

Abstract
Summary
De novo assembly is a difficult issue for heterozygous diploid genomes. The advent of high-throughput short-read and long-read sequencing technologies provides both new challenges and potential solutions to the issue. Here, we present HaploMerger2 (HM2), an automated pipeline for rebuilding both haploid sub-assemblies from the polymorphic diploid genome assembly. It is designed to work on pre-existing diploid assemblies, which are typically created by using de novo assemblers. HM2 can process any diploid assemblies, but it is especially suitable for diploid assemblies with high heterozygosity (≥3%), which can be difficult for other tools. This pipeline also implements flexible and sensitive assembly error detection, a hierarchical scaffolding procedure and a reliable gap-closing method for haploid sub-assemblies. Using HM2, we demonstrate that two haploid sub-assemblies reconstructed from a real, highly-polymorphic diploid assembly show greatly improved continuity.
Availability and Implementation
Source code, executables and the testing dataset are freely available at https://github.com/mapleforest/HaploMerger2/releases/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
There is an increasing demand for sequencing of heterozygous diploid genomes. However, since the era of Sanger sequencing, de novo assembly of heterozygous diploid genomes has been a difficult issue (Vinson et al., 2005). It becomes more challenging when using massive, short-read sequencing technologies (Zhang et al., 2012). Though several de novo assembly methods and post-assembly methods have been designed to improve heterozygous short-read assemblies (Gnerre et al., 2011; Huang et al., 2012; Kajitani et al., 2014; Pryszcz and Gabaldon, 2016; Safonova et al., 2015), these assemblies hardly reach the same level of quality as non-heterozygous assemblies. The latest high-throughput long-read sequencing technologies provide a promising approach to polymorphic assembly (Berlin et al., 2015; Chin et al., 2013; Koren et al., 2017; Xiao et al., 2016), especially when combined with heterozygosity-aware assembly algorithms (Chin et al., 2016). However, in long-read diploid assemblies, there are still assembly errors, and, more importantly, allelic relations between scaffolds might not be fully resolved. This is because, as the heterozygosity increases, alleles from the same locus are more likely to be mistaken as sequences from different loci.
Previously, we developed HaploMerger (HM), an automated pipeline to resolve allelic relations in polymorphic diploid assembly and output the reference haploid assembly (Huang et al., 2012). Since HM works on pre-existing diploid assemblies, it can be easily incorporated into any assembly pipelines. Thus far, HM has been used to create over ten published reference assemblies, including large draft genomes for amphioxus (∼450 Mb with ∼4% heterozygosity) and hookworms (∼330 Mb with <1% heterozygosity) (Huang et al., 2014; Schwarz et al., 2015).
We perceive that, because alleles often functionally complement each other in a highly polymorphic genome, both haploid sub-assemblies are necessary to represent the complete genomic landscape. Moreover, the presence of both haploid sub-assemblies enables the study of widespread heterozygosities in a highly polymorphic genome, including single nucleotide polymorphisms, indels, copy-number variation, structural variation and recent transposition.
Here, we provide HaploMerger2 (HM2), a major upgrade over the old pipeline, which we redesigned to reconstruct both haploid sub-assemblies from short-read and long-read diploid assemblies. HM2 can work with both heterozygosity-aware and -unaware genome assemblers and process both low and high heterozygosity assemblies. However, it is especially suitable for difficult tasks in which the diploid assemblies have high heterozygosity (≥3%). Compared with the old pipeline, HM2 also implements more flexible assembly error detection, a hierarchical scaffolding procedure and a reliable gap-closing method on haploid assemblies (Fig. 1). In this applications note, we describe the features and applications of HM2.
Fig. 1.
