

Abstract
Motivation
Metagenomic read classification is a critical step in the identification and quantification of microbial species sampled by high-throughput sequencing. Although many algorithms have been developed to date, they suffer significant memory and/or computational costs. Due to the growing popularity of metagenomic data in both basic science and clinical applications, as well as the increasing volume of data being generated, efficient and accurate algorithms are in high demand.
Results
We introduce MetaOthello, a probabilistic hashing classifier for metagenomic sequencing reads. The algorithm employs a novel data structure, called l-Othello, to support efficient querying of a taxon using its k-mer signatures. MetaOthello is an order-of-magnitude faster than the current state-of-the-art algorithms Kraken and Clark, and requires only one-third of the RAM. In comparison to Kaiju, a metagenomic classification tool using protein sequences instead of genomic sequences, MetaOthello is three times faster and exhibits 20–30% higher classification sensitivity. We report comparative analyses of both scalability and accuracy using a number of simulated and empirical datasets.
Availability and implementation
MetaOthello is a stand-alone program implemented in C ++. The current version (1.0) is accessible via https://doi.org/10.5281/zenodo.808941.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Metagenomics is the study of genomic content obtained in bulk from an environment of interest, such as the human body (Huttenhower and Human Microbiome Project Consortium, 2012), seawater (Venter et al., 2004), or acidic mine drainage (Tyson et al., 2004). Metagenomics studies often generate tens of millions of sequencing reads in order to capture the presence of microbial organisms and quantify their relative abundances, rendering the classification and analysis of these data a logistical challenge.
One of the major computational challenges in the analysis of metagenomic data is the classification of each sequencing read into the most-specific biological taxon to which sequence conservation supports its assignment. Specifically, a read is classified as belonging to a taxon if it has high sequence similarity with the reference genomes collected for that taxon, a process made possible by the large deposits of reference sequences collected in recent years for a variety of microbial species. In 2014 alone, more than 10 000 sequence records were newly added to the NCBI RefSeq database thanks to the accessibility of high-throughput sequencing technology.
Existing classification methods can be divided into two broad categories: alignment-based and alignment-free. The former approach, implemented most popularly as BLAST (Altschul et al., 1990), assigns each read to the taxon that affords the best alignment with its reference genomes. Several methods, including MEGAN (Huson et al., 2007), PhymmBL (Brady and Salzberg, 2009) and NBC (Rosen et al., 2011), apply additional machine-learning techniques to BLAST results to increase classification accuracy. These methods are often slower than BLAST alone, rendering them computationally prohibitive for large-scale analysis of many millions of short reads. However, the recent development of Centrifuge (Kim et al., 2016) has significantly improved the scalability of the alignment-based algorithm using FM-index. Besides using genomic sequences as reference, the recently published tool Kaiju (Menzel and Krogh, 2015) performs alignments towards protein sequences, achieving faster classification speed than existing tools.
The other line of work, including but not limited to LMAT (Ames et al., 2013), Kraken (Wood and Salzberg, 2014) and Clark (Ounit et al., 2015), classifies a read using exact k-mer matches between the read and the collection of reference sequences belonging to the target taxon, thereby avoiding inefficient base-by-base alignment while maintaining a sensitivity and specificity comparable to the alignment-based approach. This approach is generally faster than alignment-based methods and allows for greater flexibility in reference material because it requires only the collection of k-mers extracted from reference sequences belonging to each taxon. Thus k-mers extracted from DNA or RNA sequencing data can be included as reference material without being assembled, increasing the sensitivity of the algorithm in capturing natural variants that are often missed using reference genomes alone.
The above alignment-free algorithms rely on the use of indexing structures for k-mer matching. For example, Kraken indexes its lexicographically sorted k-mer database using a minimizer offset array, while Clark uses a hash table to store the mapping between a k-mer and its classification information. Both Kraken and Clark require computers with large memory to support the construction of their indexing structures (at least 120 GB RAM) and the classification of reads (at least 70 GB RAM) to achieve optimal classification accuracy. Although there exist variations on both algorithms requiring smaller memory footprints, they often afford significantly lower sensitivity or slower execution speed compared to the full version. For this reason, the ever-increasing amount of sequencing and reference-genome data call for tools with better scalability in both memory and computation.
In this paper, we present a new algorithm, dubbed MetaOthello, for taxonomic classification of sequencing reads. Our algorithm builds upon taxon-specific k-mer signatures to support direct assignment to any level in the taxonomy. It employs a novel data structure, l-Othello, to support ultra-fast k-mer classification, achieving at least an order-of-magnitude improvement in speed over the state-of-the-art methods, Kraken and Clark, and [runs] three times faster than Kaiju. In the meantime, MetaOthello also substantially reduces the memory footprint, typically requiring only one-third [the memory] of the aforementioned methods. This modest memory requirement allows our algorithm to run on typical lab servers with 32 GB RAM, rendering it more accessible to biological researchers than those with memory requirements achievable only by supercomputers. Additionally, our algorithm is capable of conducting hierarchical top-down taxonomic classification and delivers performance competitive to, if not better than, other algorithms in both sensitivity and specificity as validated by benchmarking on a variety of datasets.
