

Abstract
Pre-mRNA splicing factors recognize consensus signals within preliminary transcripts, and as cogs of the spliceosome machine, orchestrate the excision and rejoining of pre-mRNA regions for gene expression. Large-scale sequencing has demonstrated that mutations in key genes encoding pre-mRNA splicing factors are common among myeloid neoplasms and also occur in a variety of other cancers. This revelation offers new therapeutic opportunities to target pre-mRNA splicing vulnerabilities in hematologic and other malignancies. The mutated residues typically alter 3′ splice site choice for a subset of transcripts. In this review, we highlight mechanistic insights from recent 3D structures that reveal the affected residues poised for pre-mRNA recognition.
Keywords: MDS, myelodysplastic syndrome, myeloid neoplasm, spliceosome, SRSF2, U2AF1, SF3B1
Defects of pre-mRNA splicing and splicing factors in cancers
Aberrant pre-mRNA splicing is known to affect each of the widely accepted hallmarks of cancer [1]. Indeed, spliceoform changes, cryptic splice sites, and intron retention (e.g. [2,3]), could be designated an emerging hallmark. The splice sites are chosen through integrated cis- signals encoded in the pre-mRNA sequences and trans-acting protein and RNA factors (reviewed in [4]). Mutations of pre-mRNA splice sites have long been associated with at least 15% of inherited human diseases [5] and also occur as somatic mutations in cancer [6]. Altered expression of pre-mRNA splicing factors is common among cancers (reviewed in [7]). For example, SRSF1 is frequently overexpressed in solid tumors, and its overexpression increases cell proliferation [8]. On the flip side, copy number loss of the SF3B1 gene, such as observed in kidney, bladder, and breast cancers, renders a cancer cell line highly sensitive to drug-induced degradation of this splicing protein [9]. Moreover, overexpression of the MYC oncogene overloads the splicing machinery, making the MYC-driven cancer cells more vulnerable to pharmacological inhibition of the spliceosome [10,11]. Indeed, the pre-mRNA splicing changes of cancers offers many new therapeutic opportunities, ranging from chemical splicing inhibitors to anti-sense oligonucleotides [12].
Large-scale sequencing established recurrent somatic mutations of four splicing factor genes (SRSF2, SF3B1, U2AF1, and ZRSR2) in the genomic landscape of myeloid neoplasms [13–16]. Missense mutations of SRSF2, SF3B1, U2AF1 recur at specific hotspots, whereas mutations of ZRSR2, which plays an auxiliary role for the minor class of spliceosomes, are broadly distributed throughout the protein and often null. The splicing factor mutations are heterozygous and typically mutually exclusive. The splicing factor genes appear to be mutated early in the evolution of MDS [15] and SF3B1 mutations confer a favorable prognosis for MDS compared to mutations of SRSF2 or U2AF1 [17]. Although splicing factor mutations are generally less common among solid tumors, SF3B1 mutations recur among a variety of malignancies, including uveal melanomas [18–20], luminal breast tumors [6,21–25], pancreatic cancers [26,27], bladder urothelial carcinomas [28], mucosal melanomas [29], and hepatocellular carcinomas [30] as well as at high frequencies in refractory anemias with ring sideroblasts (RARS) [13,17,31] and chronic lymphocytic leukemias (CLL) [32,33]. The U2AF1 gene is mutated in pancreatic cancers [27], and lung adenocarcinomas [34,35], as well as myeloid neoplasms including MDS, hairy cell leukemia, CMML and AML [13,36,37]. Acquired mutations of other splicing factors are relatively rare among most non-hematologic malignancies.
The details of the splicing factor mutations and the field’s efforts to relate these mutations to downstream hematologic consequences have been the subject of several distinguished reviews (e.g. [38–40]). Here we focus on molecular insights derived from the emerging structures of MDS-relevant splicing factors and their hotspot mutations.
Molecular-level functions of cancer-relevant pre-mRNA splicing factors
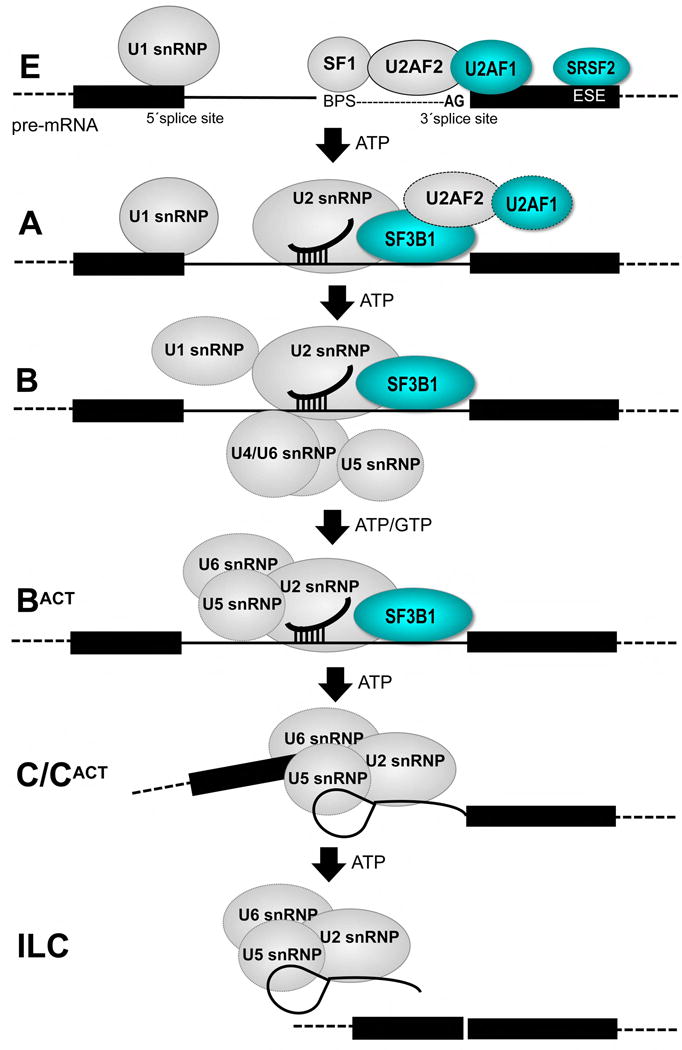
SRSF2, SF3B1, U2AF1, and ZRSR2 recognize pre-mRNA splice site signals at an early stage of the splicing process (Figure 1). The pre-mRNA splicing “machine” comprising U1, U2, U4/U5/U6 small nuclear ribonucleoproteins (snRNPs) and numerous associated factors [4] assembles through a series of discrete, ATP-dependent conformational rearrangements. SRSF2 (also called SC35) is a member of an SR protein family that recognizes “exonic splicing enhancers” (ESE) with an RNA recognition motif (RRM), and directs U1 and U2 snRNPs to the splice site via RS domain-mediated protein-protein interactions and RNA-RNA annealing (reviewed in [41]). U2AF1 (also called U2AF35) recognizes a consensus “AG” dinucleotide at the 3′ splice site as a heterodimer with a second, essential pre-mRNA splicing factor U2AF2 (also called U2AF65) that is less frequently mutated in cancers. The U2AF1–U2AF2 heterodimer first forms a ternary complex with Splicing Factor-1 (SF1) and altogether recognizes the respective AG, polypyrimidine (Py), and AG-dinucleotide consensus elements preceding the 3′ splice site. Next, RNP unwindases such as Prp5 (also called DDX46) catalyze the ATP-dependent exchange of SF1 for the SF3b1 subunit (also called SF3b155), which in turn stabilizes U2 snRNP association with the pre-mRNA. The ZRSR2 protein (also called URP) shares a similar domain structure as U2AF1, but promotes splicing by a minor, U12-type of spliceosome [42,43] that recognizes a distinct subset of splice sites.
