

Abstract
Summary
Recent advances in protein residue contact prediction algorithms have led to the emergence of many new methods and a variety of file formats. We present ConKit, an open source, modular and extensible Python interface which allows facile conversion between formats and provides an interface to analyses of sequence alignments and sets of contact predictions.
Availability and Implementation
ConKit is available via the Python Package Index. The documentation can be found at http://www.conkit.org. ConKit is licensed under the BSD 3-Clause.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Residue–residue contact predictions are becoming an increasingly popular and dynamic field of bioinformatics research as well as source of information in structural biology. Over recent years, great advances have been made to facilitate highly accurate predictions (e.g. Jones et al., 2015; Marks et al., 2011), which enabled these predicted contacts to, for example, guide accurate structure predictions (e.g. Michel et al., 2014; Ovchinnikov et al., 2015), identify functional sites (e.g. Grigolon et al., 2016; Hopf et al., 2012; Parente et al., 2015), or predict modes of protein interaction (e.g. Hopf et al., 2014; Ovchinnikov et al., 2014).
The many prediction algorithms and pipelines currently available have adopted a variety of different file format conventions. Additionally, metapredictors rely on file conversions to either combine various predictions, or standardize the output in their chosen style. These various file formats lead to a dilemma for software developers writing tools to utilize predicted contacts. Although a standardized format—Casp RR—exists, it has not been widely adopted. Therefore, software developers must either insist on a given format for their tools, or develop an extensive library of conversion algorithms to handle multiple formats.
Users of contact prediction methods are often faced with the challenge of estimating the quality of a prediction. In co-evolution based methods, the quality typically depends on the Multiple Sequence Alignment depth (Jones et al., 2015; Ovchinnikov et al., 2015). However, the quantification of this depth, commonly referred to as number of effective sequences, and other important measures, such as the sequence coverage in the alignment, often requires further software packages or manual method development.
With this motivation, ConKit was developed to satisfy many of the issues outlined earlier and to provide additional functionality useful to a variety of software developers and users.
2 Materials and methods
ConKit is a cross-platform package written in the Python programming language. It is based in part on the NumPy (Oliphant, 2015), SciPy (http://www.scipy.org), BioPython (Cock et al., 2009) and matplotlib (Hunter, 2007) packages. ConKit’s modular design enables it to have numerous applications as a standalone package. It is currently made up of four distinct packages—the data model, input/output structure, plotting facility and command-line application wrappers. ConKit also easily integrates into larger software packages and it is already distributed with CCP4 v7.0.032 (Winn et al., 2011) and CCP-EM beta (update January 7, 2017) (Wood et al., 2015).
2.1 Data model
The underlying data model in ConKit stores contact information in a three-tier hierarchy, which provides easy access to the contact information stored within. At its lowest level, ConKit stores individual contact pairs in the Contact class. All Contact instances are combined and held in a ContactMap class, which provides routine functions to handle all contacts in a single prediction. At the top level, ConKit allows users to store multiple ContactMap instances in the ContactFile class. Each tier has attributes and functions relating to the data stored within, such as the sequence attribute in the ContactMap class, which allows users to easily associate a sequence with a contact prediction.
Alongside the data model for contact information, a SequenceFile hierarchy was implemented. Although BioPython’s SeqIO and AlignIO packages (Cock et al., 2009) already provide such a data structure, ConKit's hierarchy enables customized interactions for the models. Two tiers are currently implemented, with the SequenceFile class storing one or more Sequence classes.
Both hierarchies provide storage, modification and analysis methods. For example, a ContactMap instance allows users to calculate the precision value of a given contact map when compared against the contacts extracted from a protein structure (Morcos et al., 2011). This feature is essential for assessing the quality of contact predictions when structural information is available (Jones et al., 2015; Monastyrskyy et al., 2016). It also has potential value in scoring the quality of ab initio models based on the number of long-range contacts fulfilled in a model (de Oliveira et al., 2016; Ovchinnikov et al., 2017).