2 Algorithms
2.1 k-mer taxon signatures
A k-mer is a length k subsequence of genomic sequences; for any sequence of length L, there exist a maximum of possible k-mers. Metagenomic reference material consists of one or more complete reference genomes belonging to an organism. Increasingly sophisticated sequencing techniques have permitted discovery of distinct reference genomes for a single species of organism, thereby capturing genomic variations that are often important to the functionality of the microbial species. The number of genomes (whether draft or complete) available as metagenomic reference material increases with each new discovery. If we consider each dataset as a collection of k-mers, a given taxon can be described by the set of k-mers present in the reference sequences belonging to its taxonomic subtree. The problem of classifying a metagenomic read thus simplifies to the identification of the taxon that best matches the set of k-mers associated with the target read. When k is sufficiently large (e.g. ), the majority of k-mers are unique to the species carrying them. These species-specific k-mers may serve as signatures, directly implicating the appropriate taxonomic classification. However, a significant proportion of k-mers is present in multiple species, making them unique only to higher-ranking taxa. In this paper, we formalize the taxonomic specificity of a k-mer as the signature of a taxon: A k-mer is considered to be a signature of a taxon if (i) the k-mer does not appear in any genomic references belonging to ancestors or siblings of the target taxon, but only to sequences belonging to the taxon’s subtree, and (ii) the k-mer is not a signature of any lower-ranked taxon in the subtree. Equivalently, the taxon evincing a k-mer signature is the lowest common ancestor (LCA) of all species in the taxonomy whose reference genomes contain that k-mer (Ames et al., 2013; Tu et al., 2014).
In this way, as illustrated in Figure 1A, the set of all k-mers present in the genomic references of a taxonomy can be divided into disjoint collections, each of which contains the set of signature k-mers belonging to a single node in the taxonomy tree. Formally, let S be the set of all k-mers present in genomic references annotated by the taxonomy and let T = be the taxa (nodes) present in the taxonomy. Then S can be divided into disjoint sets, , where for any node , St corresponds to the set of k-mer signatures belonging to taxon t. Thus, there exists a mapping, , such that g(s) = t if the k-mer, , is a signature of the taxon, .
Fig. 1.

Illustration of MetaOthello algorithm. (A) An example of taxonomy with reference sequences in the leaf nodes. 3-mers that are signatures to each taxon are highlighted in bold font with different shades. (B) A two-step approach to read classification (Color version of this figure is available at Bioinformatics online.)
2.2 k-mer classification with l-Othello
The core data structure of MetaOthello is called l-Othello. l-Othello is essentially a hashing classifier, first designed for fast-forwarding information base queries (Yu et al., 2016). It is capable of classifying a key to the appropriate member of a large collection of categories with high efficiency in memory and speed. In our particular application, l-Othello supports the mapping between a k-mer signature and the corresponding taxon.
2.2.1 Overview of the l-Othello data structure
l-Othello maintains a query function between any given k-mer and a taxon: , where SU is the universal set of k-mers (i.e. ). l is determined by the total number of taxa T, where . The algorithm satisfies the following properties: (i) l-Othello is always able to retrieve the correct taxon ID corresponding to a valid k-mer signature; that is, for any , where t is the ID of the taxon to which s is specific. (ii) When provided an alien k-mer (i.e. a k-mer that does not appear in the reference sequences), l-Othello is able to recognize the alien with high success rate but may carry a slight risk of assigning it to a random taxon in the taxonomy.
Figure 2 shows an example l-Othello structure. An l-Othello data structure works by maintaining a pair of hash functions, and two arrays of l-bit integers, A and B. and , where ma and mb are integer values determined during l-Othello construction. The relationships among the elements of A and B can be viewed as a bipartite graph , where nodes in U and V correspond to elements in A and B respectively. A query of k-mer s on the graph yields a node index in U and a node index in V. The classification of k-mer s is determined by the values at and , via a ⊕ (bitwise ) operation:
The bit-wise operation of integers has the following property: For any l-bit integer x, .
Fig. 2.