Figure 1.
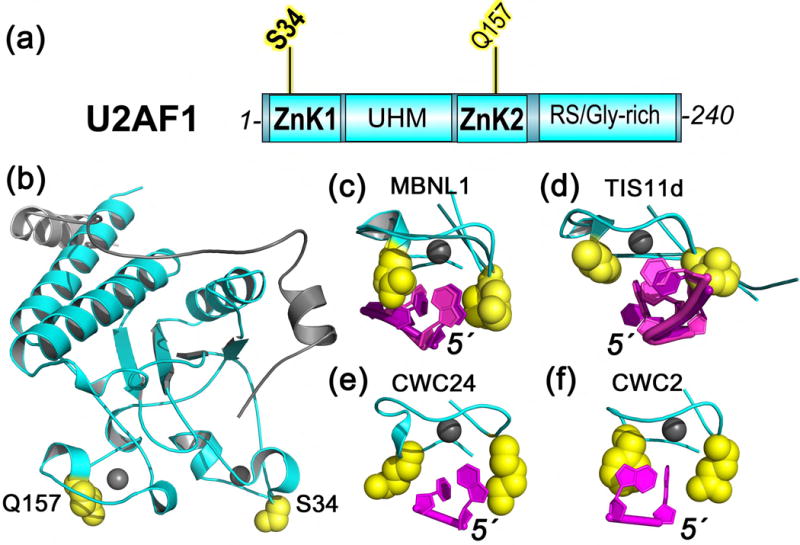
Diagram of the major pre-mRNA splicing pathway. Subunits with recurrent acquired mutations in cancers are colored cyan. BPS, branchpoint sequence; ESE, exonic splicing enhancer; ILC, intron-lariat complex.
The downstream, transcriptome-wide changes in splicing have been extensively characterized for each of the major MDS-associated splicing factor mutations, and differ from the splicing changes induced by loss-of-function knockdowns. RNAseq analyses of cell lines, mouse models and patient samples show that the hotspot mutations of SRSF2 [44,45] or U2AF1 [46–50] alter splicing of a small fraction of transcripts in a sequence-specific manner, most often promoting exon skipping/inclusion or alternative 3′ splice sites. The hotspot mutations of SF3B1 likewise alter the 3′ splice sites of a subset of transcripts analyzed by RNAseq of pancreatic cancer cell lines, chronic lymphocytic leukemias (CLL), breast cancers, or uveal melanomas [51–53]. However, unlike the altered splice site preferences of SRSF2 and U2AF1, the SF3B1 mutations promote cryptic 3′ splice sites located 10–30 bp upstream of the normal sites. Recent 3D structures and biochemical analyses of SRSF1, SF3B1, and U2AF1 and their cancer-relevant mutations suggest potential mechanisms for the emerging theme of altered 3′ splice site recognition, as we discuss below.
Structural interfaces with cancer-relevant “hotspots” of pre-mRNA splicing factors
Ground-breaking spliceosome structures have been determined through recent technical advances of cryo-electron microscopy (cryo-EM), including B- [54,55], BACT- [56,57], C- [58,59], C*- [60–63] and IL-complexes [64–66]. The B- and BACT-structures reveal the SF3B1 subunit bound to the U2 snRNA– pre-mRNA duplex. Although the dynamic, early-stage splicing factors SRSF2 and U2AF1 are lacking from current cryo-EM structures, the mutated U2AF1 domains now can be modeled based on structural homology with core spliceosome subunits [67]. Moreover, traditional NMR and X-ray methods have determined the piecewise structures of SRSF2 and U2AF1 subunits [68–70]. Below we summarize the current structural understanding of the field for each of the major cancer-relevant splicing factors.
