

Abstract
Motivation
The Human Protein Atlas (HPA) enables the simultaneous characterization of thousands of proteins across various tissues to pinpoint their spatial location in the human body. This has been achieved through transcriptomics and high-throughput immunohistochemistry-based approaches, where over 40 000 unique human protein fragments have been expressed in E. coli. These datasets enable quantitative tracking of entire cellular proteomes and present new avenues for understanding molecular-level properties influencing expression and solubility.
Results
Combining computational biology and machine learning identifies protein properties that hinder the HPA high-throughput antibody production pipeline. We predict protein expression and solubility with accuracies of 70% and 80%, respectively, based on a subset of key properties (aromaticity, hydropathy and isoelectric point). We guide the selection of protein fragments based on these characteristics to optimize high-throughput experimentation.
Availability and implementation
We present the machine learning workflow as a series of IPython notebooks hosted on GitHub (https://github.com/SBRG/Protein_ML). The workflow can be used as a template for analysis of further expression and solubility datasets.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Since its initial release in 2005, the Human Protein Atlas (Uhlén et al., 2010, 2015) has evolved into an extensive knowledge base capable of detecting 84% of the human proteome (v.15) through antibody-based proteomics. Reaching high proteome coverage requires tremendous numbers of recombinant protein fragments to be expressed and sampled, which, in turn, can lead to significant technical challenges for high-throughput experimental pipelines. Challenges, in part, stem from the complexity of producing high amounts of heterologous protein fragments in E. coli, due to a host of systems-level effects that impede expression, such as toxicity, codon bias, limiting factors in batch cultivation and formation of inclusion bodies, among others. In addition, the high-dimensionality of protein fragment expression systems makes systematically extracting biologically meaningful information for a single protein fragment, let alone thousands of protein fragments, a significant challenge. In the majority of cases, improvements in expression platforms are based on relatively few outputs (Rosano and Ceccarelli, 2014), which severely limits the capability of large-scale projects like the Human Protein Atlas. This motivates the development of in silico tools to better characterize the biological components of these complex systems, decrease the heavy reliance on iterative trial-and-error, and ultimately, bring this technology closer to other, more rational, engineering disciplines.
Relating a protein’s physical properties to its characteristic expression and solubility has paved the way for structural genomics initiatives (Williamson, 2000); analysis of standardized protein expression data (at the scale of over 16 900 protein-coding genes generated under uniform conditions) was not possible until recently. Datasets generated by the Human Protein Atlas provide new avenues for characterizing molecular-level properties of protein fragments and improving the performance of high-throughput experimental pipelines. For example, chemical and biophysical characteristics of protein fragments provide extensive insight into what factors influence a protein’s amenability to high-throughput experimentation (Goh et al., 2004).
While standardized datasets are becoming increasingly available, major impediments still prevent the realization of the potential impact of big data resources (Berger et al., 2013). Modern machine learning methods bring the promise of leveraging large-scale omics data to make accurate predictions (Angermueller et al., 2016; Berger et al., 2013), but the required skill sets to apply such methods extend outside the traditional scope of biochemistry and molecular biology. Thus, development of appropriate in silico tools and sufficient cross-disciplinary training resources are paramount for further progress in big data science (Rolfsson and Palsson, 2015). In this contribution, we hope to lower the barrier of entry into computational biology and data science by providing a computational framework, complete with IPython tutorials, upon which large-scale omics data from HPA can be analyzed and interpreted.
Here, we take advantage of two synergistic, accelerating domains of science—computational biology and machine learning—to develop a workflow that reconciles systems-level, multi-omic and computational biology with high-throughput protein expression and solubility. Using a machine learning approach, we probed the influence of biological and physical properties of over 45 000 recombinant Protein Epitope Signature Tags (PrESTs) used as antigens to generate antibodies for profiling tissue microarrays. Our workflow applies multiple machine learning-based methods, including linear regression, support vector machines (SVMs), random forest decision trees and neural networks, to characterize the diverse landscape of expression and solubility characteristics. Application of this workflow identified the roles of chemical and biophysical properties of the PrESTs in observed experimental expression and solubility levels. This characterization further facilitated the rational de novo selection of highly expressed protein tags to significantly reduce the total number of required experiments. The contributed workflow is available as an open-source tool, in the format of IPython notebooks.
2 Materials and methods
2.1 High-throughput expression of human protein fragments in E. coli
Human protein fragments were cloned, expressed in E. coli as fusion-proteins, and purified by immobilized metal affinity chromatography (IMAC). Briefly, PrEST sequences (ranging from 20 to 150 amino acids) representing a unique part of each human protein were selected (Berglund et al., 2008) and cloned (Lundqvist et al., 2015) as cDNA from human tissue lysates into a pET-vector for expression. E. coli BL21 and Rosetta were used for IPTG induced intracellular protein expression and the produced proteins were purified by IMAC (Tegel et al., 2009), and validated by mass spectrometry. Protein solubility was determined as previously published in Stenvall et al. (2005).
