

Key Points
Germ line mutations in MECOM cause a heterogeneous bone marrow failure syndrome with congenital hypomegakaryocytic thrombocytopenia.
MECOM-associated syndrome includes various organ malformations with variable penetrance, including radioulnar synostosis.
Abstract
Heterozygous mutations in MECOM (MDS1 and EVI1 complex locus) have been reported to be causative of a rare association of congenital amegakaryocytic thrombocytopenia and radioulnar synostosis. Here we report on 12 patients with congenital hypomegakaryocytic thrombocytopenia caused by MECOM mutations (including 10 novel mutations). The mutations affected different functional domains of the EVI1 protein. The spectrum of phenotypes was much broader than initially reported for the first 3 patients; we found familial as well as sporadic cases, and the clinical spectrum ranged from isolated radioulnar synostosis with no or mild hematological involvement to severe bone marrow failure without obvious skeletal abnormality. The clinical picture included radioulnar synostosis, bone marrow failure, clinodactyly, cardiac and renal malformations, B-cell deficiency, and presenile hearing loss. No single clinical manifestation was detected in all patients affected by MECOM mutations. Radioulnar synostosis and B-cell deficiency were observed only in patients with mutations affecting a short region in the C-terminal zinc finger domain of EVI1. We propose the term MECOM-associated syndrome for this heterogeneous hereditary disease and inclusion of MECOM sequencing in the diagnostic workup of congenital bone marrow failure.
Visual Abstract
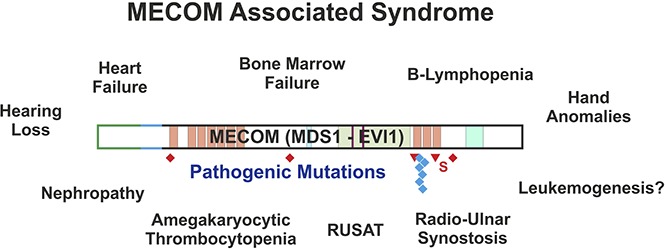
Introduction
Inherited bone marrow failure syndromes (IBMFSs) are a heterogeneous group of rare diseases that are characterized by insufficient production of blood cells of single or multiple hematopoietic lineages.1 The underlying genes can be specific for hematopoietic cells, and hence, the symptoms are restricted to hematopoiesis, as in congenital amegakaryocytic thrombocytopenia (CAMT; Mendelian Inheritance in Man [MIM] #604498) caused by mutations in MPL.2,3 However, a majority of IBMFSs are caused by defects in genes involved in common cellular pathways, such as transcriptional regulation, telomere maintenance, DNA repair, and ribosome function. The BMF in these diseases is often associated with other somatic anomalies that can be specific for particular diseases.1
The rare association of proximal radioulnar synostosis (RUS) with congenital amegakaryocytic thrombocytopenia (AT [this abbreviation is used for the hematological phenotype to distinguish it from the disease CAMT caused by mutations in MPL]) progressing to pancytopenia was first described in 3 patients from 2 families and was attributed to a heterozygous variant in the homeobox gene HOXA11 (RUSAT1; MIM #605432).4,5 No other patients with pathogenic HOXA11 variants have been reported since this first description.
In 2015, Niihori et al6 identified heterozygous mutations in MECOM (MDS1 and EVI1 complex locus) in 3 unrelated patients with RUS and AT (RUSAT2; MIM #616738). Recently, 2 other patients with RUSAT2 were described,7,8 and Bluteau et al9 identified 6 patients with MECOM mutations in a large cohort of patients with IBMFSs (supplemental Table 1).
Here we report on 12 patients with AT caused by germ line mutations in MECOM, including 10 novel mutations. We found familial as well as sporadic cases; the clinical spectrum ranged from isolated RUS with no or mild hematological involvement to severe BMF without obvious skeletal abnormalities. The clinical picture can also include RUS, clinodactyly, cardiac and renal malformations, B-cell deficiency, and presenile hearing loss. We propose considering the different disease patterns as manifestations of a MECOM-associated syndrome, with RUSAT2 as a subgroup.
Patients and methods
Patients suspected to have CAMT, but without mutations in the MPL gene, and patients with suspected hereditary thrombocytopenia were analyzed for mutations in HOXA11 and MECOM. Patient material and clinical data were provided after informed consent. The study was approved by the local ethics committee.
Sequencing of MECOM, HOXA11, MPL, and TERC genes
Genomic DNA was extracted from leukocytes, and mutational analyses were performed using Sanger sequencing with standard techniques. Primers for MPL, HOXA11, TERC, and MECOM were used as published previously.2,6,10,11 Numbering of MECOM exons and designation of mutations refer to MECOM transcript variant 3 (NM_001105078.3) unless otherwise specified.
Evaluation of pathogenicity
Sequence variants were assessed for their pathogenicity according to a consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology using several types of variant evidence (eg, computational, functional, segregation, population data).12,13 Different computational algorithms were used for prediction of the effect of MECOM mutations on protein function: PROVEAN (Protein Variation Effect Analyzer),14 SIFT (Sorting Intolerant From Tolerant),15 Polyphen2,16 and MutationTaster.17
Flow cytometric analyses
The staining of hematopoietic progenitors in fresh BM or frozen BM mononuclear cells (MNCs) was performed as described previously.18 Peripheral blood leukocyte subsets in fresh blood or frozen MNCs were analyzed with the following antibodies (all BioLegend, San Diego, CA): CD3 FITC (OKT3), CD14 AF700 (63D3), CD16 PECy7 (3G8), CD19 PE (HIB19), CD45 PerCPCy5.5 (2D1), and CD56 APCCy7 (HCD56).
Thrombopoietin plasma levels
Thrombopoietin (THPO) plasma levels were determined using a commercial enzyme-linked immunosorbent assay kit (Quantikine; R&D Systems, Minneapolis, MN).