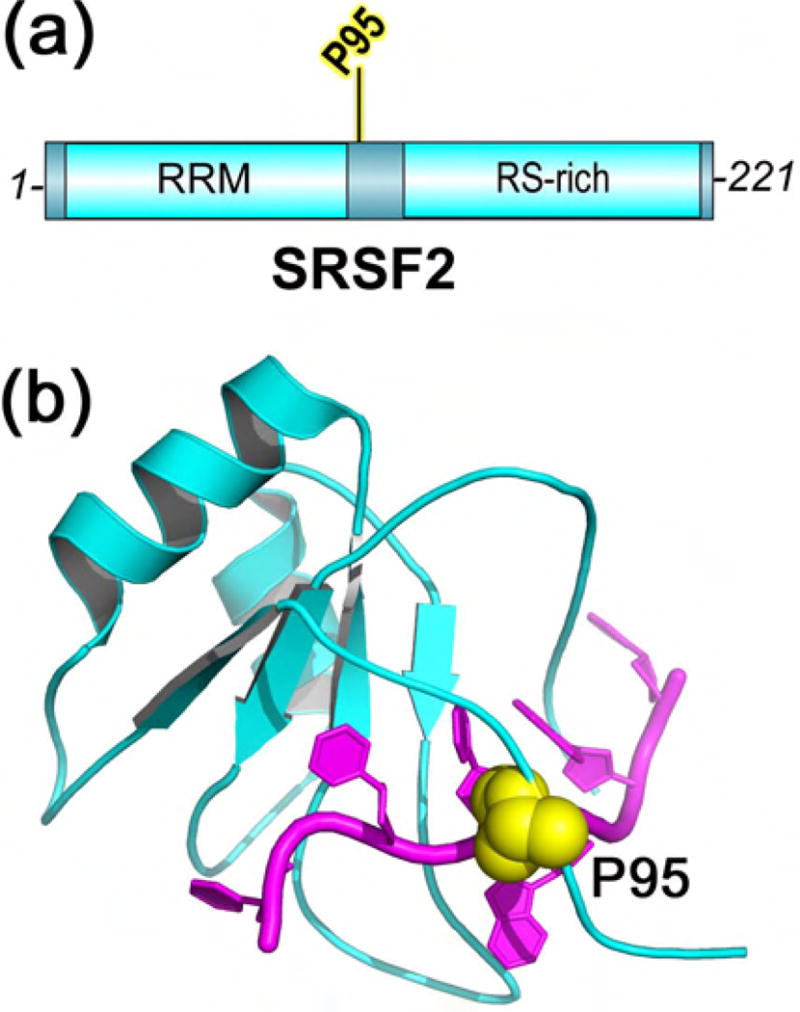
SRSF2
SRSF2 was the first frequently mutated splicing factor among myeloid malignancies for which a high resolution structure became available [68]. The SRSF2 mutations are particularly prevalent in CMML [71,72] and high-risk MDS patient samples [73–75] (~40% or 15%, respectively). All documented CMML or MDS-associated mutations change a P95 residue located between an RNA recognition motif (RRM) and arginine serine (RS)-rich domain (Figure 2A), most commonly to histidine or leucine. RNAseq analyses of knock-in mice, cell lines, and patient samples expressing the P95 mutation preferentially enrich exons bearing a CCNG motif and decreased inclusion of those with a GGNG motif [44,45]. Electrophoretic mobility shift assays and quantitative isothermal titration calorimetry of recombinant SRSF2 proteins demonstrated that the P95 mutations increased its binding to (C/G)CNG- containing RNAs and decreased binding to (C/G)GNG-containing RNAs [44,45]. In contrast, WT SRSF2 binds either CCNG or GGNG motifs equally well [68].
Figure 2.
SRSF2 (cyan) has recurrent acquired mutations in CMML and MDS. The hotspot P95 residue (yellow) is here shown mapped on the protein (a) domains and (b) structure (PDB ID 2LEB), which includes residues 1–101 and is bound to RNA sequence 5′-UCCAGU-3′ (magenta).
The high resolution solution structure of SRSF2 bound to CCNG compared to GGNG RNAs revealed that the second guanosine base adapts to the WT protein by flipping to the syn- rather than anti-conformer [68]. The WT P95 residue makes extensive base stacking contacts with the second nucleotide of the affected RNA site (Figure 2B). Following P95 mutation, large chemical shift perturbations indicate that the entire P95-containing region of mutant SRSF2 relocates [44]. In contrast with the reproducible changes in RNA binding, no differences in protein-protein interactions with U2AF1, U1-70K, or SF3B1 were detected in co-immunoprecipitations of WT vs. mutant SRSF2 [45]. Nevertheless, for a few model substrates and ~15% of the transcriptome, P95-dependent splicing outcomes did not strictly correlate with detailed variation of CCNG/GGNG motifs. For these cases, combinatorial effects may converge to determine the final consequences of the SRSF2 P95 mutation for splicing.
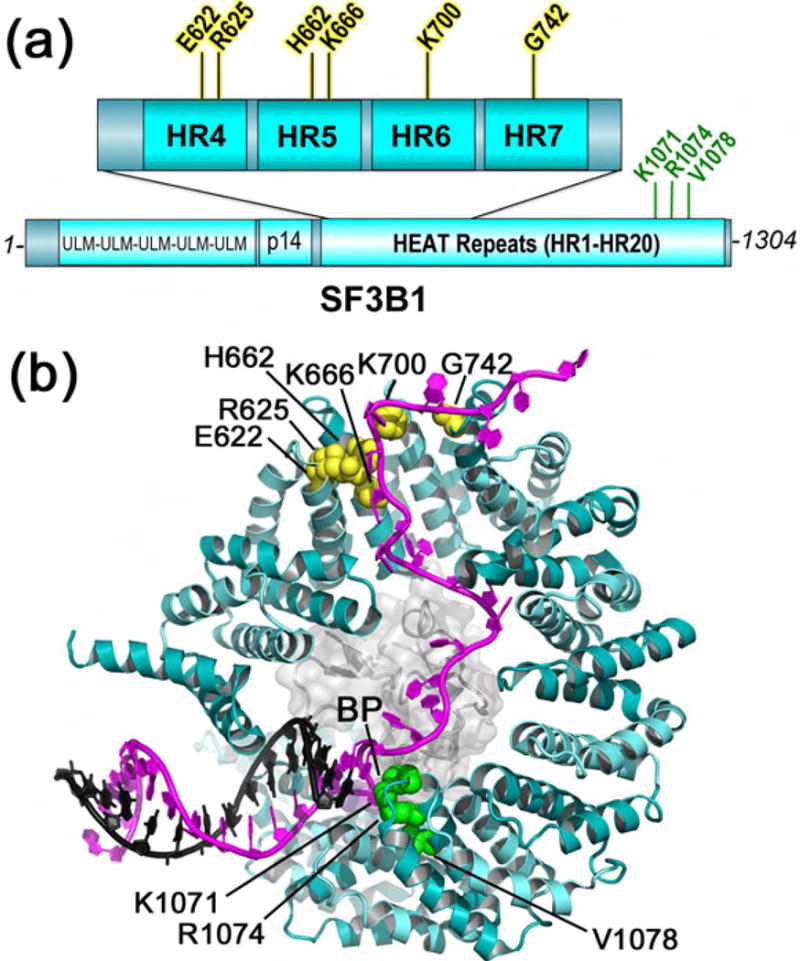
Structures of SF3B1
The most common amino acid substitutions of SF3B1 include K700E, which recurs among hematologic malignancies (up to 80% of RARS), pancreatic, and breast cancers, or R625 substitutions among uveal melanomas, among others (Figure 3A). As mentioned above, in cell lines expressing the common SF3B1 mutations (K700, R625, or K666), the cryptic upstream 3′ splice sites are associated with new BPS′ that are up to ten nucleotides closer than the normal BPS-to-3′ splice site separation [52,53]. Based on the crystal structure of human SF3B1 in the SF3b protein particle [76], coupled with recent cryoEM structures of Saccharomyces cerevisiae SF3B1 in the context of the B- and BACT-spliceosomes [54–57], the affected SF3B1 residues map to the fourth to seventh repeats of an α-helical “HEAT” repeat (HR) domain (fifth to eighth repeats of the primary protein sequence) (Figure 3A).
Figure 3.
