

Abstract
Cytochromes P450 (CYPs) oxidize alkylated amines commonly found in drugs and other biologically active molecules, cleaving them into an amine and an aldehyde. Metabolic studies usually neglect to report or investigate aldehydes, even though they can be toxic. It is assumed that they are efficiently detoxified into carboxylic acids and alcohols. Nevertheless, some aldehydes are reactive and escape detoxification pathways to cause adverse events by forming DNA and protein adducts. Herein, we modeled N-deal-kylations that produce both amine and aldehyde metabolites and then predicted the reactivity of the aldehyde. This model used a deep learning approach previously developed by our group to predict other types of drug metabolism. In this study, we trained the model to predict N-dealkylation by human liver microsomes (HLM), finding that including isozyme-specific metabolism data alongside HLM data significantly improved results. The final HLM model accurately predicted the site of N-dealkylation within metabolized substrates (97% top-two and 94% area under the ROC curve). Next, we combined the metabolism, metabolite structure prediction, and previously published reactivity models into a bioactivation model. This combined model predicted the structure of the most likely reactive metabolite of a small validation set of drug-like molecules known to be bioactivated by N-dealkylation. Applying this model to approved and withdrawn medicines, we found that aldehyde metabolites produced from N-dealkylation may explain the hepatotoxicity of several drugs: indinavir, piperacillin, verapamil, and ziprasidone. Our results suggest that N-dealkylation may be an under-appreciated bioactivation pathway, especially in clinical contexts where aldehyde detoxification pathways are inhibited. Moreover, this is the first report of a bioactivation model constructed by combining a metabolism and reactivity model. These results raise hope that more comprehensive models of bioactivation are possible. The model developed in this study is available at http://swami.wustl.edu/xenosite/.
Graphical abstract
INTRODUCTION
Alkylated amines are often important determinants of the bioactivity of organic molecules. The alkylated amine groups in biological molecules, like acetylcholine and epinephrine, often are required for their intended pharmacological functionality. As a result, alkylated amines are frequently used in medicinal chemistry.
When metabolized by cytochromes P450 (CYPs), alkylated amines usually undergo N–C bond cleavage (N-dealkylation) and give rise to an amine and an aldehyde. N-dealkylation impacts clearance as well as pharmacodynamic properties (Figure 1).1 Typically, N-dealkylation inactivates drugs and facilitates their elimination. For example, morphine undergoes N-demethylation to form the more readily excretable normorphine.2 Alternatively, N-dealkylations may activate pro-drugs, like the N-demethylation of fluoxetine (Prozac) to the more potent metabolite norfluoxetine.3,4
Figure 1.
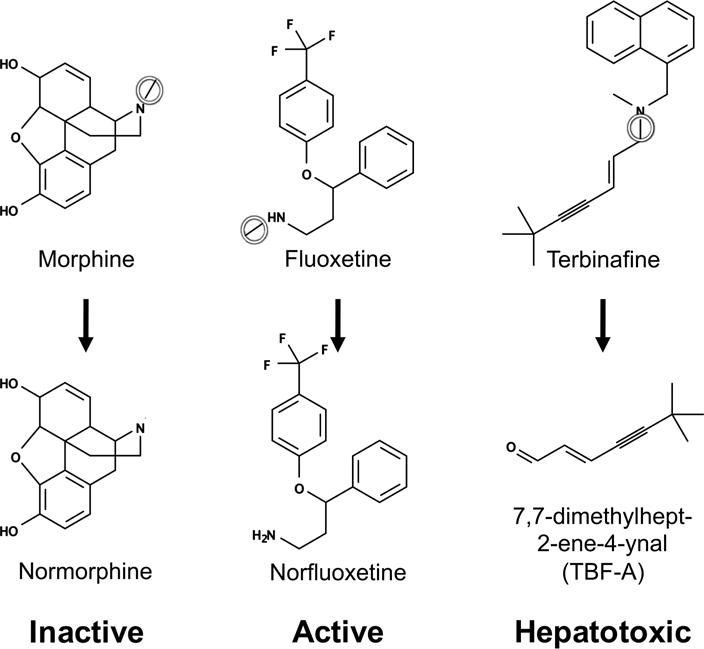
N-dealkylation can alter the efficacy and safety profile of drugs. Three drugs (with site of metabolism circled) demonstrate different outcomes of N-dealkylation. Although N-dealkylation produces two metabolites, only one observed metabolite for each reaction is shown. (Left) N-dealkylation of a morphine, an analgesic drug, produces normorphine, pharmacologically inactive and more soluble metabolite that can be easily excreted in comparison to the parent compound.30 (Middle) N-dealkylation of fluoxetine, an antidepressive drug, generates norfluoxetine, the active metabolite responsible for the majority of fluoxetine’s therapeutic effects.3,4 (Right) N-dealkylation of terbinafine gives rise to 7,7-dimethylhept-2-ene-4-ynal (TBF-A), a reactive metabolite that can conjugate to proteins and lead to toxicity.10
Frequently, metabolic studies focus on the amine metabolite, without reporting the aldehyde metabolite. Aldehydes are presumed to be detoxified to carboxylic acids or alcohols, which are suitable for conjugation and excretion. These detoxification pathways are very efficient for common aldehyde metabolites like formaldehyde, so researchers frequently ignore aldehydes and their effects on biological systems.
However, a subset of aldehydes can cause adverse events when they escape detoxification pathways. While most aldehydes form unstable adducts with protein, glutathione, or DNA, some can form stable adducts. We use the term “reactive” to refer to aldehydes that form stable adducts with biological molecules.5,6 Reactive metabolites are especially problematic in patients where the detoxification pathways are inhibited by other drugs.7,8 For example, N-dealkylation of terbinafine yields a conjugated unsaturated aldehyde (TBF-A), a reactive metabolite that forms a transiently stable glutathione adduct that appears to cause liver toxicity in patients.9,10 Biochemical studies also suggest that several drugs, including clozapine and chlorpromazine, inhibit detoxification pathways enough to increase the toxicity of aldehyde metabolites.8,11 In addition, while animal models are frequently used to study competing bioactivation and detoxification pathways, species-specific differences in these detoxification pathways make these in vivo models less reliable in predicting metabolic outcome in human.12 In summary, aldehyde formation is understudied, and their toxicity may be contingent on several additional factors that are only now being elucidated.7 As the result, aldehydes’ importance may be under-appreciated, and, at least in some cases, they might cause idiosyncratic toxicity in patients.
Data-driven models are already used in scientific and regulatory context. In lead optimization, very similar machine learning models are used to identify metabolic hotspots on molecules.13,14 Likewise, alert structures are commonly used to identify liabilities in molecule structure,15–17 and we previously demonstrated that metabolism models can improve the specificity of alerts.18 The hope is that modeling of metabolism and reactivity will improve substantially on purely statistical approaches to understanding toxicity, as are commonly relied upon within industry and regulatory agencies.19 Metabolism modeling with machine learning does not produce detailed enzyme mechanics, but they do provide finer grain information that is currently used, and for this reason are a significant step forward.
Our group has published extensively on deep learning models of metabolism and reactivity, with the ultimate goal of comprehensively predicting bioactivation pathways of drug-like molecules. These studies have included models for reaction types yielding specific metabolites such as epoxides20 and quinone species.21 We have also modeled the reactivity of metabolites toward adduction with glutathione, DNA, proteins, and cyanide, which often are biomarkers for toxicity as well as drivers of toxicological mechanisms.5,22 DNA conjugation can lead to cancer,23,24 and protein conjugation can lead to cellular dysfunction, cell death, and sometimes immune response cascades that extend the damage to organ and systemic levels.6 As insight into those possibilities, our models yield probabilities for individual sites on the molecule to undergo the specific reaction, enabling prediction of metabolite structures too. Taken together, metabolism modeling can predict if a bioactivation reaction is likely to take place, and this information can guide downstream analyses.
In this study, we aim to study a specific bioactivation pathway, the formation of reactive aldehydes by N-dealkylation. Though detailed enzyme mechanisms are not considered here, the question is mechanistic in that a specific metabolic pathway is being assessed for its mechanistic role in clinically observable toxicity. Our goal is to assess the impact of the N-dealkylation pathway, specifically. In contrast, other studies have focused on fitting statistical models to toxicity data without regard to bioactivation pathways.19 We approach this question by developing a new model of N-dealkylation, combining it with a previously reported reactivity model, and assessing drug-like molecules in the literature for bioactivation by this pathway. To develop the metabolism model, we used a deep learning algorithm, previously developed by our group to predict other types of drug metabolism.20,25–29 In this study, we trained the model to predict N-dealkylations by human liver microsomes (HLM). We also assessed the value of including isozyme-specific metabolism data alongside the HLM data, in building metabolism models. Next, we combined the metabolism and reactivity models into a bioactivation model. This combined model predicted the structure of the most likely reactive metabolites formed by N-dealkylation of an input molecule. Molecules known to form reactive metabolites by N-dealkylation were used to assess the bioactivation model. Finally, we used this model to study the importance of N-dealkylation as a bioactivation pathway.
