

Abstract
Motivation
Population genomic analyses are often hindered by difficulties in obtaining sufficient numbers of genomes for analysis by DNA sequencing. Selective whole-genome amplification (SWGA) provides an efficient approach to amplify microbial genomes from complex backgrounds for sequence acquisition. However, the process of designing sets of primers for this method has many degrees of freedom and would benefit from an automated process to evaluate the vast number of potential primer sets.
Results
Here, we present swga, a program that identifies primer sets for SWGA and evaluates them for efficiency and selectivity. We used swga to design and test primer sets for the selective amplification of Wolbachia pipientis genomic DNA from infected Drosophila melanogaster and Mycobacterium tuberculosis from human blood. We identify primer sets that successfully amplify each against their backgrounds and describe a general method for using swga for arbitrary targets. In addition, we describe characteristics of primer sets that correlate with successful amplification, and present guidelines for implementation of SWGA to detect new targets.
Availability and Implementation
Source code and documentation are freely available on https://www.github.com/eclarke/swga. The program is implemented in Python and C and licensed under the GNU Public License.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Selective whole-genome amplification (SWGA) provides a means of obtaining sufficient numbers of genomes from a target organism to perform whole-genome sequence analysis, even in the presence of overwhelming DNA from other organisms (Leichty and Brisson, 2014). Difficulties in isolating a target of interest are common in microbial population genomics, which requires acquiring adequate genomic DNA from a target while limiting the amount of non-target DNA (Mardis, 2008). Often, the genomes of interest represent only a fraction of a percent of the total nucleic acids in a sample, and so direct sequencing is inefficient and expensive. Laboratory culture of the target microbe is the traditional solution, but many microbes replicate poorly or not at all in in vitro conditions (Amann et al., 1990; Ghazanfar et al., 2010; Schmeisser et al., 2007).
SWGA allows sequence acquisition without culture of the target organism or extensive purification of target DNA. It achieves this by preferentially amplifying the target genome using a set of selective primers and 29 polymerase-based multiple displacement amplification (MDA) (Dean et al., 2002; Leichty and Brisson, 2014). Since its introduction, this method has been used to study Wolbachia pipientis in Drosophila melanogaster (Leichty and Brisson, 2014), and to understand the evolution and drug resistance of Plasmodium falciparum (Guggisberg et al., 2016; Oyola et al., 2016; Sundararaman et al., 2016) and Plasmodium vivax (Cowell et al., 2017). Further applications of SWGA to population genomics may help reconstruct epidemic transmission patterns, characterize patterns of inter-host viral transmission, detect escape from antimicrobial agents, and delineate the evolutionary dynamics of immune escape (Hume et al., 2003; Luikart et al., 2003; Martínez et al., 2012; Nelson et al., 2008; Nunes et al., 2012; Stack et al., 2012).
Implementation of SWGA has been complicated by the difficulty in identifying an effective set of selective primers, as there are many constraints and degrees of freedom in the composition of potential primer sets. These primers must reflect DNA sequence motifs common in the target genome but rare in the background DNA. They also must have binding sites sufficiently near each other to enable the branching and displacement actions of the 29 polymerase that are essential for MDA. A previously published method used a set of Perl scripts (Leichty and Brisson, 2014) to identify primers with the highest ratio of binding frequencies in the target genome versus the background DNA. However, choosing a set by the above method is suboptimal: for one, the primers may form heterodimers with each other or homodimers with themselves; they may be individually selective but in aggregate bind too frequently to the background DNA; or, they may bind to the target’s telomeric or mitochondrial DNA, and not be sufficiently evenly distributed across the genome. There are aspects of the primer sets that have an unknown effect on the efficiency of the reaction, including the annealing and melting temperature of the primer sequences, the evenness of the binding sites across the target genome, and the density of binding sites. The Perl scripts mentioned above are unable to evaluate many of these criteria, requiring extensive manual effort and trial-and-error to create workable designs.
Here we present swga, a program that identifies selective primer sets for a given target genome and background. swga evaluates all potential primer sequences and forms sets of valid primers that meet the above criteria. It automatically calculates a variety of metrics for each set that potentially affect the efficacy and selectivity of the reaction. These sets are then are ranked and presented to the user, enabling the selection of primer sets most likely to succeed. Nearly all operating parameters of the program are user-specifiable but initialized with reasonable defaults based on the target and background genomes selected, reducing the work needed to get started.
We demonstrate the use of swga to design primer sets and test them on two biological systems: Wolbachia pipientis from infected Drosophila melanogaster, and Mycobacterium tuberculosis DNA spiked into human blood. For each system, we designed multiple primer sets to explore the effect of various aspects of the primer sets on reaction efficacy, such as primer melting temperature, binding density on the target genome, and the evenness of binding sites. These experimental results clarify the relative importance of each and allow us to describe an effective workflow for using swga.
2 Materials and methods
2.1 Program overview
The swga program can be divided into four modules (Fig. 1).
Fig. 1.