SF3B1 (cyan) has recurrent acquired mutations (yellow) in myeloid neoplasms and cancers, here mapped on the (a) domains and (b) structure of the Baker’s yeast homologue in the BACT complex (PDB ID 5GM6). Residues matching human SF3B1 were identified by superposition with the SF3B particle (PDB ID 5IFE). Mutational hotspots (yellow), residues for which mutations confer resistance to splicing modulators (green), and branchpoint (BP) are labeled. The bound pre-mRNA is magenta, the U2 snRNA strand is black, and the surface of the PHF5A subunit is colored light gray.
In both B- and BACT- structures, the SF3B1 subunit adopts a nearly-circular C-shape, in which the 5′ terminus of the pre-mRNA binds near the C-terminal HR of the protein, where it engages in the U2 snRNA–BPS duplex (Figure 3B). Due to the extreme curvature of the SF3B1 structure, the U2 snRNA–BPS duplex is sandwiched on one side by the N-terminal HR1 of SF3B1. On the opposite side, the branch point nucleotide inserts near K1071, R1074, and V1078 residues in SF3B1 HR15, for which mutations confers resistance to pladienolide and related splicing inhibitors [77,78]. A globular PHF5a subunit provides the jelly filling of the SF3B1 donut. Specifically, the PHF5A protein contacts the branchpoint-containing portion of the U2 snRNA–BPS duplex and downstream pre-mRNA nucleotides, a binding site that explains the resistance of a PHF5A Y36C mutation to herboxidiene and pladienolide [78]. Together with prior findings that pladienolide and the related compound spliceostatin interfere with U2 snRNA – BPS base pairing [79,80], the locations of the K1071, R1074, V1078, and Y36 resistance mutations suggest that the pladienolide family of splicing inhibitors competes for branch site binding to a pocket comprising these residues in the SF3B1 – PHF5A complex.
The BACT-spliceosome coordinates include approximately 25 nucleotides of pre-mRNA traversing the interior of the C-shaped SF3B1 subunit [56], whereas the pre-mRNA model of the lower resolution B-spliceosome structure terminates at PH5A [54]. The SF3B1 mutational hotspots cluster surrounding the ordered 3′ nucleotides of the BACT-pre-mRNA (Figure 3B). It is conceivable that the cancer-relevant mutations shorten the BPS′-to-3′ splice site distance by disrupting SF3B1 binding to this region of the pre-mRNA. Although the K700E mutation of SF3B1 remains capable of binding a consensus splice site RNA at saturating protein concentrations [76], quantitative titrations and RNAs with distinct BPS-to-3′ splice site separations have yet to be tested for differences in binding to the mutant vs. WT SF3B1 protein.
No direct protein contacts with the affected HR regions are apparent among available SF3B1-containing structures. Nevertheless, cancer-relevant mutations of the S. cerevisiae SF3B1 homologue (HSH155) alter its physical and genetic interactions with the RNP unwindase Prp5 (yeast homologue of human DDX46) [81,82], which is required for stable U2 snRNP association with the pre-mRNA [83]. Other early-stage splicing factors also associate with SF3B1 and regulate spliceosome assembly (e.g. RBM15, U2AF2, Tat-SF1, CAPERα, SPF45) ([84,85]), and the K700E mutation alters co-immunoprecipitation of SF3B1 with RBM15 [86]. The interplay of SF3B1 hotspots and its protein partners remains an important area for future biochemical, genetic and structural research.
Structures of U2AF1
Mutations of an S34 residue to phenylalanine, or in a minority of cases, the related residue tyrosine, account for the vast majority of U2AF1 mutations in MDS (approximately 80% of mutations) [13,36] and all documented U2AF1 mutations in other cancers [27,34,35,37] (Figure 4A). Less frequently, a Q157 residue is mutated to arginine or proline in MDS [13,36]. The S34 vs. Q157 mutations of U2AF1 show distinct sequence trends among altered 3′ splice sites: the S34 mutants are more likely to skip splice junctions preceded by a -3U and include those preceded by -3C/A [46–50], whereas Q157 mutants are more likely to skip splice junctions followed by a +1A and include those followed by +1G [50] (numbered relative to the intron-exon junction). Quantitative assays of recombinant S34F vs. WT U2AF1 proteins (as a ternary complexes with U2AF2 and SF1) demonstrate that their RNA binding preferences match the sequence trends for splicing [48,49]. Similar RNA binding preferences are obtained from isothermal titration calorimetry (ITC) of Schizosaccharomyces pombe (Sp) U2AF1 in complex with the minimal (“MIN”) interacting region of SpU2AF2 [87], an apparent lack of binding by the mutant proteins is likely due to the limits of the ITC experiment [88].
Figure 4.
U2AF1 (cyan) has recurrent acquired mutations (yellow) in MDS and cancers, here mapped on the (a) domains of the human protein and (b) structure of the fission yeast homologue (PDB ID 4YH8). Four structures contain zinc knuckles (ZnK) bound to RNA: (c) MBNL1 (PDB ID 3D2S), (d) TIS11d (PDB ID 1RGO), (e) CWC24 (PDB ID 5GM6), (f) CWC2 (PDB ID 5GMK). The ZnK orientations are identical among panels (c)–(f). For MBNL1 and TIS11d, both of the two ZnK in each structure are shown following superposition of matching Cα-atoms. The residues corresponding to Q157 (left) and S34 (right) are yellow spheres. Gray spheres represent zinc ions. The 5′ termini of the bound RNAs (magenta) are labeled to indicate the different orientation of TIS11d-bound RNA.
A key structure of the SpU2AF1–U2AF2MIN heterodimer revealed the 3D locations of the S34 and Q157 hotspots on two CCCH-type zinc knuckles (ZnK1 and ZnK2) (Figure 4b). Although a tantalizing snapshot, the SpU2AF1–U2AF2MIN structure lacks bound RNA. Prior models of U2AF1– RNA complexes [50,70] have been unreliable due to discrepancies between the only available ZnK– RNA structures at the time: TIS11d and MBNL1 [89,90] (Figure 4c–d). Namely, the bound RNA strands run in opposite 5′-to-3′ directions relative to the ZnK folds of the TIS11d vs. MBNL1 structures. The residues corresponding to U2AF1 S34 interact with RNA in either orientation, but the Q157 paralogue only contacts RNA in the MBNL1 structure. The recent spliceosome structures bring to light new CCCH-type ZnK–RNA structures, including CWC24 bound to the 5′ exon of the BACT complex [56] and CWC2 bound to the U6 snRNA of the BACT, C, C*, and ILS complexes [56–59,64,65,91,92] (Figure 4e–f). The CWC24 and CWC2 ZnK bind RNA in similar 5′-to-3′ orientations as MBNL1, although the exact RNA path differs slightly for CWC2. By comparison, the well-characterized RRM class of RNA binding domain also binds RNAs in a typical 5′-to-3′ orientation relative to the core protein folds (reviewed in [93]). Considering these emerging themes of ZnK–RNA recognition, as well as the high sequence identity between U2AF1 and CWC24 or MBNL1 ZnK (45% identity between human U2AF1 ZnK1 and yeast CWC24 ZnK; 40% identity between human U2AF1 ZnK2 and the N-terminal ZnK1 of human MBNL1), it is likely that the S34 and Q157 hotpots of U2AF1 contact the bound pre-mRNA site. Moreover, the spacing of the S34 and Q157 residues in the intact SpU2AF1 structure are consistent with respective contacts at the −3 and +1 nucleotides surrounding the 3′ splice site junction [67], which would explain the sequence trends among affected splice sites [46–50].