2.2 Characterizing molecular features of protein tags
A variety of features intended to capture a broad characterization of the data were calculated from the nucleotide and amino acid sequence for each PrEST, separated into five main functional categories. The mRNA features include nucleotide and codon composition, GC content of the full sequence and the first 30 nucleotides, presence of Shine-Dalgarno and Shine-Dalgarno-like sequences, the RNA folding energy of the full sequence and the first 40 nucleotides, and the tRNA adaptation index (tAI). The folding energy was calculated using Mfold (Markham and Zuker, 2008), and the tAI was calculated from the CodonR program (dos Reis et al., 2004). The primary structure features included the amino acid composition and the fraction of various types of residues (e.g. polar, aliphatic). In addition, various physical properties were computed using Biopython’s implementation of ProtParam (Cock et al., 2009; Gasteiger et al., 2003), such as isoelectric point, grand average of hydropathy (GRAVY), fragment length and charge. We used the SCRATCH suite to predict secondary structures for each PrEST from the primary sequence, using both a 3-letter and 8-letter system (Cheng et al., 2005). In addition, we utilized SCRATCH’s solvent accessibility predictor to calculate the solvent accessibility of each protein, and the hydrophobicity of solvent accessible and inaccessible regions. The disorder was predicted through three programs, DisEMBL HOTLOOPS, COILS and REM465 (Linding et al., 2003), DISOPRED3 (Jones and Cozzetto, 2015) and RONN (Yang et al., 2005). DISOPRED3 was also used to predict protein binding for each PrEST. A total of 147 features were calculated for each PrEST in the expression dataset, and a limited set of 38 features was calculated for the solubility dataset (See Table 1). Since the PrESTs are short fragments of whole human proteins, we assumed that the PrESTs did not maintain the same biological functions as the whole proteins, such as post-translational modifications or conformational state. The IPython notebook ‘create_feature_matrix.ipynb’ guides the interested reader through the calculation of these features.
Table 1.
List of features used in the expression data analysis and their sources
| Feature category | Features | Source software | Citations | Number of features |
|---|---|---|---|---|
| mRNA features | Codon composition (64 features) | Mfold, tAI | 81 | |
| Nucleotide composition (4 features) | ||||
| Number and fraction of SD and SD-like sequences | ||||
| on forward and reverse strands (8 features) | ||||
| GC content of full sequence and first 30 nt | ||||
| RNA folding energy of full sequence and first 40 nt | ||||
| tRNA adaptation index | ||||
| Amino acid properties | Amino acid composition (20 features)a | 29 | ||
| % Aliphatic, Uncharged Polar, Polar, Hydrophobic, Positive, Negative, Sulfur- containing, Amide-containing and Alcohol- containing residuesa | ||||
| PrEST physical properties | PrEST Lengtha, Isoelectric Pointa, Molecular Weighta | Biopython ProtParam | 9 | |
| Aromaticity and Instabilitya | ||||
| Grand Average of Hydropathy (GRAVY)a | ||||
| Absolute Charge and Charge per Residuea | ||||
| Average Absolute Charge per Residuea | ||||
| Structural predictions | 3-category Secondary Structures (3 features) | SCRATCH-1D | Cheng et al. (2005) | 19 |
| 8-category Secondary Structures (8 features) | ||||
| Solvent-accessible fraction | ||||
| Mean accessibility score | ||||
| GRAVY of outer and inner residues | ||||
| % Hydrophobic solvent-accessible residues | ||||
| % Hydrophobic solvent-inaccessible residues | ||||
| % Hydrophilic solvent-accessible residues | ||||
| % Hydrophilic solvent-inaccessible residues | ||||
| Disorder predictions | Fraction of Disordered Residues as predicted by: |
|
9 | |
| −DisEMBL COILS, HOTLOOPS and REM 465 | ||||
| −RONN and DISOPRED3 | ||||
| Average Disorder Index (RONN and DISOPRED3) | ||||
| Protein-binding Fraction (DISOPRED3) | ||||
| Protein-binding Index (DISOPRED3) |
Subset of features computed for solubility data
2.3 Targets and the initial dataset
The initial expression dataset reported 45 206 unique PrESTs expressed in E. coli BL21 and Rosetta with concentrations at 0–20 mg/mL and lengths ranging from 20 to 150 amino acids. In order to coarse-grain the analysis, PrESTs with concentrations in the top 25th percentile (11 301 PrESTs) were designated as ‘highly expressed’ and the PrESTs with concentrations in the bottom 25th percentile (11 302 PrESTs) were labeled as ‘poorly expressed’. The remaining PrESTs were removed from the dataset as their labels would be highly susceptible to noise. The solubility dataset reported 16 082 unique PrESTs in five solubility classes based on percentiles. The PrESTs in the highest class were designated as highly soluble (7667 PrESTs), whereas the bottom three classes were designated as insoluble (3324 PrESTs). One solubility class was removed to improve separation.
2.4 Machine learning algorithms
We applied four machine learning algorithms to the dataset: logistic regression, random forest classification, support vector machine (SVM) classification and a deep neural network. Additionally, we used a decision tree-based approach to determine which features generally separate highly expressed PrESTs from those that are poorly expressed. The effects of these features were measured using a Mann–Whitney-U test to determine the P-value of the separations. The outcome of such an analysis pipeline is to preferentially select PrESTs based on a subset of selective features to maximize their production in high-throughput experimentation.
2.5 Machine learning workflow
Each dataset was randomly split, with 70% of the data used to train the machine learning models, and the remaining 30% of the data used to validate the results. Each algorithm has a set of hyperparameters that define the configuration of the model. We optimized these hyperparameters using a subset of the training data, and then applied the final tuned model to the holdout testing data to compare the four algorithms. An ensemble model was generated by averaging the prediction probabilities from the top two models, and the final accuracy was measured using 5-fold cross validation. The entire workflow for this project has been compiled into a series of user-friendly, scalable IPython notebooks, located at https://github.com/SBRG/Protein_ML.
3 Results and discussion
3.1 High-throughput proteomics generates a large-scale protein expression dataset
Recently, Uhlén et al. (2015) presented a map of protein expression across 32 different tissue types in the human body (Fig. 1A). The high-throughput workflow utilized a combination of RNA sequencing technology and antibody profiling to understand the dynamic expression and functioning of over 20 000 protein-coding genes. While transcriptomics data provided quantitative information on gene expression levels across the tissues and organs, the antibody-based protein profiles show the spatial distribution at a single cell level for the corresponding protein in the various substructures and cell types of the tissues. To achieve the antibody-based profiles, a high-throughput approach to human proteomics was developed and applied to streamline the production of antibodies, using tissue microarray (TMA) technology for immunohistochemistry. The high-throughput approach consists of several steps, including: (i) using informatics to select unique regions of protein-coding genes for expression (Linding et al., 2003); (ii) cloning the sequences from RNA pools using specifically designed primers (Lundqvist et al., 2015) and (iii) producing the heterologous protein fragments, expressed under an identical T7 promoter in E. coli and purified by immobilized metal affinity chromatography (IMAC) before being quantified by bicinchoninic acid (BCA) assay (Tegel et al., 2009). The expression and/or solubility of these protein fragments were assessed (Fig. 1B) to determine whether the protein fragment could serve as a reliable protein tag, or a Protein Epitope Signature Tag (PrEST). These PrESTs serve as antigens with multiple epitopes for the generation of specific polyclonal antibodies (Agaton et al., 2003). The overall pipeline is illustrated in Figure 1C.