An overview of the swga workflow. The program begins by counting all nucleotide sequences of length k (k-mer) in both the target and background genomes for a given range of k (e.g. 8–12 bp). The k-mers are then filtered by criteria that include the binding frequencies in the background and target genome, their melting temperatures, and the likelihood of hairpin or homodimer formation. The best k-mers are then used to form compatible sets, in which no k-mer would likely form a heteroduplex with any other in the set. These sets are then evaluated for multiple criteria including binding frequencies and evenness. The results can be exported into common formats for downstream use and visualization
2.1.1 Primer identification
The user starts by defining the target and background sequences using swga init. At this point, a set of sequences can be supplied that define a priori where primers should not bind, such as a mitochondrial genome or plasmids (the ‘exclusionary sequences’). The swga count command then uses DSK (Rizk et al., 2013) to identify all nucleotide sequences in the size range specified by parameters min_size and max_size that exist in the target genome and do not exist in the exclusionary sequences (if provided). These primers are used to populate a local SQLite database for later retrieval. The selectivity of these primers is determined by their frequency in the target genome versus the background DNA, so swga count saves the frequency that each primer appears in the target and background as well. Primers that appear extremely rarely in the target and overly frequently in the background (as defined by user-editable parameters, with defaults set by swga init), are not saved to help speed up downstream steps. Additionally, primers that would form internal hairpins or homodimers with themselves are omitted.
2.1.2 Primer filtering
The command swga filter ranks and filters potential primers by their melting temperature, selectivity, and evenness of binding in the target genome. First, primers that bind too sparsely to the target genome (lower than parameter min_fg_bind) or too frequently to the background (max_bg_bind) are removed. Next, the melting temperature is approximated using nearest-neighbor thermodynamics (Allawi and SantaLucia, 1997) with corrections for mono- and divalent cations. Primers with melting temperatures outside the range defined by min_tm and max_tm are removed. The evenness of binding then is calculated by finding the Gini index (Gini, 1912) of the distances between each primer binding site on the target. The Gini index varies between 1 and 0, where 1 represents extremely uneven and 0 represents perfectly even. A primer with a low Gini index has binding sites that are each separated by similar distances, whereas a primer with a high Gini index may reflect one where many of the primer binding sites are clumped together (e.g. on tandem repeat regions). Primers with Gini indices higher than max_gini are removed. Finally, primers are ranked by the ratio of target binding frequency to background binding frequency and those primers with the highest ratio are identified for downstream use (by default, this identifies the top 200 primers, and is modifiable via the max_primers parameter). The thresholds for each filter are user-editable, and the swga filter command caches results so that it can be quickly re-run to explore different results.
2.1.3 Primer set evaluation
The swga find sets command is then used to find sets of compatible primers from the ones identified in the last step of swga filter. Brute force evaluation of all primer sets is computationally infeasible: given n primers and a set size of k, the total number of possible sets is (n choose k). With the default parameters of n = 200 and , there are over possible sets. Fortunately, not all of these sets are usable for swga. A pair of primers are incompatible if they form heterodimers (calculated by the number of consecutive complimentary bases), or if one primer is a subsequence of another. swga find sets calculates the pairwise compatibility of all selected primers and stores the results as a graph. In this graph, primers are vertices and compatible primers are connected with edges. The problem of finding compatible sets then reduces to a problem of finding sets of vertices in the graph that are all interconnected (a ‘clique’ in graph theory). swga also stores the average distance between binding sites on the background as a ‘weight’ on each vertex. This allows the program to prioritize cliques that have higher total weights, representing sets of primers that bind infrequently to the background.
To find these cliques, the swga find_sets command uses a modified version of the program cliquer by Niskanen and Östergård (2003). The branch-and-bound algorithm in cliquer is a computationally efficient way of finding cliques in a graph. We have extended the algorithm to find only cliques that meet certain criteria. By specifying the desired criteria a priori the algorithm can skip sets that do not meet the requirements and save computation time. These criteria include the minimum distance between binding sites in the background (min_bg_bind_dist) and maximum distance between binding sites on the target (max_fg_bind_dist). In addition, the algorithm can explore a range of set sizes (min_size and max_size) in order to find valid sets. By specifying a broad range of set sizes, the algorithm is able to find sets with a broad range of characteristics independent of the number of primers.
Primer sets that meet these criteria are further evaluated on metrics including the average and maximum distance between primer binding sites on the target genome and the Gini index of all binding sites in the set. These sets and their accompanying metrics are then saved.
Even with the above optimizations, the number of valid sets can be quite large. For this reason, swga find_sets can be safely stopped after evaluating and storing a sufficient number of sets. In our usage, we generally stop after 1–5 million sets have been saved.
2.1.4 Primer set output and visualization
The saved primer sets can be explored and exported using swga export. This command allows the user to order the sets by any of the evaluated metrics, export all or some of the sets of interest to Excel-compatible formats, or export a set to a BedGraph or BedFile format for visualization in a genome browser (Kent et al., 2002).
2.2 Empirical primer set testing
To evaluate swga, we used it to design primer sets for amplification of W. pipientis DNA against a background of D. melanogaster and of M. tuberculosis against a background of H. sapiens. We evaluated primer sets on their ability to selectively and evenly amplify the target genome.
2.2.1 Designing primer sets for W. Pipientis
We created four primer sets for W. pipientis against D. melanogaster, varying each by melting temperature range, selectivity, and evenness of binding sites on the target genome. We first initialized swga on the W. pipientis genome with D. melanogaster as the background, and ran swga count to store all potential primers.