Conclusions and Outlook
Altogether, recent structural and biochemical results establish pre-mRNA interactions as a common theme shared among MDS-relevant hotspots of the SRSF2, SF3B1, and U2AF1 splicing factors. Less commonly, cancer-associated mutations are documented in other splicing factor genes, including ZRSR2, the U2AF1 paralogue of the minor spliceosome discussed above. The gene encoding PRPF8, a central subunit of the spliceosome, often is mutated in inherited autosomal dominant retinitis pigmentosa and has acquired mutations in approximately 3% of patients with myeloid neoplasms [94]. However, as for ZRSR2, the PRPF8 mutations are scattered throughout the protein and are in some cases null, rather than missense mutations at discrete hotspots.
For both cases in which the RNA binding affinities of the mutant vs. WT proteins (SRSF2 and U2AF1) have been quantified, the effects of the mutations have been subtle but specific (2–4-fold increase or decrease depending on the RNA sequence). This raises the question of whether the RNA binding differences are sufficient to explain the changes in splicing, or more globally, to drive the progression to disease. Indeed, the enrichments of the splice site logos correlate with the magnitudes of the mutation-dependent RNA binding changes. Nevertheless, only a few misspliced transcripts have been flagged as potential contributors to the development of myeloid neoplasms (e.g. EZH2 for SRSF2 [44], iron regulators [95,96]/erythropoiesis transcription factors for SF3B1 [86]). Already for U2AF1, the S34F mutation has been shown to alter polyadenylation with similar frequencies as pre-mRNA splicing, and the consequent decreases in an ATG7 transcript pre-disposes cells to transformation [97]. The SRSF2 and SF3B1 proteins also wear multiple hats for gene expression: SRSF2 is a component of the 7SK RNP and stimulates transcription elongation [98]; SF3B1 associates with chromatin [83] and also with polycomb group protein repressors of Hox genes [99]. Whether the hotspot mutations of SRSF2 and SF3B1 trigger epigenetic changes that contribute to disease pathogenesis remains an outstanding question.
Altogether, defective pre-mRNA splicing has emerged as a recurring vulnerability of myeloid neoplasms and cancers. The field’s increasing knowledge will guide ongoing efforts to develop new chemotherapies that exploit recurrent, cancer-associated defects in pre-mRNA splicing.
Acknowledgments
Supported by National Institutes of Health grants (GM070503 and GM117005) and by the Edward P. Evans Foundation. I am grateful to Dr. J. Jenkins for proofreading and organizing the spliceosome PDBs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Recommended Reading
* of special interest
** of outstanding interest
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, Burge CB, Gertler FB. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011;7:e1002218. doi: 10.1371/journal.pgen.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dvinge H, Bradley RK. Widespread intron retention diversifies most cancer transcriptomes. Genome Med. 2015;7:45. doi: 10.1186/s13073-015-0168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papasaikas P, Valcarcel J. The Spliceosome: The Ultimate RNA Chaperone and Sculptor. Trends Biochem Sci. 2016;41:33–45. doi: 10.1016/j.tibs.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Krawczak M, Thomas NS, Hundrieser B, Mort M, Wittig M, Hampe J, Cooper DN. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat. 2007;28:150–158. doi: 10.1002/humu.20400. [DOI] [PubMed] [Google Scholar]
- 6.Dorman SN, Viner C, Rogan PK. Splicing mutation analysis reveals previously unrecognized pathways in lymph node-invasive breast cancer. Sci Rep. 2014;4:7063. doi: 10.1038/srep07063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anczukow O, Krainer AR. Splicing-factor alterations in cancers. RNA. 2016;22:1285–1301. doi: 10.1261/rna.057919.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anczukow O, Akerman M, Clery A, Wu J, Shen C, Shirole NH, Raimer A, Sun S, Jensen MA, Hua Y, et al. SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol Cell. 2015;60:105–117. doi: 10.1016/j.molcel.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paolella BR, Gibson WJ, Urbanski LM, Alberta JA, Zack TI, Bandopadhayay P, Nichols CA, Agarwalla PK, Brown MS, Lamothe R, et al. Copy-number and gene dependency analysis reveals partial copy loss of wild-type SF3B1 as a novel cancer vulnerability. Elife. 2017;6 doi: 10.7554/eLife.23268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koh CM, Bezzi M, Low DH, Ang WX, Teo SX, Gay FP, Al-Haddawi M, Tan SY, Osato M, Sabo A, et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature. 2015;523:96–100. doi: 10.1038/nature14351. [DOI] [PubMed] [Google Scholar]
- 11.Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015;525:384–388. doi: 10.1038/nature14985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonnal S, Vigevani L, Valcarcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov. 2012;11:847–859. doi: 10.1038/nrd3823. [DOI] [PubMed] [Google Scholar]
- 13.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 14.Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119:3203–3210. doi: 10.1182/blood-2011-12-399774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013 doi: 10.1182/blood-2013-08-518886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–247. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malcovati L, Karimi M, Papaemmanuil E, Ambaglio I, Jadersten M, Jansson M, Elena C, Galli A, Walldin G, Della Porta MG, et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood. 2015;126:233–241. doi: 10.1182/blood-2015-03-633537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet. 2013;45:133–135. doi: 10.1038/ng.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin M, Masshofer L, Temming P, Rahmann S, Metz C, Bornfeld N, van de Nes J, Klein-Hitpass L, Hinnebusch AG, Horsthemke B, et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013;45:933–936. doi: 10.1038/ng.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, Turajlic S, Piperno-Neumann S, de la Grange P, Roman-Roman S, et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013;3:1122–1129. doi: 10.1158/2159-8290.CD-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maguire SL, Leonidou A, Wai P, Marchio C, Ng CK, Sapino A, Salomon AV, Reis-Filho JS, Weigelt B, Natrajan RC. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J Pathol. 2015;235:571–580. doi: 10.1002/path.4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. doi: 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 28.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, Patch AM, Kakavand H, Alexandrov LB, Burke H, et al. Whole-genome landscapes of major melanoma subtypes. Nature. 2017;545:175–180. doi: 10.1038/nature22071. [DOI] [PubMed] [Google Scholar]