In comparison, a SequenceFile instance enables users to calculate the alignment depth, a key estimate for assessing the usefulness of an alignment in co-evolution based predictions (Monastyrskyy et al., 2016). A SequenceFile instance also provides the functionality to determine the sequence coverage in the stored alignment, which can prove useful when trimming alignments to the core region of a protein domain.
2.2 Input/output
Manually constructing a data model in ConKit is generally not necessary. Four sequences and 17 contacts file format parsers have been implemented to allow read and write functionality. Importantly, the modular design of ConKit allows for an easy addition of new file format parsers in the future. In general, the two methods, read() and write(), are the access points to the parsers. To make file format conversions more accessible a third notable I/O function has been implemented, namely convert(), which acts as a wrapper encapsulating read() and write(). For a full list of available file formats, refer to ConKit’s documentation.
2.3 Data visualization
Besides the analysis functionality outlined previously, ConKit also provides an interface for data visualization. It uses the matplotlib (Hunter, 2007) package and ConKit’s data models to extract and visualize data. Using the matplotlib package enables native support for many file formats, such as Portable Network Graphics or Scalable Vector Graphics. In ConKit, all plots are created via Python classes, thus enabling full customizability via simple class attribute setting. ConKit provides plotting classes for both SequenceFile and ContactMap hierarchies, such as the SequenceCoverageFigure class to illustrate the sequence coverage (Fig. 1a) in a Multiple Sequence Alignment or the ContactMapFigure for the commonly used contact map visualization (Supplementary Fig. S1). ConKit also provides the PrecisionEvaluationFigure class for a stepwise evaluation of precision scores when comparing a predicted contact map to protein structure (Fig. 1b). For a full list of available plots alongside usage examples, refer to the documentation.
Fig. 1.
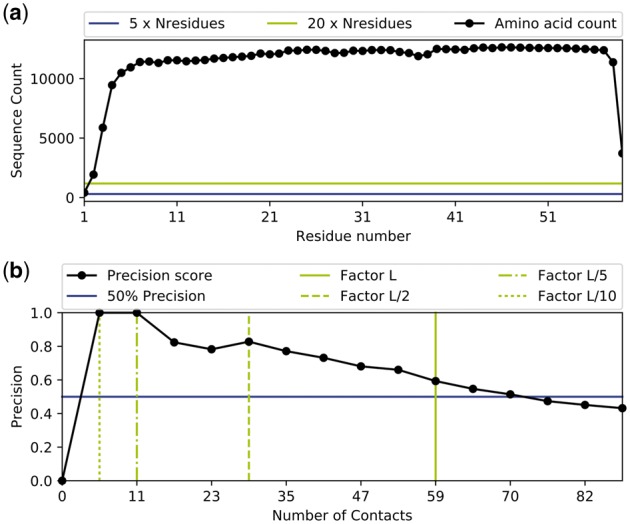
Data visualization in ConKit. (a) Sequence coverage in an example Multiple Sequence Alignment. (b) Precision score evaluation of a contact prediction file at various contact cutoff values where L = sequence length (rounded down to the nearest whole number of contacts). Data for both examples are based on PDB entry: 1DTX
2.4 Command-line application interface
Considering the number of different features of ConKit, we believe that the command-line application interface could be particularly useful to create metapredictor pipelines. To date, these wrappers comprise the following executables: HHblits (Remmert et al., 2012); Jackhmmer (Johnson et al., 2010); HHfilter (Remmert et al., 2012); CCMpred (Seemayer et al., 2014); PSICOV (Jones et al., 2012); and bbcontacts (Andreani and Söding, 2015). All wrappers are based on the AbstractCommandline class in BioPython, and thus a fully working version of the package is required for this ConKit sub-package.