Fig. 1.
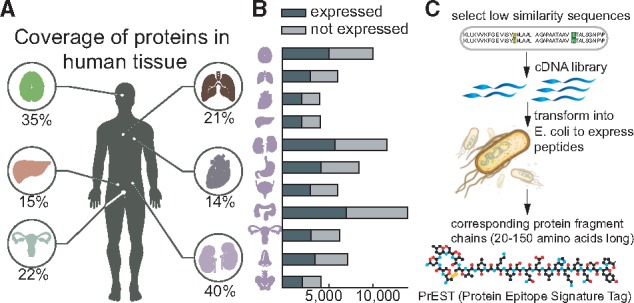
The data for this study was generated by the Human Proteome Atlas project. (A) Coverage of protein fragments (PrESTs) in the expression dataset for six (out of 44 total tissues) major biological tissues. (B) Distribution of highly and poorly expressed proteins across 11 tissue types. (C) Experimental workflow to produce PrESTs from a known protein. Low similarity sequences ranging from 20 to 150 amino acids from the protein are selected and transformed into E. coli to produce each PrEST
It became apparent that the initial selection of PrESTs determined the total number of experiments needed in later steps of the pipeline. In the first stage of this high-throughput protocol, short protein fragments are chosen from a human protein such that specific polyclonal antibodies can be generated. This is achieved by identifying a unique stretch of 20–150 amino acids with as low similarity as possible with respect to other proteins. Aside from this initial selection criteria, and avoidance of hydrophobic transmembrane regions, no other computational approaches have yet been applied to optimize the high-throughput expression of the fragments. As such, many of the PrESTs fail expression or solubility tests and require selecting different regions of the same protein. The process of selecting short amino acid sequences currently is carried out in a random manner, leading to an unnecessarily large number of trial-and-error experiments later on in the pipeline. We were therefore interested in knowing whether computational analyses of the PrEST expression profiles could aid and reduce the number of experiments needed.
The dataset used to generate the antibody-based profiles involves the tissue atlas of 44 different human tissues and organs with annotation data for 83 different cell types. To date, this is the most extensive database of proteins or protein fragments used for applications in machine learning to guide our understanding in global expression and solubility characteristics. This dataset differs from that of other datasets [for a recent review, please see (Habibi et al., 2014)] in several ways: (i) it is over 50-fold larger than that of any other reported dataset, which presents a novel challenge for both feature extraction (i.e. finding appropriate features that determine global expression behavior) as well as the learning capability of the machine learning algorithm; (ii) it is an in vivo standardized dataset, in which all expression and solubility information has been generated in the same high-throughput manner in the same laboratory; and (iii) the class sizes are similar, obviating the imbalance problem (Zhao et al., 2008a, 2008b).
3.2 Molecular characterization of protein tags
The first step in our computational workflow entails characterization of the protein fragments (PrESTs) themselves. As PrESTs are not whole proteins, we rely on a number of tools from computational biology to characterize their chemical and biophysical properties. As regulation of protein expression involves the interplay of transcription, translation, RNA degradation and protein degradation, we assessed each of these properties for 45 206 PrESTs. Different properties are computed on the basis of local and global mRNA sequence and amino acid sequence properties (Fig. 2). The descriptions of each of the properties and their biological implications for protein expression are described below. These computed properties are used as features to guide machine learning and classification of highly versus poorly expressed protein fragments. All features and the methods used to compute them are delineated in Table 1 and an IPython notebook titled ‘create_feature_matrix.ipynb’.
Fig. 2.
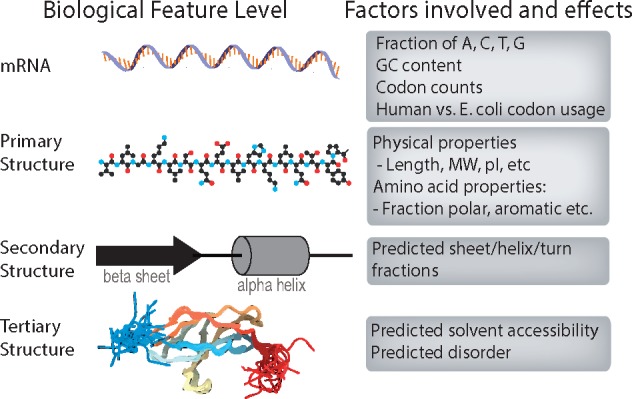
The spread of features, from mRNA to protein structure, examined to capture the many characteristics that may affect protein expression and solubility levels
3.2.1 Codon usage and mRNA sequence effects
Variation in mRNA sequence plays a key role in regulating protein expression in a range of different organisms, from E. coli to humans (Tuller et al., 2010; Sharp and Li, 1987; Li et al., 2014, 2012; Bazzini et al., 2016; Boël et al., 2016). We computed several properties related to both local and global mRNA sequence and codon usage bias. The properties include codon composition, nucleotide composition and GC content (globally and locally, within the first 30 nucleotides of the transcript) (Table 1). These properties were calculated directly from sequence information and had among the largest coefficients of variation. The nucleotide compositions all varied from <10% to 50%, and the GC content ranged from 20% to 80%.