- 30.Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017;169:1327–1341 e1323. doi: 10.1016/j.cell.2017.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–1395. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca DS, Zhang L, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365:2497–2506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, Ramsay AJ, Bea S, Pinyol M, Martinez-Trillos A, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012;44:47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 34.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, Krysiak K, Harris CC, Koboldt DC, Larson DE, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2012;44:53–57. doi: 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waterfall JJ, Arons E, Walker RL, Pineda M, Roth L, Killian JK, Abaan OD, Davis SR, Kreitman RJ, Meltzer PS. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet. 2014;46:8–10. doi: 10.1038/ng.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dvinge H, Kim E, Abdel-Wahab O, Bradley RK. RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer. 2016;16:413–430. doi: 10.1038/nrc.2016.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer. 2017;17:5–19. doi: 10.1038/nrc.2016.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joshi P, Halene S, Abdel-Wahab O. How do messenger RNA splicing alterations drive myelodysplasia? Blood. 2017;129:2465–2470. doi: 10.1182/blood-2017-02-692715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Howard JM, Sanford JR. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip Rev RNA. 2015;6:93–110. doi: 10.1002/wrna.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen H, Zheng X, Luecke S, Green MR. The U2AF35-related protein Urp contacts the 3′ splice site to promote U12-type intron splicing and the second step of U2-type intron splicing. Genes Dev. 2010;24:2389–2394. doi: 10.1101/gad.1974810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, Sanada M, Grossmann V, Sundaresan J, Shiraishi Y, et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun. 2015;6:6042. doi: 10.1038/ncomms7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44**.Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell. 2015;27:617–630. doi: 10.1016/j.ccell.2015.04.006. P95-mutant SRSF2 alters splicing of exons bearing CCNG/GGNG motifs. These splicing changes correlate with altered RNA binding affinities of mutant SRSF2. The P95 region relocates in the mutated SRSF2 structure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45*.Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, Raza A, Mukherjee S, Manley JL. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci U S A. 2015;112:E4726–4734. doi: 10.1073/pnas.1514105112. P95-mutant SRSF2 alters splicing of exons bearing CCNG/GGNG motifs. Binding to CCNG/GGNG RNAs likewise is altered in assays of mutant SRSF2. The mutant SRSF2 protein associates with known protein partners like WT SRSF2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Przychodzen B, Jerez A, Guinta K, Sekeres MA, Padgett R, Maciejewski JP, Makishima H. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. 2013;122:999–1006. doi: 10.1182/blood-2013-01-480970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brooks AN, Choi PS, de Waal L, Sharifnia T, Imielinski M, Saksena G, Pedamallu CS, Sivachenko A, Rosenberg M, Chmielecki J, et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS One. 2014;9:e87361. doi: 10.1371/journal.pone.0087361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48*.Okeyo-Owuor T, White BS, Chatrikhi R, Mohan DR, Kim S, Griffith M, Ding L, Ketkar-Kulkarni S, Hundal J, Laird KM, et al. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia. 2015;29:909–917. doi: 10.1038/leu.2014.303. The authors identify splicing changes following expression of S34F-mutant U2AF1. Altered binding to -3C/-3U RNAs could explain the sequence logos of S34F-affected splice sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49**.Fei DL, Motowski H, Chatrikhi R, Prasad S, Yu J, Gao S, Kielkopf CL, Bradley RK, Varmus H. Wild-Type U2AF1 Antagonizes the Splicing Program Characteristic of U2AF1-Mutant Tumors and Is Required for Cell Survival. PLoS Genet. 2016;12:e1006384. doi: 10.1371/journal.pgen.1006384. The S34F:WT U2AF1 ratio correlates with the sequence logos of S34F-affected splice sites. This result suggested that the S34F- and WT-U2AF1 proteins compete for a limiting factor. We compared the binding affinities of S34F- vs. WT U2AF1 for 12 splice sites. The results suggest that competitive RNA binding contributes to S34F-induced splicing changes. The S34F-mutant U2AF1 is not required for viability of the cancer cell lines. Yet, the WT U2AF1 allele is required. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, Bradley RK. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015;25:14–26. doi: 10.1101/gr.181016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, Jamieson CH, Carson D, Kipps TJ, Frazer KA. Transcriptome sequencing reveals potential mechanism of cryptic 3′ splice site selection in SF3B1-mutated cancers. PLoS Comput Biol. 2015;11:e1004105. doi: 10.1371/journal.pcbi.1004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52**.Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015;13:1033–1045. doi: 10.1016/j.celrep.2015.09.053. Hotspot mutations of SF3B1 induce cryptic upstream 3′ splice sites. These cryptic splice sites differ from splicing changes due to SF3B1 knockdown. The cryptic splice sites depend on upstream, de novo BPS. [DOI] [PubMed] [Google Scholar]
