

Abstract
Summary
BreakPoint Surveyor (BPS) is a computational pipeline for the discovery, characterization, and visualization of complex genomic rearrangements, such as viral genome integration, in paired-end sequence data. BPS facilitates interpretation of structural variants by merging structural variant breakpoint predictions, gene exon structure, read depth, and RNA-sequencing expression into a single comprehensive figure.
Availability and implementation
Source code and sample data freely available for download at https://github.com/ding-lab/BreakPointSurveyor, distributed under the GNU GPLv3 license, implemented in R, Python and BASH scripts, and supported on Unix/Linux/OS X operating systems.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The expanding scale and diversity of human sequence data present opportunities to investigate structural variants (SVs) in the human genome with unprecedented resolution (Feuk et al., 2006). Viral integration, a type of SV where the inserted sequence has a viral origin, has been implicated in the initiation and progression of a number of cancers (Martin and Gutkind, 2008). Yet, the examination, interpretation, and visualization of SVs, especially at human-virus boundaries (‘breakpoints’), is complicated by the large number of features associated with such events, including their size, orientation, state (inserted, inverted, deleted, copied, translocated), number of copies, source of inserted DNA, and whether the event results in a gene fusion.
There are a number of steps in an SV calling pipeline, including data preprocessing, SV discovery, verification, annotation and visualization (Guan and Sung, 2016). Existing SV visualization tools include linear genome browsers which indicate breakpoint positions along a single reference genome, and tools which represent breakpoints explicitly as arcs connecting linear or circular genomic segments (see Supplementary Material for references). A major limitation of the latter is that large numbers of breakpoints result in a thicket of overlapping lines, making details of breakpoint structure difficult to discern. The toolkit presented here encodes breakpoints as points on a grid, with coordinates indicating the junction position along the chromosome or virus. Such an approach provides a clear representation of breakpoint clusters and promotes efficient visual inspection of integration events and their fine structures.
BreakPoint Surveyor (BPS) is a new enterprise-level pipeline for examining breakpoint predictions, together with their associated structural variation and gene context. Its rendering engine is coupled with a flexible pipeline to detect structural variants, and it accommodates a variety of toolsets and analyses of whole genome sequencing (WGS) and RNA-sequencing (RNA-Seq) data. We illustrate the ability of BPS to combine multiple data types from The Cancer Genome Atlas (TCGA), including breakpoint predictions, copy number, gene/exon annotation and relative gene expression, into a comprehensive visual representation of the structural variation associated with viral integration in tumors and their effects on expression of genes adjacent to the event. Online documentation includes two additional reference workflows to illustrate functionality with human/human breakpoints.
2 Description of pipeline
Starting with WGS and RNA-Seq BAM files, BPS multi-stage workflow yields illustrations of inter- and intra-chromosomal SV events through a series of sequential data processing steps (Supplementary Fig. S1a). The pipeline consists of a series of Unix shell scripts that invoke core apps written primarily in R and Python (Supplementary Fig. S1b).
Preliminary steps include realignment and breakpoint detection: (i) If investigating human-virus breakpoints, WGS data must be realigned to a reference which includes both human and specific virus sequences so that virus reads are properly mapped and used to detect the presence of viruses. (ii) Context-specific breakpoint detection (i.e. human-virus) must be performed on the sequence alignment data using any suitable algorithm (Guan and Sung, 2016) to yield breakpoint positions (see Supplementary Material), with discordant read pair, Pindel and contig assembly techniques illustrated.
Breakpoint analysis and impact evaluation then involves several tasks: (iii) We combine any breakpoints which occur within a given distance of one another (50 Kbp in this example) on the same chromosome pair into a single integration event; positions of such events can be further refined manually. This clustering process defines the list of regions (‘PlotList’) for further analysis and visualization. (iv) Optionally, RNA-Seq-based expression values are calculated for genes near integration events, or preprocessed (e.g. TCGA RSEM) expression data are used. The algorithm compares expression of genes in cases against their corresponding controls to discern over- or under-expression (see Supplementary Material). (v) Breakpoints may be more precisely defined with other SV callers or contig assembly (see Supplementary Material).