3.2.2 mRNA folding and degradation
One of the codes embedded in mRNA specifies how the genetic code is translated into an amino acid sequence, whereas another code shapes mRNA stability (Bazzini et al., 2016; Kozak, 2005; Shakin-Eshleman and Liebhaber, 1988; Goodman et al., 2013; Kudla et al., 2009). Furthermore, translational regulatory information is contained in the codon code itself, influencing transcript decay (Bazzini et al., 2016) and the dynamics of ribosomal elongation (Boël et al., 2016). To address these properties, we have computed RNA folding energy, considering both the entire length of the transcript as well as the first 40 nucleotides (Table 1). The free energy of the most stable structure represents the folding energy of the sequence, which ranged from −4 to −221 kcal/mol.
3.2.3 tRNA and amino acid availability
The concentration of the cognate transfer RNA (tRNA) is known to correlate with codon usage frequency (Ikemura, 1981; Dong et al., 1996), and these parameters potentially play key roles in influencing in vitro protein elongation rates (Spencer et al., 2012; Caskey et al., 1968) and protein yield in vivo (Chen and Inouye, 1994; Deana et al., 1996). In this case, more frequent codons are thought to be translated more accurately, as their levels of cognate tRNAs are systematically higher.
3.2.4 Translational efficiency
The transient pausing of ribosomes has been shown to affect a variety of co-translational processes, including protein targeting and folding. Using ribosome profiling, we previously found that Shine-Dalgarno (SD) like sequences account for 20–22% of ribosome density at pause sites, which is consistent with recent studies (Mohammad et al., 2016; Ebrahim et al., 2016), and four times less frequent than what is found previous studies (Li et al., 2012). Thus, we included the number and fraction of SD and SD-like sequences on the forward and reverse strands of all transcripts encoding PrESTs in our dataset (Table 1).
3.2.5 Biophysical composition of protein fragments
Amino acid composition has been shown to influence mRNA stability, based on the stabilizing and destabilizing effects of certain synonymous codons (Bazzini et al., 2016). Furthermore, predictions of protein solubility routinely find strong correlations with the chemical properties of peptides and proteins (Smialowski et al., 2007; Diaz et al., 2010; Habibi et al., 2014). To this end, we computed both sequence-based and 3D structure-based properties of PrESTs in the dataset. Sequence-based properties include fragment length, percentage of physical composition (e.g. aliphatic, charged, polar, etc.), isoelectric point, molecular weight, aromaticity, hydropathy, charge distribution and others (Table 1). Structural-based properties include composition of predicted secondary structural content (e.g. alpha helix, coil, beta sheet), fraction of the PrEST that is predicted to be disordered, predicted solvent-accessible regions, and protein binding fraction. It is important to note that our workflow does not incorporate structural characterization (i.e. folding and 3D structure prediction) of protein fragments, as it is computationally demanding, requiring long-timescale molecular dynamics simulations (Piana et al., 2014).
3.3 Machine learning-based classification of protein expression
Once the dataset of molecular properties has been built, it is used to guide learning which PrESTs express well and to pinpoint which characteristics distinguish them from the lowly expressed counterparts. Our overall approach to identifying which properties affect expression and solubility is 2-fold. First, we employ four types of machine learning algorithms to determine the spread of sequence-, structure- and biophysical-based features that influence whether a protein fragment is expressed and/or soluble. Here, we have applied logistic regression, support vector machines (SVMs), random forest classifiers and a neural networks approach. Each of these methods has advantages and disadvantages when considering large-scale data mining applications. For example, logistic regression is a simple classification algorithm that produces results that are straightforward to interpret, however it is not capable of learning higher-order interactions between protein properties, which is often crucial for complex biological data. In contrast, SVMs and random forest ensemble classifiers introduce more complexity to the learning process and accommodate features that interact in unintuitive ways. While these approaches tend to achieve better results (i.e. prediction accuracy) than logistic regression, their predictions are more challenging to interpret (i.e. what specific features lead to high versus low expression), due to the effect of permutations of features. Finally, a deep neural network approach provides the most unbiased method of feature learning, which is especially useful when the important features are unknown. For more details, we direct the interested reader to a detailed review discussing the scope of these methods in predicting solubility of recombinant proteins in E. coli (Habibi et al., 2014). A general pipeline, common to most machine learning approaches, was applied in each of the four models consisting of three stages: (i) feature matrix construction; (ii) model training; and (iii) model testing (Fig. 3A). The outcome of this pipeline is a model prediction accuracy which indicates how predictive a subset or combination of features is with respect to the expression level of all PrESTs (See IPython notebook titled ‘classification_workflow.ipynb’).
Fig. 3.
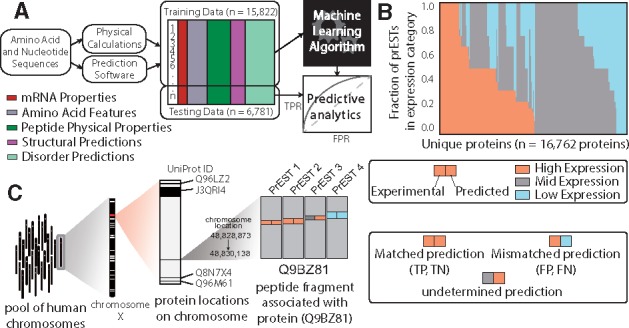
Machine learning-based approach to classify expression and solubility of protein fragments. (A) Classification workflow, starting with mRNA and amino acid sequences. The features described in Table 1 are generated for each PrEST and compiled into a feature matrix. Some of the data is used to train the models, and the rest is used to validate them. (B) The fraction of PrESTs in each expression level for each protein. (C) Multi-scale illustration of a protein in this study. Each protein is coded in a chromosome, and contains a number of PrESTs. Each PrEST has an experimental expression level and a predicted expression level from the machine learning algorithm
3.3.1 Ensemble classifiers outperform other machine learning-based approaches
Sequence and expression level were collected for 45 206 PrESTs, which were linked back to their representative proteins. We performed four separate machine learning analyses, as well as an ensemble approach (which combines the top two methods), on all the PrESTs in this dataset to discover features, described above, that are the strongest predictors for high versus low expression. We grouped the PrESTs into categories based on their overall expression level: high, moderate, or poor expression (Fig. 3B). To ensure the largest separation of features and most selective rules, we focused on the classification of highly and poorly expressed protein fragments, discarding the moderately expressed fragments. Each model was trained on 15 822 PrESTs (70% of the resulting dataset) to predict which features most accurately separated the highly expressed PrESTs from the poorly expressed PrESTs. The combination of features leading to the highest reported prediction accuracy was then tested on the remaining subset of PrESTs (30%) and the predicted expression levels were compared to the true expression levels (Fig. 3B).