MATERIALS AND METHODS
Training Data
We collected a data set from the literature-derived Accelrys Metabolite Database (AMD) as a foundation for modeling efforts. A total of 1290 N-dealkylation reactions were extracted, each of which catalyzed by human liver microsome (HLM) or one of the nine most common human P450 isozymes: CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4 (Figure 2). We implemented an automated labeling algorithm that used the structure of each N-dealkylation product to identify the bond within the reactant that breaks, as the site of N-dealkylation. Duplicate molecules were merged into a single representation, with all metabolized bonds labeled. After merging, our final data set contained 883 unique substrates with each nitrogen–carbon (N–C) bond assigned 10 binary values (including missing if not-known), corresponding to metabolism by HLM and the individual P450 isozymes. There were 25,506 bond-level training labels in the data set. Under the AMD license agreement, we were not allowed to disclose the structures of molecules in the data set. Nevertheless, to enable reproduction of our results, we provided the AMD reaction and molecule registry numbers and the metabolism status of every compound in the final data set in the Supporting Information.
Figure 2.
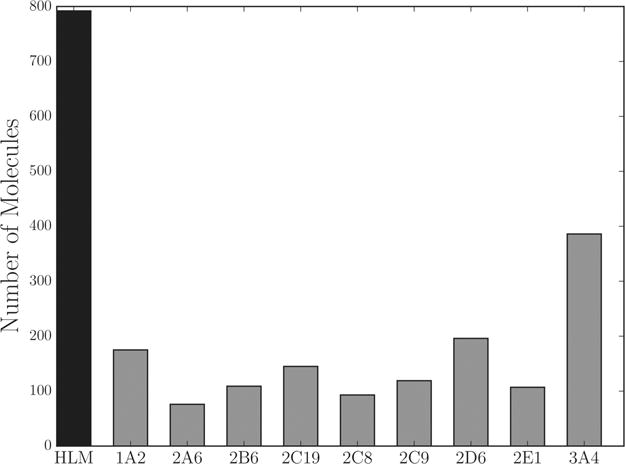
Size of each data set. Each data set contains unique N-dealkylation substrates metabolized by the corresponding cytochromes P450 (CYP) isozyme or in human liver microsome (HLM). The size of each data set reflects the contribution of each enzymatic entity to N-dealkylation metabolism. Many of the substrates are metabolized by multiple CYP isozymes and HLM. The combined data set contains 883 molecules.
External Validation Data
An external data set was used to assess the generalizability of models built on the training data. This testing set contained 108 unique HLM-mediated N-dealkylation substrates recently added to AMD (Jan, 2017 version) that are not in our training data set. We provided the AMD reaction and molecule registry numbers and the metabolism status of every compound in the test data set in the Supporting Information.
Descriptors
To model N-dealkylation, we encoded chemical information in numerical descriptors. Each bond was represented by a vector of 381 descriptors that described its properties at the atom, bond, and molecule levels. All of these descriptors were calculated by in-house software from Open Babel 2D SDF files.31 Specifically, the 381 descriptors included two sets of 179 atom descriptors, 7 bond descriptors (e.g., bond type, bond length), and 16 molecule descriptors (e.g., molecular weight, topological polar surface area, or molar refractivity). Atom-level descriptors (e.g., atom identity, charge, or hybridization), representing atoms on either side of each bond, were developed for our previous models of metabolism and reactivity.20,25,26 The complete list of descriptors used in this study is provided in the Supporting Information.
Heuristic Model
We constructed a simple heuristic model using overall data set statistics to provide a baseline of performance against which to compare to more complex methods.27 In this heuristic model, we identified all N–C bonds as potential sites of N-dealkylation in a test molecule. Based on the carbon chain attached to the nitrogen, potential site of N-dealkylation could be classified into four groups: methyl, short alkyl chains, in nonaromatic ring, and the remaining N–C bonds. The probability of being labeled as metabolized in the combined data set across all members of each group was calculated. This strategy yielded probabilities of 65.32%, 64.51%, 5.41%, and 16.98% for methyl, short alkyl chains, in nonaromatic ring, and the remaining N–C bonds, respectively (Table 1). Each potential site was initially assigned with the probability of its group. All other bonds were assigned initial scores of zero. Next, these scores were linearly scaled so that they summed to one in molecules that have at least one potential site. The bonds of a molecule with no potential sites all received a score of zero. A python implementation of this model and an excel file containing the detailed statistics are included in the Supporting Information to facilitate future studies.
Table 1.
Classes of N–C Bonds in the Heuristic Model and the Probability of Being N-Dealkylated by HLM
Multitarget Modular Model
For each input molecule, atom-level, bond-level, and molecule-level descriptors were calculated for all bonds between pair of heavy atoms. These descriptors were inputs to a neural network with two hidden layers that generates vectors of 10 scores corresponding to one for HLM and one for each P450 isozyme. Each element in the vector represented the probability that a bond is metabolized, mediated by the corresponding isozyme or HLM. The weights of the model were calibrated during training by performing gradient descent on the cross-entropy error of the difference between the predicted and actual response values of each bond.
For assessment, predictions were computed using 10-fold cross-validation. Cross-validation is a jackknifing technique widely used in machine learning for providing an estimate of generalization accuracy. In our case, a model generated by nine-tenths of the data set was used to predict the remaining one-tenth of the training data. The process was then repeated until all data were used in a test set exactly one time. The aggregated accuracy was tracked and compiled to assess the cross-validated accuracy as a reasonable estimate of model accuracy toward new data.
The model predicted N-dealkylation at the bond level using a multitarget, modular network36–40 with one input layer, three hidden layers, and one output layer (Figure 3). This architecture had several advantages over a standard neural network. First, it mirrors the local structure of some of the input descriptors. The contribution of each descriptor to the final model depended on the distance from the atom or bond it described.20,25,26 Accordingly, the modular network combined descriptors by their distances to the atom of interest into neighborhood groups (Table S4). Second, the multitask architecture solves related problems simultaneously. In this case, P450 share the same catalytic mechanism while differing in substrate specificity. This information from the isozyme specific data sets could then be used to improve the accuracy of HLM predictions in our model design. Finally, the multitask model architecture has nearly the same number of parameters as the single-task HLM model, but makes use of much more data.
Figure 3.
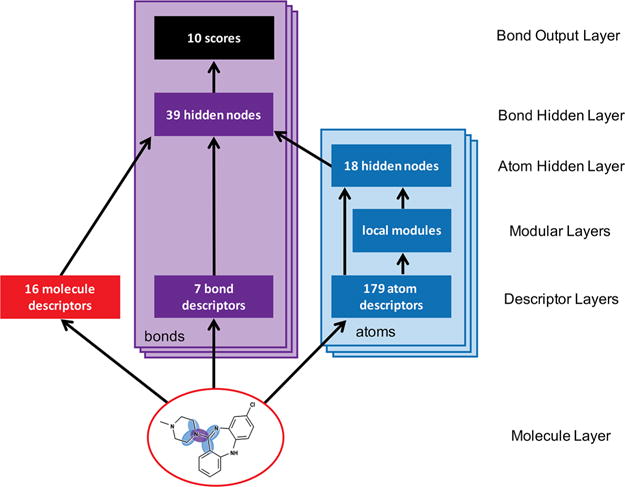
The structure of the N-dealkylation model. This diagram shows how information flows through the model, which is composed of one input layer, three hidden layers, and one output layer. This model computes predictions for each N–C bond in a test molecule. From the 2D structure of an input molecule, 16 molecule-level, 7 bond-level, and two sets of 179 atom-level descriptors are calculated. All nodes in the second hidden layer feed to the output layer of 10 nodes (each corresponds to a cytochrome P450 isozyme or human liver microsome). In the diagram, bars represent vectors of numbers. Layers are colored red, purple, and blue corresponding to, respectively, molecules, bonds, and atoms. The model has 6375 weights.
Single Target Models
Multitargeted models were more effective than single target models for predicting molecule reactivity.5 We tested if this was true also for N-dealkylation by training 10 single-target neural network models, each on a P450 isozyme or HML data set to provide a baseline of performance against which to compare to the multitarget modular deep neural network. A matrix of descriptor encoded bonds between heavy atoms in a data set was presented to a neural network with 20 hidden nodes. During training, the model learned a mapping between the descriptor values of each bond and the binary experimental response of that bond, metabolized or not metabolized by HLM or each P450 isozyme. The weight calibration and cross-validation procedures were identical to those of the multitarget modular deep neural network.