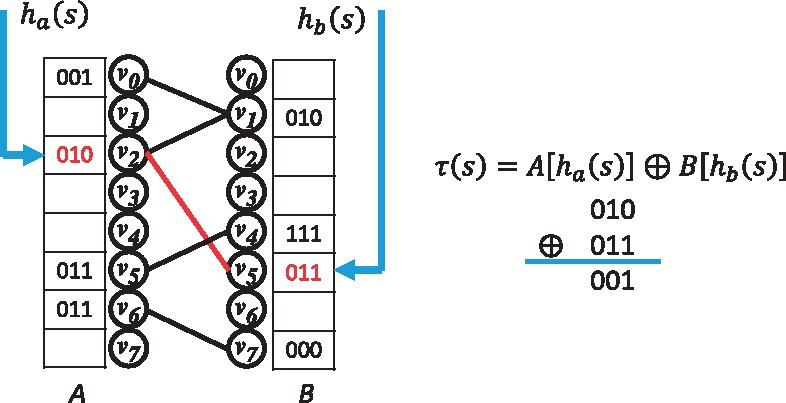
An example of l-Othello with l = 3 classifying n = 5 k-mers. Left: Bipartite graph G and corresponding bitmaps A and B. Each edge in G represents a k-mer. Hash functions ha and hb map the k-mer s into corresponding locations in A and B. Right: Query of s returns
When l-Othello is properly constructed, for any k-mer specific to taxon t, i.e. . The success of l-Othello relies on assigning bitmap values on both sides of the bipartite graph, where the ⊕ operation between two nodes can directly generate the class membership. Setting the bitmap of two nodes with no initial values is fairly simple and can be achieved in multiple ways. For example, when l = 1 and assuming the membership of a key is 1, we can assign the bit values of the two nodes ui and vj as either or . However, if the value of one node has already been determined by another key, then only the remaining value is altered. For example, if is already set as 0, given , then . In the worst-case scenario, both and have already been determined by their involvement with other keys. This situation creates a cycle in the graph when edge ui and vj is added, possibly resulting in a conflict and failed assignment of a bit value. When a conflict arises, we have two options: remove the k-mer or select a different hash function. Theorem 1 shows that for a randomly selected pair of hash functions , the probability of G being cyclic is extremely low. In our experiment, fewer than 100 k-mers among 6 billion were removed due to conflicts. Additionally, because multiple k-mers manifest in one read, losing one k-mer does not significantly affect the accuracy of the algorithm, so we may omit k-mers whose inclusion would cause a conflict.
Theorem 1 Suppose ha, hb are randomly selected from a family of random hash functions such that and . Given a set of k-mers , let . Construct a bipartite graph , where an edge if and only if there is a k-mer such that and . Let χ be the number of cycles in G. Then χ converges to a Poisson distribution with parameter , where .
The proof of Theorem 1 can be found in Supplementary Section S1. We recommend the values of ma and mb as follows: Let ma and mb be powers of 2, where ma is the smallest value such that , and mb is the smallest value such that . As a result, we have and . In practice, we can always expect the number of cycles in an l-Othello to be less than 2 with probability 97%, and smaller than 9 with probability higher than 99.999%.
2.2.2 Time and space complexity for l-Othello construction and query
Construction Complexity: The construction of the l-Othello data structure generally follows a depth-first traversal of the bipartite graph; thus the complexity is O(n), where n is the total number of k-mers. The memory complexity is O(ln), where l is the number of bits to encode the number of categories. We can further reduce the memory cost by dividing the set of k-mers into smaller groups based on prefixes, commonly of length 3, corresponding to g = 64 groups. Each group contains approximately k-mers. For each group, we build an l-Othello, using time with memory cost . In total, constructing g l-Othello structures still takes O(n) time.
Query Complexity: For each query , l-Othello computes two hash values and , and accesses two memory locations and . The time complexity is O(1). This procedure only includes a few basic arithmetic operations, resulting in an extremely fast execution speed.
2.2.3 On alien k-mers
An alien k-mer is defined as a k-mer that is not included during the construction of an l-Othello. In the context of taxonomic classification, they are those k-mers that are not included in any of the reference materials. Alien k-mers often arise due to noise or genomic sequences belonging to a novel species sampled by a sequence read. We have designed two strategies to detect alien k-mers. First, we would like to increase the randomness of assignments in the case of alien k-mers; secondly, we may add another bit in the bitmap (i.e. let ), doubling the number of categories, so that an alien k-mer has a much higher chance of being assigned to alien categories (i.e., categories not used by existing taxon labels).
Here we show how we may leverage the randomness of alien assignment to predict an alien k-mer within the l-Othello itself. We first discuss the query result for l = 1, and then we extend it to l-Othello. When l = 1, Othello classifies k-mers () into S0 and S1. Each element in A or B is a 1-bit value. For a query of an alien k-mer , l-Othello still returns a value . For alien keys, . Let a0 and a1 be the fraction of 0s and 1s in the bitmaps A respectively, i.e. . Similarly b0 and b1 are the fractions in B. Suppose ha and hb are uniformly distributed random hash functions, and is an arbitrary k-mer in the universal set; then returns 1 with probability . Similarly, returns 0 with probability .