For the first two sets, we used swga filter with the ‘standard’ temperature range established in Leichty and Brisson (2014), and default in swga, of 15–45°C. This range we named Tm Low, or TmL. After running swga find_sets and storing 1 million sets, we used swga export to output the set with the lowest target to background binding distance ratio, which we called Set TmL/Selective. We then used swga export again to output the set with the lowest Gini index, which we called Set TmL/Even.
The next two sets were designed with a higher melting temperature range. We re-ran swga filter with a Tm range of 35–55°C, which we named Tm High, or TmH. As above, we then re-ran swga find_sets on the new primers and chose the most selective and most even sets from the results. These are called TmH/Selective and TmH/Even, respectively. The complete parameter listing is included in Supplementary File 1. The primers belonging to each set are given in Supplementary Table 1.
2.2.2 Designing primer sets for M. tuberculosis
We created ten primer sets for M. tuberculosis using swga. Our target genome was M. tuberculosis strain H37Rv (NC_000962.3) and our background was the human genome, version GRCh38. For this system, we ran swga filter with a temperature range constant at 15–45°C, and imposed a maximum per-primer Gini index of 0.6. We stopped swga find_sets after storing five million sets and exported all of them to CSV format using swga export. The sets were filtered to only sets with mean distance between target binding sites <5000 bases. We selected ten sets with the most extreme combinations of mean target binding distance and evenness (via the metrics fg_dist_mean and fg_dist_gini, respectively). These sets we named Mtb1 through Mtb10. The distribution of these sets in the pool is visualized in Supplementary Figure S1. In addition, we selected from the original five million the set with the highest Gini index (most uneven) and highest mean target binding distance as negative comparisons, named MtbUneven and MtbSparse, respectively. The full parameter listing is included in Supplementary File 2. The primers belonging to each set are given in Supplementary Table 2.
2.2.3 Selective whole-genome amplification and sequencing
The Wolbachia-specific primer sets were tested on pooled genomic DNA extracted from 10 Wolbachia-infected D. melanogaster (strain ). Pooling was performed to eliminate inter-fly variability in Wolbachia infection levels, and each primer set was tested in triplicate using 40 ng of input DNA per reaction, except as noted for additional tests of the TmL/Even Wolbachia primer set. For consistency with the approach used in Leichty and Brisson (2014), the pooled genomic extract was digested with NarI (NEB, New England Biolabs, Inc., Ipswich, MA, USA) at 37°C for 30 minutes, in order to suppress mitochondrial amplification. This step is likely unnecessary in the general case because swga includes an option to omit mitochondrial sequences from primer formation.
Mycobacterium primer sets were tested on purified M. tuberculosis DNA (strain H37Rv, ATCC 27294D-2), diluted to 1% in human genomic DNA extracted from cultured CD4+ T cells. Primer sets were tested in triplicate.
Selective whole-genome amplification was performed as previously described (Sundararaman et al., 2016), with slight modifications. Reactions were performed in a volume of 50 μL using input DNA, 3.5 mM total of SWGA primers, 1× phi29 buffer (New England Biolabs), 1 mM dNTPs and 30 units phi29 polymerase (New England Biolabs). Amplification conditions included a 1 h ramp-down step (35–30°C), followed by a 16 h amplification step at 30°C. Phi29 was then denatured for 10 min at 65°C.
Amplified samples were purified using AmpureXP beads (Beckman Coulter), prepared for Illumina sequencing as described in Kryazhimskiy et al. (2014), and sequenced on an Illumina MiSeq (150 bp, paired end). We also sequenced the unamplified pool to establish a baseline for amplification efficiency. Illumina-specific adapter and primer sequences were removed from the reads using Cutadapt (Martin, 2011). In both systems, reads were first aligned to the background (D. melanogaster or human) using smalt (Ponstingl and Ning, 2010). Unmapped reads were then mapped to the target genome (W. pipientis or M. tuberculosis, respectively), also using smalt. Analysis of sequence coverage of the target genome and sequencing rarefaction analyses were performed using R (R Core Team, 2015). All code used in the analysis and to generate the figures is available online at https://github.com/eclarke/swga_paper.
3 Results
We used swga to design four primer sets for amplifying Wolbachia against a background of D. melanogaster, which tested the effect of melting temperature ranges, selectivity, and evenness. We designed twelve primer sets for amplifying M. tuberculosis against a background of human DNA with varying primer binding evenness and density on the target. Ten sets tested were various combinations of high density and evenness Two, for comparison, were the most uneven and most sparse. For M. tuberculosis, we compared amplification using random hexamers (e.g. standard MDA) to the swga-designed primer sets.
3.1 Evaluation of primer sets for W. pipientis
The four primer sets for W. pipientis were designed with two different temperature ranges (TmL: 15–45°C, TmH: 35–55°C). From the sets identified in each temperature range, we chose the set with the highest selectivity, defined by the lowest target to background binding distance ratio (TmL/Selective and TmH/Selective). We also chose the sets with the most even distribution of binding sites (TmL/Even and TmH/Even). As a control, we included the primer set from Leichty and Brisson (2014). The composition and metrics for each of these five sets is shown in Table 1.
