

Abstract
Motivation
Loss-of-function genetic variants are frequently associated with severe clinical phenotypes, yet many are present in the genomes of healthy individuals. The available methods to assess the impact of these variants rely primarily upon evolutionary conservation with little to no consideration of the structural and functional implications for the protein. They further do not provide information to the user regarding specific molecular alterations potentially causative of disease.
Results
To address this, we investigate protein features underlying loss-of-function genetic variation and develop a machine learning method, MutPred-LOF, for the discrimination of pathogenic and tolerated variants that can also generate hypotheses on specific molecular events disrupted by the variant. We investigate a large set of human variants derived from the Human Gene Mutation Database, ClinVar and the Exome Aggregation Consortium. Our prediction method shows an area under the Receiver Operating Characteristic curve of 0.85 for all loss-of-function variants and 0.75 for proteins in which both pathogenic and neutral variants have been observed. We applied MutPred-LOF to a set of 1142 de novo vari3ants from neurodevelopmental disorders and find enrichment of pathogenic variants in affected individuals. Overall, our results highlight the potential of computational tools to elucidate causal mechanisms underlying loss of protein function in loss-of-function variants.
Availability and Implementation
1 Introduction
Genetic data-driven approaches to human health have resulted in the implication of loss-of-function (LOF) variants in phenotypes ranging from complex neuropsychiatric diseases to Mendelian blood groups (Stenson et al., 2014). Loss-of-function variants include frameshifting and stop variants and are of particular interest because of their potentially profound impact on the mRNA transcript and translated protein. Frameshifting variants are insertions and deletions of nucleotides (indels) not divisible by three, causing a change in the mRNA coding frame. Stop variants, on the other hand, entail the gain or loss of stop codons in mRNA; stop-gain or nonsense variants introduce a premature termination codon that truncates the protein, whereas stop-loss or nonstop variants alter the termination codon and lead to elongated proteins.
Altered transcripts resulting from LOF variants are either degraded at the mRNA or protein levels or rendered non-functional, often leading to disease phenotypes. For example, disease-causing stop variants are significantly enriched for alterations which activate nonsense mediated decay, resulting in haploinsufficiency (Mort et al., 2008). However, LOF variants can also be functionally and phenotypically neutral; in fact, each human genome may contain hundreds of frameshifting indels and dozens of stop variants with little or no observable impact upon phenotype (Kircher et al., 2014; MacArthur and Tyler-Smith, 2010; MacArthur et al., 2012; Sulem et al., 2015; Thousand Genomes Project Consortium, 2010). Robustness in the genome through gene duplication (Hsiao and Vitkup, 2008) and compensatory mechanisms (Hu and Ng, 2012) can result in the toleration of many LOF variants. Furthermore, gene loss in regions under relaxed selection, including olfactory and taste receptor genes, is typically tolerated (Risso et al., 2014).
Recently, there has been a growing interest in the phenotypic and clinical roles of de novo LOF variants. For example, an estimated one out of every hundred de novo LOF mutations contributes to autism spectrum disorders (Ronemus et al., 2014). Data from the Exome Aggregation Consortium (ExAC) found that the exomes of 60 706 individuals contain nearly sixty thousand non-singleton nonsense variants while 3230 genes exhibit near-complete depletion (Lek et al., 2016); therefore, a major challenge remains in understanding the nature and quantifying the impact of LOF variants in a given genome. The lack of annotation for previously unseen genetic variants in many such cases further highlights the need for sophisticated prediction models specifically designed for LOF variants.
Unlike single nucleotide variants (Cline and Karchin, 2011), LOF variants are not as well-studied. Early approaches used in the work of Zia and Moses (2011), SIFT Indel (Hu and Ng, 2012) and NutVar (Rausell et al., 2014) have primarily utilized conservation features. However, these features are limited in distinguishing between LOF variants of different classes in the same protein. Moreover, these methods restrict their training sets to core proteins that have high quality annotations, thereby reducing their utility on less well-studied genes. To circumvent this issue, CADD (Kircher et al., 2014), a method designed to predict the impact of all classes of genetic variation, was trained on simulated de novo and ancestral mutations. DDIG-in further supplemented conservation-based features with intrinsic disorder predictions from the region affected by stop gain and frameshifting variants (Folkman et al., 2015), whereas VEST-Indel evaluates frameshifting indels via a random forest model, yet restricts functional and structural features to stability, solvent accessibility and the temperature factor (Douville et al., 2016). However, there are numerous other structural and functional properties of proteins that could potentially be impacted by LOF variants. The incorporation of such information alongside features indicative of functional redundancy of the protein into more complex predictive models is expected to not only increase discriminative power but also suggest the specific nature of functional impact in a principled manner.
To address these challenges, we study the evolutionary, structural and functional signatures of loss-of-function genetic variants. We subsequently develop machine learning methods for the discrimination of pathogenic from tolerated frameshifting and stop gain variants and hypothesizing specific molecular alterations. Our results provide evidence that there is significant potential to analyze the causal mechanisms for loss of function in these classes of variants and point out potential difficulties in developing computational tools for automated prioritization of loss-of-function variants.
2 Materials and methods
2.1 Training data sets
Pathogenic (disease-causing) stop gain and frameshifting variants were obtained from the July 2016 version of the Human Gene Mutation Database (HGMD) (Stenson et al., 2014) and ClinVar (Landrum et al., 2016). The set of putatively neutral variants was composed of frameshifting and stop gain variants from ExAC which were annotated to have passed all quality filters. To remove potential biases, we did not perform filtering based upon population frequency and only retained canonical isoforms in subsequent analyses. Table 1 summarizes all data sets.
Table 1.