For l-Othello, a similar property also holds. Let p(t) be the probability that the query of an alien k-mer returns exactly t. indicates . Note that ha and hb are uniform random hash functions and are not correlated. Hence,
where ax is the fraction of elements has value x in a and is the fraction of elements has value in B.
Given a particular l-Othello, we can always compute p(t) values for all using time . These p(t) values are affected by the occurrence frequency of each l-bit integer, namely ax and bx for all . In some cases, these values are not uniformly distributed, which may result in imbalance among p(t). Under such circumstances, we can tune these values by flipping the bitmaps of a connected component in the bipartite graph without changing for . In practice, we can always tune the values so that p(t) is of the same order of magnitude for all t, and all of them are approximately . Two tuning approaches are described in Supplementary Section S2.
We can also explicitly detect alien k-mers by by increasing l, thus intentionally expanding the number of targeted categories, where the majority of them are dummy (alien). Due to the randomness of class assignment in the presence of an alien, many alien k-mers are likely to fall in these dummy categories, and are thus recognized as alien. Formally, if for some is an alien. Thus, alien k-mers are recognized in this stage with probability .
2.3 Taxonomic classification of sequencing reads
As illustrated in Figure 1B, given any sequencing read, our algorithm iterates over each k-mer from the beginning of the read and, for each k-mer, retrieves the taxon to which it is specific using l-Othello. Taxonomic classification of the read is determined by assembling the taxa for all k-mers in the read. The classification is straightforward when all k-mers indicate the same taxon, but this is not often the case. Disparate taxa are considered to be consistent if they belong to the same path in the taxonomy, meaning that one assignment is the higher rank of the other. When these taxa belong to different branches, they represent conflicting information. The issue is further complicated by the possibility of false taxonomic information returned from querying alien k-mers, where the k-mer in the read does not appear in any of the reference sequences.
To tackle this challenge, we have designed a window-based classification approach. A window is defined as a sequence of consecutive k-mers that are assigned to the same taxon of a given level. The window-based approach guards against false-positive assignments due to alien k-mers. Assuming that the taxon ID returned by an alien k-mer is random, the chance of having two consecutive alien k-mers return the same taxon ID is
This value is very small, regardless of k. Additionally, each window corresponds to a maximum read subsequence that matches the reference sequences. Thus, the longer the window, the longer the subsequence match, and the less likely the match is random. In comparison, other algorithms such as Kraken and Clark count the total number of k-mer matches, regardless of their spatial distribution across the read.
If multiple taxon windows are available, MetaOthello scores each of them using the summed squares of window sizes as in the following formula; the taxon with the maximum score will be selected:
where denotes the number of k-mers in the ith window classified to taxon t.
A k-mer signature belonging to a taxon is also specific to its higher-ranking taxa, so at higher taxonomic ranks, there exist more k-mers to distinguish a taxon from its siblings. Thus, longer k-mer windows and more-accurate classifications are expected at higher taxonomic ranks. Under this assumption, a ‘top-down’ strategy is adopted during read classification. Given a read sequence, MetaOthello starts the classification at the top rank and continues the classification down the ranks until there does not exist a sufficiently large k-mer window supporting the level. Based on the k-mer distribution in each taxon, MetaOthello establishes a threshold on minimum window size when the classification on that taxon requires. Theorem 2 shows that the minimum window-size threshold can be precomputed for each taxon prior to read classification. The minimum window size required for a taxon is determined by the probability of an alien k-mer query on l-Othello returning a taxon rooted in t and the acceptable false-positive rate. The larger the size of the taxon subtree, the higher the probability that a random alien k-mer may match to t and thus the longer the window required for reliable classification. Additionally, a larger window size will be required in order to lower the false-positive rate.
Theorem 2. Given a user-defined false-positive rate λ and the total read number M, the minimum window-size threshold required for a taxon t can be computed as , where pt denotes the probability that an alien k-mer query on l-Othello returns a value in the taxon subtree with root t.
The proof is presented in Supplementary Section S3. For example, when t is a genus-level node, supposing l = 12, then . Given 10 million reads and suppose , then , and only windows larger than three will be taken into consideration when determining the read assignment.
3 Experimentation and evaluation
3.1 Classification accuracy and the relative abundance of taxon-specific k-mers
Accurate classification of a read to a taxon is largely dependent upon the presence of k-mer signatures. Thus the abundance of these signature k-mers (i.e. the proportion of taxon-specific k-mers among all k-mers present in the reference sequences for the taxon) becomes an important indicator of the capability of our algorithm. Thus we first investigate the correlation between classification accuracy and the relative abundance of taxon-specific k-mers.