Results
Sequencing
We analyzed DNA from 151 patients whose material had been sent to us for molecular confirmation of CAMT in which we had not found any MPL mutations. MECOM variants were found in 20 (Table 1; Figure 1). In 6 patients (P1-P6), we found heterozygous MECOM mutations in the previously described small region coding for the eighth zinc finger, including 1 missense mutation previously reported for RUSAT2 patients (P5, P6),6,7,9 3 novel missense mutations (P1-P3), and 1 novel frameshift deletion (P4). Seven more patients were identified with heterozygous mutations (7 novel) in other regions of the MECOM gene (P7-P13). Rare single-nucleotide variations reported in public databases were identified in 7 other patients (P14-P20). All patients tested (n = 19; P1-P11, P13-P20) had no alterations in TERC excluding a cooperative effect of this gene, which is located in the immediate vicinity of MECOM.
Table 1.
MECOM variations in patients with hypomegakaryocytic thrombocytopenia
| Patient | Genomic (GRCh38.p7) variation (refSNP) | Transcript variant 3 (NM_001105078.3) | PROVEAN14 prediction/score (cutoff −2.5) | SIFT15 prediction/score (cutoff 0.05) | PolyPhen-216 prediction/score | MutationTaster17 prediction/probability | Found in ExAC or 1000G (MAF/MAC) | Pathogenicity (ACMG Consensus12,13) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E/I No. | CDS/protein | Type | ||||||||||
| P1 | chr3:g.169100919G>A | E11 | c.2251C>T p.His751Tyr | Missense | Deleterious −5.48 | Damaging 0.000 | Probably damaging 1.000 | Disease causing 1.000 | N | Likely pathogenic | ||
| P2 | chr3:g.169100894T>A | E11 | c.2276A>T p.Gln759Leu | Missense | Deleterious −6.27 | Damaging 0.001 | Probably damaging 0.996 | Disease causing 1.000 | N | Likely pathogenic | ||
| P3 | chr3:g.169100892G>A | E11 | c.2278C>T p.Pro760Ser | Missense | Deleterious −6.97 | Damaging 0.006 | Probably damaging 1.000 | Disease causing 1.000 | N | Likely pathogenic | ||
| P4 | Chr3:g.169100962_ 169100963delTC | E11 | c.2208-1_2208delGA p.Arg736Ser fs*13 | Deletion frameshift | NA | NA | NA | Disease causing 1.000 | N | Pathogenic | ||
| P5/P6 | chr3:g.169100922G>A (rs864309724) | E11 | c.2248C>T p.Arg750Trp | Missense | Deleterious −6.98 | Damaging 0.000 | Probably damaging 1.000 | Disease causing 1.000 | N | Pathogenic | ||
| P7 | chr3:g.169093016G>A | E13 | c.2542C>T p.Arg848Ter | Nonsense | NA | NA | NA | Disease causing 1.000 | N | Likely pathogenic | ||
| P8 | chr3:g.169128041G>T | E4 | c.69C>A p.Cys23Ter | Nonsense | NA | NA | NA | Disease causing 1.000 | N | Likely pathogenic | ||
| P9 | chr3:g.169095075C>G | I12 | c.2455+1G>C Loss of splice site | Splice site mutation | NA | NA | NA | Disease causing 1.000 | N | Pathogenic | ||
| P10 | chr3:g.169116194G>A | E7 | c.1114C>T p.Gln372Ter | Nonsense | NA | NA | NA | Disease causing 1.000 | N | Likely pathogenic | ||
| P11 | Chr3:g.169095111_ 169095112insT | E12 | c.2419_2420insAp.Arg807Lys fs*7 | Insertion frameshift | NA | NA | NA | Disease causing 1.000 | N | Likely pathogenic | ||
| P12 | chr3:g.169095235A>G | E12 | c.2296T>C p.Cys766Arg | Missense | Deleterious −10.12 | Damaging 0.000 | Possibly damaging 0.845 | Disease causing 1.000 | N | Likely pathogenic | ||
| P13 | chr3:g.169116402G>C | E7 | c.906C>G p.Ser302Arg | Missense | Neutral −1.53 | Tolerated 0.071 | Probably damaging 0.999 | Disease causing 1.000 | N | Uncertain significance | ||
| P14/P15 | chr3:g.169095082T>C (rs200049869) | E12 | c.2449A>G p.Met817Val | Missense | Neutral 0.16 | Tolerated 0.706 | Benign 0.000 | Disease causing 0.949 | ExAc: Y (0.0004/45) 1000G: N | Uncertain significance | ||
| P16/P17/ P18 | chr3:g.169143748C>A (rs116535717) | E2 | c.-105G>T?c | 5′UTR | NA | NA | NA | Disease causing 0.529 | ExAc: Y (0.0047/561) 1000G: Y (0.0068/34) | Benign | ||
| P19 | chr3:g.169122674T>C (rs34896995) | E5 | c.320A>G p.Gln107Arg | Missense | Neutral −2.26 | Damaging 0.030 | Possibly damaging 0.946 | Disease causing 1.000 | ExAc: (0.0026/319) 1000G: (0.0010/5) | Uncertain significance | ||
| P20 | chr3:g.169145034T>C (rs370795924) | 5′UTR | 5′UTR | NA | NA | NA | Disease causing 1.000 | N (1 individual48) | Uncertain significance | |||
| P16/P17/ P18 | chr3:g.169143748C>A (rs116535717) | E3* | c.88G>T p.Ala30Ser* | Missense* | Neutral −1.11 | Damaging 0.003 | Possibly damaging 0.548 | Disease causing 0.529 ExAc: 561/120542 1000G: 34 |
Benign | |||
The table lists all mutations found in this study, together with the results from different algorithms analyzing the potential effects on protein structure and function (bold font indicates novel mutations). The last column lists the results of the classification of the mutations using the recommendations of the American College of Medical Genetics and the Association for Molecular Pathology (supplemental Table 2).12 Only patients P1 to P12 with mutations judged to be pathogenic or likely pathogenic were included in further phenotypic analysis.
ACMG, American College of Medical Genetics and Genomics; CDS, coding sequence; MAC, minor allele count; MAF, minor allele frequency; N, no; NA, not applicable; UTR, untranslated region; Y, yes.
All designations in the table except for the last row refer to MECOM transcript variant 3 (NM_001105078.3, NP_001098548.2). Designations for transcript variant 1 (NM_001105077.3, NP_001098547.3) are added for mutation rs116535717, located in the 5′-UTR of transcript variant 3 (Figure 1).
Figure 1.