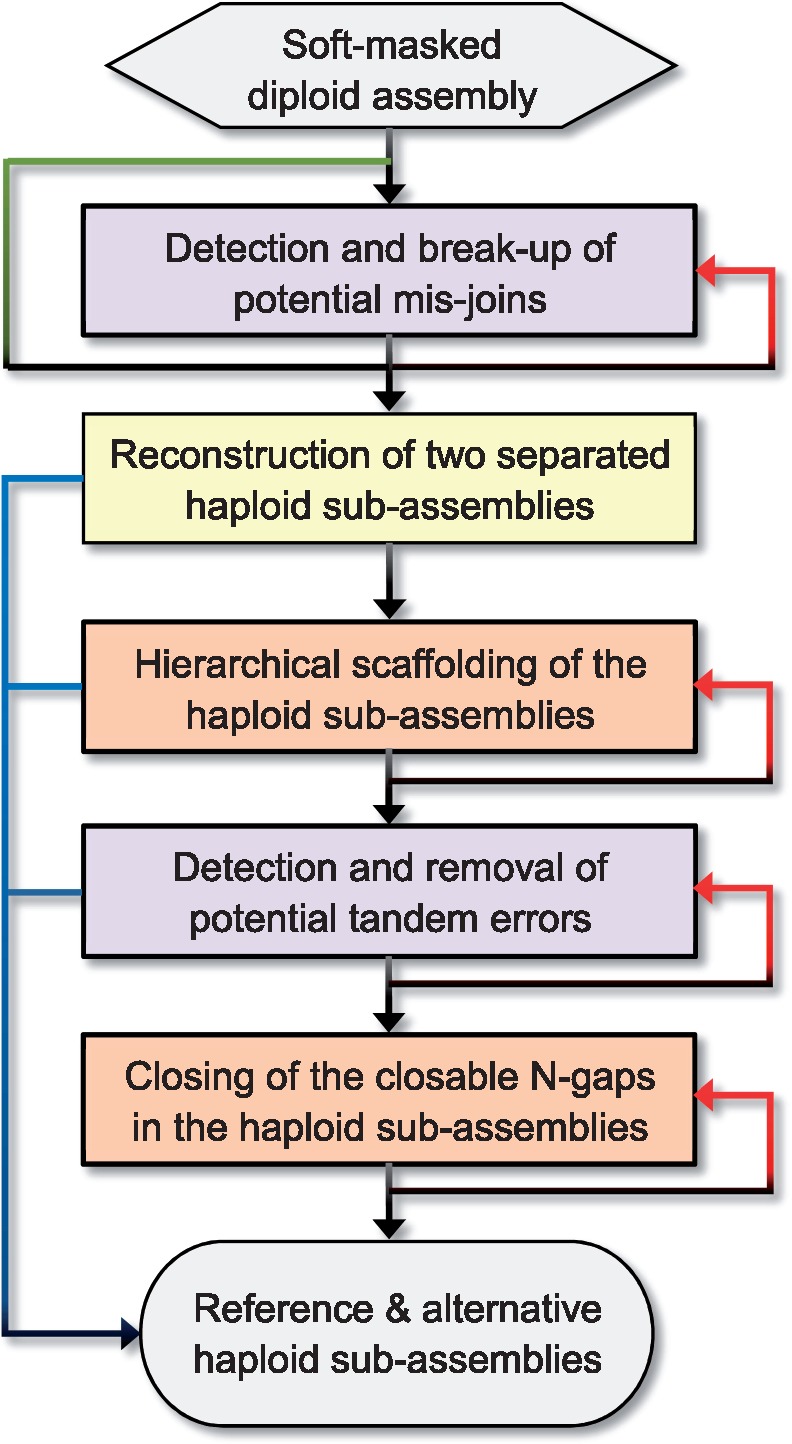
A flowchart of the HaploMerger2 (HM2) pipeline. HM2 comprises five functionally independent modules. Each module can work on its own. The users can run any module separately or choose some of them to form a specific pipeline, as suggested in the flowchart. In this specific pipeline, all but the second module are optional and can be iterated to achieve better results
2 Software description
2.1 Preparation and requirements
An initial diploid assembly should first be generated by using de novo assemblers. To include as many alleles as possible into the diploid assembly, the de novo assembler should be run with stringent parameters (e.g. low error rates) or in the heterozygosity-aware mode, which forces alleles from the same locus to be assembled and outputted separately. To avoid false alignments, repetitive sequences in the diploid assembly, including simple repeats, transposable elements and highly duplicated coding sequences, should be soft-masked using WindowMasker and/or RepeatMask (Morgulis et al., 2006; Tarailo-Graovac and Chen, 2009). To achieve optimal specificity and sensitivity, knowledge of the allelic polymorphism rate and mutational biases is very important. For example, if the heterozygosity is 1% and the alignment threshold is set to 10%, many sequences will be falsely removed as alleles. On the other hand, if the heterozygosity is 10% and the alignment threshold is set to 1%, many true allele pairs will be undetected and remain in the haploid sub-assemblies. HM2 provides tools to infer proper parameters to handle these situations. Finally, due to algorithmic limitation, HM2 is more suitable to process diploid assemblies with an initial scaffold N50 size >100 Kb.