Number of variants (proteins) present in each data set
| Disease | Neutral | Total | |
|---|---|---|---|
| Frameshift | 18 116 (1545) | 90 135 (13 427) | 108 251 (13 713) |
| Stop gain | 14 318 (1681) | 7960 (4990) | 22 278 (6137) |
| Total | 32 434 (1995) | 98 095 (13 605) |
The set of canonical sequences was derived from UniProt (Suzek et al., 2007). The number of available stop-loss variants was too small to be included in this work.
2.2 Neurodevelopmental disorders dataset
We applied MutPred-LOF to a data set, curated from the published literature, consisting of 970 de novo LOF variants identified through whole-exome or whole-genome sequencing of individuals diagnosed with six neurodevelopmental disorders, including autism spectrum disorder (ASD), schizophrenia, intellectual disability, bipolar disorder, developmental delay and epileptic encephalopathy (de Ligt et al., 2012; De Rubeis et al., 2014; EuroEPINOMICS-RES Consortium, Epilepsy Phenome/Genome Project and Epi4K Consortium, 2014; Epi4K Consortium and Epilepsy Phenome/Genome Project, 2013; Fromer et al., 2014; Gilissen et al., 2014; Girard et al., 2011; Guipponi et al., 2014; Gulsuner et al., 2013; Hashimoto et al., 2016; Iossifov et al., 2012, 2014; Jiang et al., 2013; Kong et al., 2012; McCarthy et al., 2014; Neale et al., 2012; O’Roak et al., 2011, 2012a, b; Rauch et al., 2012; Sanders et al., 2012; Turner et al., 2016; Xu et al., 2011; Xu et al., 2012; Yuen et al., 2015, 2016; S.Jonathan et al., unpublished data, doi: https://doi.org/10.1101/102327) and a control set of 172 de novo LOF mutations from healthy siblings (Gulsuner et al., 2013; Iossifov et al., 2012, 2014; O’Roak et al., 2011, 2012b; Rauch et al., 2012; Sanders et al., 2012; Xu et al., 2011, 2012; S.Jonathan et al., unpublished data, doi: https://doi.org/10.1101/102327).
2.3 Feature engineering
We created features for the description of each variant based upon the properties of wildtype protein sequence, with features divided into those representing portions of the protein sequence affected and unaffected by the alteration. Amino acids occurring prior to the variant were considered to constitute the unaffected portion of the protein and are referred to in the text as the amino side (Fig. 1). Residues that occur after the variant are likewise denoted as the carboxyl side, either wildtype or mutant. The amino acid sequences of the carboxyl side in mutant variants were not reconstructed from the genomes for use in structural and functional property features; incorporation of features based upon predicted mutant amino acid sequence did not lead to improved performance and the exclusion of these features greatly simplifies predictor development and its application because the variants could be scored based on the wildtype sequence only.
Fig. 1.

Illustration of the impacted portions of the protein for loss-of-function variants. The impacted region can be shorter or longer for the mutant protein (if translated); its length is zero for the stop gain variants
The feature space covers three classes: general sequence features, evolutionary features and predicted structural and functional features. Sequence-based features include the relative position of the variant, the number of amino acids affected by the variant, binary nonsense-mediated decay prediction based upon the 50 nucleotide rule (variant is more than 50 nucleotides upstream of the final exon–exon junction (Maquat, 2004), number of amino acids from the variant to the final exon–exon junction, and a two-element binary vector indicating the type of LOF variant (stop gain or frameshifting insertion/deletion).
2.3.1 Evolutionary features
Evolutionary features involved general sequence conservation indexes for amino/carboxyl sides of the protein as well as counts of close homologs of the wildtype protein in the human and mouse genomes. Conservation features for the wildtype sequence were extracted from two sources. First, we generated a position-specific scoring matrix (PSSM) by running PSI-BLAST against the "nr" database (Altschul et al., 1997). Second, we used AL2CO (Pei and Grishin, 2001) to derive nine conservation indexes from the UCSC Genome Browser 46-species alignment (Karolchik et al., 2014) for each position in the sequence. Both normalized and unnormalized versions of these scores were calculated for the whole alignment and two sub-alignments (mammals only and primates only). To capture the relative conservation of affected and unaffected regions of the protein, we took the maximum conservation over the amino and carboxyl sides of the protein as well as the difference between these regions. This resulted in 3 × 42 = 126 PSSM features and 3 × 2 × 3 × 9 = 162 AL2CO conservation features.
2.3.2 Structural and functional property features
The structural and functional features included both gene-based and residue-based features. The gene-based features contained 2132-dimensional vectors of predicted Gene Ontology terms using FANN-GO (Clark and Radivojac, 2011), where each variant in the same protein received the same set of features. The predicted features were used in order to mitigate biases that could arise due to the fact that known disease genes are generally better studied and contain more functional information than the remaining genes.
We extended the gene-based feature space via the prediction of a variety of residue-level structural and functional properties in the wildtype protein that convert its amino acid sequence into a series of real-valued vectors of the same length. We then used the methodology behind vector quantization kernels (Clark and Radivojac, 2014) to encode these property vectors into a fixed-length feature representation. Vector quantization features address an important challenge in encoding LOF variants as they facilitate encoding of variable-length amino acid sequences into a fixed-length representation, beyond simple summary statistics. At the same time, they allow for more effective use of external biological data to be incorporated into method development via a series of prediction models previously constructed for given structural and functional properties.
