrine from human ovary: coupling to steroidogenic response. Endocrine. 2001;15:187–192. doi: 10.1385/ENDO:15:2:187. [DOI] [PubMed] [Google Scholar]
- 18.Lara HE, Dorfman M, Venegas M, Luza SM, Luna SL, Mayerhofer A, Guimaraes MA, Rosa ESAA, Ramirez VD. Changes in sympathetic nerve activity of the mammalian ovary during a normal estrous cycle and in polycystic ovary syndrome: Studies on norepinephrine release. Microsc Res Tech. 2002;59:495–502. doi: 10.1002/jemt.10229. [DOI] [PubMed] [Google Scholar]
- 19.Lutgendorf SK, Cole S, Costanzo E, Bradley S, Coffin J, Jabbari S, Rainwater K, Ritchie JM, Yang M, Sood AK. Stress-related mediators stimulate vascular endothelial growth factor secretion by two ovarian cancer cell lines. Clin Cancer Res. 2003;9:4514–4521. [PubMed] [Google Scholar]
- 20.Keiper M, Stope MB, Szatkowski D, Bohm A, Tysack K, Vom Dorp F, Saur O, Oude Weernink PA, Evellin S, Jakobs KH, Schmidt M. Epac- and Ca2+-controlled activation of Ras and extracellular signal-regulated kinases by Gs-coupled receptors. The Journal of biological chemistry. 2004;279:46497–46508. doi: 10.1074/jbc.M403604200. [DOI] [PubMed] [Google Scholar]
- 21.Kim E, Herbst R, Wistuba I, Lee J, Blumenschein G, Tsao A, Stewart D, Hicks M, Erasmus J, Gupta S, Alden C, Liu S, Tang X, Khuri F, Tran H, Johnson B, Heymach J, Mao L, Fossella F, Kies M, Papadimitrakopoulou V, Davis S, Lippman S, Hong W. The BATTLE trial (Biomarker-integrated Approaches of Targeted Therapy for Lung Cancer Elimination): Personalizing therapy for lung cancer. Cancer Discovery. 2011;1:43–51. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng B, Jeong JH, Asara JM, Yuan YY, Granter SR, Chin L, Cantley LC. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Molecular cell. 2009;33:237–247. doi: 10.1016/j.molcel.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Memmott RM, Dennis PA. LKB1 and mammalian target of rapamycin as predictive factors for the anticancer efficacy of metformin. J Clin Oncol. 2009;27:e226. doi: 10.1200/JCO.2009.25.3963. author reply e227. [DOI] [PubMed] [Google Scholar]
- 25.Oestreich EA, Malik S, Goonasekera SA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac and phospholipase Cepsilon regulate Ca2+ release in the heart by activation of protein kinase Cepsilon and calcium-calmodulin kinase II. The Journal of biological chemistry. 2009;284:1514–1522. doi: 10.1074/jbc.M806994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 27.House C, Kemp BE. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science. 1987;238:1726–1728. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- 28.Strulovici B, Cerione RA, Kilpatrick BF, Caron MG, Lefkowitz RJ. Direct demonstration of impaired functionality of a purified desensitized beta-adrenergic receptor in a reconstituted system. Science. 1984;225:837–840. doi: 10.1126/science.6089331. [DOI] [PubMed] [Google Scholar]
- 29.Hausdorff WP, Bouvier M, O’Dowd BF, Irons GP, Caron MG, Lefkowitz RJ. Phosphorylation sites on two domains of the beta 2-adrenergic receptor are involved in distinct pathways of receptor desensitization. The Journal of biological chemistry. 1989;264:12657–12665. [PubMed] [Google Scholar]
- 30.Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori M, Merritt WM, Lin YG, Mangala LS, Kim TJ, Coleman RL, Landen CN, Li Y, Felix E, Sanguino AM, Newman RA, Lloyd M, Gershenson DM, Kundra V, Lopez-Berestein G, Lutgendorf SK, Cole SW, Sood AK. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12:939–944. doi: 10.1038/nm1447. [DOI] [PubMed] [Google Scholar]