Table 1.
Characteristics of primer sets chosen for selective whole-genome amplification of Wolbachia from infected Drosophila DNA
| Ratio | # Primers | Gini | Mean target dist | Mean bg. dist | |
|---|---|---|---|---|---|
| TmL/Selective | 0.0544 | 9 | 0.654 | 5.33E+03 | 9.78E+04 |
| TmL/Evena | 0.1050 | 7 | 0.537 | 6.85E+03 | 6.53E+04 |
| TmH/Even | 0.0075 | 2 | 0.537 | 1.31E+04 | 1.73E+06 |
| TmH/Selective | 0.0005 | 2 | 0.66 | 1.21E+04 | 2.43E+07 |
| Leichty 2014 | 0.0163 | 2 | 0.712 | 5.31E+03 | 3.25E+05 |
The set that most effectively amplified Wolbachia. ‘Ratio’ is ratio of the average distance between binding distances in the target and background.
The pooled genomic DNA contained 4.7% W. pipientis DNA, as determined by sequencing of the unamplified control. We recovered ∼200 Mbp of sequence for each amplicon. The proportion of sequencing reads that were derived from W. pipientis was at least 2.5 times greater in all amplified samples than the sequencing reads from the unamplified genomic extract (Supplementary Fig. S2). We found that the primer sets with the higher melting temperatures (TmH/Selective and TmH/Even) yielded more Wolbachia reads as a total percentage, with some replicates as high as 77.8%. However, these primer sets failed to reach 10× coverage on even 10% of the W. pipientis genome (Fig. 2). This was most likely due to uneven amplification of the target genome, as shown in Supplementary Figure S3.
Fig. 2.
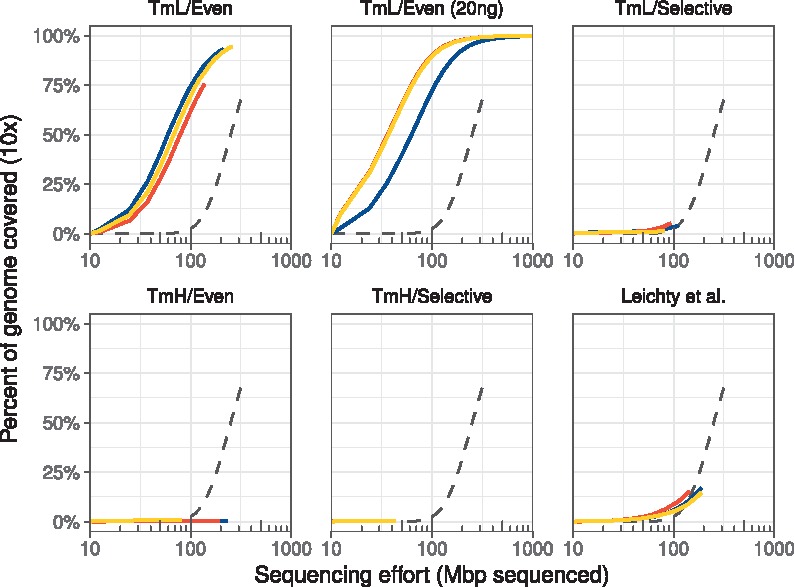
Selective whole genome amplification reduces the sequencing effort necessary to achieve at least ×10 coverage across the W. pipientis genome. Each color represents an individual technical replicate; dashed lines represent the unamplified control. Lines above the unamplified control represent better sequencing efficiency in that they yielded greater coverage of the target genome with less sequencing effort. Sequencing 100 million bases from unamplified genomic DNA extracted from 10 flies resulted in 10-fold or greater sequencing coverage in only 2.8% of the W. pipientis genome. In contrast, the TmL/Even primer set resulted in 10-fold or greater coverage of 60–75% of the W. pipientis genome with similar sequencing effort. This fraction was increased further to 72–91% when the TmL/Even primer set was used to amplify W. pipientis from 20 ng (rather than 40 ng) of total fly extract DNA (empirically, using lower total starting DNA can yield higher relative amplification when using phi29). The TmL/Selective primer set and the manually chosen set (Leichty and Brisson, 2014) improved W. pipientis sequence coverage relative to the unamplified sample. However, both of these sets failed to improve sequencing efficiency due an unevenness of coverage. The high Tm sets enriched only small portions of the genome and thus did not improve the genome coverage relative to the control
In contrast, the sets designed with the standard, lower melting temperature range (TmL) yielded more even coverage across the genome (Supplementary Fig. S3). The TmL/Even primer set, selected for having the most even distribution of primer sites across the Wolbachia genome, gave high, even coverage across the target (Fig. 3; Supplementary Fig. S3). Moreover, the TmL/Even set reduced the sequencing effort required to achieve 10× coverage across 90% of the genome by 10-fold relative to the unamplified control (Fig. 2), extrapolating from the still-rising unamplified control’s rarefaction curve. While the final two sets—TmL/Selective and the Leichty set—provided more even coverage of the genome than the TmH sets, they ultimately did not outperform the unamplified control. The previously-published primer set from Leichty and Brisson (2014) yielded low total amplification efficiency (12.1–27.7%) and uneven coverage, while the TmL/Selective set had high amplification efficiency (50–60%) but similarly uneven coverage.