Specifically, the vector quantization procedure involved a data preprocessing step and a feature construction step. In the data preprocessing step, we first defined the universe of human protein sequences as the UniRef50 database (Suzek et al., 2007), where each is a string composed of amino acids from . For each , we mapped the protein sequence into a real-valued property vector , where l is length of the string s, using any particular feature mapping described in Table 2. For example, predicting the helical propensity for a given sequence s results in one such vector p of numbers between 0 and 1. Next, we decomposed the property vector into n-dimensional overlapping subvectors , where represents the first n elements of p. The process was repeated for each protein sequence to create a large database of n-dimensional fragments for a particular property. This database of fragments was then clustered using the K-means algorithm into m groups to generate a partition to m regions , represented by centroids . Each centroid is associated with a Voronoi region, Ri, such that
| (1) |
where d(x, c) is the Euclidean distance between fragment x and centroid c. One such clustering was created for each property and a set of m centroids per property was stored. In the feature construction step, a particular property was first predicted for the wildtype sequence and a set of length-n segments within the amino and carboxyl regions relative to the variant position were extracted. A set of features was then created for each side by counting the segments closest to each of the UniRef50-derived centroids; i.e., the count at position i corresponds to the number of fragments closest to the i-th centroid. Two vectors of counts, one for the amino and one for the carboxyl side, were created for each property. Based on previous work, and with minimal experimentation, we chose n = 16 and m = 16 (Clark and Radivojac, 2014). This vector quantization framework was utilized on both the carboxyl and amino side sequence fragments for the 57 structural and functional features to derive a total of 57 × 16 × 2 = 1824 features. Overall, the data set contained 4 270 features.
Table 2.
Predicted structural and functional features
| Property category | Predicted features |
|---|---|
| Structure and dynamics | Three classes*—Helix, strand, loop; Intrinsic disorder (Peng et al., 2006); B-factor (Radivojac et al., 2004); Relative solvent accessibility*; Coiled-coil region* |
| Signal peptide and transmembrane regions* | Seven classes—N- and C-termini of signal peptide, signal helix, signal peptide cleavage site, transmembrane segment, cytoplasmic and non-cytoplasmic loops |
| Enzyme activity* | Catalytic residues |
| Regulation* | Allosteric residues |
| Macromolecular binding | DNA*; RNA*; Protein-protein interaction (PPI)*; PPI hotspots*; Molecular Recognition Features (MoRFs)*; Calmodulin-binding (Radivojac et al., 2006) |
| Metal-binding* | Cd; Ca; Co; Cu; Fe; Mg; Mn; Ni; K; Na; Zn |
| Post-translational modification (PTM) (Pejaver et al., 2014) | Acetylation, ADP-ribosylation, Amidation, Carboxylation, Disulfide linkage, Farnesylation, Geranylgeranylation, Glycosylation (C-linked, N-linked and O-linked), GPI anchor amidation, Hydroxylation, Methylation, Myristoylation, N-terminal acetylation, Palmitoylation, Phosphorylation, Proteolytic cleavage, Pyrrolidone carboxylic acid, Sulfation, SUMOylation, Ubiquitylation |
| Motifs | From PROSITE (Sigrist et al., 2013) and ELM (Dinkel et al., 2014) |
Indicates in-house predictors.
2.4 Predictor development
An ensemble of one hundred bagged two-layer feed-forward neural networks was trained for all loss-of-function variants collectively utilizing the Matlab Neural Network Toolbox. The number of hidden units in each network was fixed at 10. To eliminate features very likely to be uninformative, we applied a two-sample t-test with a high P-value threshold of 0.5. Furthermore, we applied principal component analysis with 99% retained variance on z-score normalized data to reduce dimensionality and eliminate (near-)colinear features. The resilient propagation method was used for training with 25% of the training data used as validation (Riedmiller and Braun, 1993). All parameters were set prior to training and were not varied. Finally, all models were trained on balanced training sets, where the majority class was subsampled.
2.5 Predictor evaluation
Performance of MutPred-LOF is represented by the area under the ROC curve (AUC) derived from scores generated in 10-fold per-protein cross-validation. All pre-processing steps (normalization, dimensionality reduction) were carried out on the training partition only and applied on the test partition. To ensure that the model did not suffer from over-reliance on protein-based features, we performed per-protein cross-validation such that for each fold all variants in a protein were either entirely in the training or test set. We illustrate the influence of gene-based features on estimated performance of MutPred-LOF using three types of cross-validation protocols: (1) per-variant, (2) per-protein and (3) per-cluster. Per-variant cross-validation considers all variants as independent data points and partitions variants into ten-folds without consideration of the proteins from which the set is derived. In contrast, per-protein cross-validation ensures in each fold that all variants from the same protein are either in the training or in the test partition. This evaluation protocol ensures better performance estimation when the model is presented with a variant in protein that was not in the training set or that contained only one type of variant (e.g., disease-associated). In per-cluster cross-validation, variants within proteins with at least 50% sequence identity were included in either training or test sets, and can be used to estimate performance when the model is presented with a protein for which no protein within 50% sequence identity was available in the training set. We also used this evaluation protocol to assess the overfitting potential of the per-protein performance assessment.
Next, we compared the performance of MutPred-LOF against three currently available methods, each of which was evaluated using a set of frameshifting and stop gain variants from the MutPred-LOF training set. Deleterious variants from HGMD professional version 2016 that were not present in HGMD 2015 were extracted to represent a set of deleterious variants that are unlikely to be included in the training data of these methods (2,409 variants from 745 genes). The neutral test set consisted of variants from the ExAC training data (43,534 variants from 11,098 genes). The test set was filtered to remove the training data from DDIG-in and Vest-Indel; this procedure, however, could not be replicated for the CADD training data. Further, we created a for-comparison-only version of MutPred-LOF, MutPred-LOFʹ, with these test set variants removed from the training data, to ensure that for every model in this comparison the test variants were not included in the training set.
2.6 Assessing significance of functional alterations
To identify loss-of-function variants with significant functional impact, we defined a P-value for each feature listed in Table 2 to assign significance and rank prospective mechanisms. In the development of MutPred (Li et al., 2009), a method that predicts the impact of single amino acid substitutions, we constructed empirical P-values as follows. The null distribution was first defined using the scores of functional disruption for all variants present in the set of putatively neutral substitutions. Then, given a score of functional disruption for a new variant, the P-value is determined as the fraction of neutral substitutions with disruption scores at least as high as the observed value. This method relies on two strong assumptions: (1) that each functional mechanism is equally disrupted in the set of neutral substitutions, and (2) that each functional mechanism is equally likely.