- 31.Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M, Su WC, Bennouna J, Kato T, Gorbunova V, Lee KH, Shah R, Massey D, Zazulina V, Shahidi M, Schuler M. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31:3327–3334. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 32.Wong HP, Ho JW, Koo MW, Yu L, Wu WK, Lam EK, Tai EK, Ko JK, Shin VY, Chu KM, Cho CH. Effects of adrenaline in human colon adenocarcinoma HT-29 cells. Life sciences. 2011;88:1108–1112. doi: 10.1016/j.lfs.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 33.Yao H, Duan Z, Wang M, Awonuga AO, Rappolee D, Xie Y. Adrenaline induces chemoresistance in HT-29 colon adenocarcinoma cells. Cancer genetics and cytogenetics. 2009;190:81–87. doi: 10.1016/j.cancergencyto.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 34.Madden KS, Szpunar MJ, Brown EB. beta-Adrenergic receptors (beta-AR) regulate VEGF and IL-6 production by divergent pathways in high beta-AR-expressing breast cancer cell lines. Breast cancer research and treatment. 2011;130:747–758. doi: 10.1007/s10549-011-1348-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang XY, Wang HC, Yuan Z, Huang J, Zheng Q. Norepinephrine stimulates pancreatic cancer cell proliferation, migration and invasion via beta-adrenergic receptor-dependent activation of P38/MAPK pathway. Hepato-gastroenterology. 2012;59:889–893. doi: 10.5754/hge11476. [DOI] [PubMed] [Google Scholar]
- 36.Sloan EK, Priceman SJ, Cox BF, Yu S, Pimentel MA, Tangkanangnukul V, Arevalo JM, Morizono K, Karanikolas BD, Wu L, Sood AK, Cole SW. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer research. 2010;70:7042–7052. doi: 10.1158/0008-5472.CAN-10-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le CP, Nowell CJ, Kim-Fuchs C, Botteri E, Hiller JG, Ismail H, Pimentel MA, Chai MG, Karnezis T, Rotmensz N, Renne G, Gandini S, Pouton CW, Ferrari D, Moller A, Stacker SA, Sloan EK. Chronic stress in mice remodels lymph vasculature to promote tumour cell dissemination. Nature communications. 2016;7:10634. doi: 10.1038/ncomms10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monami M, Filippi L, Ungar A, Sgrilli F, Antenore A, Dicembrini I, Bagnoli P, Marchionni N, Rotella CM, Mannucci E. Further data on beta-blockers and cancer risk: observational study and meta-analysis of randomized clinical trials. Current medical research and opinion. 2013 doi: 10.1185/03007995.2013.772505. [DOI] [PubMed] [Google Scholar]
- 39.Barron TI, Connolly RM, Sharp L, Bennett K, Visvanathan K. Beta blockers and breast cancer mortality: a population- based study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:2635–2644. doi: 10.1200/JCO.2010.33.5422. [DOI] [PubMed] [Google Scholar]
- 40.Grytli HH, Fagerland MW, Fossa SD, Tasken KA, Haheim LL. Use of beta-blockers is associated with prostate cancer-specific survival in prostate cancer patients on androgen deprivation therapy. The Prostate. 2013;73:250–260. doi: 10.1002/pros.22564. [DOI] [PubMed] [Google Scholar]
- 41.Laag E, Majidi M, Cekanova M, Masi T, Takahashi T, Schuller HM. NNK activates ERK1/2 and CREB/ATF-1 via beta-1-AR and EGFR signaling in human lung adenocarcinoma and small airway epithelial cells. Int J Cancer. 2006;119:1547–1552. doi: 10.1002/ijc.21987. [DOI] [PubMed] [Google Scholar]