Fig. 3.
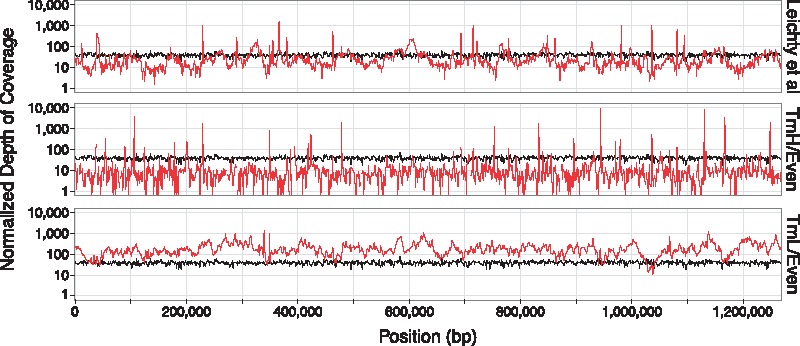
Sequencing coverage of two swga-chosen sets, and the set from Leichty and Brisson (2014), across the W. pipientis genome. The depth of sequencing coverage per 1 Mb of sequencing effort (1 Mb * coverage depth/total bp sequenced) is shown for representative replicates of? TmH/Even and TmL/Even (red lines) relative to the unamplified control (black lines). SWGA using the TmL/Even primer set improves depth of coverage across the majority of the W. pipientis genome by 10- to 100-fold, relative to the unamplified control. SWGA using the Leichty and Brisson (2014) (top panel) or TmH/Even (middle panel) sets also improve depth of coverage but over smaller regions of the genome, with the TmH/Even set resulting in high but localized amplification. Depth of coverage plots for all primer sets and replicates are shown in Supplementary Figure S3
We had originally expected that high numbers of primer binding sites in local regions of the genome would provide better coverage of that region. This was not seen in any of the sets tested (Supplementary Fig. S4). In each of the five sets tested, we did not detect a correlation between the number of primer binding sites and coverage. However, in primer sets with an overall higher density of binding sites on the target (as measured by a low average distance between binding sites), we had generally higher coverage across more of the Wolbachia genome (compare Table 1 and Fig. 2).
In summary, the primer set with the lowest Gini index and standard melting temperature (TmL/Even) was the best at selectively and evenly amplifying Wolbachia. While other sets provided a higher percentage of Wolbachia DNA (Supplementary Fig. S3), the overall coverage of these sets was low and amplification mostly occurred in specific regions (Fig. 3). This suggests that evenness of primer binding sites on the target is a major factor in the efficacy of the primer set.
3.2 Evaluation of primer sets for M. tuberculosis
For M. tuberculosis, we restricted the primer pool to only those with a low Gini index (<0.6). We let the program identify five million primer sets and then selected only those sets whose mean distance between binding sites on the M. tuberculosis genome was <5 kb. From the resulting pool of primer sets, we selected ten sets with the most extreme combinations of primer set binding evenness and density to test the contributions of each. These ten will be referred to as our positive tests (Mtb1-10), and the distribution of these points on the total pool of sets is shown in Supplementary Figure S1. We also selected the least selective set and the most uneven set from the five million set pool as negative controls (MtbSparse and MtbUneven, respectively). The composition and metrics for each of these 12 sets is shown in Table 2.
Table 2.
Characteristics of primer sets chosen for selective whole-genome amplification of M. tuberculosis from human DNA, ordered by ratio
| Ratio | # Primers | Gini | Mean target dist. | Mean bg. dist. | |
|---|---|---|---|---|---|
| Mtb6a | 0.0057 | 7 | 0.501 | 1.95E+03 | 3.41E+05 |
| Mtb9a | 0.0058 | 7 | 0.538 | 1.78E+03 | 3.05E+05 |
| Mtb4a | 0.0062 | 7 | 0.512 | 1.88E+03 | 3.04E+05 |
| Mtb8a | 0.0062 | 7 | 0.533 | 1.80E+03 | 2.92E+05 |
| Mtb7 | 0.0066 | 7 | 0.499 | 2.03E+03 | 3.09E+05 |
| Mtb2 | 0.0095 | 7 | 0.484 | 3.29E+03 | 3.45E+05 |
| Mtb5 | 0.0155 | 6 | 0.480 | 5.00E+03 | 3.22E+05 |
| Mtb1 | 0.0171 | 7 | 0.476 | 4.97E+03 | 2.90E+05 |
| Mtb3 | 0.0172 | 7 | 0.478 | 4.99E+03 | 2.90E+05 |
| Mtb10 | 0.0181 | 7 | 0.479 | 4.29E+03 | 2.37E+05 |
| MtbUneven | 0.0140 | 2 | 0.623 | 1.14E+04 | 8.10E+05 |
| MtbSparse | 0.0387 | 3 | 0.505 | 2.60E+04 | 6.71E+05 |
‘Ratio’ indicates the ratio between the average distance between binding distances in the target and background. Primer sequences are listed in Supplementary Table 2.
The sets that most effectively amplified Mycobacterium for SWGA.