2.2 Detection and break-up of potential mis-joins
Mis-joins of unrelated genomic portions can be detected by examining the alignments between allelic scaffolds. In the old HM pipeline, mis-join processing and haploid assembly rebuilding were inseparable. In HM2, mis-join processing is redesigned as the first independent module, which allows for choosing optimal parameters and running iterations of the module to maximize error detection. It is worth noting that false detection of mis-joins due to repetitive sequences has been suppressed by the initial repeat-masking procedure. In a pair of allelic scaffolds involved in a mis-join, it is difficult to determine which has the error. Additionally, it is hard to discriminate between mis-joins from natural inversions and translocations. Therefore, HM2 breaks up both scaffolds involved in a potential mis-join. The correct connection can be restored later by the scaffolding module.
2.3 Reconstruction of two separated haploid sub-assemblies
The old version of HM reconstructs allelic relations based on the best reciprocal, mirrored whole-genome alignments of the diploid assembly. Then, a heuristic method is employed to elect the best allele into the reference haploid assembly, whereas another allele is used to fill the N-gaps or is simply discarded. This procedure might cause the loss of the alternative alleles and excessive switches between two haplotypes. In HM2, we revised the algorithm to reconstruct both haploid sub-assemblies: the reference sub-assembly and the alternative sub-assembly. Specifically, if two alleles are available for a locus, HM2 separates them into two different sub-assemblies, with the better-quality allele placed in the reference sub-assembly. If only one allele is available for a locus (often due to haplotype collapsing or the allele is simply discarded by the de novo assembler), HM2 puts this allele into both sub-assemblies. In the sub-assemblies, the allelic scaffolds are given the same scaffold name. Finally, because there are switches between haplotypes in the rebuilt haploid sub-assemblies, the sub-assemblies are not haplotype phased.
2.4 Hierarchical scaffolding of the haploid sub-assemblies
In polymorphic diploid assembly, scaffolding with mate-pairs is ineffective because reads of the same pair are often aligned to different haplotypes. This is the major factor that causes heavy fragmentation and excessive assembly errors in polymorphic assembly (Huang et al., 2012; Kajitani et al., 2014; Safonova et al., 2015; Vinson et al., 2005). In HM2, we implement hierarchical scaffolding in the haploid sub-assemblies. Without interference of the different alleles, this re-scaffolding procedure can dramatically improve the sequence continuity. Currently, HM2 invokes a third-party program, SSPACE v3.0, to implement scaffolding (Boetzer et al., 2011). In our experience, SSPACE v3.0 implements a fast, straightforward greedy scaffolding algorithm. However, HM2 also supports other scaffolders, as long as the scaffolders do not remove or add sequences to the sub-assembly. Continuity could be further improved by invoking multiple rounds of the scaffolding module. Additionally, only the reference sub-assembly needs re-scaffolding because HM2 will update the alternative sub-assembly according to the new scaffolding layout of the reference sub-assembly.
2.5 Detection and removal of potential tandem assembly errors
HM2 utilizes an updated module with several fixed bugs and new configurable options. For example, the module now can scan tandems as small as 100 bp, and detect tandems of unequal length (option ‘XvY’). In addition, multiple rounds of tandem removal can be performed, usually with decreased tandem sizes and increased sensitivity. Then, the tandem-assembled sequences that have been removed are collected in an output file rather than discarded as was done before. Finally, the users should be careful with this module because it is the only module in HM2 that may lose genomic information.
2.6 N-gap closing
HM2 invokes a third-party software, GapCloser (Luo et al., 2012), to implement N-gap closing. Since gap-filling sequences generated by GapCloser are not always reliable, HM2 will re-examine all the gap-filling sequences and choose to retain the reliable ones. Because this examination is specific to the GapCloser output, HM2 currently does not support other gap-filling software. It is possible to run multiple rounds of gap-filling with different datasets. All gap-filling sequences are annotated in an AGP-formatted file (v.1.1).
3 Sample applications
We provide three examples for testing HM2. The first two examples use an artificial diploid assembly (∼100 Kb) to test if HM2 is installed successfully and functions properly. Both examples can be finished in a minute.