- 53**.Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun. 2016;7:10615. doi: 10.1038/ncomms10615. Mutant SF3B1 induces cryptic upstream 3′ splice sites in uveal melanomas. The cryptic splice site junctions typically overlap the prior, normal BPS. The spacing between each cryptic splice site and its BPS is typically 10 nucleotides less than normal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54**.Plaschka C, Lin PC, Nagai K. Structure of a pre-catalytic spliceosome. Nature. 2017;546:617–621. doi: 10.1038/nature22799. The S. cerevisiae B-spliceosome was determined at an average 7.5 Å resolution. SF3a/SF3b proteins (including SF3B1) chaperone the U2 snRNA and intron conformations. Specific B-complex proteins stabilize the Brr2 helicase and U6 snRNA structure. The snRNA and pre-mRNA structures undergo large changes for spliceosome activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55*.Bertram K, Agafonov DE, Dybkov O, Haselbach D, Leelaram MN, Will CL, Urlaub H, Kastner B, Luhrmann R, Stark H. Cryo-EM Structure of a Pre-catalytic Human Spliceosome Primed for Activation. Cell. 2017;170:701–713 e711. doi: 10.1016/j.cell.2017.07.011. The human B-spliceosome is determined at an average 4.5 Å resolution. Most subunit and RNA positions of this complex are identified (including SF3B1). However most of the SF3a complex is lacking from the model. The U4/U5/U6 snRNP rearranges in the B-complex compared to the isolated tri-snRNP. [DOI] [PubMed] [Google Scholar]
- 56**.Yan C, Wan R, Bai R, Huang G, Shi Y. Structure of a yeast activated spliceosome at 3.5 A resolution. Science. 2016;353:904–911. doi: 10.1126/science.aag0291. The S. cerevisiae BACT-spliceosome is determined at an average 3.5 Å resolution. The SF3B1 and PHF5A homologues shield the BPS–U2 snRNA duplex. The pre-mRNA traverses the concave interior of the SF3B1 helical repeat domain. The 3′ end of the pre-mRNA interacts with its mutational hotspots in cancers. The Cwc24 and Cwc2 subunits provide new structural examples of ZnK–RNA recognition. [DOI] [PubMed] [Google Scholar]
- 57.Rauhut R, Fabrizio P, Dybkov O, Hartmuth K, Pena V, Chari A, Kumar V, Lee CT, Urlaub H, Kastner B, et al. Molecular architecture of the Saccharomyces cerevisiae activated spliceosome. Science. 2016;353:1399–1405. doi: 10.1126/science.aag1906. [DOI] [PubMed] [Google Scholar]
- 58.Wan R, Yan C, Bai R, Huang G, Shi Y. Structure of a yeast catalytic step I spliceosome at 3.4 A resolution. Science. 2016;353:895–904. doi: 10.1126/science.aag2235. [DOI] [PubMed] [Google Scholar]
- 59.Galej WP, Wilkinson ME, Fica SM, Oubridge C, Newman AJ, Nagai K. Cryo-EM structure of the spliceosome immediately after branching. Nature. 2016;537:197–201. doi: 10.1038/nature19316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yan C, Wan R, Bai R, Huang G, Shi Y. Structure of a yeast step II catalytically activated spliceosome. Science. 2017;355:149–155. doi: 10.1126/science.aak9979. [DOI] [PubMed] [Google Scholar]
- 61.Bertram K, Agafonov DE, Liu WT, Dybkov O, Will CL, Hartmuth K, Urlaub H, Kastner B, Stark H, Luhrmann R. Cryo-EM structure of a human spliceosome activated for step 2 of splicing. Nature. 2017;542:318–323. doi: 10.1038/nature21079. [DOI] [PubMed] [Google Scholar]
- 62.Fica SM, Oubridge C, Galej WP, Wilkinson ME, Bai XC, Newman AJ, Nagai K. Structure of a spliceosome remodelled for exon ligation. Nature. 2017;542:377–380. doi: 10.1038/nature21078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang X, Yan C, Hang J, Finci LI, Lei J, Shi Y. An Atomic Structure of the Human Spliceosome. Cell. 2017;169:918–929 e914. doi: 10.1016/j.cell.2017.04.033. [DOI] [PubMed] [Google Scholar]
- 64.Yan C, Hang J, Wan R, Huang M, Wong CC, Shi Y. Structure of a yeast spliceosome at 3.6-angstrom resolution. Science. 2015;349:1182–1191. doi: 10.1126/science.aac7629. [DOI] [PubMed] [Google Scholar]
- 65.Hang J, Wan R, Yan C, Shi Y. Structural basis of pre-mRNA splicing. Science. 2015;349:1191–1198. doi: 10.1126/science.aac8159. [DOI] [PubMed] [Google Scholar]
- 66.Wan R, Yan C, Bai R, Lei J, Shi Y. Structure of an Intron Lariat Spliceosome from Saccharomyces cerevisiae. Cell. 2017 doi: 10.1016/j.cell.2017.08.029. [DOI] [PubMed] [Google Scholar]
- 67.Jenkins JL, Kielkopf CL. Splicing Factor Mutations in Myelodysplasias: Insights from Spliceosome Structures. Trends Genet. 2017;33:336–348. doi: 10.1016/j.tig.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Daubner GM, Clery A, Jayne S, Stevenin J, Allain FH. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J. 2012;31:162–174. doi: 10.1038/emboj.2011.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kielkopf CL, Rodionova NA, Green MR, Burley SK. A novel peptide recognition mode revealed by the X-ray structure of a core U2AF35/U2AF65 heterodimer. Cell. 2001;106:595–605. doi: 10.1016/s0092-8674(01)00480-9. [DOI] [PubMed] [Google Scholar]
- 70*.Yoshida H, Park SY, Oda T, Akiyoshi T, Sato M, Shirouzu M, Tsuda K, Kuwasako K, Unzai S, Muto Y, et al. A novel 3′ splice site recognition by the two zinc fingers in the U2AF small subunit. Genes Dev. 2015;29:1649–1660. doi: 10.1101/gad.267104.115. S. pombe U2AF1 bound to a minimal U2AF2 ligand is determined at 1.7 Å resolution. The structure shows the S34/Q157 hotspot locations on the ZnK folds. The residues align on the same surface of the protein. The hotspot S34F mutant lacks detectable binding to a consensus 3′ splice site by ITC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patnaik MM, Lasho TL, Finke CM, Hanson CA, Hodnefield JM, Knudson RA, Ketterling RP, Pardanani A, Tefferi A. Spliceosome mutations involving SRSF2, SF3B1, and U2AF35 in chronic myelomonocytic leukemia: prevalence, clinical correlates, and prognostic relevance. Am J Hematol. 2013;88:201–206. doi: 10.1002/ajh.23373. [DOI] [PubMed] [Google Scholar]
- 72.Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V, Kohlmann A, Alpermann T, Yoshida K, Ogawa S, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML) Blood. 2012;120:3080–3088. doi: 10.1182/blood-2012-01-404863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J, Wlodarski MW, Kolking B, Wichmann M, Gorlich K, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood. 2012;119:3578–3584. doi: 10.1182/blood-2011-12-399337. [DOI] [PubMed] [Google Scholar]
- 74.Zhang SJ, Rampal R, Manshouri T, Patel J, Mensah N, Kayserian A, Hricik T, Heguy A, Hedvat C, Gonen M, et al. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood. 2012;119:4480–4485. doi: 10.1182/blood-2011-11-390252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75*.Wu SJ, Kuo YY, Hou HA, Li LY, Tseng MH, Huang CF, Lee FY, Liu MC, Liu CW, Lin CT, et al. The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood. 2012;120:3106–3111. doi: 10.1182/blood-2012-02-412296. The human SF3b subunit arrangement is revealed at 3.1 Å resolution. [DOI] [PubMed] [Google Scholar]