MECOM mutations in patients with congenital BMF and consequences for protein isoforms of MECOM. Exon structure of the MECOM locus together with the main transcript variants are shown together with constitutive mutations in MECOM found in this study. The transcript variants use different start codons and alternatively spliced amino termini. Truncating mutations are labeled in red, missense mutations in blue, and 5′ untranslated region mutations in black. Patient identifiers for those affected by RUSAT are labeled in green. Pathogenic mutations (P1-P12) are labeled in full color; mutations of uncertain significance of pathogenicity or benign variations (P13-P20) are labeled in pale colors. Diamonds indicate nonsense mutations; triangles indicate frameshift mutations. AD, acidic domain; NLS, nuclear localization sequence; RD, repression domain; S, splice site mutation; ZF, zinc finger motif.
Evaluation of pathogenicity
We assessed the probability of pathogenicity of the variants found in the patients by analyzing the data from public databases, family analyses, and prediction algorithms12,13 (Table 1; supplemental Table 2).
Population data.
Mutations found in patients P1 to P13 were not listed in publicly available population databases (including ExAc, covering >60 000 exomes), supporting a pathogenetic significance. Rare variants listed in the ExAc database were found in patients P14 to P19. The allele frequencies of variations observed in patients P16 to P19 in our cohort were in the range of expectance from the ExAc data, arguing for a benign interpretation. In contrast, the allele frequency of the variation found in P14 and P15 in our cohort was significantly higher than expected from the frequency reported in ExAc (2 of 298 vs 45 of 12 1406; Fisher’s exact test P < .01), suggesting pathogenic involvement.
Family analyses.
We were able to analyze DNA from family members for 11 of the patients; segregation of the phenotype with the mutation was observed in 3 families (P1, P2, P10), and de novo mutations were found in 4 patients (P3-P5, P9). For 4 patients, we observed the mutation in an unaffected family member (P8, P13, P15, P16).
In silico analyses.
Analysis of the mutations using different algorithms revealed deleterious and disease-causing effects for all missense mutations in patients P1, P2, P3, P5, P6, and P12. Mutations in patients P1 to P6 affected the previously described highly conserved region of the eighth zinc finger motif; the mutation in P12 affected the adjacent ninth zinc finger motif (Figure 1). Mutations in P7 to P11 were all predicted to lead to a truncated protein. The mutation in P13 and the alterations in P14 to P20, which have been listed in public databases as rare polymorphisms, were predicted to have less effect on MECOM function (Table 1).
On the basis of information (Table 1; supplemental Table 2), 11 of the 16 different variations could be judged as pathogenic (n = 3) or likely pathogenic (n = 8; supposed certainty >90%12). For 4 variations, the significance remains uncertain because of a lack of data or because of contradictory criteria. Only the variation rs116535717 found in patients P16 to P18 was judged as benign on the basis of the available data. For the further phenotypic analysis of the patients with MECOM mutations, we included patients P1 to P12 bearing mutations with a high likelihood of pathogenicity. Clinical information about patients P13 to P20 is listed in supplemental Table 3.
Patient phenotypes
Patients with RUSAT.
P1, characterized by a novel heterozygous missense mutation affecting the same amino acid residue as a previously described mutation6 (c.2251C>T, p.His751Tyr), was born with severe thrombocytopenia and bilateral RUS. There was a history of a similar malformation of the forearms in this family; the father of the patient, the paternal grandfather, and a paternal half-brother were all affected by a similar deformity of the forearms, leading to limited pronation and supination, which could be attributed to proximal RUS in the patient and his father (Figure 2A). No detailed information was available for the other family members. DNA from peripheral blood cells of P1’s father revealed only a minor signal for the MECOM mutation that could have resulting from somatic mosaicism.
Figure 2.

Pedigrees and radiographs of upper limbs of patients P1, P2, and P5. Pedigrees of the familial cases P1 (A) and P2 (B) showing the occurrence of RUS (left area black) and CAMT/congenital aplastic anemia (right area black); radiographs of the upper limbs demonstrating proximal RUS in family members (small symbols indicate stillborn fetuses). (C) Pedigree and radiograph of sporadic case P5; radiograph of the forearm of P5 in pronated state, ruling out RUS. Genotype is included for the analyzed individuals of the pedigrees. M, wild-type allele; m, mutated allele.
The family of P2 (c.2276A>T, p.Gln759Leu) presented after 2 unexplained intrauterine deaths. Postmortem analysis revealed hypocellular BM in both fetuses. The older brother of P2 suffered an unexplained neonatal stroke. Blood counts, coagulation studies, and marrow morphology were normal. P2 developed symptomatic AT, which progressed to pancytopenia and required hematopoietic stem-cell transplantation (HSCT). The mother, both children, and both stillborn fetuses had RUS (Figure 2B). P2 was also deaf. All members of the family affected with RUS carried the MECOM mutation.
In P3 and P4, RUS was diagnosed some years after birth. P3 (c.2278C>T, p.Pro760Ser) was born with severe pancytopenia, and a BM examination revealed a lack of megakaryocytes. Physical examination was inconspicuous. Pancytopenia was ameliorated during the first weeks of life, and the child was temporarily lost to follow-up. At age 25 months, the patient presented with limited supination of the forearms and reduced extension of the elbow joint, hindering the protective arm movements during falling. RUS was diagnosed by radiography.
P4, characterized by a loss of splice acceptor site at the 5′ border of exon 11 (c.2208-1_2208delGA), postnatally presented with severe pancytopenia and severely hypoplastic BM. She underwent allogeneic HSCT with the preliminary diagnosis of CAMT. RUS was first detected at the age of 3 years by radiography.
Patient P12, with a missense mutation affecting the ninth zinc finger motif (c.2296T>C, p.Cys766Arg), presented at age 1 year with bilateral RUS and mild thrombocytopenia, which gradually progressed to pancytopenia and BMF.
BMF without RUS.
In 2 patients (P5, P6) with severe congenital pancytopenia, we detected the mutation c.2248C>T (p.Arg750Trp) previously reported for 3 other patients with RUSAT2.6,7,9 Both were platelet and erythrocyte transfusion dependent from birth and experienced severe recurrent bacterial and fungal infections. Radiographs for upper extremities revealed no anomalies; RUS could be excluded in P5 (Figure 2C). P5 died as a result of catheter-related sepsis at the age of 8 months. Of the 3 elder sisters of P6, 1 presented with Pierre Robin sequence and another with craniosynostosis. P6 and another sister had no physical stigmata. The girl died after HSCT at the age of 5 months.