The third example uses a real, highly-polymorphic diploid assembly for a wild-type amphioxus. This assembly was created from a mixture of 454 and Illumina reads (∼60X) using the Celera assembler CABOG v6.1 (Miller et al., 2008). A copy of this assembly can be downloaded from GenBank (accession: AYSR00000000.1), or from our HM2 release website (named ‘bbv18wm.fa.gz’). It has been soft-masked and is ready to use. This assembly contains ∼708 M bases and has a scaffold/contig N50 size of 264 Kb/30 Kb, exhibiting an average rate of allelic polymorphism of ∼4%. After a single round of HM2, we can obtain two separated haploid sub-assemblies of ∼406 Mb with a scaffold/contig N50 size of 2.2 Mb/40 Kb. This takes <3 hours to finish on a machine with 12-cores and 64 Gb of memory. The results and performance are highly reproducible. A full description of this application is provided in the Supplementary information.
4 Discussion
HM2 works in the post-assembly stage. Its performance is bound by the quality of the initial diploid assembly. For example, if one of the haplotypes is largely missing in the diploid assembly, HM2 cannot recover it. However, a reference haploid sub-assembly is always guaranteed. HM2 has algorithmic limitations, which offer little help if the diploid assembly is too fragmented (i.e. <100 kb).
In essence, HM2 is a tool kit comprising a set of executables of independent function, as well as wrappers for winMasker, Lastz, chainNet, SSPACE and GapCloser. The intermediate information and running messages are tracked and documented for each step and function. The pipeline presented here is a special organization of a selection of tools from this kit. Therefore, HM2 can be used for other applications in post-assembly analysis. For example, it can be used to create self-versus-self whole-genome alignments or pairwise alignments between two genome assemblies to detect tandem duplication, further scaffold an assembly and close some N-gaps.
Funding
This work was supported by the 973 Project [grant number 2013CB835305], the National Nature Science Fund [grand number 31171193] and by the National Supercomputer Center in Guangzhou and the Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund.
Conflict of Interest: none declared.
Supplementary Material
References
- Berlin K. et al. (2015) Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol., 33, 623–630. [DOI] [PubMed] [Google Scholar]
- Boetzer M. et al. (2011) Scaffolding pre-assembled contigs using SSPACE. Bioinformatics, 27, 578–579. [DOI] [PubMed] [Google Scholar]
- Chin C.S. et al. (2013) Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods, 10, 563–569. [DOI] [PubMed] [Google Scholar]
- Chin C.S. et al. (2016) Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods, 13, 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnerre S. et al. (2011) High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U. S. A., 108, 1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S. et al. (2012) HaploMerger: reconstructing allelic relationships for polymorphic diploid genome assemblies. Genome Res., 22, 1581–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S. et al. (2014) Decelerated genome evolution in modern vertebrates revealed by analysis of multiple lancelet genomes. Nat. Commun., 5, 5896.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajitani R. et al. (2014) Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res., 24, 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S. et al. (2017) Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R. et al. (2012) SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience, 1, 18.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.R. et al. (2008) Aggressive assembly of pyrosequencing reads with mates. Bioinformatics, 24, 2818–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgulis A. et al. (2006) WindowMasker: window-based masker for sequenced genomes. Bioinformatics, 22, 134–141. [DOI] [PubMed] [Google Scholar]
- Pryszcz L.P., Gabaldon T. (2016) Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res., 44, e113.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safonova Y. et al. (2015) dipSPAdes: assembler for highly polymorphic diploid genomes. J. Comput. Biol., 22, 528–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz E.M. et al. (2015) The genome and transcriptome of the zoonotic hookworm Ancylostoma ceylanicum identify infection-specific gene families. Nat. Genet., 47, 416–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo-Graovac M., Chen N. (2009) Using RepeatMasker to identify repetitive elements in genomic sequences, Curr Protoc Bioinformatics, Chapter 4, Unit 4 10. [DOI] [PubMed]
- Vinson J.P. et al. (2005) Assembly of polymorphic genomes: algorithms and application to Ciona savignyi. Genome Res., 15, 1127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C.-L. et al. (2016) MECAT: an ultra-fast mapping, error correction and de novo assembly tool for single-molecule sequencing reads. bioRxiv. [DOI] [PubMed] [Google Scholar]
- Zhang G. et al. (2012) The oyster genome reveals stress adaptation and complexity of shell formation. Nature, 490, 49–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.