Results from the ensemble classifiers approach indicated that this method outperforms all other machine learning methods, with a prediction accuracy of 70% and an AUC score of 0.77. Various performance metrics are reported in Table 2, and receiver operating characteristic (ROC) curves were constructed for all five models (Fig. 4A). Individually, the deep learning neural network and random forest algorithms outperformed the other two models, both with final accuracies of 69%, and the area under the ROC curve (AUC) of 0.76 and 0.75, respectively. We report accuracies in the range of 70–72%, which are similar to the reported accuracies of similar studies (Magnan et al., 2009; Smialowski et al., 2007, 2012; Hirose and Noguchi, 2013; Kumar et al., 2007; Idicula-Thomas et al., 2006; Diaz et al., 2010).
Table 2.
Various scoring metrics for all five models applied to the expression dataset
| Deep neural network | Ensemble model | Logistic regression | Random forest classifier | Support vector classifier | |
|---|---|---|---|---|---|
| Accuracy | 0.690 ± 0.007 | 0.700 ± 0.008 | 0.672 ± 0.007 | 0.686 ± 0.010 | 0.673 ± 0.008 |
| Precision | 0.649 ± 0.010 | 0.671 ± 0.011 | 0.670 ± 0.015 | 0.674 ± 0.014 | 0.670 ± 0.016 |
| F1 score | 0.728 ± 0.012 | 0.723 ± 0.012 | 0.674 ± 0.010 | 0.696 ± 0.013 | 0.674 ± 0.011 |
| Recall | 0.829 ± 0.017 | 0.785 ± 0.013 | 0.678 ± 0.010 | 0.719 ± 0.014 | 0.679 ± 0.010 |
| AUC | 0.764 ± 0.010 | 0.767 ± 0.011 | 0.738 ± 0.008 | 0.750 ± 0.010 | 0.738 ± 0.008 |
Note: Error values refer to standard deviation from 5-fold cross validation
Fig. 4.
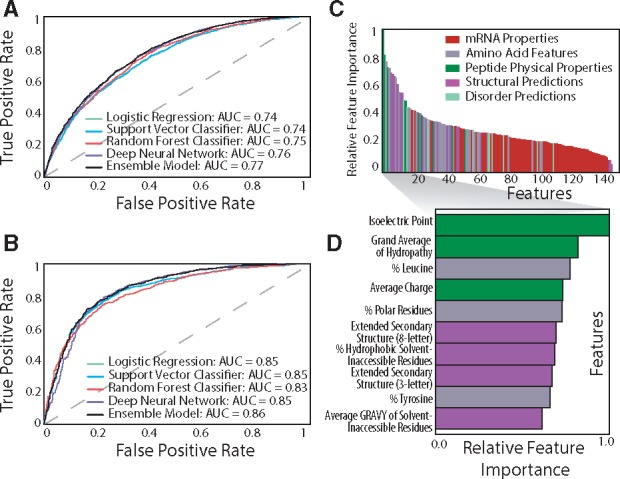
Result of machine learning workflow. Receiver Operating Curve (ROC) of the machine learning model for (A) expression data and (B) solubility data displaying the trade-off between the True Positive Rate and False Positive Rate. (C) Relative importance of protein features for the expression data, colored by property class. (D) Identities and relative feature importance of the top 10 features (Color version of this figure is available at Bioinformatics online.)
Solubility is another important factor in determining whether a protein fragment is amenable to affinity purification. A separate analysis was performed on a standardized dataset of 10 991 PrESTs with solubility characteristics to understand which properties influence solubility. The deep learning approach achieved an accuracy of 82% (Fig. 4B), which is significantly higher than accuracies reported by other machine learning studies (Magnan et al., 2009; Smialowski et al., 2007, 2012; Hirose and Noguchi, 2013; Kumar et al., 2007; Idicula-Thomas et al., 2006). In this case, aromaticity and hydropathy are the major determining factors for protein fragment solubility. Over 50% of insoluble PrESTs have a high fraction of aromatic amino acids, are negatively charged, and have a grand average of hydropathy above −0.69, compared to only 16% of soluble proteins (Supplementary Fig. S1a). Similar to protein expression, average charge and isoelectric point play an important role in protein solubility, which suggests that such properties can be adjusted to simultaneously optimize both expression and solubility during high-throughput experimentation.
3.3.2 Main protein features that separate protein fragment expression
Results from the random forest and decision tree analysis suggest that, for heterologous protein fragments, PrEST properties such as isoelectric point, the leucine content and various hydrophobicity indicators tend to correlate with protein expression level (Fig. 4C,D). Previous studies have found that the isoelectric point, molecular weight, charge and prevalence of specific amino acids were most important in both solubility and expression of whole proteins (Diaz et al., 2010; Mehlin et al., 2006). Surprisingly, none of the mRNA properties significantly influence protein expression in this dataset. This finding contrasts with evidence from recent studies that suggests codon content and mRNA-folding properties (in the initial 16 codons) influences protein expression (Boël et al., 2016). Although the tRNA adaptation index had the highest level of variation among all the features, it did not have much predictive power for the expression levels of the PrESTs (Supplementary Table S1). These findings highlight that the properties that influence expression or solubility of heterologous short sequence protein fragments are likely different than those that affect recombinant (or endogenous) full length proteins. Thus, our pipeline will likely identify an entirely different set of properties to predict expression level from protein fragments as compared to full proteins that are either endogenous or heterologously expressed. However, this pipeline can be adapted to whole proteins given a sufficiently large, standardized dataset.