Classification accuracy is computed as the fraction of reads assigned correctly. Using a next-generation sequencing (NGS) read simulator called ART (Huang et al., 2012), we simulated 10 000 reads for each of 2629 reference genomes in the NCBI RefSeq bacterial genome database, for a total of 26 290 000 reads. The database is available at ftp://ftp.ncbi.nih.gov/genomes/archive/old_refseq/Bacteria/. Each read is paired-end and of length 100 bp with a fragment size of 250 bp, generated using the default error profile for the HiSeq platform. Figure 3 shows the read-classification accuracy for each species as a function of species-specific k-mer proportion, where read assignments were generated by the MetaOthello algorithm using 20-mers or 31-mers, respectively. The scatter plot for either k-mer size demonstrates that the vast majority of all species manifest more than 50% 20-mers that are species-specific, and almost all species have 75% species-specific 31-mers. Although in general more species-specific k-mers afford better classification accuracy, these results suggest that the presence of 50% or more species-specific k-mers affords suitably high classification accuracy, thereby demonstrating the utility of k-mer signatures in classifying metagenomic reads.
Fig. 3.

Correlation between species-specific k-mer signatures and classification accuracy when k = 20 (A) and k = 31 (B). In each panel, the central figure depicts the correlation between species-specific k-mer proportion and read-classification accuracy for all species; the top histogram shows the distribution of species as a function of species-specific k-mer proportion, and the right histogram shows the distribution of classification accuracy for all species
To investigate how the window-based approach improves MetaOthello’s performance over the widely adopted count-based approach as in Kraken and Clark, we implemented a count-based version of MetaOthello, and compared its performance against that of the window-based version executed on the same datasets. The results demonstrate the clear advantages of a window-based approach over count-based, especially on assignment precision. This suggests that the window-based approach is more effective at eliminating the false positives caused by alien k-mers. Detailed results are reported in Supplementary Section S5.
3.2 Comparison with state-of-the-art tools
We now assess the performance of MetaOthello in comparison to three state-of-the-art tools: Kraken (version 0.10.5 beta), Clark (version 1.2.3) and Kaiju (version 1.4.4). Besides the newly published tool Kaiju, Kraken and Clark were chosen based on the recommendation of a recent benchmarking paper (Lindgreen et al., 2016), which evaluated 14 tools using six datasets and subsequently declared Kraken and Clark the best performers over Genometa (Davenport et al., 2012), GOTTCHA (Freitas et al., 2015), LMAT (Ames et al., 2013), MEGAN (Huson et al., 2007, 2011), MG-RAST (Meyer et al., 2008), the One Codex webserver, taxator-tk (Dröge et al., 2015), MetaPhlAn (Segata et al., 2012), MetaPhyler (Liu et al., 2010), mOTU (Sunagawa et al., 2013) and QIIME (Caporaso et al., 2010). The comparison was benchmarked against three publicly available datasets: HiSeq, MiSeq and SimBA5. The same datasets have been used multiple times to evaluate a number of metagenomic classification tools, including Kraken in previous studies (Wood and Salzberg, 2014). All tools were executed using the same reference database (NCBI RefSeq as of October 1st, 2016), and all other parameters follow the default settings.
3.2.1 Classification accuracy
We first compare classification accuracy. Three different k-mer lengths (20-mer, 25-mer and 31-mer) are used to assess the performance relationship with k-mer size for Kraken, Clark and MetaOthello; Kaiju is not a k-mer-based algorithm. To facilitate the comparison and to mimic the sequencing data generated by current platforms, we discarded reads shorter than 36 bp and those whose taxon is not included in the reference taxonomy.
Reads were classified by each algorithm at three taxonomic levels: phylum, genus and species. MetaOthello, Kraken and Kaiju were able to classify reads at the three levels simultaneously, while Clark required three separate runs to conduct similar classifications. Thus for Clark, results from these runs were merged for the purpose of direct comparison. Precision and sensitivity were computed at each of the three classification levels. Precision is defined as the ratio between correctly assigned reads and the total number of reads in an assignment; sensitivity is calculated as the fraction of total reads assigned correctly. F-score (i.e. the harmonic mean of precision and sensitivity) was also calculated to quantify the balance between these two metrics. Results of the comparison are shown in Table 1.
Table 1.