Finally, figures are generated: (vi) Structure plots consist of four panels (Fig. 1a, Supplementary Fig. S2) illustrating breakpoint positions, copy number, and gene annotations for a selected target region. (vii) The expression levels of genes in the vicinity of integration events, relative to a population of controls, are illustrated in a gene expression plot (Fig. 1b, Supplementary Fig. S3). Figure 1 provides a comprehensive visual summary of several important features of virus integration events (see also Supplementary Figs S2–S8 for examples from additional samples and workflows).
Fig. 1.
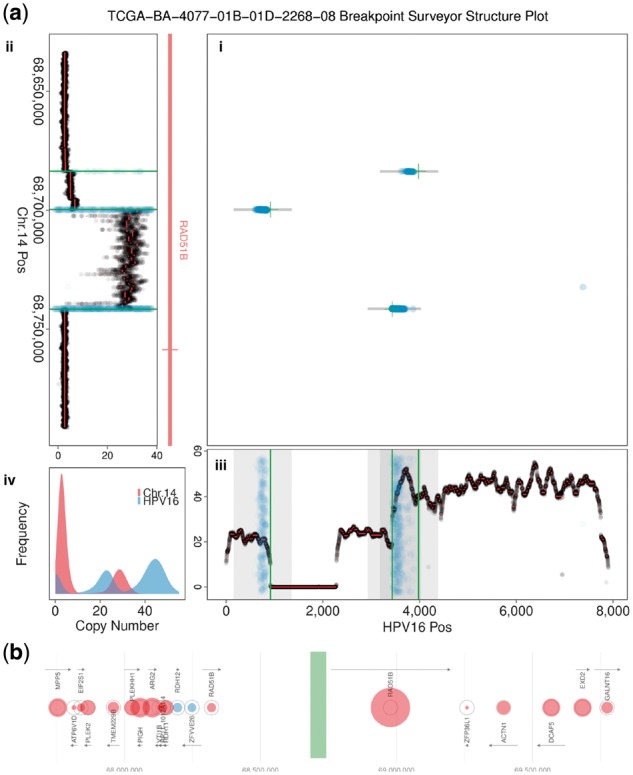
BPS plots illustrating HPV16 virus integration in a head and neck cancer sample. (a) Structure plot consists of four panels (see also Supplementary Fig. S2): (i) Breakpoint positions from three different methods, with X, Y coordinates given by the virus and human chromosome positions of the breakpoint, respectively. (ii, iii) Copy number near the integration event along the virus and human chromosome, aligned to match the axes of the breakpoint panel. Genes and exons near the integration event provide context. Here, integration occurs in an intronic section of gene RAD51B and copy number changes are coincident with breakpoints. (iv) Distribution of copy number for the virus and human chromosome. (b) Relative gene expression near integration event is indicated by circle size (see key in Supplementary Fig. S3), with gene position, size, orientation and name shown. Large red circles correspond to significantly upregulated gene expression. Here, expression of RAD51B exons downstream of integration event is significantly upregulated
BPS is a pipeline for integrating large, complex data sets with a scalable architecture supporting analysis from individual samples on a laptop to very large data sets on compute clusters. Additional example workflows provided with the online distribution demonstrate how BPS can be applied to the analysis of non-viral structural variants.
Supplementary Material
Acknowledgements
We acknowledge critical reading by Kuan-lin Huang and Robert Jay Mashl and valuable discussions with members of the TCGA Research Network.
Funding
This work was supported by the National Cancer Institute [R01CA178383, R01CA180006, 1U24CA211006-01, and 1U24CA210972-01 to L.D., R01CA172652 to K.C.]; and National Human Genome Research Institute [U01HG006517 to L.D.].
Conflict of Interest: none declared.
References
- Feuk L. et al. (2006) Structural variation in the human genome. Nat. Rev. Genet., 7, 85–97. [DOI] [PubMed] [Google Scholar]
- Guan P., Sung W.-K. (2016) Structural variation detection using next-generation sequencing data: A comparative technical review. Methods, 102, 36–49. [DOI] [PubMed] [Google Scholar]
- Martin D., Gutkind J.S. (2008) Human tumor-associated viruses and new insights into the molecular mechanisms of cancer. Oncogene, 27(Suppl 2), S31–S42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.