While our analysis does not point to any one specific property capable of separating highly expressed PrESTs and poorly expressed PrESTs, we can observe how interactions between various properties affect expression (Supplementary Fig. S2), and identify general patterns that have implications to guide future engineering efforts. Increased separation of high and low expression can be achieved by constructing multi-dimensional rules; combining a low isoelectric point with a low hydropathy score and a higher fraction of polar residues correlates with high expression (Supplementary Fig. S1b). A decision tree analysis suggests that protein fragments meeting this multi-dimensional rule have a much higher chance of being highly expressed compared to those without these properties (65% compared to 43%, P-value < 0.001). This suggests that an optimal combination of such properties can promote higher chances of recombinant protein fragment expression.
3.3.3 Potential bottlenecks occurring during protein expression or solubility
The next step of our pipeline deals with identifying potential bottlenecks to high-throughput expression and solubility. In general, we find that the properties that have the most influence on expression and solubility and thus are the most amenable to further engineering are: (i) the number of hydrophobic, polar and charged residues; (ii) residues that influence isoelectric point; (iii) and aromatic content. Our decision tree analysis suggests that several bottlenecks exist that prevent a PrEST from being highly expressed. The first bottleneck occurs when a PrEST has low isoelectric point (≤9.4). The second bottleneck is related to PrESTs with lower hydropathy scores (≤−0.0328), in which high expression can be selected for if PrESTs have a minimum fraction of polar residues (35%). For solubility, the bottleneck appears to mainly lie in the degree of aromaticity (≤0.07). For PrESTs that meet this criteria, the majority (84%) are highly expressed. For cases that do not meet this criteria, the average charge and grand average of hydropathy of the PrEST determines whether or not it is likely to be highly expressed. The decision trees for both expression and solubility indicate that these features are strong determinants for whether a protein fragment can be purified. In general, the expressed proteins that are not soluble have a higher number of hydrophobic residues (43% compared to 41%, P-value < 0.001) and they have more extreme hydropathy scores (−0.59 compared to −0.33, P-value < 0.001). Similarly, the protein fragments that are soluble have lower aromaticity compared to their counterparts (6% compared to 9%, P-value < 0.001).
3.4 Computer-aided prediction of highly expressed proteins accelerates experimental throughput
The last stage of our computational pipeline entails applying the knowledge of what causes a bottleneck in expression or solubility to accelerate high-throughput experimentation. We do this by selecting PrESTs that are predicted to express well, based on the computational profile of the most important properties (e.g. isoelectric point, etc.). As mentioned before, PrESTs are currently selected at random, (i.e. without knowing their amenability to expression a priori), which results in a trial-and-error approach to express and purify PrESTs for each individual protein. In this case, numerous experiments must be attempted to express multiple PrESTs before successfully generating a soluble fragment. Here, we demonstrate that our pipeline to select fragments based on likelihood of expression reduces the total number of required experiments by 39%.
In order to test the utility of our pipeline, we generated predictions that (i) informed which PrESTs would most likely be highly expressed and (ii) determined the economization of experiments (i.e. the number of experiments that would be saved by running our pipeline before experimentation). First, we selected a subset of PrESTs belonging to the same protein that had a range of different expression outcomes (Fig. 5A). Based on the current dataset, we could link the PrESTs to over 16 000 proteins, where, in some cases, several PrESTs map to the same protein. An example is that of human metallo-beta-lactamase protein, which is associated with four distinct PrESTs from different parts of the protein, ranging from residues 135 to 263. In total, our prediction dataset consisted of 11 533 PrESTs, which are linked to 4759 total human proteins (on average 2.4 PrESTs per protein). From this set, we iteratively selected the PrESTs that were most likely to be highly expressed for each protein, based on our machine learning predictions (Fig. 5B).
Fig. 5.
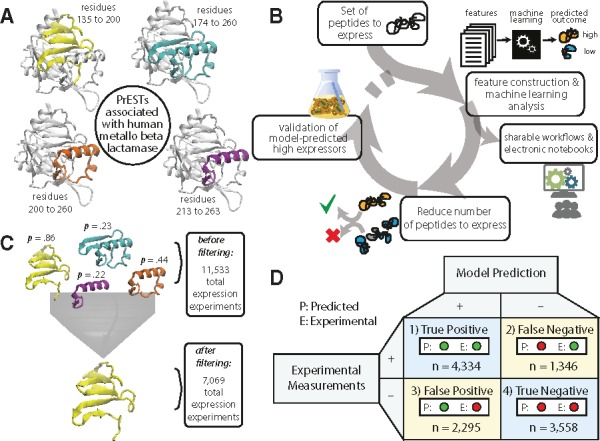
Computer-aided selection of highly expressed PrESTs. (A) The four possible PrESTs for human metallo-β-lactamase cover varying regions of the protein. (B) Prediction of expression levels reduces the number of experiments required to produce valid PrESTs, while enabling shareable workflows and electronic notebooks. Any dataset of proteins or peptides can be fed to the workflow to generate predictions on expression or solubility classes. Proteins with higher probabilities of expression can be selected for experimentation. (C) Retrospective validation on the dataset shows that computer-aided design could have reduced the number of experiments to generate PrESTs for a set of proteins from 11 533 experiments to 7069 experiments. (D) Confusion matrix for the results of the retrospective validation, comparing the amounts of true and false results
Once the PrESTs were selected, their expression was validated by comparison with experimental measurements (Fig. 5C). The proteins with low expressing PrESTs were submitted back into the workflow for four total iterations, when the prediction dataset was exhausted. We find that our computer-aided prediction pipeline spared a total of 4464 experiments (a 39% reduction) (See Table 3 and an IPython notebook titled ‘retrospective_analysis.ipynb’). Our findings correctly predicted the expression groups for 7892 out of 11 533 cases (Fig. 5D).