In this work, we rank functional disruption scores by modifying MutPred’s approach so as to mitigate the latter assumption. Specifically, we rank the molecular mechanisms by adjusting the P-values as
| (2) |
where P is the P-value assigned in a way identical to MutPred’s approach and α is the frequency of a particular functional mechanism. We refer to this quantity as prior-corrected P-value. The rationale for this adjustment comes from the definition of the false discovery rate (FDR); i.e.,
| (3) |
where TPR is the true positive rate and FPR is the false positive rate. Here, we use the P-value as an approximation of the false positive rate and ignore the denominator. Ideally, molecular alterations would be prioritized based on the posterior probability that a particular mechanism (e.g., DNA binding, or protein binding) is disrupted. However, this step either requires a data set of disrupted and non-disrupted mechanisms that can be used to estimate true positive and false positive rates or further assumptions on how to probabilistically reason on the disruption of structural/functional propensity for entire protein regions based on such propensities on a single-residue basis.
The scoring function that was used to determine the empirical null distribution and assign P-values was the number of residues with high structural and functional propensities. The thresholds for these high propensity scores were determined separately for each individual model described in Table 2 during the training phase (low false positive rates; here, 10%). On the other hand, the prior probabilities that a particular residue has a specific property were estimated using the AlphaMax algorithm (Jain et al., 2016).
2.7 MutPred-LOF output
For every mutation input into MutPred-LOF, the model returns a score between zero and one, where variants with higher scores are more likely to be pathogenic. In addition, MutPred-LOF returns up to five structural and functional mechanisms that are impacted in the affected region of the protein, and have significant prior-corrected P-values (less than 0.05). For the purposes of classification, we provide three score thresholds to aid users in discriminating between pathogenic and neutral variation, at different levels of false positive rate (FPR): 0.40 (10% FPR), 0.50 (5% FPR, recommended), 0.70 (1% FPR).
3 Results
3.1 Evolutionary conservation of loss-of-function variants
Conservation has consistently been shown to be a key signature of pathogenic variants (Ng and Henikoff, 2003). Unsurprisingly, these features were also shown to be informative across all variant types in our data. To highlight the differences in conservation between pathogenic and neutral loss-of-function variants, Figure 2 shows a representative plot created from one of the conservation metrics. The plots show the approximate probability density function of average unnormalized unweighted entropy in the vertebrate alignment for amino and carboxyl sides of the wildtype sequences. While the affected region in disease-associated variants is more conserved than that of the neutral variants, the similarity between distributions of conservation between amino and carboxyl sides indicates that the conservation of the protein as a whole plays a dominant role. Therefore, for such a feature to be sufficiently effective in discriminating between pathogenic and neutral variants, the training set would need to contain both types of variants for most proteins, which is currently not the case.
Fig. 2.
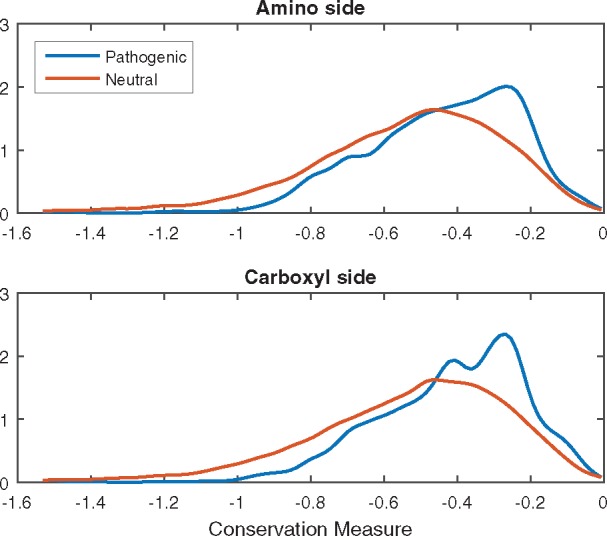
Approximate probability density functions of an average conservation measure of the wildtype protein (A) at the amino side of the variant and (B) at the carboxyl side of the variant for pathogenic (blue) and neutral (orange) variants. To ensure clarity, we omit proteins that contain both pathogenic and neutral variants from this figure. Proteins harboring disease variants are generally more conserved at both sides of the variant
An alternative metric to analyze sequence conservation between variation is the number of closely related proteins. Functional redundancy in the genome, as measured by the number of closely related paralogs has been shown to be associated with neutral loss-of-function variants (MacArthur et al., 2012) and genes with homologs that have 90% sequence identity are three times less likely to harbor disease variants (Hsiao and Vitkup, 2008). To test if our data were consistent with this trend, we counted sequences with high sequence identity in human and mouse proteomes. The average number of homologs in overlapping sequence identity regions for each class of loss-of-function variation are shown in Figure 3. In the human-human comparison, proteins containing pathogenic variants tend to have fewer similar proteins across all levels of sequence identity than proteins containing neutral variants. Broadly speaking, this suggests increased robustness of the system through functional redundancy; i.e., it allows very similar proteins to compensate for one another. On the other hand, human-mouse protein comparisons show the opposite trend. Proteins with disease-associated variants tend to have more homologs in the mouse genome than proteins with neutral variants. Although trends remain consistent, the magnitude between the average homolog count differs between the types of variant. This discrepancy may partially be due to biases in the data sets but may also reflect evolutionary constraints underlying sequences susceptible to each variant type.
Fig. 3.