- 42.Sindhu S, Thomas R, Shihab P, Sriraman D, Behbehani K, Ahmad R. Obesity Is a Positive Modulator of IL-6R and IL-6 Expression in the Subcutaneous Adipose Tissue: Significance for Metabolic Inflammation. PLoS One. 2015;10:e0133494. doi: 10.1371/journal.pone.0133494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qu D, Liu J, Lau CW, Huang Y. IL-6 in diabetes and cardiovascular complications. Br J Pharmacol. 2014;171:3595–3603. doi: 10.1111/bph.12713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aldaham S, Foote JA, Chow HH, Hakim IA. Smoking Status Effect on Inflammatory Markers in a Randomized Trial of Current and Former Heavy Smokers. Int J Inflam. 2015;2015:439396. doi: 10.1155/2015/439396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hara MR, Kovacs JJ, Whalen EJ, Rajagopal S, Strachan RT, Grant W, Towers AJ, Williams B, Lam CM, Xiao K, Shenoy SK, Gregory SG, Ahn S, Duckett DR, Lefkowitz RJ. A stress response pathway regulates DNA damage through beta2-adrenoreceptors and beta-arrestin-1. Nature. 2011;477:349–353. doi: 10.1038/nature10368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kose M. GPCRs and EGFR - Cross-talk of membrane receptors in cancer. Bioorg Med Chem Lett. 2017;27:3611–3620. doi: 10.1016/j.bmcl.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 47.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 48.Drube S, Stirnweiss J, Valkova C, Liebmann C. Ligand-independent and EGF receptor-supported transactivation: lessons from beta2-adrenergic receptor signalling. Cellular signalling. 2006;18:1633–1646. doi: 10.1016/j.cellsig.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 49.Koivunen JP, Kim J, Lee J, Rogers AM, Park JO, Zhao X, Naoki K, Okamoto I, Nakagawa K, Yeap BY, Meyerson M, Wong KK, Richards WG, Sugarbaker DJ, Johnson BE, Janne PA. Mutations in the LKB1 tumour suppressor are frequently detected in tumours from Caucasian but not Asian lung cancer patients. British journal of cancer. 2008;99:245–252. doi: 10.1038/sj.bjc.6604469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsuchiya Y, Denison FC, Heath RB, Carling D, Saggerson D. 5′-AMP-activated protein kinase is inactivated by adrenergic signalling in adult cardiac myocytes. Bioscience reports. 2012;32:197–213. doi: 10.1042/BSR20110076. [DOI] [PubMed] [Google Scholar]
- 51.Kaufman JM, Amann JM, Park K, Arasada RR, Li H, Shyr Y, Carbone DP. LKB1 Loss induces characteristic patterns of gene expression in human tumors associated with NRF2 activation and attenuation of PI3K-AKT. J Thorac Oncol. 2014;9:794–804. doi: 10.1097/JTO.0000000000000173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, Buczkowski KA, Liu Y, Awad MM, Denning WL, Diao L, Wang J, Parra Cuentas ER, Wistuba, Soucheray M, Thai TC, Asahina H, Kitajima S, Altabef A, Cavanaugh JD, Rhee K, Gao P, Zhang H, Fecci PE, Shimamura T, Hellmann M, Heymach JV, Hodi FS, Freeman GJ, Barbie DA, Dranoff G, Hammerman PS, Wong KK. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T cell activity in the lung tumor microenvironment. Cancer Res. 2016 doi: 10.1158/0008-5472.CAN-15-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nanjundan M, Byers LA, Carey MS, Siwak DR, Raso MG, Diao L, Wang J, Coombes KR, Roth JA, Mills GB, Wistuba II, Minna JD, HRV Proteomic profiling identifies pathways dysregulated in non-small cell lung cancer and an inverse association of AMPK and adhesion pathways with recurrence. Journal of Thoracic Oncology. 2010 doi: 10.1097/JTO.0b013e3181f2a266. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A, Pratilas CA, Rosen N, Gray NS, Wong KK, Janne PA. Combined EGFR/MEK Inhibition Prevents the Emergence of Resistance in EGFR-Mutant Lung Cancer. Cancer discovery. 2015;5:960–971. doi: 10.1158/2159-8290.CD-15-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ercan D, Xu C, Yanagita M, Monast CS, Pratilas CA, Montero J, Butaney M, Shimamura T, Sholl L, Ivanova EV, Tadi M, Rogers A, Repellin C, Capelletti M, Maertens O, Goetz EM, Letai A, Garraway LA, Lazzara MJ, Rosen N, Gray NS, Wong KK, Janne PA. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer discovery. 2012;2:934–947. doi: 10.1158/2159-8290.CD-12-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmidt C, Kraft K. Beta-endorphin and catecholamine concentrations during chronic and acute stress in intensive care patients. Eur J Med Res. 1996;1:528–532. [PubMed] [Google Scholar]