Table 3.
Number of proposed experiments in each iteration of the computer-aided prediction workflow compared to the total number of performed experiments
| Number of experiments | Expressed proteins | |
|---|---|---|
| Iteration 1 | 4759 | 2947 |
| Iteration 2 | 1812 | 543 |
| Iteration 3 | 416 | 75 |
| Iteration 4 | 82 | 7 |
| Total | 7069 | 3572 |
| Actual Experiments | 11 533 | 3572 |
4 Conclusion
The Human Protein Atlas provides a wealth of data informing the molecular details of whole cell proteomes. The high-throughput nature of the expression and solubility platforms used to generate these datasets provide invaluable resources from which to discover insights into structure-function properties of protein fragments and heterologously expressed peptides. Combining these large-scale datasets with advanced methods in machine learning, we can move beyond trial-and-error-based experimental approaches to heterologous expression and incorporate computer-aided predictions to guide the rational design of experiments.
As the amount of data for tissue-specific proteome-wide studies increases in scope and scale, data collected from these efforts can continually aid in developing and improving optimization platforms that accelerate high-throughput approaches like microarray-based immunohistochemistry. This study suggests that several key properties can already guide the design of better experiments: hydrophobicity, charge, aromaticity and secondary structure. A series of rules, or criteria, have been constructed to determine the likelihood that a recombinant peptide or protein fragment is expressed and/or soluble at a high or low concentration, based on each of these features. Such rules can be used to determine a priori whether a recombinant protein fragment will advance successfully through a cell factory pipeline. As demonstrated in the application of our computational workflow, mining sequences based on their expected properties not only optimizes the PrESTs that advance through the pipeline, but economizes the number of experiments needed to achieve an optimal PrEST-per-protein ratio. This study opens new avenues for selecting protein fragments in proteins without prior detection information based on sequence- and structure-based characteristics.
Combining computational biology and machine learning brings promise to finding meaning in large-scale biological datasets. A large dataset is generally required to accurately analyze protein expression (principal component analysis shows that over 55 dimensions are required to capture 90% of the variability in the dataset; Supplementary Fig. S3). The fact that an ensemble classifier approach outperforms all other machine learning approaches used in this study indicates the highly non-linear nature and complexity of the dataset as well as the limit of other approaches to deal with the size of the current dataset. Advanced machine learning algorithms, such as tree-based models and deep neural networks are able to find high-dimensional patterns in noisy data, such as those encountered in heterologous protein expression. Using these analyses to select optimal sequences will improve the efficiency and reliability of high-throughput pipelines and extensively decrease the cost and time associated with naive experimentation.
Supplementary Material
Acknowledgements
The authors acknowledge the entire Human Protein Atlas team for their efforts, Dr. David Heckmann for his advice on neural networks, Marc Abrams for editing the manuscript, and the NERSC computer facilities.
Funding
This work is supported by the National Institutes of Health [R01 GM057089], the US Department of Energy [DE-FG02-02ER63445], the Knut and Alice Wallenberg Foundation and funding through the Novo Nordisk Foundation Center for Biosustainability at DTU [NNF10CC1016517].
Conflict of Interest: none declared.
References
- Agaton C. et al. (2003) Affinity proteomics for systematic protein profiling of chromosome 21 gene products in human tissues. Mol. Cell. Proteomics, 2, 405–414. [DOI] [PubMed] [Google Scholar]
- Angermueller C. et al. (2016) Deep learning for computational biology. Mol. Syst. Biol., 12, 878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini A.A. et al. (2016) Codon identity regulates mRNA stability and translation efficiency during the maternal-to-zygotic transition. EMBO J., 35, 2087–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger B. et al. (2013) Computational solutions for omics data. Nat. Rev. Genet., 14, 333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund L. et al. (2008) A whole-genome bioinformatics approach to selection of antigens for systematic antibody generation. Proteomics, 8, 2832–2839. [DOI] [PubMed] [Google Scholar]
- Boël G. et al. (2016) Codon influence on protein expression in e. coli correlates with mRNA levels. Nature, 529, 358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caskey C.T. et al. (1968) RNA codons and protein synthesis. 15. dissimilar responses of mammalian and bacterial transfer RNA fractions to messenger RNA codons. J. Mol. Biol., 37, 99–118. [DOI] [PubMed] [Google Scholar]
- Chan P.P., Lowe T.M. (2009) GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res., 37, D93–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.T., Inouye M. (1994) Role of the AGA/AGG codons, the rarest codons in global gene expression in Escherichia coli. Genes Dev., 8, 2641–2652. [DOI] [PubMed] [Google Scholar]
- Cheng J. et al. (2005) SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res., 33, W72–W76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock P.J.A. et al. (2009) Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics, 25, 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deana A. et al. (1996) Synonymous codon selection controls in vivo turnover and amount of mRNA in Escherichia coli bla and ompa genes. J. Bacteriol., 178, 2718–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz A.A. et al. (2010) Prediction of protein solubility in Escherichia coli using logistic regression. Biotechnol. Bioeng., 105, 374–383. [DOI] [PubMed] [Google Scholar]
- Dong H. et al. (1996) Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol., 260, 649–663. [DOI] [PubMed] [Google Scholar]