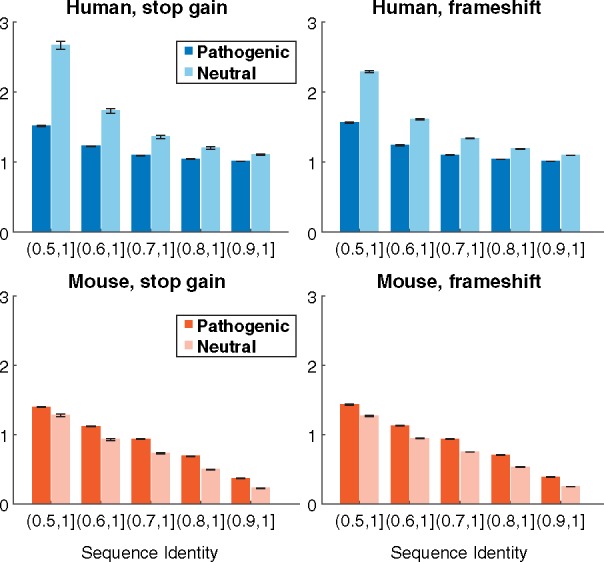
Homology profiles for the types of loss-of-function variants. Each plot shows the average number of sequences affected by pathogenic and putatively neutral variants, within a particular range of global sequence identity against human and mouse genomes
Generally, our results agree with previous observations suggesting the importance of evolutionary conservation for identifying disease-associated variants. However, we also find that disease variants are often located in more evolutionarily conserved proteins compared to the neutral variants suggesting limitations in using such information as a sole discriminator of pathogenicity of variants. The homology profiles from Figure 3 also support previous observations that proteins harboring disease variants tend to have fewer homologs in the human genome but more homologs in the genomes of model organisms compared to other proteins (Hsiao and Vitkup, 2008; Mushegian et al., 1997).
3.2 Prediction evaluation
Classifier performance is reported as the area under the ROC curve (AUC) and shown in Figure 4. In panel A, we show the AUC of alternative models based upon per-variant, per-protein and per-cluster cross-validation. The per-variant version of MutPred-LOF outperforms per-gene and per-cluster methods as a result of the model exhibiting over reliance on gene-specific features. The per-gene and per-cluster versions show comparable performance, and thus further analyses are carried out using the per-gene evaluation. Additionally, we assessed the performance of an alternative model utilizing per-cluster cross-validation with 25% sequence similarity and found similar performance to the 50% sequence identity threshold (data not shown). Figure 4B shows the performance of MutPred-LOF on the two types of loss-of-function variants separately. We see that the performance of stop gain variants is significantly lower than frameshifting variants, which have higher collective performance. We observed that models developed specifically for each variant type show similar, but worse collective performance than a single unified model (data not shown). Finally, we compared the performance of MutPred-LOF against three currently available tools in Figure 4C. We define a set of proteins which contain both neutral and pathogenic variants as bi-class proteins, and similarly derive a set of variants contained in those proteins as the bi-class subset. We observe that MutPred-LOF and CADD show reduced performance on the bi-class subset compared to the full set of variants whereas the other methods show significantly degraded performance on subset of variants in bi-class proteins. This suggests that MutPred-LOF is less dependent on the global protein-specific attributes that may simply be reflective of the signatures of disease genes. Although we have carried out as stringent a comparison as possible, community-wide assessments such as the Critical Assessment of Genome Interpretation (CAGI) will be able to further establish performance of all available methods on a common set of variants in the future.
Fig. 4.

Receiver operating characteristic (ROC) curves and Areas Under the ROC Curves (AUC). (A) Cross-validation performance of MutPred-LOF with per-variant, per-protein, and per-cluster cross-validation; (B) Cross-validation performance of MutPred-LOF for frameshifting and stop gain variants separately; (C) The performance for other methods based upon the testing set. Black curves represent the performance of MutPred-LOFʹ. The dotted line represents performance of each model on the subset of variants from bi-class proteins; (D) Proportion of high-scoring de novo variants implicated in neurodevelopmental disorders in the case and control datasets based upon 5 and 10% false positive rate thresholds. The P-value derived from Fishers exact test is shown above
3.3 Performance of feature sets
We investigated the predictive capacity of each individual feature subset on the performance of MutPred-LOF for frameshifting and stop-gain variants. To accomplish this, we generated a neural network model with the same parameters as the full MutPred-LOF but with a reduced feature set, shown in Table 3. Values in Table 3 correspond to features discussed in Section 2.3. Here we also observe metal binding and predicted GO terms show performance on par with conservation-based features. Any individual feature subset, particularly vector quantized functional features, are not sufficient to discriminate between pathogenic and neutral variants. However, if any feature set is removed from the training data then the AUC of the full model drops by several points (data not shown).
Table 3.
Per-feature evaluation: top ten performing feature sets
| Feature set | Full model |
|---|---|
| Predicted GO Terms | 0.729 |
| Maximum conservation | 0.707 |
| Metal binding | 0.660 |
| Structure and dynamics | 0.652 |
| Enzyme activity | 0.645 |
| Regulation | 0.641 |
| Macromolecular binding | 0.633 |
| Homology counts | 0.614 |
| Post-translational modification | 0.611 |
| Signal peptide and transmembrane | 0.610 |
For each set of features we train ensembles of neural networks with the same parameters in all models. The performance (AUC) of a model trained on a feature set is used to estimate the performance of each feature separately.
3.4 Phenotype-specific impact on structural and functional features
We identified structural and functional features that exhibit differences between pathogenic variants and the neutral background, reflected in a P-value with respect to a given feature. To ascertain the discriminative capacity of these P-values for individual proteins, we identified and analyzed several proteins that can represent typical use cases. For these examples, we selected proteins which contain both pathogenic and putatively neutral variants, to highlight functional differences underlying proteins with both disease-causing and neutral loss-of-function variants.