- 76.Cretu C, Schmitzova J, Ponce-Salvatierra A, Dybkov O, De Laurentiis EI, Sharma K, Will CL, Urlaub H, Luhrmann R, Pena V. Molecular architecture of SF3b and structural consequences of Its cancer-related mutations. Mol Cell. 2016;64:307–319. doi: 10.1016/j.molcel.2016.08.036. [DOI] [PubMed] [Google Scholar]
- 77*.Yokoi A, Kotake Y, Takahashi K, Kadowaki T, Matsumoto Y, Minoshima Y, Sugi NH, Sagane K, Hamaguchi M, Iwata M, et al. Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J. 2011;278:4870–4880. doi: 10.1111/j.1742-4658.2011.08387.x. A Y36C mutation of PHF5A is identified that confers resistance to splicing inhibitors. Resistance mutations also are identified at K1071, V1078, and R1074 residues of SF3B1. These PHF5A and SF3B1 residues cluster at an interface with pre-mRNA branch site. [DOI] [PubMed] [Google Scholar]
- 78.Teng T, Tsai JH, Puyang X, Seiler M, Peng S, Prajapati S, Aird D, Buonamici S, Caleb B, Chan B, et al. Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nat Commun. 2017;8:15522. doi: 10.1038/ncomms15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Corrionero A, Minana B, Valcarcel J. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev. 2011;25:445–459. doi: 10.1101/gad.2014311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Folco EG, Coil KE, Reed R. The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev. 2011;25:440–444. doi: 10.1101/gad.2009411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tang Q, Rodriguez-Santiago S, Wang J, Pu J, Yuste A, Gupta V, Moldon A, Xu YZ, Query CC. SF3B1/Hsh155 HEAT motif mutations affect interaction with the spliceosomal ATPase Prp5, resulting in altered branch site selectivity in pre-mRNA splicing. Genes Dev. 2016;30:2710–2723. doi: 10.1101/gad.291872.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Carrocci TJ, Zoerner DM, Paulson JC, Hoskins AA. SF3b1 mutations associated with myelodysplastic syndromes alter the fidelity of branchsite selection in yeast. Nucleic Acids Res. 2017 doi: 10.1093/nar/gkw1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kfir N, Lev-Maor G, Glaich O, Alajem A, Datta A, Sze SK, Meshorer E, Ast G. SF3B1 association with chromatin determines splicing outcomes. Cell Rep. 2015;11:618–629. doi: 10.1016/j.celrep.2015.03.048. [DOI] [PubMed] [Google Scholar]
- 84.Zhang L, Tran NT, Su H, Wang R, Lu Y, Tang H, Aoyagi S, Guo A, Khodadadi-Jamayran A, Zhou D, et al. Cross-talk between PRMT1-mediated methylation and ubiquitylation on RBM15 controls RNA splicing. Elife. 2015;4 doi: 10.7554/eLife.07938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Loerch S, Kielkopf CL. Unmasking the U2AF homology motif family: a bona fide protein-protein interaction motif in disguise. RNA. 2016;22:1795–1807. doi: 10.1261/rna.057950.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jin S, Su H, Tran NT, Song J, Lu SS, Li Y, Huang S, Abdel-Wahab O, Liu Y, Zhao X. Splicing factor SF3B1K700E mutant dysregulates erythroid differentiation via aberrant alternative splicing of transcription factor TAL1. PLoS One. 2017;12:e0175523. doi: 10.1371/journal.pone.0175523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yoshida H, Park SY, Oda T, Akiyoshi T, Sato M, Shirouzu M, Tsuda K, Kuwasako K, Unzai S, Muto Y, et al. A novel 3′ splice site recognition by the two zinc fingers in the U2AF small subunit. Genes Dev. 2015;29:1649–1660. doi: 10.1101/gad.267104.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 89.Hudson BP, Martinez-Yamout MA, Dyson HJ, Wright PE. Recognition of the mRNA AU-rich element by the zinc finger domain of TIS11d. Nat Struct Mol Biol. 2004;11:257–264. doi: 10.1038/nsmb738. [DOI] [PubMed] [Google Scholar]
- 90.Teplova M, Patel DJ. Structural insights into RNA recognition by the alternative-splicing regulator muscleblind-like MBNL1. Nat Struct Mol Biol. 2008;15:1343–1351. doi: 10.1038/nsmb.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yan C, Wan R, Bai R, Huang G, Shi Y. Structure of a yeast step II catalytically activated spliceosome. Science. 2016 doi: 10.1126/science.aak9979. [DOI] [PubMed] [Google Scholar]
- 92.Fica SM, Oubridge C, Galej WP, Wilkinson ME, Bai XC, Newman AJ, Nagai K. Structure of a spliceosome remodelled for exon ligation. Nature. 2017 doi: 10.1038/nature21078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Clery A, Blatter M, Allain FH. RNA recognition motifs: boring? Not quite. Curr Opin Struct Biol. 2008;18:290–298. doi: 10.1016/j.sbi.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 94.Kurtovic-Kozaric A, Przychodzen B, Singh J, Konarska MM, Clemente MJ, Otrock ZK, Nakashima M, Hsi ED, Yoshida K, Shiraishi Y, et al. PRPF8 defects cause missplicing in myeloid malignancies. Leukemia. 2015;29:126–136. doi: 10.1038/leu.2014.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dolatshad H, Pellagatti A, Liberante FG, Llorian M, Repapi E, Steeples V, Roy S, Scifo L, Armstrong RN, Shaw J, et al. Cryptic splicing events in the iron transporter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes. Leukemia. 2016;30:2322–2331. doi: 10.1038/leu.2016.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Conte S, Katayama S, Vesterlund L, Karimi M, Dimitriou M, Jansson M, Mortera-Blanco T, Unneberg P, Papaemmanuil E, Sander B, et al. Aberrant splicing of genes involved in haemoglobin synthesis and impaired terminal erythroid maturation in SF3B1 mutated refractory anaemia with ring sideroblasts. Br J Haematol. 2015;171:478–490. doi: 10.1111/bjh.13610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Park SM, Ou J, Chamberlain L, Simone TM, Yang H, Virbasius CM, Ali AM, Zhu LJ, Mukherjee S, Raza A, et al. U2AF35(S34F) Promotes Transformation by Directing Aberrant ATG7 Pre-mRNA 3′ End Formation. Mol Cell. 2016;62:479–490. doi: 10.1016/j.molcel.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ji X, Zhou Y, Pandit S, Huang J, Li H, Lin CY, Xiao R, Burge CB, Fu XD. SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell. 2013;153:855–868. doi: 10.1016/j.cell.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Isono K, Mizutani-Koseki Y, Komori T, Schmidt-Zachmann MS, Koseki H. Mammalian polycomb-mediated repression of Hox genes requires the essential spliceosomal protein Sf3b1. Genes Dev. 2005;19:536–541. doi: 10.1101/gad.1284605. [DOI] [PMC free article] [PubMed] [Google Scholar]