Reactivity Model
Previously, we developed a model to predict reactivity to biological molecules.5 This model was a deep neural network that predicted, given a molecule’s structure, its probabilities of conjugating to protein, DNA, glutathione (GSH), and cyanide. The model was trained on 1364 electrophilic and 1439 nonreactive molecules. On the original training data, the reactivity model predicted reactive atoms within reactive molecules with average site AUC of 96.6%, 89.8%, 92.8%, and 94.4% for cyanide, DNA, GSH, and protein, respectively. It is unreported how accurately the reactivity model predicts the reactivity of aldehydes in particular, and the cross-validated predictions were used to assess the performance on subset of molecules with aldehydes.
Metabolite Structures
The metabolite algorithm constructed a pair of amine and aldehyde/ketone products for every possible N–C bond cleavage. However, N-demethylations were excluded because the formaldehydes produced by these reactions are rapidly detoxified.42 A python implementation of this N-dealkylation metabolite structure generator is included in the Supporting Information to facilitate future studies.
When a molecule is N-dealkylated at a N–C bond, this reaction generates two metabolites, that is, a lower order amine from the nitrogen-side and aldehyde/ketone from the carbon-side.1 Usually only the nitrogen side of the product is reported (Figure 4). Even when reported, carbon-side products are only described as aldehydes/ketones 48.5% of the time in the AMD. In some cases, the reported reactions instead included downstream metabolites of the aldehydes, that is, alcohols (26.1%) and acids (25.4%), generated by subsequent detoxification reactions by aldehyde reductases and aldehyde dehydrogenases.42–44 For this reason, the metabolite algorithm always inferred the formation of aldehydes (or ketones) and does not model subsequent transformations.
Figure 4.
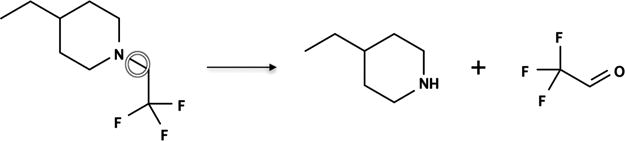
Metabolites formed by N-dealkylation. C–N bond breakage during N-dealkylation creates two metabolites, one from each side of the bond. The nitrogen-side metabolite (R2NH) has a hydrogen in place of the leaving alkyl group. The carbon-side metabolite is an aldehyde/ketone (RCOH/RCOR). Trifluoroacetaldehyde (TFALD) precursor’s site of N-dealkylation is circled.41
N-Dealkylation Bioactivation Model
We built a bioactivation model that linked metabolism, metabolite structure prediction, and reactivity models. This combined model predicted the most likely reactive metabolites generated by N-dealkylation from precursor molecules. For each potential metabolite, the combined model defined its output as the product of two predictions, the probability of formation times the probability of being reactive. The first prediction was the probability of formation as computed by HLM N-dealkylation model. The second prediction was the maximum of the probabilities of reactivity toward protein and DNA as computed by the reactivity model. The metabolite with the highest score (the product of both predictions) was predicted to be the most likely reactive metabolite generated by N-dealkylation.
■ RESULTS AND DISCUSSION
The following sections study the performance of the models and then use them to assess N-dealkylation as a bioactivation pathway. First, we evaluated the N-dealkylation model in its accuracy of identifying metabolized sites within a metabolized molecule. Second, we analyzed the model predictions at the global level by calculating classification performance on N–C bonds in the entire data set. Third, we evaluated the accuracy of the N-dealkylation model on an external testing data set. Fourth, we assessed the reactivity model predictions on aldehyde containing molecules. Fifth, we tested the bioactivation model’s prediction of reactive metabolites from molecules known to produce reactive aldehydes. Finally, we present three case studies of drugs known to produce reactive aldehydes by N-dealkylation.
Metabolism Model Performance
A key objective was to identify the specific bond within a substrate that undergoes N-dealkylation: its site of N-dealkylation. For every bond in a test molecule, the N-dealkylation model generated 10 scores, each ranging from 0 to 1 and corresponding to the probability that the bond was metabolized by HLM or a certain P450 isozyme. The HLM model predicts most sites correctly (Figure 5).
Figure 5.
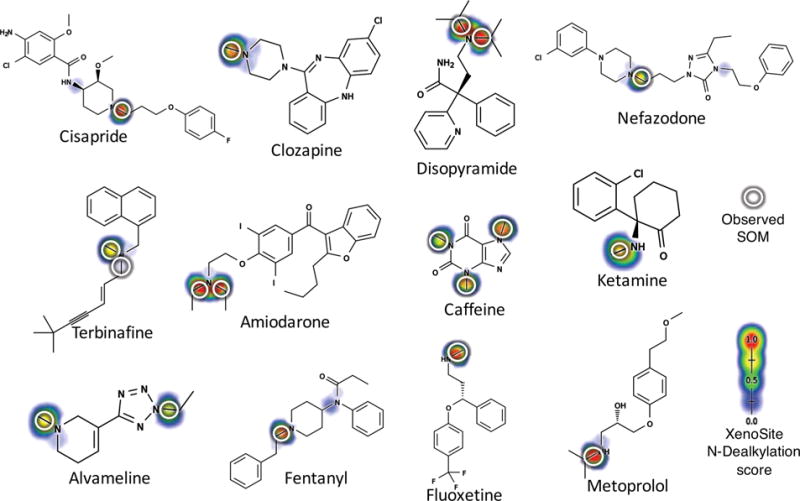
The HLM accurately predicts metabolism of most molecules. Cross-validated human liver microsome (HLM) N-dealkylation scores on example drugs from our training data are shown. Experimentally observed HLM sites of N-dealkylation are circled. The model gives the highest score to the correct sites in all these cases.
An informative model should assign metabolized sites with higher scores than nonmetabolized sites within each molecule. We evaluated the model’s success at this objective using two metrics. First, we calculated “top-two” performance, where a substrate was considered to be correctly predicted if any of its experimentally observed sites of metabolism were predicted in the top two rank-positions out of all potential sites in the substrate. The top-two metric is commonly used to evaluate CYP site of metabolism models.14,45 Second, we calculated “average N–C AUC. This metric is computed by measuring the area under the ROC curves (AUC),46 for all N–C bonds in the dataset.20,26
The model accurately predicted metabolized sites within metabolized molecules. HLM model had cross-validated top-two and average N–C AUC accuracies of 96.6% and 93.7%, respectively. Across all 10 targets, the model had cross-validated top-two and average N–C AUC accuracies of 97.2% and 95.3%, respectively (Tables 2 and 3). As an additional test for overfitting, we constructed a negative control where the metabolism targets were permuted randomly. The model was trained and tested, in cross-validation, on the permuted data. In this negative control, performance was poor, with an average AUC across isozymes of 49.4%, worse than a random classifier. The poor performance on this negative control further confirms the model is not overfitting and the high performances observed are not artifacts.
Table 2.
Scores and Dataset Sizes for Reactive and Nonreactive Moleculesa
| mean score
|
number of compounds
|
|||
|---|---|---|---|---|
| reactive | nonreactive | reactive | nonreactive | |
| protein | 0.23 | 0.06 | 20 | 222 |
| GSH | 0.32 | 0.03 | 48 | 1082 |
| DNA | 0.31 | 0.03 | 10 | 195 |
| cyanide | 0.16 | 0.02 | 1 | 128 |
For the HLM model, a reasonable cutoff might be a score of 0.1–0.15. However, our model uses the score itself as a probability in downstream analysis.
Table 3.