P7 to P11 had nonsense, frameshift, or loss of splice site mutations predicted to lead to termination of the MECOM protein (Table 1). These patients showed severe pancytopenia from birth or isolated thrombocytopenia with rapid development of pancytopenia during the first weeks of life.
Additional phenotypes observed in patients with MECOM-associated syndrome.
Besides BMF and RUS, other malformations were observed in some of the patients (Table 2). Skeletal anomalies other than RUS were mainly observed in the hands (clinodactyly or brachydactyly in patients P1, P3, P7, P9, P10, P12; Table 2). Malformations in other organ systems included congenital heart failure (P7: ventricular septal defect plus aortic coarctation; P8: ASD; P10: tetralogy of Fallot) and renal malformations (P4: cystic kidney with megaureter left and functional duplex kidney right; P5: mild bilateral renal calyceal dilatation). Congenital deafness was observed in 3 patients in this study (P2, P10, P12). Interestingly, the father of P10, from whom the MECOM mutation was inherited, was also affected by deafness.
Table 2.
Clinical characteristics of patients with MECOM mutations with high certainty of pathogenicity
| Patient | Sex | Hematological course | HSCT (age in months) | RUS | Other skeletal malformations | Other malformations | Hearing | Other/remarks | Family history | MECOM mutation (transcript variant 3) |
|---|---|---|---|---|---|---|---|---|---|---|
| P1 | M | Congenital TP and anemia, progressive BMF | MUD (11) | Y | Hypoplasia of middle and end phalanx D5 | N | Normal | RUS in family members | c.2251C>T, p.His751Tyr | |
| P2 | M | Congenital TP, progressive BMF | First: MFD (8); second: MUD (21) | Y | N | N | Impaired (hearing aids) | RUS and RUSAT in family members | c.2276A>T, p.Gln759Leu | |
| P3 | M | Severely pancytopenic at birth but spontaneous amelioration during first weeks of life, persisting mild TP | N | Y | Clinodactyly D5, hypoplasia of middle phalanx D5 | N | ND | c.2278C>T, p.Pro760Ser | ||
| P4 | F | Congenital TP and anemia, progressive BMF | MUD (7) | Y | Toe malposition D2 bilateral | Cystic kidney (left), duplex kidney (right), urinary obstruction with megaureter (left) | Normal | Hypogammaglobulinemia | N | c.2208-1_2208delGA, loss of splice acceptor site |
| P5 | F | Severe congenital pancytopenia, progressive BMF | Died as a result of sepsis before HSCT | N | N | Hepatomegaly and mild bilateral renal calyceal dilatation | ND | Severe infections, B-cell deficiency | N | c.2248C>T, p.Arg750Trp |
| P6 | F | Severe congenital pancytopenia, extremely hypocellular BM | MUD (5), TRD | N | N | N | ND | Severe bacterial and fungal infections, GI bleeding | Healthy parents, 1 sister with Pierre Robin sequence, 1 sister with craniosynostosis | c.2248C>T, p.Arg750Trp |
| P7 | M | Congenital TP and anemia, progressive BMF | MUD (4), TRD | N | Brachymesophalangy D5 | Subpulmonary VSD, aortic coarctation | Normal | c.2542C>T, p.Arg848Ter | ||
| P8 | F | Congenital TP and anemia, progressive BMF | UCB (3) | N | N | ASD, small cleft palate | ND | Precocious puberty, cognitive deficits | c.69C>A, p.Cys23Ter | |
| P9 | M | Fetal adrenal hemorrhage (w30) resulting from TP, fast progression to pancytopenia and BMF | MUD (10.5), TRD | N | Thumb under D2 | N | Severe bacterial and fungal infections | c.2455+1G>C, loss of splice donor site | ||
| P10 | M | Pancytopenia | MFD (156) | N | Floating elbow, clinodactyly D5 | Tetralogy of Fallot | Congenital hearing loss | Niece with severe BMF (HSCT: MUD at age 8 mo); father hearing impaired | c.1114C>T, p.Gln372Ter | |
| P11 | F | Congenital TP, progressive BMF | MUD (16) | N | Hip dysplasia (left) | N | Inconspicuous | Gynecomastia in infancy (Tanner stage B3) | N | c.2419_2420insA, p.Arg807Kfs*7 |
| P12 | M | Mild TP at age 1 y, progressive BMF | MUD (85) | Y | Clinodactyly D5, brachydactyly D1 and D2, small patellae | N | Bilateral deafness | N | c.2296T>C, p.Cys766Arg |
HSCT outcome was positive unless otherwise indicated.
ASD, atrial septal defect; F, female; GI, gastrointestinal; M, male; MFD, matched family donor; MUD, matched unrelated donor; ND, no data; TP, thrombocytopenia; TRD, transplantation-related death; UCB, unrelated cord blood.
Developmental anomalies in some of the patients may hint at hormonal disturbances. Precocious puberty was observed in P8, and gynecomastia in infancy (Tanner stage B3) occurred in P11.
Chromosome breakage analysis or G2-arrest analysis for diagnosis of Fanconi anemia revealed normal results for all patients. None of the patients showed typical symptoms of dyskeratosis congenita (eg, nail dystrophy, abnormal pigmentation, or leukoplakia)
Analysis of hematopoietic progenitor cells and THPO plasma levels
Because Evi1 has been reported to regulate Mpl expression in mice,19 it has been speculated that the hematological phenotype of MECOM haploinsufficiency might be related to altered MPL expression on hematopoietic progenitors.20 By means of flow cytometry, we were able to analyze hematopoietic progenitors in the BM of P1. We found a percentage of CD34+ in the total nucleated cells of the BM; it was within the normal range (2.8%), but there was only a small population of CD34hiCD38lo progenitor cells, which may reflect an early exhaustion of early hematopoietic progenitors (0.06% of total nucleated cells and 2% of CD34+ cells; Figure 3A). Although rare, these CD34hiCD38lo cells showed a low but distinct MPL (CD110+) expression (Figure 3B), distinguishing them clearly from CD34hiCD38lo cells from patients with CAMT.18 Retrospective analysis of cryopreserved BM MNCs from patients P3, P11, P15, and P18 showed normal MPL expression on early CD34hiCD38lo cells.