3.4.1 pcdh19
Several mutations in Protocadherin-19 (PCDH19) are included in the training data, including two putatively neutral variants from ExAC and dozens of pathogenic variants from HGMD, all of which have been predicted accurately in cross-validation. Mutations in PCDH19 have been identified as causative for early infantile epileptic encephalopathy 9, a disease shown to exhibit variable expressivity (Depienne and LeGuern, 2012). In this case, the two neutral variants occur towards the carboxyl-terminus representing an ideal use case of MutPred-LOF on a bi-class protein. In particular, the neutral variants occur in the final 50 amino acids of the protein and do not directly impact the primary Protocadherin-19 domain in the protein. Additionally, MutPred-LOF uncovers several molecular mechanisms significantly disrupted by these pathogenic variants including calcium binding, phosphorylation and palmitoylation, that are not identified in the neutral variants.
3.4.2 sim1
The Single-minded homolog 1 (SIM1) protein similarly contains both a pathogenic variant and several putatively neutral variants. Previously discovered loss-of-function mutations in SIM1 result in haploinsuffciency and subsequent hypodevelopment of paraventricular nuclei in the hypothalamus, and have been associated with severe early-onset obesity and Prader-Willi-like syndrome features (Bonnefond et al., 2013). The pathogenic variant abolishes the majority of a PAS domain, including a ubiquitination site (), whereas the neutral variants impact a portion of the C-terminal single-minded domain. These putatively neutral variants may still be associated with obesity if derived from obese ExAC participants, depending upon the inclusion criteria for particular studies. The neutral variants impact the C-terminal Single Minded domain, which has proposed relationship to transcriptional regulation of SIM1 and therefore may still have some clinical relevance (Ramachandrappa et al., 2013).
3.5 Performance on de novo variants in neurodevelopmental disorders
We applied MutPred-LOF to de novo loss-of-function variants that have been observed in whole exome and whole genome sequencing of families affected by neurodevlopmental disorder (de novo variant set and MutPred-LOF scores are available on the website). In this setting, the case variants may include a large fraction of non-pathogenic variants and so the performance of MutPred-LOF on this set cannot be accurately assessed in a binary classification framework (Jain et al., 2017). To this end, we utilized a Fisher’s exact test to determine if there is a significantly higher proportion of LOF variants predicted to be pathogenic in the case samples than in the control samples, shown in Figure 4D. For the threshold associated with 5% false positive rate, we find that 56% (547/970) of the case LOF variants are scored with high confidence to be pathogenic compared to only 46% (79/172) of the control variants (P = 0.0071). For the threshold associated with 10% false positive rate, we find that 86% (839/970) of the case variants are scored with high confidence to be pathogenic compared to only 80% (137/172) of the control variants (P = 0.0152). The excess of LOF variants has been previously observed in the patients with autism compared to their healthy siblings (Iossifov et al., 2012). The fact that we observe a significantly higher fraction of predicted pathogenic LOFs in the patients compared to controls suggests that LOF variants may have an important role in neurodevelopmental diseases.
4 Discussion
Understanding the repertoire of molecular alterations consequent to genetic variation is essential to advancing personalized medicine (Rost et al., 2016). Severe alterations in mRNA transcripts resulting from stop variants and frameshifting indels are particularly challenging because of their potentially major impact on the sequence of translated proteins as well as on their structure and function. To address these challenges, we assembled a large data set of human genetic variants and analyzed specific types of molecular alterations that could potentially be causative of underlying diseases. Using insights from this analysis, we developed a computational model MutPred-LOF, an extension to our variant predictor MutPred (Li et al., 2009), to discriminate between pathogenic and tolerated putatively loss-of-function variants. MutPred-LOF exploits detailed evolutionary and functional information to classify LOF variation from different and disparate contexts including severely pathogenic variation, pathogenic recessive variants in a heterozygous state, variants tolerated due to gene redundancy and sequencing errors (MacArthur et al., 2012). In addition to providing a classification score for pathogenic vs. tolerated variants, MutPred-LOF provides hypotheses regarding affected molecular events that might be responsible for pathogenicity of the variant.
4.1 Evolutionary conservation vs. structure and function
In the course of our study, we were particularly interested in understanding the influence of evolutionary conservation as well as structural and functional impact of the variant. Because pathogenic loss-of-function variants cover a relatively small subset of human genes, we found that a straightforward analysis may be misleading and is likely to recover signatures of known disease genes (Dalkilic et al., 2008), instead of properly accounting for the combined influence of the gene and variant features. To effectively include protein structure and function, we incorporated over 50 computational models that output positional propensities for several major types of structural and functional features. These models were integrated via a vector quantization-like approach into a rich feature representation. The estimated accuracy of MutPred-LOF suggests that the inclusion of additional features was beneficial in the modeling process.
4.2 Positive-unlabeled learning framework
Technically, our classification setting falls under the category of positive-unlabeled learning (Denis et al., 2005), with a further caveat that a fraction of positive data might be incorrectly labeled. Recent advances in machine learning suggest that, under mild assumptions, the traditional supervised models trained on positive vs. negative examples provide the same ranking as the (non-traditional) models trained on positive vs. unlabeled examples (Blanchard et al., 2010; Elkan and Noto, 2008; Menon et al., 2015). Moreover, if the class prior probability is known or can be estimated, one may further exploit the following: (1) there exists a monotonic relationship between the traditional and non-traditional posterior class distributions (Jain et al., 2016) and (2) the performance accuracy in the non-traditional setting can be corrected to reflect the performance accuracy in the traditional setting (Jain et al., 2017). Given these theoretical results, we decided to use the entire ExAC database as a set of ‘negative’ examples in our training procedure, with the reasoning that filtering out a subset of variants (e.g., rare variants) is more likely to be harmful by biasing the sample than class label noise.