The Model Accurately Identifies Sites of N-Dealkylationa
| isozyme | top-two
|
average N–C AUC
|
global N–C AUC
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| self | MT | ST | HR | self | MT | ST | HR | self | MT | ST | HR | |
| HLM | 98.9 | 96.6 | 95.7 | 80.8 | 96.1 | 93.7 | 92.9 | 81.4 | 97.5 | 95.6 | 95.4 | 87.1 |
| CYP1A2 | 98.9 | 96.6 | 94.9 | 86.5 | 97.0 | 95.0 | 91.5 | 87.2 | 95.1 | 90.1 | 90.5 | 83.4 |
| CYP2A6 | 100 | 98.7 | 98.7 | 88.4 | 96.2 | 94.8 | 86.4 | 80.9 | 96.3 | 88.2 | 87.7 | 84.4 |
| CYP2B6 | 99.1 | 99.1 | 96.3 | 90.2 | 99.7 | 98.9 | 94.4 | 89.9 | 97.3 | 92.4 | 90.8 | 87.1 |
| CYP2C19 | 100 | 98.6 | 95.9 | 86.3 | 98.7 | 97.3 | 96.4 | 91.4 | 97.0 | 92.8 | 92.3 | 84.8 |
| CYP2C8 | 100 | 97.8 | 96.8 | 85.4 | 98.3 | 96.4 | 93.9 | 88.2 | 95.4 | 89.3 | 88.6 | 82.9 |
| CYP2C9 | 99.2 | 97.5 | 95.8 | 88.7 | 98.4 | 97.4 | 96.5 | 93.4 | 96.3 | 90.6 | 89.9 | 85.8 |
| CYP2D6 | 100 | 99.0 | 98.5 | 89.6 | 98.2 | 97.0 | 97.0 | 91.9 | 95.7 | 91.4 | 90.7 | 84.4 |
| CYP2E1 | 98.1 | 97.2 | 96.3 | 84.8 | 95.8 | 93.9 | 89.6 | 81.2 | 96.7 | 91.6 | 89.4 | 83.9 |
| CYP3A4 | 98.2 | 96.1 | 96.6 | 80.1 | 98.2 | 95.8 | 95.5 | 86.8 | 95.9 | 90.9 | 90.2 | 82.8 |
The table contains 10-fold cross-validated top-two, average N–C AUC, and global N–C AUC performance of the multitask (MT), single target (ST), and heuristic (HR) models. Accuracies of the MT model on the training data set (self) are also included for reference. For each metric, the highest cross-validated performance is bolded. Any scores not statistically different from the best performance are italicized. In all cases, the neural networks are significantly better than the heuristic model. The performance difference between the HLM single target and the multitask models by top-two and average N–C AUC is not significant (P = 0.352 and 0.425, respectively, by Mann–Wittney U test). The performance difference by global N–C AUC is statistically significant (P = 0.021 by paired permutation test).
As another control designed to test the value of multitask training, we trained 10 single-target models, each on a CYP isozyme or HML data set, for comparison. The multitask model performed slightly better than the single target models, but this improvement was not statistically significant (P = 0.351 and 0.425 for top-two and average N–C AUC, respectively). Both multitask and single target models significantly outperformed the heuristic model, which had top-two and average N–C AUC accuracies of 80.8% and 81.4%, respectively.
An effective model should distinguish metabolized sites globally in the data set across all N–C bonds. To quantify this performance, we computed the area under the ROC curve globally across all metabolized and nonmetabolized N–C bonds (Table 3).27,47
The model accurately predicted sites of N-dealkylation by this metric. Across all 10 targets, the model had an average cross-validated global N–C AUC of 92.6% (Table 3). Notably, the multitask model predicted HLM-mediated sites with global N–C AUC of 95.6%, significantly outperforming the single target model (P = 0.021 by paired permutation test).46 Both multitask and single target models significantly outperformed the heuristic model, which had an average global N–C AUC accuracy of 85.2%.
The output of the model is a well-scaled probability. When we binned N–C bonds by HLM prediction score, the proportion of sites of HLM-mediated N-dealkylation in each bin closely correlates with the bin’s score (Figure 6, R2 = 0.975). Thus, the model’s output is interpretable as a probability and can be combined with other probabilistic outputs.
Figure 6.
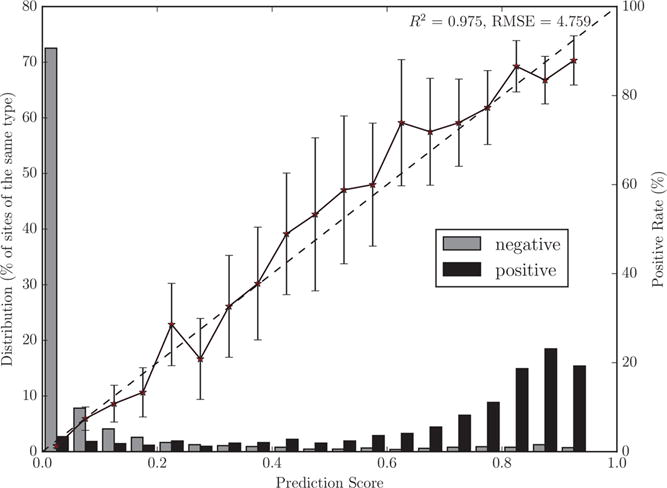
The model makes well-scaled predictions, corresponding to probabilities. The bar graphs plot the distributions of scores across 4071 dealkylated and nondealkylated N–C bonds. The solid lines plot the percentage of N–C bonds that are dealkylated by human liver microsome (HLM) (using non-normalized frequencies) in each bin. The diagonal dashed lines indicate a hypothetical perfectly scaled prediction. HLM N-dealkylation score has a strong correlation to a perfectly scaled prediction (R2 value of 0.975 and RMSE of 4.759%). This means that the score is interpretable as the probability that a N–C bond is metabolized in HLM. Reliability diagrams for all cytochrome P450 isozymes in the multitarget model are in the Supporting Information.
External Validation of N-Dealkylation
The multitask N-dealkylation model performed well on the external testing data set of 108 HLM-metabolized molecules. Prediction accuracies of our model on this external testing data set were 95.5%, 94.1%, and 94.8% for top-two, average N–C AUC, and global N–C AUC, respectively. Overall, performance on the testing data set was comparable to the cross-validated performance on the training data set.
Reactivity Predictions on Aldehydes
The accuracy of the reactivity model on aldehydes, specifically, was not assessed. A effective reactivity model for aldehydes is important for this study and assessed here.
We find that the reactivity model accurately predicted aldehydes reactivity. We identified 746 aldehyde containing molecules and 1128 aldehyde sites from the reactivity model’s training data set and used their cross-validated predictions to access the model performance on this subset of data. First, we assessed the ability of model to identify the correct atom in the molecule as reactive. The average site AUC is computed by averaging the AUC of sites computed within each molecule separately. The reactivity model predicted reactive atoms of 746 aldehyde containing molecules with average site AUC accuracies of 97.0%, 91.7%, 95.7%, and 94.9% for cyanide, DNA, GSH, and protein, respectively. Next, we assessed the ability of the model to separate reactive and nonreactive aldehyde molecules. Across the full database 1128 aldehydes, the model can accurately separate reactive and nonreactive substructures with AUCs of 93.0%, 80.3%, 89.1%, and 71.0% for cyanide, DNA, GSH, and protein, respectively. The model’s performance was similarly strong on the 313 α–β unsaturated aldehyde in this same data set (Supporting Information). These assessments demonstrate that the reactivity model can accurately model the reactivity of aldehyde containing compounds.
A Census of Reactive Aldehyde Metabolites
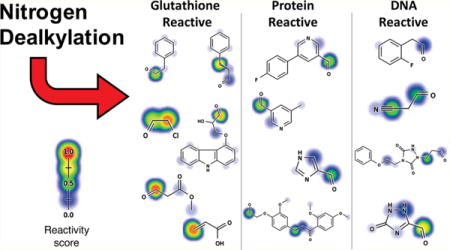
The aldehyde products from the carbon-side of N-dealkylation reactions are frequently omitted in the literature. The training data set contained 1290 N-dealkylation producing aldehyde metabolites, yet only 26 of these reactions were reported with aldehyde metabolites. Of these 26 aldehydes, the literature reports 16 and 21 conjugate to GSH and DNA, respectively. Moreover, several of the aldehydes unreported from these reactions appear to be reactive (Figure 7). This omission underscores the large gaps in the literature and suggests the potential importance of aldehydes in toxicity pathways too.
Figure 7.
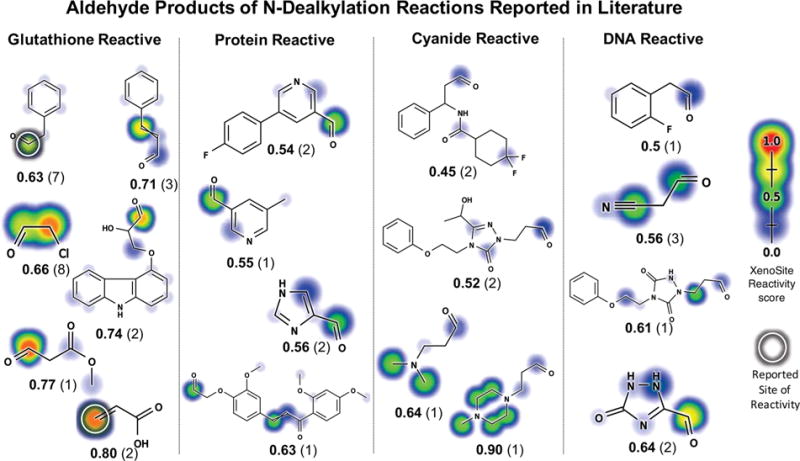
Reactive aldehyde metabolites. We computed the structures of all aldehyde metabolites generated from observed N-dealkylation reactions. Next, we applied the reactivity model to these predicted metabolites to assess their reactivity.5 Examples of the aldehyde metabolites and their reactivity predictions are shown. The molecular reactivity scores (MRS) are bolded. The number of molecules known to produce this metabolite are shown in parentheses. Experimentally determined sites of reactivity are circled.