- dos Reis M. et al. (2004) Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res., 32, 5036–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahim A. et al. (2016) Multi-omic data integration enables discovery of hidden biological regularities. Nat. Commun., 7, 13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E. et al. (2003) ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res., 31, 3784–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh C.-S. et al. (2004) Mining the structural genomics pipeline: identification of protein properties that affect high-throughput experimental analysis. J. Mol. Biol., 336, 115–130. [DOI] [PubMed] [Google Scholar]
- Goodman D.B. et al. (2013) Causes and effects of n-terminal codon bias in bacterial genes. Science, 342, 475–479. [DOI] [PubMed] [Google Scholar]
- Habibi N. et al. (2014) A review of machine learning methods to predict the solubility of overexpressed recombinant proteins in Escherichia coli. BMC Bioinformatics, 15, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose S., Noguchi T. (2013) ESPRESSO: a system for estimating protein expression and solubility in protein expression systems. Proteomics, 13, 1444–1456. [DOI] [PubMed] [Google Scholar]
- Idicula-Thomas S. et al. (2006) A support vector machine-based method for predicting the propensity of a protein to be soluble or to form inclusion body on overexpression in Escherichia coli. Bioinformatics, 22, 278–284. [DOI] [PubMed] [Google Scholar]
- Ikemura T. (1981) Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J. Mol. Biol., 151, 389–409. [DOI] [PubMed] [Google Scholar]
- Jones D.T., Cozzetto D. (2015) DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics, 31, 857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. (2005) Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene, 361, 13–37. [DOI] [PubMed] [Google Scholar]
- Kudla G. et al. (2009) Coding-sequence determinants of gene expression in Escherichia coli. Science, 324, 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P. et al. (2007). Granular support vector machine based method for prediction of solubility of proteins on overexpression in Escherichia coli In: Ghosh A., De R. K., Pal S. K. (eds.) Pattern Recognition and Machine Intelligence, Lecture Notes in Computer Science. Springer Berlin Heidelberg, pp. 406–415. [Google Scholar]
- Li G.-W. et al. (2012) The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature, 484, 538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.-W. et al. (2014) Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell, 157, 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linding R. et al. (2003) Protein disorder prediction: implications for structural proteomics. Structure, 11, 1453–1459. [DOI] [PubMed] [Google Scholar]
- Lundqvist M. et al. (2015) Solid-phase cloning for high-throughput assembly of single and multiple DNA parts. Nucleic Acids Res., 43, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnan C.N. et al. (2009) SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics, 25, 2200–2207. [DOI] [PubMed] [Google Scholar]
- Markham N.R., Zuker M. (2008) UNAFold: software for nucleic acid folding and hybridization. Methods Mol. Biol., 453, 3–31. [DOI] [PubMed] [Google Scholar]
- Mehlin C. et al. (2006) Heterologous expression of proteins from Plasmodium falciparum: results from 1000 genes. Mol. Biochem. Parasitol., 148, 144–160. [DOI] [PubMed] [Google Scholar]
- Mohammad F. et al. (2016) Clarifying the translational pausing landscape in bacteria by ribosome profiling. Cell Rep., 14, 686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piana S. et al. (2014) Assessing the accuracy of physical models used in protein-folding simulations: quantitative evidence from long molecular dynamics simulations. Curr. Opin. Struct. Biol., 24, 98–105. [DOI] [PubMed] [Google Scholar]
- Rolfsson Ó., Palsson B.O. (2015) Decoding the jargon of bottom-up metabolic systems biology. Bioessays, 37, 588–591. [DOI] [PubMed] [Google Scholar]
- Rosano G.L., Ceccarelli E.A. (2014) Recombinant protein expression in Escherichia coli: advances and challenges. Front. Microbiol., 5, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakin-Eshleman S.H., Liebhaber S.A. (1988) Influence of duplexes 3′ to the mRNA initiation codon on the efficiency of monosome formation. Biochemistry, 27, 3975–3982. [DOI] [PubMed] [Google Scholar]
- Sharp P.M., Li W.H. (1987) The codon adaptation index: a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res., 15, 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smialowski P. et al. (2007) Protein solubility: sequence based prediction and experimental verification. Bioinformatics, 23, 2536–2542. [DOI] [PubMed] [Google Scholar]
- Smialowski P. et al. (2012) PROSO II: a new method for protein solubility prediction. FEBS J., 279, 2192–2200. [DOI] [PubMed] [Google Scholar]
- Spencer P.S. et al. (2012) Silent substitutions predictably alter translation elongation rates and protein folding efficiencies. J. Mol. Biol., 422, 328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenvall M. et al. (2005) High-throughput solubility assay for purified recombinant protein immunogens. Biochim. Biophys., 1752, 6–10. [DOI] [PubMed] [Google Scholar]
- Tegel H. et al. (2009) High-throughput protein production--lessons from scaling up from 10 to 288 recombinant proteins per week. Biotechnol J., 4, 51–57. [DOI] [PubMed] [Google Scholar]
- Tuller T. et al. (2010) Translation efficiency is determined by both codon bias and folding energy. Proc. Natl. Acad. Sci. U. S. A., 107, 3645–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M. et al. (2010) Towards a knowledge-based human protein atlas. Nat. Biotechnol., 28, 1248–1250. [DOI] [PubMed] [Google Scholar]
- Uhlén M. et al. (2015) Proteomics. Tissue-based map of the human proteome. Science, 347, 1260419.. [DOI] [PubMed] [Google Scholar]
- Williamson A.R. (2000) Creating a structural genomics consortium. Nat. Struct. Biol., 7 Suppl, 953. [DOI] [PubMed] [Google Scholar]
- Yang Z.R. et al. (2005) RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics, 21, 3369–3376. [DOI] [PubMed] [Google Scholar]
- Zhao X.-M. et al. (2008a) Gene function prediction using labeled and unlabeled data. BMC Bioinformatics, 9, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.-M. et al. (2008b) Protein classification with imbalanced data. Proteins, 70, 1125–1132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.