4.3 A note on terminology
Loss-of-function variants are defined here as frameshifting and stop gain variants. The term can be considered a misnomer, due to the potential for the interpretation that ‘loss of function’ implies axiomatic loss of functional activity. Instead, we use the term to refer to a class of variants that are likely to result in profound impact on the protein, similar to the term ‘protein disrupting variant’. Loss-of-function variants, although frequently causing disease, are likely present in every human genome. Misinterpretation of the impact of a variant that would appear to result in loss of molecular function can lead to attributing phenotype to the wrong root molecular cause.
By using this terminology, we sought to emphasize that the extent of impact on function lies on a spectrum from the absolute abolition of function, to reduction of functional capacity and, finally, no phenotypic consequence. Adding an additional layer of complexity, we allowed for the fact that functional impact may or may not result in pathogenicity. One of the objectives behind the development of MutPred-LOF is to clarify the relationship between molecular function and pathogenicity by allowing potential users to make a more informed assessment based upon both pathogenicity prediction and impacted molecular function. To this end, we provided two separate scoring mechanisms, and embrace the complexity underlying loss-of-function variation.
4.4 Limitations
While MutPred-LOF showed good performance, shortcomings in the training data and method development may be a limitation. Context-based genetic information such as zygosity or haplotype are typically not known since publicly available databases are commonly stripped of this information to maintain participant anonymity. The mutational context including rescuing frameshifting mutations and relevant variation within the gene or pathway is important to consider in causative variant discovery. Population bias and undiscovered pathogenic variation in the neutral data set may also have unintended impact on the final model. Finally, in the method development step, we do not encode properties of the mutant protein sequence, thereby excluding outcomes such as splice site disruptions that may not damage the entire protein sequence downstream of the variant. Reduced performance on bi-class variants highlights difficulties in discrimination between variants in bi-class genes will continue to be of particular difficulty and should be further emphasized.
4.5 Final thoughts
We believe that our analysis provides new insights into the understanding of loss-of-function variants, especially the interplay between protein-specific and variant-specific features. MutPred-LOF encodes both types of features and shows the ability to differentiate between disease-causing and tolerated loss-of-function mutations, especially those occurring in the bi-class proteins. As such, MutPred-LOF allows for the specialized interpretation of one of the most impactful forms of genetic variation to facilitate variant and genome interpretation.
Funding
This work has been supported by the National Institutes of Health through the awards R01LM009722 (SDM), R01MH105524 (LMI and PR), R21MH104766 (LMI), R01MH109885 (LMI), RO1MH076431 (JS) and the Indiana University Precision Health Initiative. MM and DNC received financial support from Qiagen Inc. through a License Agreement with Cardiff University.
Conflict of Interest: none declared.
References
- Altschul S.F. et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard G. et al. (2010) Semi-supervised novelty detection. J. Mach. Learn. Res., 11, 2973–3009. [Google Scholar]
- Bonnefond A. et al. (2013) Loss-of-function mutations in sim1 contribute to obesity and prader-willi-like features. J. Clin. Invest., 123, 3037–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark W.T., Radivojac P. (2011) Analysis of protein function and its prediction from amino acid sequence. Proteins, 79, 2086–2096. [DOI] [PubMed] [Google Scholar]
- Clark W.T., Radivojac P. (2014) Vector quantization kernels for the classification of protein sequences and structures. Pac. Symp. Biocomput., 19, 316–327. [PubMed] [Google Scholar]
- Cline M., Karchin R. (2011) Using bioinformatics to predict the functional impact of snvs. Bioinformatics, 27, 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalkilic M. et al. (2008) From protein-disease associations to disease informatics. Front Biosci., 13, 3391–3407. [DOI] [PubMed] [Google Scholar]
- de Ligt J. et al. (2012) Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med., 367, 1921–1929. [DOI] [PubMed] [Google Scholar]
- De Rubeis S. et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature, 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis F. et al. (2005) Learning from positive and unlabeled examples. Theor Comput. Sci., 348, 70–83. [Google Scholar]
- Depienne C., LeGuern E. (2012) PCDH19-related infantile epileptic encephalopathy: an unusual X-linked inheritance disorder. Hum. Mutat., 33, 627–634. [DOI] [PubMed] [Google Scholar]
- Dinkel H. et al. (2014) The eukaryotic linear motif resource ELM: 10 years and counting. Nucleic Acids Res., 42, D259–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douville C. et al. (2016) Assessing the pathogenicity of insertion and deletion variants with the Variant Effect Scoring Tool (VEST-Indel). Hum. Mutat., 37, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkan C., Noto K. (2008) Learning classifiers from only positive and unlabeled data. In Proceedings of the 14th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, KDD 2008, pages 213–220, New York, NY, USA. ACM.
- Epi4K Consortium and Epilepsy Phenome/Genome Project (2013) De novo mutations in epileptic encephalopathies. Nature, 501, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EuroEPINOMICS-RES Consortium, Epilepsy Phenome/Genome Project and Epi4K Consortium (2014) De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am. J. Hum. Genet., 95, 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman L. et al. (2015) DDIG-in: detecting disease-causing genetic variations due to frameshifting indels and nonsense mutations employing sequence and structural properties at nucleotide and protein levels. Bioinformatics, 31, 1599–1606. [DOI] [PubMed] [Google Scholar]
- Fromer M. et al. (2014) De novo mutations in schizophrenia implicate synaptic networks. Nature, 506, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilissen C. et al. (2014) Genome sequencing identifies major causes of severe intellectual disability. Nature, 511, 344–347. [DOI] [PubMed] [Google Scholar]
- Girard S.L. et al. (2011) Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet., 43, 860–863. [DOI] [PubMed] [Google Scholar]
- Guipponi M. et al. (2014) Exome sequencing in 53 sporadic cases of schizophrenia identifies 18 putative candidate genes. PLoS One, 9, e112745.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulsuner S. et al. (2013) Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell, 154, 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto R. et al. (2016) Whole-exome sequencing and neurite outgrowth analysis in autism spectrum disorder. J. Hum. Genet., 61, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao T.L., Vitkup D. (2008) Role of duplicate genes in robustness against deleterious human mutations. PLoS Genet., 4, e1000014.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Ng P.C. (2012) Predicting the effects of frameshifting indels. Genome Biol., 13, R9.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I. et al. (2012) De novo gene disruptions in children on the autistic spectrum. Neuron, 74, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I. et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature, 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S. et al. (2016). Estimating the class prior and posterior from noisy positives and unlabeled data. In Proceedings of the 30th Advances in Neural Information Processing Systems, NIPS 2016, pages 2693–2701. Curran Associates, Inc.