Next, we studied the aldehyde metabolites of the reported N-dealkylation reactions of approved and withdrawn drugs. The complete list of these drugs, their aldehyde metabolites, and reactivity scores are provided in the Supporting Information. Several hepatotoxic drugs with unknown mechanisms of hepatotoxicity,48 such as indinavir, piperacillin, verapamil, and ziprasidone, are predicted by the reactivity model to have aldehyde metabolites more reactive than the parent drug and all the rest of their reported metabolites in the AMD (Figure 8). Although piperacillin toxicity has been recently associated with β-lactam ring opening and subsequent protein conjugation, metabolic studies on this antibiotic did not assess the reactivity of its acetyl aldehyde metabolite produced via N-demethylation, a major in vitro and in vivo metabolic pathway of piperacillin.49 This omission adds to the evidence that N-dealkylation may be an under-appreciated bioactivation pathway. Reactive aldehydes could contribute to hepatotoxicity of these drugs. In future work, we plan to study these pathways and confirm them in appropriate in vitro studies.
Figure 8.
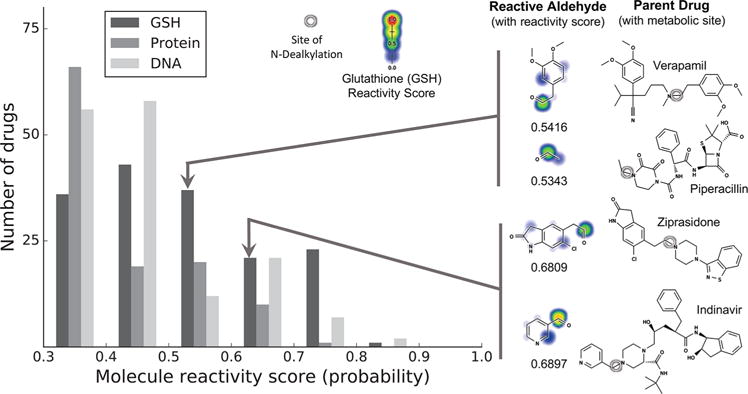
Reactive aldehyde metabolites formed by N-dealkylation reactions. Of 1925 approved and withdrawn drugs, the literature reports N-dealkylation reactions for 380. The aldehydes formed by these N-dealkylation reactions were inferred, and their reactivity was assessed with a previously published reactivity model.5 (Left) The distribution of reactivity scores above 0.3 for glutathione (GSH), protein, and DNA molecule reactivity. For reference, the reactivity scores of N-acetyl-p-benzoquinone imine, a well-known electrophile responsible for acetaminophen’s toxicity, are 0.75, 0.54, and 0.38 for GSH, protein, and DNA, respectively. (Right) Several drugs that are (1) known to be hepatotoxic (2) by unknown mechanisms also (3) appear to form reactive aldehydes. These aldehyde metabolites were predicted to be more reactive than the parent drug as well as their other observed metabolites, so these aldehydes might be the mechanism of toxicity.
When a N-dealkylation reaction is reported in the literature without reporting the aldehyde metabolite, it is unclear if the aldehyde is unreported because (1) it was observed but not deemed important enough to report, (2) it was not observed because experiments were not tuned to detect them, or (3) it was not observed because it is a reactive metabolite that conjugates to protein. This final case is most concerning because these aldehydes could contribute to the toxicity of a large number of drugs. N-dealkylation appears to be a neglected bioactivation pathway that merits systematic study, especially in cases with evidence of idiosyncratic adverse events typically associated with reactive metabolites.
Modeling Bioactivation Into Reactive Aldehydes
We tested the bioactivation model on molecules that are known to produce reactive metabolites by N-dealkylation. The metabolism model’s training data set contains 112, 52, and 3 molecules with N-dealkylation metabolites reported to be reactive with DNA, GSH, and protein, respectively. No training is done in this experiment, so the molecules here are just used for validation. In this task, the aldehyde metabolites that are not formed or are not reactive are intrinsic negative controls. For each molecule, the goal is to predict the known reactive first, above all possible N-dealkylation metabolites.
The bioactivation model can predict reactive metabolite generated through N-dealkylation with high accuracy (Figure 9). The bioactivation model can rank the metabolites observed to be reactive with DNA, GSH, and protein at the first position 79.4%, 75.0%, and 66.6% of the time. Averaging the AUC of the metabolites associated with each molecule, the bioactivation model ranking predicts observed reactive metabolites with accuracies of 95.4%, 95.7%, and 86.6% for DNA, GSH, and protein, respectively. The Supporting Information includes the metabolite structures and their bioactivation model scores, with the reactive aldehyde labeled.
Figure 9.
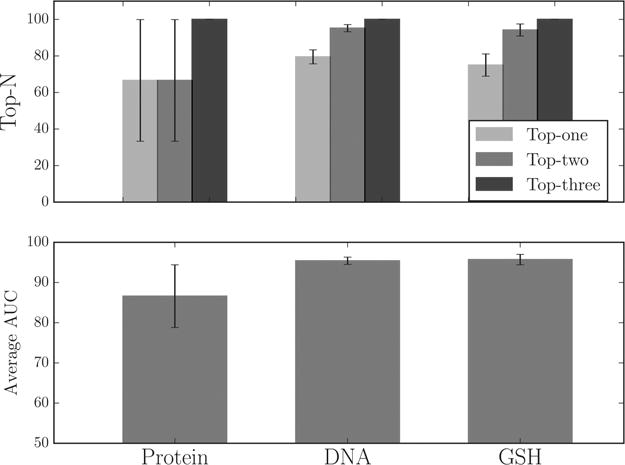
The bioactivation model accurately identifies reactive metabolites produced by N-dealkylation. The training data set contains 112, 52, and 3 molecules that produce through N-dealkylation metabolites reported to conjugate to DNA, GSH, and protein, respectively. (Top) Top-N are the percentages of molecules which have their observed reactive metabolite predicted in the first-, second-, or third-rank positions by the bioactivation model. (Bottom) AUC for predictions of all metabolites produced via N-dealkylation from each molecule is computed and then averaged across the whole data set, measuring the per-molecule performance.
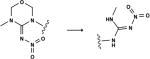
The three molecules known to have toxicity associated with reactive metabolites generated by N-dealkylation: a trifluor-oacetaldehyde (TFALD) precursor,41 isfosamide,50,51 and terbinafine (TBF)10 illustrate the model’s predictions (Figures 10 and 11). The bioactivation model correctly identifies the toxic metabolite in all three cases (Figures 10 and 11). In the first case, the TFALD precursor, TFALD is identified with high probability and is known to cause cytotoxicity52,53 The second case, ifosamide is an anticancer prodrug that is extensively metabolized by P450.50 Ifosamide’s hepato- and nephrotoxicity-profile has been attributed to chloroacetaldehyde, a metabolite formed through N-dealkylation of ifosamide.51 For both cases of TFALD precursor41 and ifosfamide,50,51 the combined model correctly ranks known reactive metabolites (TFALD and chloroacetaldehyde) as the most likely reactive metabolites produced by N-dealkylation from the parent molecules (Figure 10). The antifungal TBF, the third case, is more complicated. The correct metabolite is identified (Figure 11), however it is assigned a low probability, just 6%. As we will see, this is because the reactive metabolite may be formed by double rather than single dealkylation(s) from TBF.
Figure 10.
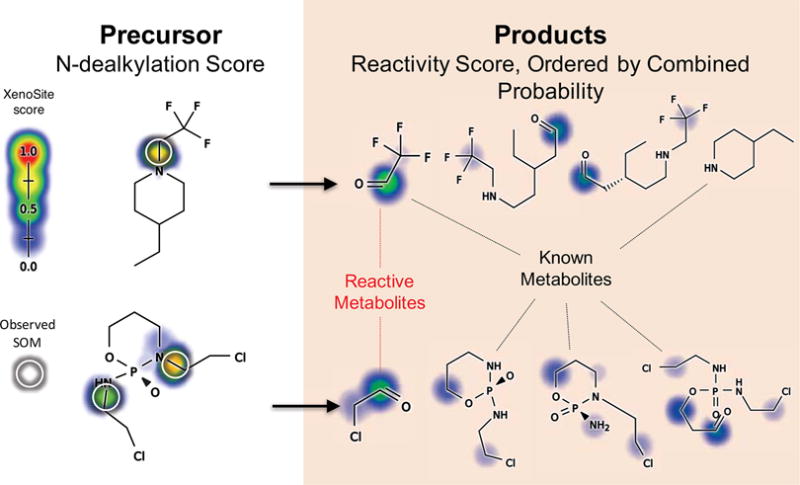
Combination of metabolism, metabolite structure prediction, and reactivity models to predict reactive metabolites. The combined model predicts the most likely reactive metabolites produced by N-dealkylation from precursor molecules. Example molecules are trifluoroacetaldehyde precursor41 and ifosfamide.50,51 Terbinafine is discussed in the next section and figure.