- 57.Tang H, Xiao G, Behrens C, Schiller J, Allen J, Chow CW, Suraokar M, Corvalan A, Mao J, White MA, Wistuba, Minna JD, Xie Y. A 12-gene set predicts survival benefits from adjuvant chemotherapy in non-small cell lung cancer patients. Clin Cancer Res. 2013;19:1577–1586. doi: 10.1158/1078-0432.CCR-12-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba, Fong KM, Lee H, Toyooka S, Shimizu N, Fujisawa T, Feng Z, Roth JA, Herz J, Minna JD, Gazdar AF. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–346. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 60.Lim SO, Li CW, Xia W, Lee HH, Chang SS, Shen J, Hsu JL, Raftery D, Djukovic D, Gu H, Chang WC, Wang HL, Chen ML, Huo L, Chen CH, Wu Y, Sahin A, Hanash SM, Hortobagyi GN, Hung MC. EGFR Signaling Enhances Aerobic Glycolysis in Triple-Negative Breast Cancer Cells to Promote Tumor Growth and Immune Escape. Cancer Res. 2016;76:1284–1296. doi: 10.1158/0008-5472.CAN-15-2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Byers LA, Sen B, Saigal B, Diao L, Wang J, Nanjundan M, Cascone T, Mills GB, Heymach JV, Johnson FM. Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin Cancer Res. 2009;15:6852–6861. doi: 10.1158/1078-0432.CCR-09-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hu J, He X, Baggerly KA, Coombes KR, Hennessy BT, Mills GB. Non-parametric quantification of protein lysate arrays. Bioinformatics. 2007;23:1986–1994. doi: 10.1093/bioinformatics/btm283. [DOI] [PubMed] [Google Scholar]
- 63.Arbiser JL, Moses MA, Fernandez CA, Ghiso N, Cao Y, Klauber N, Frank D, Brownlee M, Flynn E, Parangi S, Byers HR, Folkman J. Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proc Natl Acad Sci U S A. 1997;94:861–866. doi: 10.1073/pnas.94.3.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sheridan JF, Feng NG, Bonneau RH, Allen CM, Huneycutt BS, Glaser R. Restraint stress differentially affects anti-viral cellular and humoral immune responses in mice. Journal of neuroimmunology. 1991;31:245–255. doi: 10.1016/0165-5728(91)90046-a. [DOI] [PubMed] [Google Scholar]
- 65.Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W, Diao L, Wang J, Roybal JD, Patel M, Ungewiss C, Peng D, Antonia S, Mediavilla-Varela M, Robertson G, Jones S, Suraokar M, Welsh JW, Erez B, Wistuba, Wang S, Ullrich SE, Heymach JV, Kurie JM, Qin FX. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5:5241. doi: 10.1038/ncomms6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Frequency of T790M EGFR secondary mutations in erlotinib resistant cells.
Fig. S2. Resistance of HCC827 ER and HCC4006 ER cells to EGFR TKIs.
Fig. S3. ADRB expression and EGFR status in NSCLC cell lines.
Fig. S4. Induction of IL-6 following β-AR activation.
Fig. S5. Differential effects of β2-AR signaling on EGFR mutant and wild-type cells.
Fig. S6. Stress hormone-induced effects on LKB1, PKC and CREB.
Fig. S7. Effect of EGFR inhibition on β-AR-induced p-LKB1, p-CREB, and IL-6.
Fig. S8. Effect of β-AR signaling on NF-κB activity.
Fig. S9. Effect of chronic stress on IL-6 expression and MVD in NSCLC xenografts.
Fig. S10. Stress hormones promote EGFR inhibitor resistance in NSCLC.
Table S1. PFS and OS of NSCLC patients with high or low IL-6 treated with erlotinib.
Table S2. Smoking status of patients with high or low circulating IL-6 in the ZEST trial.
Table S3. Lung cancer cell lines used in ADRB gene expression analysis.