Figure 3.

Analysis of hematopoietic progenitors from P1. Flow cytometric analysis of BM cells of patient P1 at age 2.3 months. Cells were stained with CD38-FITC, CD34-APC, CD110-PE (or PE-labeled isotype control), and 4',6-diamidino-2-phenylindole as a viability marker. Gated on viable nucleated cells (A) and on CD34+CD38lo cells (B). Red indicates isotype control; blue indicates CD110 (MPL).
Plasma levels of THPO were exceptionally high in samples available from P1, P4, and P11, pointing to an advanced exhaustion of hematopoiesis (supplemental Table 4). Only a moderately elevated THPO level was seen in patient P8, which was not in accordance with the advanced state of BMF but which could have resulted from the long shipping time of the sample.
Lymphocyte subpopulations
Some of the patients in this study experienced severe recurrent infections (P4, P5, P6), as reported earlier for other patients with RUSAT.21,22 Clinical reevaluation revealed hypogammaglobinemia and B-cell deficiency in patients P4 and P5. For some of the patients, flow cytometric determination of lymphocyte subsets was initially performed during diagnostic workup; for others, we were able to analyze cryopreserved samples from the patients (Table 3). We found low absolute and/or relative numbers of B cells in the peripheral blood of patients P1, P3, P4, and P5, all with mutations affecting the eighth zinc finger. Relative and absolute B-cell numbers were below the 90% prediction range in P1 and P4. Interestingly, the father of P1, affected by RUS but not by thrombocytopenia, also showed low relative and absolute B-cell counts. For P5, absolute B-cell numbers were not available, but a severe B-cell deficiency was obvious in light of the severe pancytopenia and the B-cell percentage of 1% in the lymphocytes. P3 showed low absolute B-cell count at the age of 9 months; at age 7 years, B cells were within the normal range. This was in line with the amelioration of anemia and thrombocytopenia in this patient. In all patients with mutations in other regions of the MECOM gene, we found normal percentages of B cells in the lymphocytes.
Table 3.
Lymphocyte subpopulations in patients
| Patient | Age, mo | CD3 relative, % of lymphocytes (reference value49: mean; 90% range) | CD3, µL−1 (reference value50: predicted; 90% range) | CD19, % of lymphocytes (reference value49: mean; 90% range) | CD19, µL−1 (reference value50: predicted; 90% range) |
|---|---|---|---|---|---|
| P1 | 2.3 | NA | NA | 5.5 (17; 8-33) | 356 (1589; 740-2574) |
| P1 father | 480 | 89.6 (67; 50-91) | 2292 (1373; 679-2214) | 3.72 (10; 4-28) | 95 (268; 125-434) |
| P3 | 8.7 | 72.2 (68; 49-95) | 2203 (3845; 1906-6195) | 17.7 (16; 4-54) | 540 (1446; 674-2343) |
| P3 | 87 | 77.3 (73; 55-97) | 1640 (1734; 854-2794) | 16.6 (12; 4-33) | 352 (559; 260-905) |
| P4 | 2.7 | 95 (69; 49-97) | 3469 (4274; 2121-6887) | 5 (17; 8-33) | 200 (1581; 736-2561) |
| P4 | 4.9 | 92 (69; 49-97) | 10 326 (4107; 2038-6617) | 1 (17; 8-33) | 78 (1529; 712-2477) |
| P5 | 4.0 | 97 (69; 49-97) | NA | 1 (17; 8-33) | NA |
| P8 | 1.5 | 85.0 (70; 55-90) | NA | 13.9 (14; 3-60) | NA |
| P11 | 9.5 | 60.7 (70; 56-87) | NA | 37.4 (15; 3-77) | NA |
Discussion
The association of RUS and AT caused by mutations in HOXA11 (RUSAT1) was first described by Thompson and Nguyen.4 An association of RUS with late-onset progressive BMF was described earlier by Dokal et al.23 As in the pedigrees with HOXA11 variants, the skeletal defect segregated as an autosomal dominant trait and the hematological phenotype of progressive marrow failure showed a variable expression. Niihori et al6 found mutations in MECOM in 3 sporadic cases of RUSAT2 with early BMF, suggesting a specifically severe phenotype of RUSAT2. Recently, 2 other families with RUSAT2 were described.7,8 A study investigating germ line mutations in a group of 179 patients with IBMFS identified 6 more patients with MECOM mutations; interestingly, only 1 of them had RUS (supplemental Table 1).9
In this study, we report on 12 patients with yet unclassified AT from different families caused by mutations in MECOM. We found a continuous spectrum of clinical manifestations in the patients and affected relatives.
MECOM codes for a zinc finger transcription factor with important roles in normal development and oncogenesis. The MECOM locus encodes a number of differentially spliced transcripts yielding the MDS1, MDS1-EVI1, and EVI1 protein isoforms. From different animal models, it has become obvious that the physiological role of the gene lies in the regulation of embryonic development and regulation of hematopoietic stem-cell renewal.24-29 Spatiotemporal expression patterns were observed during mouse embryonic development, with expression in the developing lung, heart, urogenital system, and emerging limb buds.30 MECOM is involved in numerous rearrangements leading to overexpression or formation of fusion transcripts with RUNX1 or ETV6 in myeloid malignancies.31
The role of MECOM in human hematopoiesis is emphasized by the phenotype of the patients in this study; all patients were affected by thrombocytopenia, and most of them developed hypoplastic BM and pancytopenia. Our experiments, together with published data, strongly suggest an impaired maintenance of hematopoietic stem cells as the reason; we were able to demonstrate a severe reduction of early CD34+CD38lo progenitors in the BM, which closely resembled the strong reduction of early hematopoietic cells in the BM of mice with a heterozygous deletion of Evi1 exon 4.28 Other experiments have suggested that Evi1 expression leads to upregulation of stemness genes and enhanced survival.29 The influence of EVI1 expression on MPL, another important player in stem-cell regulation, is not clear. Whereas expression of MPL was positively correlated with that of EVI1 in patients with acute myeloid leukemia,32 Evi1 repressed the expression of Mpl in a mouse model.19 In our patients, we found highly elevated THPO plasma levels, which were in the range of those measured in patients with CAMT.2,33 Nevertheless, MPL expression was detectable on CD34+CD38lo cells, making downregulation of MPL unlikely. The high THPO plasma levels can be explained by the marked loss of MPL-expressing cells in blood and BM.