Accuracy of read taxonomic classification in terms of precision, sensitivity and F-score
| Phylum | Genus | Species | |||
|---|---|---|---|---|---|
| Prec/Sens/F-score | Prec/Sens/F-score | Prec/Sens/F-score | |||
| 20mer | 98.4/95.0/.967 | 97.2/92.5/.948 | 82.0/69.4/.751 | ||
| MetaOthello | 25mer | 99.4/92.2/.957 | 99.1/91.2/.950 | 84.2/69.1/.760 | |
| 31mer | 99.4/89.0/.939 | 99.3/88.2/.934 | 85.7/68.0/.758 | ||
| 20mer | 97.8/94.8/.963 | 96.1/92.0/.940 | 80.2/69.2/.743 | ||
| HiSeq | Kraken | 25mer | 99.7/92.3/.959 | 99.1/91.4/.951 | 83.7/69.4/.759 |
| 31mer | 99.7/88.3/.937 | 99.3/87.6/.931 | 85.4/67.6/.745 | ||
| 20mer | 97.7/95.5/.966 | 95.1/92.6/.939 | 76.4/69.5/.728 | ||
| Clark | 25mer | 99.7/92.1/.958 | 99.1/91.2/.950 | 83.5/69.2/.757 | |
| 31mer | 99.7/88.8/.940 | 99.3/88.1/.934 | 85.4/68.0/.757 | ||
| Kaiju | 99.4/68.7/.812 | 98.6/65.1/.785 | 89.2/34.7/.499 | ||
| 20mer | 99.2/97.5/.983 | 96.2/92.2/.942 | 91.8/78.6/.846 | ||
| MetaOthello | 25mer | 99.6/95.4/.975 | 97.4/91.4/.943 | 93.0/78.3/.850 | |
| 31mer | 99.6/92.9/.961 | 98.0/89.7/.937 | 93.8/77.2/.847 | ||
| 20mer | 99.0/97.5/.983 | 95.8/92.2/.939 | 91.0/78.9/.845 | ||
| MiSeq | Kraken | 25mer | 99.8/95.1/.974 | 97.4/91.2/.942 | 92.7/78.3/.849 |
| 31mer | 99.9/92.3/.960 | 98.0/89.3/.935 | 93.6/76.8/.844 | ||
| 20mer | 98.8/97.8/.983 | 94.4/92.5/.934 | 86.9/78.8/.826 | ||
| Clark | 25mer | 99.8/95.2/.975 | 97.1/91.5/.942 | 91.9/78.5/.847 | |
| 31mer | 99.9/92.7/.962 | 98.0/89.8/.937 | 93.4/77.3/.846 | ||
| Kaiju | 99.5/75.7/.860 | 98.5/68.0/.805 | 95.2/40.6/.570 | ||
| 20mer | 99.9/99.7/.998 | 99.6/95.8/.977 | 99.3/84.2/.911 | ||
| MetaOthello | 25mer | 99.9/98.2/.990 | 99.8/94.6/.971 | 99.5/83.1/.906 | |
| 31mer | 99.5/92.2/.957 | 99.5/88.7/.938 | 99.4/77.9/.873 | ||
| 20mer | 99.8/99.5/.996 | 99.4/95.9/.976 | 98.8/84.6/.912 | ||
| simBA5 | Kraken | 25mer | 99.9/98.5/.992 | 99.8/95.0/.974 | 99.5/83.8/.909 |
| 31mer | 99.9/94.2/.970 | 99.9/90.9/.952 | 99.7/80.0/.887 | ||
| 20mer | 99.8/99.6/.997 | 98.5/95.8/.971 | 94.4/84.2/.890 | ||
| Clark | 25mer | 99.9/98.4/.992 | 99.8/94.8/.973 | 99.4/83.4/.907 | |
| 31mer | 99.9/93.5/.966 | 99.9/90.2/.948 | 99.7/79.2/.883 | ||
| Kaiju | 99.6/75.6/.860 | 97.9/65.9/.788 | 96.5/46.7/.630 | ||
In general, longer k-mers enhance the precision of read classification but decrease sensitivity, as observed in MetaOthello, Kraken and Clark. Within each dataset, the overall winner (in bold) was the one with highest F1-score when considering across all three k-mer sizes. In phylum-level classification, MetaOthello outperforms the other algorithms in all three datasets all using 20-mers. At both genus and species levels, MetaOthello exhibits the best performance on two out of the three datasets using either 25-mers or 20-mers. Kraken performs the best in the remaining comparisons, followed closely by MetaOthello in both cases. In general, Kaiju delivered much lower (20–30%) sensitivity compared to the other three tools, due to its lack of capability in classifying reads from non-protein coding regions. We also ran Kaiju using two additional databases, nr and proGenomes, as recommended in Kaiju’s manual. Although higher sensitivities were achieved in some datasets at some taxonomic levels, the overall accuracy is still considerably lower than the other algorithms. Detailed results of the two additional runs are reported in Supplementary Section S6.