Figure 11.
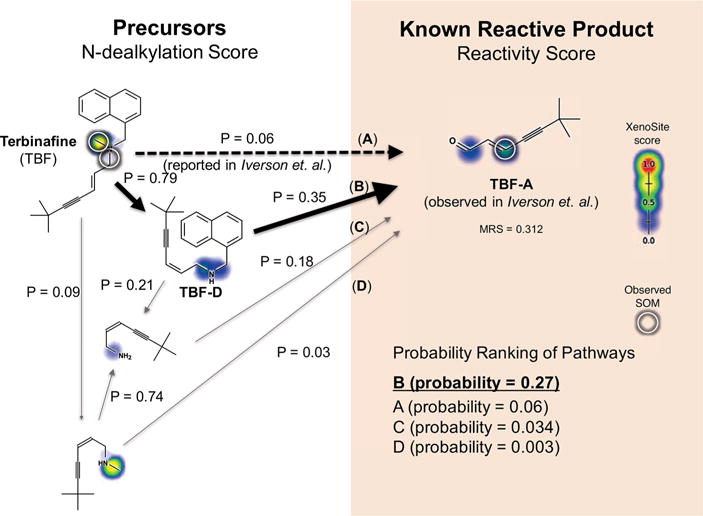
Terbinafine is predicted to form its reactive metabolite by two sequential dealkylations. Multiple metabolites generated by N-dealkylation of terbinafine have been observed.10,54 The reactive metabolite 7,7-dimethylhept-2-ene-4-ynal (TBF-A, depicted) was identified as a key mediator of hepatotoxicity.10 It is proposed that TBF-A is formed directly from terbinafine single dealkylation (dashed arrows, P = 0.06), but several alternate pathways are possible. The model computes the probability of each pathway, finding that TBF-A is most likely formed by two sequential dealkylations (path B, P = 0.27) through the intermediate desmethyl terbinafine (TBF-D). Supporting this prediction, TBF-D is reported as a metabolite of TBF by some studies.54 This suggests a revision of the single dealkylation pathway reported in the literature (path A, P = 0.06).
These results are encouraging. No training is need to sequentially apply the metabolism and reactivity models. So without fitting an parameters, it appears the bioactivation model can correctly identify reactive aldehydes in drug-like molecules. A better analysis might include more molecules, but aldehyde toxicity is not studied enough to be sure any aldehydes safe.
Bioactivation of Terbinafine
TBF is a widely used antifungal agent. The most common adverse side effects associated with oral terbinafine treatment are mild to severe gastrointestinal and cutaneous reactions.55 However, TBF causes transient liver injury in at least 1% of patients, which may progress to death or fulminant liver failure.56,57 Consequently, oral TBF prescriptions require regular monitoring of liver enzymes.9,56 Hepatotoxicity is thought to be caused by 7,7-dimethylhept-2-ene-4-ynal (TBF-A, Figure 11). This reactive metabolite was only demonstrated 15 years after marketing TBF worldwide and was not identified by standard reactive metabolite screens.10
Although TBF-A was eventually identified as a reactive metabolite, the exact mechanism of its formation is unclear.10,40,54 TBF-A has been proposed as a metabolite of TBF,10 but there are several pathways possible, and the precise pathway was not identified. TBF possesses three N-alkyl groups that are susceptible to oxidative cleavage. These metabolic reactions may generate reactive TBF-A in a single dealkylation (path A, Figure 11) or multiple dealkylations (paths B–D, Figure 11). The model identifies the most probable pathway (path B, Figure 11). Though beyond the scope of this computational study, preliminary experimental data suggest the double-dealkylation pathway is more important.
A sequential dealkylation pathway to forming TBF-A might explain why it was not detected in the glutathione trapping experiments commonly used to screen for reactive metabolites. Likewise, TBF-A may be more reactive with proteins than with GSH, as many aldehydes are, and therefore not be reliably trapped by GSH. These are all reasons TBF-A’s role as a reactive metabolite might have been missed for so long and highlight the promise of using computational modeling alongside experimental screens. We envision a joint approach. Computational models might identify reactive metabolites missed in the experimental screens. Further study would then verify or rule out these metabolites, so the risk factors of a molecule can be more reliably understood.
At the same time, the TBF case study demonstrated the limitations of the current bioactivation model, which only considers single-transformation bioactivation even though multiple-transformation bioactivation is important too. Our immediate goals are to expand the range of metabolism that can be modeled and then use these models to predicting multiple generations of metabolites. Combining reactivity models with this comprehensive metabolism model might enable bioactivation modeling for most drugs.
■ LIMITATIONS AND FUTURE WORK
The most prominent limitation of the current approach is that it does not consider competing metabolic pathways and multiple metabolic transformations. For example, a molecule can undergo either hydroxylation and then dehydrogenation at one site or N- dealkylation and then reduction at another site. We are currently developing comprehensive models to predict competing Phase I and II metabolic pathways. Our next goal is being able to predict the complete metabolic network for a test molecule. Ultimately, we aim to build comprehensive metabolic and reactivity models. Perhaps a first step might extend this approach to O-dealkylation, another reaction that produces aldehydes. Alternatively, we could include epoxidation or quinone formation, both of which often produce reactive molecules and are predicted by models previously published by our group.
Just as important, this model does not account for all important factors. Dosage, co-administered medicines, and patient factors (age, co-morbidities, genetic variations) also play important roles in adverse drug reactions (ADR). For example, limiting daily dose to under 20 mg/day can significantly decrease ADR.58 Similarly, understanding the factors governing which specific proteins and amino acid are bound by reactive metabolites may be important to understanding toxicity.59 While taking all of these factors in to account will be a daunting task, we believe that models solving smaller problem like ours are steady steps toward this goal.
Finally, as with any machine learning approaches, there is no guarantee that the models’ applicability domain will extend beyond their training domain of metabolically studied small molecules. Proprietary chemical domains such as natural products and peptide-based drugs that are actively explored by pharmaceutical companies may not be well suited to the current models trained on literature-derived data. However, this approach could be easily applied to new data to expand its domain of applicability.
■ CONCLUSION
This study demonstrated a deep network model for predicting N-dealkylation leading to the formation of reactive aldehydes. The model predicted sites of N-dealkylation for HLM substrates with top-two, average N–C AUC, and global N–C AUC accuracies of 96.6%, 93.7%, and 95.6%, respectively. We also predicted reactive and potentially toxic metabolites by combining metabolism and reactivity models into a bioactivation model. One limitation to the current approach is that it does not automatically consider multiple metabolic steps. More comprehensive modeling of bioactivation pathways is on the horizon and actively being developed by our group. Even so, we identified multistep pathways for terbinafine. Likewise, we identified several drugs (indinavir, piperacillin, verapamil, and ziprasidone) that are hepatotoxic by unknown mechanisms of hepatotoxicity and produce reactive aldehyde metabolites by N-dealkylation. This suggests that N-dealkylation may be an under-appreciated bioactivation pathway and that it should be more carefully reported and assessed in metabolic studies. These results also encourage continued work toward building comprehensive models of bioactivation, to consider the full range of metabolic transformations.
Supplementary Material
Acknowledgments
The authors thank Matthew Matlock for previous development of the XenoSite algorithm, which is expanded in this study. We also thank the developers of the open-source cheminformatics tools Open Babel and RDKit, of which we made significant use.
Funding
Research reported in this publication was supported by the National Library Of Medicine of the National Institutes of Health under award numbers R01LM012222 and R01LM012482. Computations were performed using the facilities of the Washington University Center for High Performance Computing, which were partially funded by National Institutes of Health (NIH) grant numbers 1S10RR022984-01A1 and 1S10OD018091-01. Na Le Dang was partially supported by NIH Medical Scientist Training Program grant GM07200. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also thank both the Department of Immunology and Pathology at the Washington University School of Medicine and the Washington University Center for Biological Systems Engineering for their generous support of this work.
ABBREVIATIONS
- ADR
adverse drug reaction
- AMD
Accelrys Metabolite Database
- AUC
area under the receiver operating characteristic curve
- CYP
cytochromes P450
- HLM
human liver microsomes
- N–C
nitrogen–carbon
- N-dealkylation
nitrogen dealkylation
- TBF
terbinafine
- TBF-A
7,7-dimethylhept-2-ene-4-ynal
- TBF-D
desmethyl terbinafine
- TFALD
trifluoroacetaldehyde
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemrestox.7b00191.