3 Usage
ConKit can be used in two distinct ways. To access all features, users do require some familiarity with the Python programming language. All packages outlined earlier are available via Python’s import system and relevant classes are exposed. To circumvent this requirement for non-programmers and make ConKit a more general tool, pre-defined routines in the form of scripts are automatically installed giving the general user access to ConKit’s key features from the command line. All scripts are written in Python making them operating system independent. All scripts have the prefix conkit and a one-word suffix based on its function. For example, conkit-msatool and conkit-convert can be used to analyse alignments and convert contact prediction files, respectively, while conkit-precision calculates the precision value given a contact prediction, the corresponding sequence and a protein structure. The conkit-plot script exposes ConKit’s plotting package for simple figure generation. For a full list of available scripts, refer to ConKit’s documentation.
4 Conclusions
We present ConKit, an extensible and modular Python interface for handling and manipulating residue–residue contact predictions, multiple sequence alignments and contact maps. Its core functionality is enhanced by the provision of command line scripts and application wrappers.
Supplementary Material
Acknowledgements
The authors would like to acknowledge Stefan Seemayer for the provision of helpful software libraries.
Funding
This work was supported by the Biotechnology and Biological Sciences Research Council grant [BB/L008696/1]. CCP4 co-funds FS’s studentship.
Conflict of Interest: none declared.
References
- Andreani J., Söding J. (2015) bbcontacts: prediction of β-strand pairing from direct coupling patterns. Bioinformatics, 31, 1729–1737. [DOI] [PubMed] [Google Scholar]
- Cock P.J.A. et al. (2009) Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics, 25, 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Olivera S.H. et al. (2016) Comparing co-evolution methods and their application to template-free protein structure prediction. Bioinformatics, 33, 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigolon S. et al. (2016) Identifying relevant positions in proteins by Critical Variable Selection. Mol. Biosyst., 12, 2147–2158. [DOI] [PubMed] [Google Scholar]
- Hopf T.A. et al. (2012) Three-dimensional structures of membrane proteins from genomic sequencing. Cell, 149, 1607–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf T.A. et al. (2014) Sequence co-evolution gives 3D contacts and structures of protein complexes. Elife, 3, e03430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter J.D. (2007) Matplotlib: a 2D graphics environment. Comput. Sci. Eng., 9, 90–95. [Google Scholar]
- Johnson L.S. et al. (2010) Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics, 11, 431.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D.T. et al. (2012) PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics, 28, 184–190. [DOI] [PubMed] [Google Scholar]
- Jones D.T. et al. (2015) MetaPSICOV: combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins. Bioinformatics, 31, 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks D.S. et al. (2011) Protein 3D structure computed from evolutionary sequence variation. PLoS One, 6, e28766.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel M. et al. (2014) PconsFold: improved contact predictions improve protein models. Bioinformatics, 30, i482–i488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monastyrskyy B. et al. (2016) New encouraging developments in contact prediction: assessment of the CASP11 results. Proteins, 84 (Suppl 1), 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morcos F. et al. (2011) Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. USA, 108, E1293–E1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant T.E. (2015) A Guide to NumPy, 2nd edn.Continuum Press. Austin, USA. [Google Scholar]
- Ovchinnikov S. et al. (2014) Robust and accurate prediction of residue–residue interactions across protein interfaces using evolutionary information. Elife, 3, e02030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovchinnikov S. et al. (2015) Large-scale determination of previously unsolved protein structures using evolutionary information. Elife, 4, e09248.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovchinnikov S. et al. (2017) Protein structure determination using metagenome sequence data. Science, 355, 294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parente D.J. et al. (2015) Amino acid positions subject to multiple coevolutionary constraints can be robustly identified by their eigenvector network centrality scores. Proteins, 83, 2293–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmert M. et al. (2012) HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods, 9, 173–175. [DOI] [PubMed] [Google Scholar]
- Seemayer S. et al. (2014) CCMpred—fast and precise prediction of protein residue–residue contacts from correlated mutations. Bioinformatics, 30, 3128–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M.D. et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr., 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood C. et al. (2015) Collaborative computational project for electron cryo-microscopy. Acta Crystallogr. D Biol. Crystallogr, 71, 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.