3.2.2 Runtime and memory
Speed benchmarks were performed using the servers from Lipscomb High-Performance Computing at the University of Kentucky. The servers are equipped with Dell R820, Quad Intel E5-4640 8-core (Sandy Bridge) @ 2.4 GHz and 512 GB/node of 1600 Mhz RAM. Each algorithm was executed using eight threads and k-mer lengths as specified previously; all other parameters follow the default settings. The speed for each tool is presented in Figure 4. In general, MetaOthello achieved the highest processing speed, clocking roughly 1 billion bases per minute. This figure represents an order-of-magnitude improvement over Kraken and Clark, the two most-rapid state-of-the-art tools within the category of alignment-free classifiers. Impressively, the high speed does not entail a compromise in the memory requirement. MetaOthello only consumes about one-third (peak memory 27 GB) of the RAM required by Kraken and Clark (peak memory 73 GB).
Fig. 4.
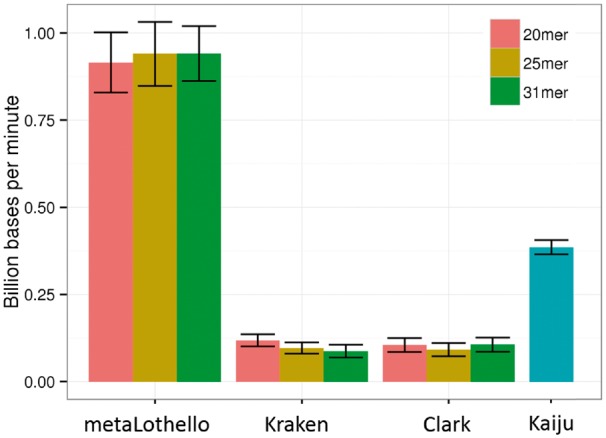
Performance comparison in terms of billion bases processed per minute using different k-mer lengths
The construction of the MetaOthello index from the NCBI RefSeq bacterial genome sequence database requires roughly 6 hours with peak memory usage up to 40 GB using 16 threads. In contrast, Kraken and Clark used 164 GB and 120 GB respectively for index construction, but both finished under 4 hours with 16 threads.
In summary, MetaOthello achieves a significant speedup with much smaller memory footprint in comparison with Kraken and Clark while delivering competitive or even superior performance in classification accuracy. While Kaiju is relatively scalable, it suffers from low sensitivity in classification.
3.3 Metagenomic classification of real datasets
3.3.1 Human Microbiome Project data
To assess the performance on real datasets of MetaOthello relative to Kraken, Clark and Kaiju, the three algorithms were run on sequencing data from three saliva samples (NCBI SRA accessions: SRS015055, SRS019120 and SRS014468) used in the Human Microbiome Project (Human and Project, 2012). We ran the three k-mer-based algorithms at each of three different k-mer length settings (20-mer, 25-mer and 31-mer) as with the simulated data. The three samples were analyzed separately, and the results were pooled together to assess the relative abundances of species. The top five most-abundant genera are presented in Table 2. The four tools reported the same five most-abundant genera: Streptococcus, Haemophilus, Prevotella, Veillonella and Neisserlia, all of which are known to be associated with human saliva. Interestingly, although the absolute abundance (i.e. the fraction of total reads assigned to a given genus) varies with k-mer size, the relative abundances remain stable except for Kaiju. The false-positive rate, however, cannot be assessed in this case.
Table 2.
The proportion of reads classified into the top-five genera by each algorithm using different k-mer lengths
| MetaOthello |
Kraken |
Clark |
Kaiju | |
|---|---|---|---|---|
| k-mer length | 20 25 31 | 20 25 31 | 20 25 31 | |
| Streptococcus | 14.32 13.24 12.12 | 14.58 13.29 12.18 | 14.25 13.15 12.02 | 10.05 |
| Haemophilus | 6.999 6.626 5.938 | 7.089 7.134 6.005 | 6.952 6.411 5.894 | 4.773 |
| Prevotella | 5.621 4.893 4.226 | 5.774 5.012 4.270 | 5.590 4.567 4.193 | 7.653 |
| Veillonella | 3.321 2.668 1.924 | 3.441 2.931 1.976 | 3.231 2.587 1.891 | 5.168 |
| Neisseria | 2.215 1.860 1.525 | 2.279 1.941 1.552 | 2.213 1.779 1.510 | 3.113 |
4 Conclusion and discussion
In this paper, we present MetaOthello, a novel metagenomic sequencing read classifier. MetaOthello leverages a novel probabilistic hashing structure, l-Othello, to conduct taxonomic classification using taxon-specific k-mer signatures. The algorithm delivers ultra-fast and memory-efficient solutions to k-mer-based taxonomic classification. Within the set of alignment-free approaches, MetaOthello achieves an order-of-magnitude improvement in classification speed relative to the fastest algorithms, Kraken and Clark, while reducing the RAM requirement from 73 GB to 27 GB. MetaOthello exhibits high sensitivity and precision competitive with Kraken and Clark, and in most cases achieves a better balance between the sensitivity and specificity (as quantified by F-score). It is also three times faster than the protein alignment-based method Kaiju and delivers much higher classification accuracy.