A recent publication suggested MECOM as a candidate gene for hereditary hematological malignancies.8 In a family with 4 members affected by MECOM mutations and RUS, 2 developed myelodysplastic syndrome at older ages (73 and 48 years). Although we did not observe any malignancies in our patients or relatives, a continuous disturbance of hematopoiesis in patients with mild hematologic phenotypes may account for a higher risk of malignant transformation.
Previous findings have suggested proximal RUS as a pathognomonic sign of MECOM-associated syndrome.6-8 However, most of our patients (as well as those in the IBMFS study by Bluteau et al9) were not affected by RUS, including 2 patients bearing a mutation reported in the 3 patients with RUSAT2.6,7,9 For a short time in embryonic development, the radius and ulna share a common perichondrium, and a perturbation of the process of segmentation by abnormal genetic or teratogenic factors can lead to a more or less distinct synostosis,34 a fact which might explain the differences in the penetrance of this trait. MECOM is specifically expressed in the emerging limb buds in embryogenesis, but insufficient production seems to have no effect on bone development; skeletal abnormalities were not observed in any of the Evi1 knockout models.35 Furthermore, 2 patients with heteroinsufficiency of the MECOM gene resulting from deletion at chromosome 3q26 did not display RUS.20,36,37 Niihori et al6 suggested that the missense mutations identified in their study might act as gain-of-function or partial loss-of-function mutations rather than complete loss-of-function mutations. This hypothesis is supported by the fact that the only MECOM-associated skeletal malformation (polydactyly in the forelimbs) in a mouse model was observed in mice with a dominant missense mutation in the C-terminal zinc finger domain of MECOM.38 Our findings of diverse MECOM mutations leading to RUS, including the mutation in P4 leading to a complete loss of the carboxy terminus including the eighth zinc finger, support the hypothesis of a partial loss of function of the C-terminal zinc finger domain vs gain of function. RUS was observed only in patients with mutations affecting a small region including the eighth and ninth zinc finger motifs. Other skeletal anomalies in our group, like hypoplastic middle phalanxes of the fifth digit (also described in patients with RUSAT26) or hip dysplasia (also described in patients with RUSAT15 and RUSAT27), were not necessarily associated with mutations in this region, but all affected the carboxy terminal part of the protein.
B-cell deficiency also seems to be a part of the clinical spectrum of MECOM-associated syndrome. In some of the patients, we saw B-cell deficiency that might have been causative of severe recurrent bacterial and fungal infections. A pancytopenia with total B-cell deficiency was also reported for a patient with a heterozygous deletion of 3q26, including the 3′ part of MECOM.37 It is not clear whether B-cell deficiency in MECOM-associated disease is due to a common stem-cell defect or to the specific involvement of MECOM in B-cell development. A specific role in B-cell development is suggested by the fact that a gene amplification of MECOM seems to play a role in persistent polyclonal binucleated B-cell lymphocytosis.39 Furthermore, Mecom has been identified as 1 of the shared targets of Menin and MLL1 in murine hematopoietic stem cells and B cells, and hence, it seems to be involved in critical pathways for regulation of regenerative hematopoiesis and B-cell differentiation.40 Additional studies in patients with MECOM-associated AT are necessary to establish a specific B-cell defect.
Two of our patients and 2 of the reported patients (supplemental Table 1) with MECOM mutations had renal phenotypes. MECOM has been found to be strongly expressed in the kidney,41 and a correlation between MECOM and kidney development has been established in different animal models: Mecom has a pivotal role in Xenopus and zebrafish nephrogenesis24,42 and is expressed in the urinary system of embryonic mice.43 However, no phenotype in the renal or urinary system has been reported from various mice knockout models.35
Although congenital heart defects are not rare, the frequency of heart defects in our patient group and in the previously reported patients as well as the similarity to the congenital heart defects observed in mice carrying a hypomorphic Evi1 allele resulting from a deletion of exon 344 argue in favor of the causative involvement of the MECOM mutation in this phenotype.
Sensorineural deafness was reported for 1 of the patients with RUSAT with HOXA11 variants5 as well as for patients with MECOM mutations.6,8 Deafness was observed in 3 patients in this study (P2, P10, P12), confirming the association of deafness with RUS and BMF in this disease. Deafness does not seem to be related to an increased susceptibility to otitis media, as described for mice with a dominant missense change in the C-terminal zinc finger region of Mecom.38 A middle ear bone dysplasia observed in 1 family with MECOM mutations8 could provide a hint regarding the pathomechanism of deafness in MECOM-associated syndrome.
From the synopsis of phenotypic, genotypic, and family analyses, public databases, and prediction algorithms, we conclude that MECOM mutations were causative of BMF syndrome in patients P1 to P12. The incomplete penetrance of certain phenotypes, even within families 1 and 2, may be explained either by stochastic factors modulating gene expression45 or by di- or oligogenic inheritance (eg, joint effects of rare and common variants46). Revertant mosaicism also cannot be excluded in family members with no hematological phenotype or in patients with amelioration of BMF.47 Additional studies are necessary to prove a possible impact of rare sequence variations with uncertain significance (P13-P15, P19, P20) in the pathogenicity of BMF.
On the basis of the phenotypic data from the patients in this and recent reports,6,9,20,36,37 we conclude that heterozygous MECOM mutations or haploinsufficiency of MECOM resulting from microdeletions cause a syndromic disease with a variable phenotypic pattern, varying from severe BMF with multiple organ manifestations to isolated RUS without additional manifestations. Although RUS and BMF are the most common manifestations, none of these symptoms are mandatory for diagnosis of a MECOM-associated syndrome. The phenotypic spectrum overlaps with that described for HOXA11-associated RUSAT. Both disease patterns include families with dominant inheritance of RUS and variable expression of a hematological phenotype and patients with clinodactyly, presenile hearing loss, and amelioration of thrombocytopenia in the first weeks of life. There are some hints of genotype-phenotype correlations; for example, the phenotype of RUS and B-cell lymphopenia has been observed only in patients with mutations in a small region of the eighth and ninth zinc fingers. However, we found large variations in the phenotypes caused by similar mutations. Major phenotypic differences existed within families (eg, P1 and P2), and in 2 of our patients without RUS, we found a MECOM mutation identical to that previously described for 3 patients with RUSAT2.6,7,9 Other genetic or epigenetic factors presumably influence the phenotypic characteristics.