The python scripts “Heuristic.py” and “get_Dealkylation_metabolite_structure.py” make heuristic predictions and generate dealkylation metabolite structures from a substrate structure provided in SDF format. The “AMD_Registry_Numbers.csv” file lists unique molecules IDs, associated molecule and reaction registry numbers, and their CYP isozymes- and HML-mediated metabolic status. The “External_Testing_set_AMD_Registry_Numbers.csv” file lists unique molecules IDs, associated reaction registry numbers of the external HML-mediated testing data set. The “Reactivity_Score_of_Observed_Drug_Aldehyde_Metabolites.csv” file contains N-dealkylated drugs, their predicted aldehyde metabolites, and their GSH, protein, cyanide and DNA molecule reactivity scores. The “Predicted_reactive_metabolites_DNA.tsv”, “Predicted_reactive_metabolites_Protein.tsv”, “Predicted_reactive_metabolites_GSH.tsv”, “Predicted_reactive_metabolites_Cyanide.tsv” files in the “Bioaction_model” folder contain the bioactivation model predictions (ZIP)
Full list of molecule, bond and atom descriptors are in Table S1, Table S2, and Table S3 respectively. The modular descriptor groups are in Table S4 (PDF)
ORCID
Tyler B. Hughes: 0000-0001-6221-9014
S. Joshua Swamidass: 0000-0003-2191-0778
Notes
The authors declare no competing financial interest.
References
- 1.Guengerich F, Okazaki O, Seto Y, Macdonald T. Radical cation intermediates in N-dealkylation reactions. Xenobiotica. 1995;25:689–709. doi: 10.3109/00498259509061886. [DOI] [PubMed] [Google Scholar]
- 2.Hand C, Moore R, McQuay H, Allen M, Sear J. Analysis of morphine and its major metabolites by differential radioimmunoassay. Ann Clin Biochem. 1987;24:153–160. doi: 10.1177/000456328702400205. [DOI] [PubMed] [Google Scholar]
- 3.Wong DT, Bymaster FP, Reid LR, Mayle DA, Krushinski JH, Robertson DW. Norfluoxetine enantiomers as inhibitors of serotonin uptake in rat brain. Neuropsychopharmacology. 1993;8:337–344. doi: 10.1038/npp.1993.33. [DOI] [PubMed] [Google Scholar]
- 4.Hiemke C, Härtter S. Pharmacokinetics of selective serotonin reuptake inhibitors. Pharmacol Ther. 2000;85:11–28. doi: 10.1016/s0163-7258(99)00048-0. [DOI] [PubMed] [Google Scholar]
- 5.Hughes TB, Dang NL, Miller GP, Swamidass SJ. Modeling Reactivity to Biological Macromolecules with a Deep Multitask Network. ACS Cent Sci. 2016;2:529–537. doi: 10.1021/acscentsci.6b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adams DH, Ju C, Ramaiah SK, Uetrecht J, Jaeschke H. Mechanisms of immune-mediated liver injury. Toxicol Sci. 2010;115:307–321. doi: 10.1093/toxsci/kfq009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanoh S, Tayama Y, Sugihara K, Kitamura S, Ohta S. Significance of aldehyde oxidase during drug development: Effects on drug metabolism, pharmacokinetics, toxicity, and efficacy. Drug Metab Pharmacokinet. 2015;30:52–63. doi: 10.1016/j.dmpk.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 8.Barr JT, Jones JP. Evidence for substrate-dependent inhibition profiles for human liver aldehyde oxidase. Drug Metab Dispos. 2013;41:24–29. doi: 10.1124/dmd.112.048546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta A, del Rosso J, Lynde C, Brown G, Shear N. Hepatitis associated with terbinafine therapy: three case reports and a review of the literature. Clin Exp Dermatol. 1998;23:64–67. doi: 10.1046/j.1365-2230.1998.00321.x. [DOI] [PubMed] [Google Scholar]
- 10.Iverson SL, Uetrecht JP. Identification of a reactive metabolite of terbinafine: insights into terbinafine-induced hepatotoxicity. Chem Res Toxicol. 2001;14:175–181. doi: 10.1021/tx0002029. [DOI] [PubMed] [Google Scholar]
- 11.Obach RS, Huynh P, Allen MC, Beedham C. Human liver aldehyde oxidase: inhibition by 239 drugs. J Clin Pharmacol. 2004;44:7–19. doi: 10.1177/0091270003260336. [DOI] [PubMed] [Google Scholar]
- 12.Vassallo JD, Hicks SM, Daston GP, Lehman-McKeeman LD. Metabolic detoxification determines species differences in coumarin-induced hepatotoxicity. Toxicol Sci. 2004;80:249–257. doi: 10.1093/toxsci/kfh162. [DOI] [PubMed] [Google Scholar]
- 13.Zaretzki J, Rydberg P, Bergeron C, Bennett KP, Olsen L, Breneman CM. RS-Predictor models augmented with SMARTCyp reactivities: robust metabolic regioselectivity predictions for nine CYP isozymes. J Chem Inf Model. 2012;52:1637–1659. doi: 10.1021/ci300009z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rydberg P, Gloriam DE, Zaretzki J, Breneman C, Olsen L. SMARTCyp: a 2D method for prediction of cytochrome P450-mediated drug metabolism. ACS Med Chem Lett. 2010;1:96–100. doi: 10.1021/ml100016x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalgutkar AS, Gardner I, Obach RS, Shaffer CL, Callegari E, Henne KR, Mutlib AE, Dalvie DK, Lee JS, Nakai Y, O’Donnell JP, Boer J, Harriman SP. A comprehensive listing of bioactivation pathways of organic functional groups. Curr Drug Metab. 2005;6:161–225. doi: 10.2174/1389200054021799. [DOI] [PubMed] [Google Scholar]
- 16.Kalgutkar AS, Dalvie D, Obach RS, Smith DA, Bioactivation and Natural Products . Reactive Drug Metabolites. Wiley-VCH; Weinheim, Germany: 2012. pp. 203–224. 10.1002/9783527655748.ch9. [Google Scholar]
- 17.Stepan AF, Walker DP, Bauman J, Price DA, Baillie TA, Kalgutkar AS, Aleo MD. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem Res Toxicol. 2011;24:1345–1410. doi: 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- 18.Dang NL, Hughes TB, Miller GP, Swamidass SJ. Computational Approach to Structural Alerts: Furans, Phenols, Nitroaromatics, and Thiophenes. Chem Res Toxicol. 2017;30:1046–1059. doi: 10.1021/acs.chemrestox.6b00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu K, Geng X, Chen M, Zhang J, Wang B, Ilic K, Tong W. High Daily Dose and Being a Substrate of Cytochrome P450 Enzymes Are Two Important Predictors of Drug-Induced Liver Injury. Drug Metab Dispos. 2014;42:744–750. doi: 10.1124/dmd.113.056267. [DOI] [PubMed] [Google Scholar]
- 20.Hughes TB, Miller GP, Swamidass SJ. Modeling epoxidation of drug-like molecules with a deep machine learning network. ACS Cent Sci. 2015;1:168–180. doi: 10.1021/acscentsci.5b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hughes TB, Swamidass SJ. Deep Learning to Predict the Formation of Quinone Species in Drug Metabolism. Chem Res Toxicol. 2017;30:642–656. doi: 10.1021/acs.chemrestox.6b00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hughes TB, Miller GP, Swamidass SJ. Site of Reactivity Models Predict Molecular Reactivity of Diverse Chemicals with Glutathione. Chem Res Toxicol. 2015;28:797–809. doi: 10.1021/acs.chemrestox.5b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller EC, Miller JA. The presence and significance of bound aminoazo dyes in the livers of rats fed p-dimethylaminoazobenzene. Cancer Res. 1947;7:468–480. [Google Scholar]
- 24.Miller EC, Miller JA. In vivo combinations between carcinogens and tissue constituents and their possible role in carcinogenesis. Cancer Res. 1952;12:547–556. [PubMed] [Google Scholar]
- 25.Zaretzki J, Matlock M, Swamidass SJ. XenoSite: Accurately predicting CYP-mediated sites of metabolism with neural networks. J Chem Inf Model. 2013;53:3373–3383. doi: 10.1021/ci400518g. [DOI] [PubMed] [Google Scholar]