The four sets with the lowest mean binding distance (sets Mtb4, Mtb6, Mtb8 and Mtb9) on the M. tuberculosis genome performed better than the unamplified controls, six other positive tests, both negative tests and the random hexamers (Fig. 4 and Table 2). These sets reached 1× coverage across 38–60% of the M. tuberculosis genome with 200 megabases of sequence, while the remaining six positive tests did not perform better than the negative controls (Fig. 4; Supplementary Fig. S5). These four sets yielded higher coverage across most of the Mycobacterium genome than the unamplified controls, while the remaining sets either only amplified certain regions or did no better than unamplified (Supplementary Fig. S6). Deeper sequencing of these four sets’ amplicons showed that the sets reached 10× coverage over 29–50% of the target by 1.5 Gbp of sequencing effort, with the unamplified controls only reaching 10× coverage on 2.5% of the target for the same sequencing effort (Fig. 5).
Fig. 4.
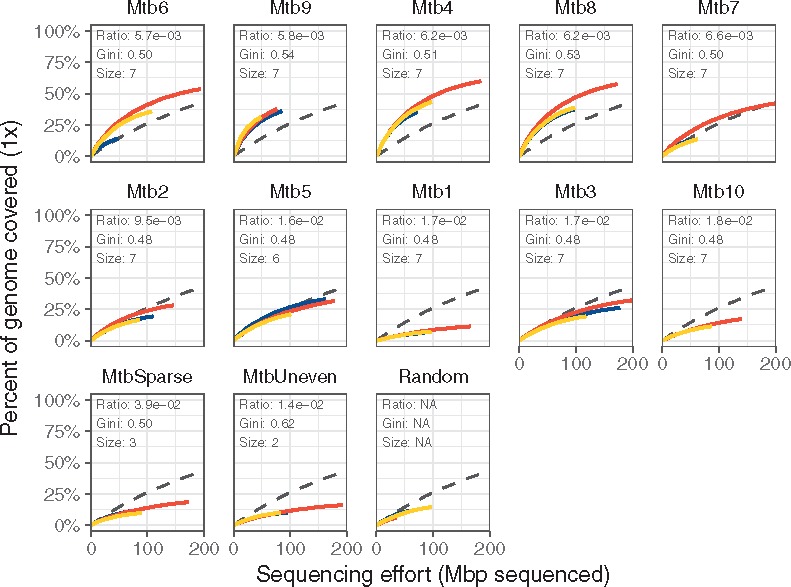
Selective amplification of Mycobacterium using swga-designed sets that prioritized primer-level evenness and set-level binding density and selectivity. Curves indicate the percent of the target covered at × 1 depth. Sets are ordered by the ratio of average distance between primer binding sites on the target to average binding distance on the background. The coloring indicates individual replicates, and the black dashed line indicates the unamplified control. The sets with the lowest ratios returned greater coverage of the target genome compared to unamplified controls than those with higher ratios, as shown by the rarefaction curves of these sets being higher than the dashed lines
Fig. 5.
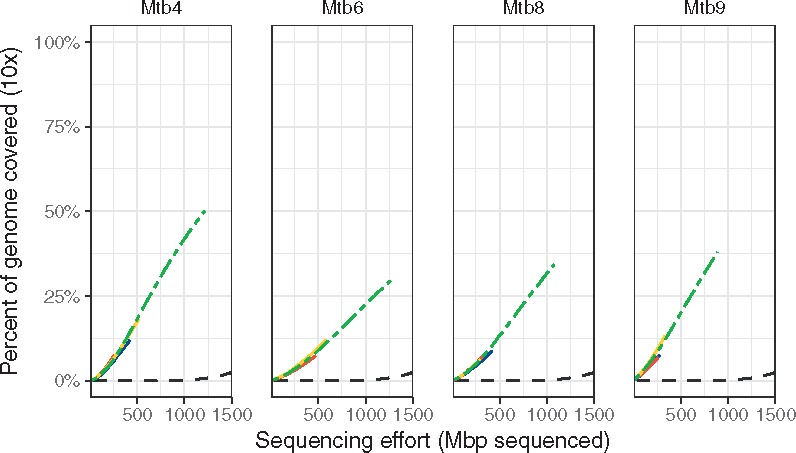
Deeper sequencing of four primer sets yields greater coverage of M. tuberculosis genome. The colored lines indicate individual replicates and the green dashed line is the pooled total. All four sets yield ∼10-fold increases in efficiency over the unamplified samples (black dashed line). The primer sets reach ×10 coverage on between 28 and 50% of the target genome while the unamplified controls were at < 2.5% ×10 coverage with 1.5 Gbp of sequencing
For Mycobacterium, we found that sets with smaller distances between primer binding sites on the target genome outperformed those optimized for lower Gini index. Nine out of the ten positive test Mycobacterium sets, including the four best sets, had lower Gini indices than the sets for Wolbachia. This suggests that after a certain threshold the Gini index becomes secondary to the primer binding site density. Therefore, pre-selecting primers with a low Gini index during swga filter and then choosing sets with high binding density in swga export allows the optimization of both attributes, and should yield effective primer sets.