To include the different manifestations of the disease, namely BMF, RUS, and other skeletal anomalies of the forearms and hands, B-cell deficiency, renal malformations, and presenile hearing loss, we propose the term MECOM-associated syndrome. This definition would also include the BMF observed in patients with haploinsufficiency of MECOM resulting from 3q26 deletions.20,36,37 MECOM mutations should be taken into account as a cause of AT or congenital aplastic anemia even if RUS has not been established.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank all patients and families who participated in this study and the referring physicians. They also acknowledge the excellent technical assistance of Yvonne Peter and Christina Struckmann.
This work was supported in part by grants of the Federal Ministry of Education and Research (German Network on Congenital Bone Marrow Failure Syndromes) and by the transnational ERA-NET funding European Platelet Network (EUPLANE). The study was supported by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children, NHS Foundation Trust, and University College London.
Authorship
Contribution: M.G. and M.B. designed and performed research, analyzed data, and wrote the manuscript; P.A., J.E., M.M., E.P., H.R., D.S., R.H.S., S.U., A.W., and B.Z. provided patients’ material and data, suggested experiments, and analyzed data; and all authors read, revised, and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Manuela Germeshausen, Central Research Facility Cell Sorting, Medizinische Hochschule Hannover, Carl-Neuberg-Str 1, 30625 Hannover, Germany; e-mail: germeshausen.manuela@mh-hannover.de; and Matthias Ballmaier, Central Research Facility Cell Sorting, Medizinische Hochschule Hannover, Carl-Neuberg-Str 1, 30625 Hannover, Germany; e-mail: ballmaier.matthias@mh-hannover.de.
References
- 1.Adam S, Melguizo Sanchis D, El-Kamah G, et al. . Concise review: getting to the core of inherited bone marrow failures. Stem Cells. 2017;35(2):284-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ballmaier M, Germeshausen M, Schulze H, et al. . c-mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood. 2001;97(1):139-146. [DOI] [PubMed] [Google Scholar]
- 3.Ballmaier M, Germeshausen M. Advances in the understanding of congenital amegakaryocytic thrombocytopenia. Br J Haematol. 2009;146(1):3-16. [DOI] [PubMed] [Google Scholar]
- 4.Thompson AA, Nguyen LT. Amegakaryocytic thrombocytopenia and radio-ulnar synostosis are associated with HOXA11 mutation. Nat Genet. 2000;26(4):397-398. [DOI] [PubMed] [Google Scholar]
- 5.Thompson AA, Woodruff K, Feig SA, Nguyen LT, Schanen NC. Congenital thrombocytopenia and radio-ulnar synostosis: a new familial syndrome. Br J Haematol. 2001;113(4):866-870. [DOI] [PubMed] [Google Scholar]
- 6.Niihori T, Ouchi-Uchiyama M, Sasahara Y, et al. . Mutations in MECOM, encoding oncoprotein EVI1, cause radioulnar synostosis with amegakaryocytic thrombocytopenia. Am J Hum Genet. 2015;97(6):848-854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lord SV, Jimenez JE, Kroeger ZA, et al. . A MECOM variant in an African American child with radioulnar synostosis and thrombocytopenia. Clin Dysmorphol. 2018;27(1):9-11. [DOI] [PubMed] [Google Scholar]
- 8.Ripperger T, Hofmann W, Koch JC, et al. . MDS1 and EVI1 complex locus (MECOM): a novel candidate gene for hereditary hematological malignancies. Haematologica. 2018;103(2):e55-e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bluteau O, Sebert M, Leblanc T, et al. . A landscape of germline mutations in a cohort of inherited bone marrow failure patients. Blood. 2018;131(7):717-732. [DOI] [PubMed] [Google Scholar]
- 10.Fleischman RA, Letestu R, Mi X, et al. . Absence of mutations in the HoxA10, HoxA11 and HoxD11 nucleotide coding sequences in thrombocytopenia with absent radius syndrome. Br J Haematol. 2002;116(2):367-375. [DOI] [PubMed] [Google Scholar]
- 11.Du HY, Pumbo E, Manley P, et al. . Complex inheritance pattern of dyskeratosis congenita in two families with 2 different mutations in the telomerase reverse transcriptase gene. Blood. 2008;111(3):1128-1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kleinberger J, Maloney KA, Pollin TI, Jeng LJ. An openly available online tool for implementing the ACMG/AMP standards and guidelines for the interpretation of sequence variants. Genet Med. 2016;18(11):1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745-2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073-1081. [DOI] [PubMed] [Google Scholar]
- 16.Adzhubei IA, Schmidt S, Peshkin L, et al. . A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361-362. [DOI] [PubMed] [Google Scholar]
- 18.Ballmaier M, Holter W, Germeshausen M. Flow cytometric detection of MPL (CD110) as a diagnostic tool for differentiation of congenital thrombocytopenias. Haematologica. 2015;100(9):e341-e344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buonamici S, Li D, Chi Y, et al. . EVI1 induces myelodysplastic syndrome in mice. J Clin Invest. 2004;114(5):713-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nielsen M, Vermont CL, Aten E, et al. . Deletion of the 3q26 region including the EVI1 and MDS1 genes in a neonate with congenital thrombocytopenia and subsequent aplastic anaemia. J Med Genet. 2012;49(9):598-600. [DOI] [PubMed] [Google Scholar]
- 21.Staba SL, Sola MC, William SB. Unrelated donor umbilical cord blood transplantation in two children with amegakaryocytic thrombocytopenia with radio-ulnar synostosis [abstract]. Biol Blood Marrow Transplant. 2006;12(2 Suppl 1):129-130. Abstract 373. [Google Scholar]
- 22.Yoshida H, Hashii Y, Okuda T, et al. . A case of congenital bone marrow failure with radio-ulnar synostosis. Int J Hematol. 2010;91(2):331-332. [DOI] [PubMed] [Google Scholar]