- 26.Hughes TB, Miller GP, Swamidass SJ. Site of Reactivity Models Predict Molecular Reactivity of Diverse Chemicals with Glutathione. Chem Res Toxicol. 2015;28:797–809. doi: 10.1021/acs.chemrestox.5b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dang NL, Hughes TB, Krishnamurthy V, Swamidass SJ. A simple model predicts UGT-mediated metabolism. Bioinformatics. 2016;32:3183–3189. doi: 10.1093/bioinformatics/btw350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dang NL, Hughes TB, Miller GP, Swamidass SJ. Computational Approach to Structural Alerts: Furans, Phenols, Nitroaromatics, and Thiophenes. Chem Res Toxicol. 2017;30:1046. doi: 10.1021/acs.chemrestox.6b00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hughes TB, Swamidass SJ. Deep Learning to Predict the Formation of Quinone Species in Drug Metabolism. Chem Res Toxicol. 2017;30:642–656. doi: 10.1021/acs.chemrestox.6b00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith HS. Opioid metabolism. Mayo Clin Proc. 2009;84:613–624. doi: 10.1016/S0025-6196(11)60750-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminf. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bellec G, Dréano Y, Lozach P, Ménez JF, Berthou F. Cytochrome P450 metabolic dealkylation of nine N-nitro-sodialkylamines by human liver microsomes. Carcinogenesis. 1996;17:2029–2034. doi: 10.1093/carcin/17.9.2029. [DOI] [PubMed] [Google Scholar]
- 33.Cho TM, Rose RL, Hodgson E. The effect of chlorpyrifos-oxon and other xenobiotics on the human cytochrome P450-dependent metabolism of naphthalene and deet. Drug Metab Drug Interact. 2007;22:235. doi: 10.1515/dmdi.2007.22.4.235. [DOI] [PubMed] [Google Scholar]
- 34.Green T, Toghill A, Lee R, Waechter F, Weber E, Noakes J. Thiamethoxam induced mouse liver tumors and their relevance to humans part 1: mode of action studies in the mouse. Toxicol Sci. 2005;86:36–47. doi: 10.1093/toxsci/kfi124. [DOI] [PubMed] [Google Scholar]
- 35.Shearer TW, Kozar MP, O’Neil MT, Smith PL, Schiehser GA, Jacobus DP, Diaz DS, Yang YS, Milhous WK, Skillman DR. In vitro metabolism of phenoxypropoxybiguanide analogues in human liver microsomes to potent antimalarial dihydrotriazines. J Med Chem. 2005;48:2805–2813. doi: 10.1021/jm049683+. [DOI] [PubMed] [Google Scholar]
- 36.Azam F. Ph D Thesis. Virginia Polytechnic Institute and State University; Blacksburg, VA: 2000. Biologically inspired modular neural networks. [Google Scholar]
- 37.Anand R, Mehrotra K, Mohan CK, Ranka S. Efficient classification for multiclass problems using modular neural networks. IEEE Trans Neural Networks. 1995;6:117–124. doi: 10.1109/72.363444. [DOI] [PubMed] [Google Scholar]
- 38.Lu BL, Ito M. Biological and Arti ficial Computation: From Neuroscience to Technology. Springer Verlag; Berlin, Germany: 1997. pp. 330–339. [Google Scholar]
- 39.Auda G, Kamel M. Modular neural network classifiers: A comparative study. Journal of Intelligent and Robotic Systems. 1998;21:117–129. [Google Scholar]
- 40.Balakin KV, Ekins S, Bugrim A, Ivanenkov YA, Korolev D, Nikolsky YV, Ivashchenko AA, Savchuk NP, Nikolskaya T. Quantitative structure-metabolism relationship modeling of metabolic N-dealkylation reaction rates. Drug Metab Dispos. 2004;32:1111–1120. doi: 10.1124/dmd.104.000364. [DOI] [PubMed] [Google Scholar]
- 41.Dowty ME, Hu G, Hua F, Shilliday FB, Dowty HV. Drug Design Structural Alert Formation of Trifluoroacetaldehyde Through N-Dealkylation is Linked to Testicular Lesions in Rat. Int J Toxicol. 2011;30:546–550. doi: 10.1177/1091581811413833. [DOI] [PubMed] [Google Scholar]
- 42.Dhareshwar SS, Stella VJ. Your prodrug releases formaldehyde: should you be concerned? No! J Pharm Sci. 2008;97:4184–4193. doi: 10.1002/jps.21319. [DOI] [PubMed] [Google Scholar]
- 43.Marchitti SA, Brocker C, Stagos D, Vasiliou V. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol. 2008;4:697–720. doi: 10.1517/17425250802102627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oppermann U. Carbonyl reductases: the complex relationships of mammalian carbonyl-and quinone-reducing enzymes and their role in physiology. Annu Rev Pharmacol Toxicol. 2007;47:293–322. doi: 10.1146/annurev.pharmtox.47.120505.105316. [DOI] [PubMed] [Google Scholar]
- 45.Singh SB, Shen LQ, Walker MJ, Sheridan RP. A model for predicting likely sites of CYP3A4-mediated metabolism on drug-like molecules. J Med Chem. 2003;46:1330–1336. doi: 10.1021/jm020400s. [DOI] [PubMed] [Google Scholar]
- 46.Swamidass SJ, Azencott CA, Daily K, Baldi P. A CROC stronger than ROC: measuring, visualizing and optimizing early retrieval. Bioinformatics. 2010;26:1348–1356. doi: 10.1093/bioinformatics/btq140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng J, Lu J, Shen Q, Zheng M, Luo X, Zhu W, Jiang H, Chen K. In silico site of metabolism prediction for human UGT-catalyzed reactions. Bioinformatics. 2014;30:398–405. doi: 10.1093/bioinformatics/btt681. [DOI] [PubMed] [Google Scholar]
- 48.Clinical and Research Information on Drug-Induced Liver Injury. https://livertox.nlm.nih.gov (accessed Decermber, 2017)
- 49.Ghibellini G, Bridges AS, Generaux CN, Brouwer KL. In vitro and in vivo determination of piperacillin metabolism in humans. Drug Metab Dispos. 2007;35:345–349. doi: 10.1124/dmd.106.012278. [DOI] [PubMed] [Google Scholar]
- 50.Chen CS, Jounaidi Y, Waxman DJ. Enantioselective metabolism and cytotoxicity of R-ifosfamide and S-ifosfamide by tumor cell-expressed cytochromes P450. Drug Metab Dispos. 2005;33:1261–1267. doi: 10.1124/dmd.105.004788. [DOI] [PubMed] [Google Scholar]
- 51.Choucha-Snouber L, Aninat C, Grsicom L, Madalinski G, Brochot C, Poleni PE, Razan F, Guillouzo CG, Legallais C, Corlu A, Eric L. Investigation of ifosfamide nephrotoxicity induced in a liver-kidney co-culture biochip. Biotechnol Bioeng. 2013;110:597–608. doi: 10.1002/bit.24707. [DOI] [PubMed] [Google Scholar]
- 52.Lloyd S, Blackburn D, Foster P. Trifluoroethanol and its oxidative metabolites: comparison of in vivo and in vitro effects in the rat testis. Food Chem Toxicol. 1986;24:653–654. doi: 10.1016/0041-008x(88)90179-2. [DOI] [PubMed] [Google Scholar]
- 53.Lloyd S, Blackburn D, Foster P. Trifluoroethanol and its oxidative metabolites: Comparison of in vivo and in vitro effects of rat testis. Toxicol Appl Pharmacol. 1988;92:390–401. doi: 10.1016/0041-008x(88)90179-2. [DOI] [PubMed] [Google Scholar]
- 54.Vickers AE, Sinclair JR, Zollinger M, Heitz F, Glänzel U, Johanson L, Fischer V. Multiple cytochrome P-450s involved in the metabolism of terbinafine suggest a limited potential for drug-drug interactions. Drug Metab Dispos. 1999;27:1029–1038. [PubMed] [Google Scholar]
- 55.Hall M, Monka C, Krupp P, O’Sullivan D. Safety of oral terbinafine: results of a postmarketing surveillance study in 25 884 patients. Arch Dermatol. 1997;133:1213–1219. doi: 10.1001/archderm.133.10.1213. [DOI] [PubMed] [Google Scholar]
- 56.Gupta AK, Shear NH. Terbinafine: an update. J Am Acad Dermatol. 1997;37:979–988. doi: 10.1016/s0190-9622(97)70076-8. [DOI] [PubMed] [Google Scholar]
- 57.Abdel-Rahman S, Nahata M. Oral terbinafine: a new antifungal agent. Ann Pharmacother. 1997;31:445–456. doi: 10.1177/106002809703100412. [DOI] [PubMed] [Google Scholar]
- 58.Kalgutkar AS, Didiuk MT. Structural alerts, reactive metabolites, and protein covalent binding: how reliable are these attributes as predictors of drug toxicity? Chem Biodiversity. 2009;6:2115–2137. doi: 10.1002/cbdv.200900055. [DOI] [PubMed] [Google Scholar]
- 59.Ikehata K, Duzhak TG, Galeva NA, Ji T, Koen YM, Hanzlik RP. Protein targets of reactive metabolites of thiobenzamide in rat liver in vivo. Chem Res Toxicol. 2008;21:1432–1442. doi: 10.1021/tx800093k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.