Besides the application of metagenomics, we also expect that our data structure l-Othello may benefit other k-mer-based sequencing-matching methods with its advantages in memory and speed efficiency.
Supplementary Material
Acknowledgements
This work was previously submitted to and accepted by The Seventh RECOMB Satellite Workshop on Massively Parallel Sequencing (RECOMB-Seq 2017). We thank the reviewers for their valuable comments and suggestions.
Funding
This work was supported by National Science Foundation [CAREER award grant number 1054631 to J.L.; grant CNS-1701681 to C.Q.]; and the National Institutes of Health [grant number P30CA177558 and 5R01HG006272-03 to J.L.].
Conflict of Interest: none declared.
References
- Altschul S.F. et al. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Ames S.K. et al. (2013) Scalable metagenomic taxonomy classification using a reference genome database. Bioinformatics (Oxford, England), 29, 2253–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady A., Salzberg S.L. (2009) Phymm and PhymmBL: metagenomic phylogenetic classification with interpolated Markov models. Nat. Methods, 6, 673–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J.G. et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat. Methods, 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport C.F. et al. (2012) Genometa – a fast and accurate classifier for short metagenomic shotgun reads. PLoS ONE, 7, e41224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröge J. et al. (2015) Taxator-tk: Precise taxonomic assignment of metagenomes by fast approximation of evolutionary neighborhoods. Bioinformatics, 31, 817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas T.A.K. et al. (2015) Accurate read-based metagenome characterization using a hierarchical suite of unique signatures. Nucleic Acids Res., gkv180.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W. et al. (2012) ART: A next-generation sequencing read simulator. Bioinformatics, 28, 593–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human T., Project M. (2012) A framework for human microbiome research. Nature, 486, 215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson D.H. et al. (2007) MEGAN analysis of metagenomic data. Genome Res., 17, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson D.H. et al. (2011) Integrative analysis of environmental sequences using MEGAN4. Genome Res., 21, 1552–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhower C. and Human Microbiome Project Consortium (2012) Structure, function and diversity of the healthy human microbiome. Nature, 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. et al. (2016) Centrifuge: rapid and sensitive classification of metagenomic sequences. bioRxiv, (054965). [DOI] [PMC free article] [PubMed]
- Lindgreen S. et al. (2016) An evaluation of the accuracy and speed of metagenome analysis tools. Sci. Rep., 6, 19233.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B. et al. (2010) MetaPhyler: Taxonomic profiling for metagenomic sequences. In: Proceedings – 2010 IEEE International Conference on Bioinformatics and Biomedicine, BIBM 2010, pp. 95–100.
- Menzel P., Krogh A. (2015) Kaiju: fast and sensitive taxonomic classification for metagenomics. bioRxiv, 7, 1–9. [DOI] [PMC free article] [PubMed]
- Meyer F. et al. (2008) The metagenomics RAST server a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics, 9, 386.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ounit R., Lonardi S. (2016) Higher classification sensitivity of short metagenomic reads with CLARK-S. Bioinformatics, 32, 3823–3825. [DOI] [PubMed] [Google Scholar]
- Ounit R. et al. (2015) CLARK: fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genomics, 16, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen G.L. et al. (2011) NBC: the Naive Bayes Classification tool webserver for taxonomic classification of metagenomic reads. Bioinformatics (Oxford, England), 27, 127–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N. et al. (2012) Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods, 9, 811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunagawa S. et al. (2013) Metagenomic species profiling using universal phylogenetic marker genes. Nat. Methods, 10, 1196–1199. [DOI] [PubMed] [Google Scholar]
- Tu Q. et al. (2014) Strain/species identification in metagenomes using genome-specific markers. Nucleic Acids Res., 42, e67–e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson G.W. et al. (2004) Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature, 428, 37–43. [DOI] [PubMed] [Google Scholar]
- Venter J.C. et al. (2004) Environmental genome shotgun sequencing of the Sargasso Sea. Science (New York, N.Y.), 304, 66–74. [DOI] [PubMed] [Google Scholar]
- Wood D.E., Salzberg S.L. (2014) Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol., 15, R46.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y. et al. (2016) A Concise Forwarding Information Base for Scalable and Fast Flat Name Switching. arXiv, (1608.05699).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.