- 23.Dokal I, Ganly P, Riebero I, et al. . Late onset bone marrow failure associated with proximal fusion of radius and ulna: a new syndrome. Br J Haematol. 1989;71(2):277-280. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Cheng CN, Verdun VA, Wingert RA. Zebrafish nephrogenesis is regulated by interactions between retinoic acid, mecom, and Notch signaling. Dev Biol. 2014;386(1):111-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mead PE, Parganas E, Ohtsuka S, et al. . Evi-1 expression in Xenopus. Gene Expr Patterns. 2005;5(5):601-608. [DOI] [PubMed] [Google Scholar]
- 26.Yuasa H, Oike Y, Iwama A, et al. . Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA-2 expression. EMBO J. 2005;24(11):1976-1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kataoka K, Sato T, Yoshimi A, et al. . Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J Exp Med. 2011;208(12):2403-2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goyama S, Yamamoto G, Shimabe M, et al. . Evi-1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell. 2008;3(2):207-220. [DOI] [PubMed] [Google Scholar]
- 29.Kustikova OS, Schwarzer A, Stahlhut M, et al. . Activation of Evi1 inhibits cell cycle progression and differentiation of hematopoietic progenitor cells. Leukemia. 2013;27(5):1127-1138. [DOI] [PubMed] [Google Scholar]
- 30.Wieser R. The oncogene and developmental regulator EVI1: expression, biochemical properties, and biological functions. Gene. 2007;396(2):346-357. [DOI] [PubMed] [Google Scholar]
- 31.Hinai AA, Valk PJ. Review: Aberrant EVI1 expression in acute myeloid leukaemia. Br J Haematol. 2016;172(6):870-878. [DOI] [PubMed] [Google Scholar]
- 32.Nishikawa S, Arai S, Masamoto Y, et al. . Thrombopoietin/MPL signaling confers growth and survival capacity to CD41-positive cells in a mouse model of Evi1 leukemia. Blood. 2014;124(24):3587-3596. [DOI] [PubMed] [Google Scholar]
- 33.Germeshausen M, Ballmaier M, Welte K. MPL mutations in 23 patients suffering from congenital amegakaryocytic thrombocytopenia: the type of mutation predicts the course of the disease. Hum Mutat. 2006;27(3):296. [DOI] [PubMed] [Google Scholar]
- 34.Simmons BP, Southmayd WW, Riseborough EJ. Congenital radioulnar synostosis. J Hand Surg Am. 1983;8(6):829-838. [DOI] [PubMed] [Google Scholar]
- 35.Blake JA, Eppig JT, Kadin JA, Richardson JE, Smith CL, Bult CJ; the Mouse Genome Database Group. Mouse Genome Database (MGD)-2017: community knowledge resource for the laboratory mouse. Nucleic Acids Res. 2017;45(D1):D723-D729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bouman A, Knegt L, Gröschel S, et al. . Congenital thrombocytopenia in a neonate with an interstitial microdeletion of 3q26.2q26.31. Am J Med Genet A. 2016;170A(2):504-509. [DOI] [PubMed] [Google Scholar]
- 37.van der Veken LT, Maiburg MC, Groenendaal F, et al. . Lethal neonatal bone marrow failure syndrome with multiple congenital abnormalities including limb defects due to a constitutional deletion of 3′ MECOM [published online ahead of print 8 February 2018]. Haematologica. doi:10.3324/haematol.2017.185033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parkinson N, Hardisty-Hughes RE, Tateossian H, et al. . Mutation at the Evi1 locus in Junbo mice causes susceptibility to otitis media. PLoS Genet. 2006;2(10):e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cornet E, Mossafa H, Courel K, Lesesve JF, Troussard X. Persistent polyclonal binucleated B-cell lymphocytosis and MECOM gene amplification. BMC Res Notes. 2016;9:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li BE, Gan T, Meyerson M, Rabbitts TH, Ernst P. Distinct pathways regulated by menin and by MLL1 in hematopoietic stem cells and developing B cells. Blood. 2013;122(12):2039-2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morishita K, Parganas E, Parham DM, Matsugi T, Ihle JN. The Evi-1 zinc finger myeloid transforming gene is normally expressed in the kidney and in developing oocytes. Oncogene. 1990;5(9):1419-1423. [PubMed] [Google Scholar]
- 42.Van Campenhout C, Nichane M, Antoniou A, et al. . Evi1 is specifically expressed in the distal tubule and duct of the Xenopus pronephros and plays a role in its formation. Dev Biol. 2006;294(1):203-219. [DOI] [PubMed] [Google Scholar]
- 43.Perkins AS, Mercer JA, Jenkins NA, Copeland NG. Patterns of Evi-1 expression in embryonic and adult tissues suggest that Evi-1 plays an important regulatory role in mouse development. Development. 1991;111(2):479-487. [DOI] [PubMed] [Google Scholar]
- 44.Bard-Chapeau EA, Szumska D, Jacob B, et al. . Mice carrying a hypomorphic Evi1 allele are embryonic viable but exhibit severe congenital heart defects. PLoS One. 2014;9(2):e89397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135(2):216-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gibson G. Rare and common variants: twenty arguments. Nat Rev Genet. 2012;13(2):135-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osumi T, Tsujimoto SI, Nakabayashi K, et al. . Somatic MECOM mosaicism in a patient with congenital bone marrow failure without a radial abnormality [published online ahead of print 22 January 2018]. Pediatr Blood Cancer. doi:10.1002/pbc.26959. [DOI] [PubMed] [Google Scholar]
- 48.Dogan H, Can H, Otu HH. Whole genome sequence of a Turkish individual. PLoS One. 2014;9(1):e85233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schatorjé EJ, Gemen EF, Driessen GJ, et al. . Age-matched reference values for B-lymphocyte subpopulations and CVID classifications in children. Scand J Immunol. 2011;74(5):502-510. [DOI] [PubMed] [Google Scholar]
- 50.Huenecke S, Behl M, Fadler C, et al. . Age-matched lymphocyte subpopulation reference values in childhood and adolescence: application of exponential regression analysis. Eur J Haematol. 2008;80(6):532-539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.