- Jain S. et al. (2017). Recovering true classifier performance in positive-unlabeled learning. In Proceedings of the 31st AAAI Conference on Artificial Intelligence, AAAI 2017, pages 2066–2072. AAAI.
- Jiang Y.H. et al. (2013) Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet., 93, 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D. et al. (2014) The UCSC Genome Browser database: 2014 update. Nucleic Acids Res., 42, D764–D770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M. et al. (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet., 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A. et al. (2012) Rate of de novo mutations and the importance of father’s age to disease risk. Nature, 488, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum M.J. et al. (2016) ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res., 44, D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M. et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B. et al. (2009) Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics, 25, 2744–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur D.G. et al. (2012) A systematic survey of loss-of-function variants in human protein-coding genes. Science, 335, 823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur D.G., Tyler-Smith C. (2010) Loss-of-function variants in the genomes of healthy humans. Hum. Mol. Genet., 19, R125–R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat L.E. (2004) Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol., 5, 89–99. [DOI] [PubMed] [Google Scholar]
- McCarthy S.E. et al. (2014) De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry, 19, 652–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon A.K. et al. (2015) Learning from corrupted binary labels via class-probability estimation. In Proceedings of the 32nd International Conference on Machine Learning, ICML 2015, pages 125–134.
- Mort M. et al. (2008) A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat., 29, 1037–1047. [DOI] [PubMed] [Google Scholar]
- Mushegian A.R. et al. (1997) Positionally cloned human disease genes: patterns of evolutionary conservation and functional motifs. Proc. Natl. Acad. Sci. USA, 94, 5831–5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale B.M. et al. (2012) Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature, 485, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P.C., Henikoff S. (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res., 31, 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak B.J. et al. (2011) Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet., 43, 585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak B.J. et al. (2012a) Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science, 338, 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak B.J. et al. (2012b) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature, 485, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J., Grishin N.V. (2001) AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics, 17, 700–712. [DOI] [PubMed] [Google Scholar]
- Pejaver V. et al. (2014) The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci., 23, 1077–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng K. et al. (2006) Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics, 7, 208.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radivojac P. et al. (2004) Protein flexibility and intrinsic disorder. Protein Sci., 13, 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radivojac P. et al. (2006) Calmodulin signaling: analysis and prediction of a disorder-dependent molecular recognition. Proteins, 63, 398–410. [DOI] [PubMed] [Google Scholar]
- Ramachandrappa S. et al. (2013) Rare variants in single-minded 1 (sim1) are associated with severe obesity. J. Clin. Invest., 123, 3042–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A. et al. (2012) Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet, 380, 1674–1682. [DOI] [PubMed] [Google Scholar]
- Rausell A. et al. (2014) Analysis of stop-gain and frameshift variants in human innate immunity genes. PLoS Comput. Biol., 10, e1003757.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedmiller M., Braun H. (1993) A direct adaptive method for faster backpropagation learning: the RPROP algorithm. Proc. IEEE Internat’l. Conf. Neural Netw, 1, 586–591. [Google Scholar]
- Risso D. et al. (2014) Genetic variation in taste receptor pseudogenes provides evidence for a dynamic role in human evolution. BMC Evol. Biol., 14, (198.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronemus M. et al. (2014) The role of de novo mutations in the genetics of autism spectrum disorders. Nat. Rev. Genet., 15, 133–141. [DOI] [PubMed] [Google Scholar]
- Rost B. et al. (2016) Protein function in precision medicine: deep understanding with machine learning. FEBS Lett., 590, 2327–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders S.J. et al. (2012) De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature, 485, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist C.J. et al. (2013) New and continuing developments at PROSITE. Nucleic Acids Res., 41, D344–D347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson P.D. et al. (2014) The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. Mar. 27. doi: 10.1007/s00439-017-1779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulem P. et al. (2015) Identification of a large set of rare complete human knockouts. Nat. Genet., 47, 448–452. [DOI] [PubMed] [Google Scholar]
- Suzek B. et al. (2007) Uniref: comprehensive and non-redundant uniprot reference clusters. Bioinformatics, 23, 1282–1288. [DOI] [PubMed] [Google Scholar]
- Thousand Genomes Project Consortium (2010) A map of human genome variation from population-scale sequencing. Nature, 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner T.N. et al. (2016) Genome sequencing of autism-affected families reveals disruption of putative noncoding regulatory DNA. Am. J. Hum. Genet., 98, 58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B. et al. (2011) Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat. Genet., 43, 864–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B. et al. (2012) De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat. Genet., 44, 1365–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen R.K. et al. (2015) Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med., 21, 185–191. [DOI] [PubMed] [Google Scholar]
- Yuen R.K. et al. (2016) Genome-wide characteristics of de novo mutations in autism. NPJ Genom. Med., 1, 160271–1602710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zia A., Moses A.M. (2011) Ranking insertion, deletion and nonsense mutations based on their effect on genetic information. BMC Bioinformatics, 12, 299.. [DOI] [PMC free article] [PubMed] [Google Scholar]