4 Discussion
Selective whole-genome amplification provides a way to preferentially amplify a target genome from a complex background. However, implementation of the SWGA method has been limited due to the difficulties in designing an effective set of primers. Assembling a primer set where all of the primers are compatible with each other, selective for the target genome, and rare in the background is a problem with many degrees of freedom. The swga program addresses this difficulty by automatically identifying and evaluating primer sets by specified criteria, allowing the user to select only those sets most likely to succeed in selective amplification of the target.
We used swga to design primer sets for W. pipientis and M. tuberculosis that selectively amplified each in the presence of their host’s genome. These sets had varying binding evenness and selectivity for the target genome, allowing us to compare these attributes to the performance of each set. In addition, we demonstrated potential clinical utility of the swga program by amplifying DNA from the M. tuberculosis pathogen spiked into human blood. While in these experiments we used target/background pairs with clearly defined genomes, there is no reason the background cannot be a heterogenous mixture of DNA, such as stool or soil. In this case, the background could be approximated by whole-genome shotgun sequencing of the mixture, and subtracting any reads belonging to the target, if present.
Based on these results, it appears that primer binding evenness (as measured by the Gini index), primer set binding selectivity, and the density of binding sites on the target genome each play an important role in the set’s efficacy. In the W. pipientis study, we established that the temperature range of 15–45°C for the primers and prioritizing evenness of binding led to more even amplification of the target genome. In addition, the swga-designed sets performed better at selectively amplifying the target than the hand-designed set in Leichty and Brisson (2014). In fact, the Leichty primer set was not generated by swga because the maximum distance between primer sites on the Wolbachia genome was greater than the specified cutoff. In M. tuberculosis, by starting with a pool of primers that bind relatively evenly to the target, we constrained the range of set binding evenness by removing primers that cluster on repeat regions. After controlling the range of binding evenness at the primer level, the sets with the highest target binding density (i.e. lowest mean distance between binding sites) achieved highest coverage, suggesting that further refinements of the sets for evenness is not necessary. These sets consequently had the lowest ratio of target to background average binding distances. This ratio, as a more complete representation of the set’s selectivity than just the binding density on the target, had a strongly inverse correlation with the amount of the genome covered after sequencing (Fig. 6). Because both attributes are closely related, it is difficult to disentangle the effects of binding density from the effects of a low ratio, and it may be that either or both of these attributes contribute to the success of these primer sets. Furthermore, some sets had relatively similar ratios (e.g. Mtb7 versus Mtb8), but Mtb8 yielded greater genome coverage. This indicates that there are likely other set attributes not considered here that also contribute to set efficacy. To compensate for this, we suggest selecting five to ten sets with low ratios to test experimentally, and then selecting the best-performing of those sets.
Fig. 6.
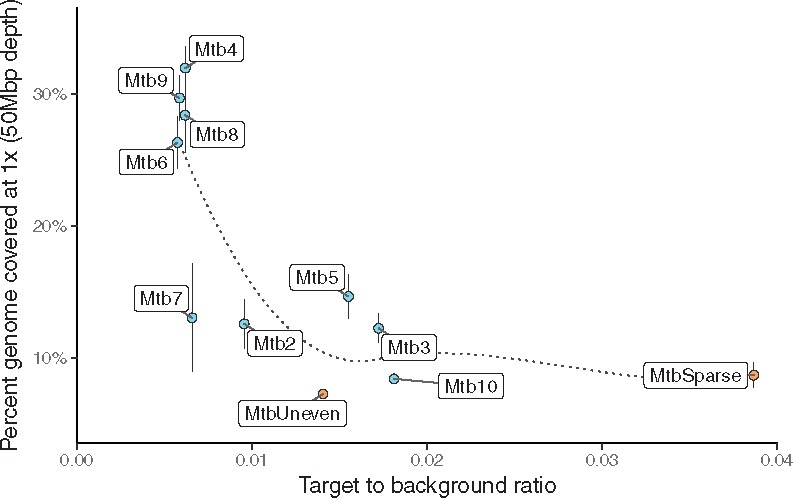
The percentage of the Mycobacterium genome covered by each set at ×1 coverage after 50 Mbp of sequencing is inversely correlated to the set’s target to background binding distance ratio (e.g. selectivity). The smoothed line of best fit (LOESS) is shown by the dotted line. The points and whiskers represent the median and standard deviation of the technical replicates. Positive tests are in blue, while negative controls are in orange. The random hexamers did not have a definable ratio and are not displayed
The swga program does not consider a specific number of primers for each set. Instead, swga considers primer sets of different sizes, and reports suggested sets. By exploring a range of sizes, the swga program allows the user to find sets with desirable attributes without having to guess what the ideal set size will be in advance.
SWGA is best suited to large scale population genomics studies and may not be cost effective in some smaller studies. Developing the SWGA primer set requires up-front costs that need to be recovered in later applications for the method to be cost effective. A detailed cost-benefit analysis over multiple applications is presented in Supplementary File 2. SWGA is most useful when large numbers of samples are to be sequenced, when the target genome is rare in the unamplified sample, and when higher sequencing coverage of the target genome is desired.
Our experiments so far suggest a general workflow that can be used to design primer sets for other systems. In particular, we recommend the following guidelines:
During swga filter, set the max_gini parameter as low as possible while still yielding 200 or more primers.
For swga find_sets, set the max_sets to 1–5 million to explore a wide range of set attributes.
Use swga export to export the sets ordered by the distance between binding sites on the target (attribute fg_mean_dist).
Pick the five to ten sets with lowest fg_mean_dist to test experimentally. Barcode each amplicon separately, then pool and sequence with low depth to assess performance. Once a high-performing set is identified, sequence that amplicon more deeply.
Once a high-performing set is identified, it is usable in any samples that have similar target/background combinations.
We expect best practices to evolve as SWGA is used more frequently. To facilitate this, we have set up a web page on the project’s source repository and a user mailing list. A tutorial on the program’s operation and more extensive documentation on each parameter and module is available on the web page as well.
Supplementary Material
Acknowledgements
We thank Michael Parisi for his generous donation of Wolbachia-infected Drosophila strains, as well as Alex Berry and other early swga users for their feedback.
Funding
This work was supported by grants from the National Institutes of Health [R01 AI097137, R01 AI076342, R01 AI091595, R37 AI050529, T32 AI007532, P30 AI045008, R01 AI100877, R01 HL113252 and R01 HL087115]; and the Burroughs Wellcome Fund [1012376].
Conflict of Interest: none declared.
References
- Allawi H.T., SantaLucia J. (1997) Thermodynamics and NMR of internal GT mismatches in DNA. Biochemistry, 36, 10581–10594. [DOI] [PubMed] [Google Scholar]
- Amann R.I. et al. (1990) Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol., 56, 1919–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell A.N. et al. (2017) Selective whole-genome amplification is a robust method that enables scalable whole-genome sequencing of plasmodium vivax from unprocessed clinical samples. mBio, 8, e02257–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean F.B. et al. (2002) Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. U.S.A., 99, 5261–5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazanfar S. et al. (2010) Metagenomics and its application in soil microbial community studies: biotechnological prospects. Malar. J., 6, 611–622. [Google Scholar]
- Gini C. (1912). Variabilità e mutuabilità. contributo allo studio delle distribuzioni e delle relazioni statistiche (variability and mutability. contribution to the study of the … . Tipogr. di Cupini.
- Guggisberg A.M. et al. (2016) Whole-genome sequencing to evaluate the resistance landscape following antimalarial treatment failure with fosmidomycin-clindamycin. J.Infect. Dis., 214, 1085–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume J.C.C. et al. (2003) Human migration, mosquitoes and the evolution of Plasmodium falciparum. Trends Parasitol., 19, 144–149. [DOI] [PubMed] [Google Scholar]
- Kent W.J. et al. (2002) The human genome browser at UCSC. Genome Res., 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryazhimskiy S. et al. (2014) Global epistasis makes adaptation predictable despite sequence-level stochasticity. Science, 344, 1519–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leichty A.R., Brisson D. (2014) Selective whole genome amplification for resequencing target microbial species from complex natural samples. Genetics, 198, 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart G. et al. (2003) The power and promise of population genomics: from genotyping to genome typing. Nat. Rev. Genet., 4, 981–994. [DOI] [PubMed] [Google Scholar]
- Mardis E.R. (2008) Next-generation DNA sequencing methods. Dx.doi.org, 9, 387–402. [DOI] [PubMed] [Google Scholar]
- Martin M. (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J., 17, p 10–12. [Google Scholar]
- Martínez F. et al. (2012) Ultradeep sequencing analysis of population dynamics of virus escape mutants in RNAi-mediated resistant plants. Mol. Biol. Evol., 29, 3297–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M.I. et al. (2008) Molecular Epidemiology of A/H3N2 and A/H1N1 Influenza Virus during a Single Epidemic Season in the United States. PLoS Pathogens, 4, e1000133.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niskanen S., Östergård P.R.J. (2003). Cliquer User’s Guide, Version 1.0.
- Nunes M.R.T. et al. (2012) Phylogeography of dengue virus serotype 4, Brazil, 2010-2011. Emerg. Infect. Dis., 18, 1858–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyola S.O. et al. (2016) Whole genome sequencing of Plasmodium falciparum from dried blood spots using selective whole genome amplification. Malar. J., 15, 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponstingl H., Ning Z. (2010). SMALT. page GNU Public License v3.
- R Core Team (2015). R: A Language and Environment for Statistical Computing Technical Report, R Foundation for Statistical Computing, Vienna. [Google Scholar]
- Rizk G. et al. (2013) DSK: k-mer counting with very low memory usage. Bioinformatics (Oxford, England), 29, btt020–btt653. [DOI] [PubMed] [Google Scholar]
- Schmeisser C. et al. (2007) Metagenomics, biotechnology with non-culturable microbes. Appl. Microbiol. Biotechnol., 75, 955–962. [DOI] [PubMed] [Google Scholar]
- Stack J.C. et al. (2012) Inferring the inter-host transmission of influenza A virus using patterns of intra-host genetic variation. Proc. R. Soc. Lond. B: Biol. Sci., 280, rspb20122173–20122173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararaman S.A. et al. (2016) Genomes of cryptic chimpanzee Plasmodium species reveal key evolutionary events leading to human malaria. Nat. Commun., 7, [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.