

Abstract
Transparent wood composites, with their high strength and toughness, thermal insulation, and excellent transmissivity, offer a route to replace glass for diffusely transmitting windows. Here, conjugated‐polymer‐based electrochromic devices (ECDs) that switch on‐demand are demonstrated using transparent wood coated with poly(3,4‐ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) as a transparent conducting electrode. These ECDs exhibit a vibrant magenta‐to‐clear color change that results from a remarkably colorless bleached state. Furthermore, they require low energy and power inputs of 3 mWh m−2 at 2 W m−2 to switch due to a high coloration efficiency (590 cm2 C−1) and low driving voltage (0.8 V). Each device component is processed with high‐throughput methods, which highlights the opportunity to apply this approach to fabricate mechanically robust, energy‐efficient smart windows on a large scale.
Keywords: electrochemistry, electrochromism, polymers, thin films, wood
Introduction
The drive to reduce energy costs and incorporate renewable materials in the building sector has motivated the development of lightweight and thermally insulating transparent wood (TW) composites.1, 2, 3 TW is composed of delignified wood or lignin‐retaining bleached wood impregnated with a polymer matrix [typically, poly(methyl methacrylate) (PMMA) or epoxy], which provides a substantial improvement in mechanical toughness compared to glass.4 The inclusion of the wood skeleton in these composites gives rise to a high degree of optical anisotropy that depends on the wood fiber orientation2 and has enabled multifunctional modifications by leveraging the hierarchical porous structure and chemical functionality of cellulose.5, 6, 7, 8, 9, 10 The combination of robust mechanical properties, optical transmittance, and a low thermal conductivity makes these composites ideal for diffusely transmitting, energy‐efficient building materials.
Consistent with the motivation to improve a building's energy efficiency is the idea of modulating transmitted light through electrochromic “smart windows”11, 12, 13 that can control incident irradiation into a building. There are three main classes of electrochromic materials that have been studied for smart windows and mirrors: metal oxides (WO3, NbOx, VOx), molecular electrochromes (e.g., viologen derivatives), and electrochromic polymers (ECPs).11, 13, 14 The working principle for electrochromic materials is the generation of new electronic states resulting from a redox reaction. These states give rise to new electronic transitions, which cause a shift in the absorption profile providing light modulation over wavelengths from the ultraviolet to the infrared.15, 16 Among these electrochromic materials, ECPs offer the unique advantage of fine color control through synthetic modifications of the polymer structure. Additionally, colors of subtly varying hues, as well as blacks and browns, have been accessed by blending primary and secondary colors.17, 18, 19, 20 Long electrochemical cycle lifetimes of 105 cycles and stability to prolonged light exposure with suitable encapsulation have also been demonstrated.21, 22 Importantly, the solubility of ECPs makes the polymers amenable for various high‐throughput processing methods. The side‐chain structure on the polymer can be readily modified for solubility in polar solvents (water or alcohols) or in nonpolar organic solvents.21, 23 Additionally, the compatibility of ECPs with large‐scale processing techniques is critical to realize practically sized devices.24, 25
To develop electrochromic devices on TW, a transparent conductor with sufficient conductivity and redox stability must be employed. Materials that do not require energy‐intensive processing such as metallic nanostructures,26, 27, 28, 29 carbon nanomaterials,30 and conducting polymers31, 32, 33 have been demonstrated as high‐performance transparent conductive electrodes (TCEs). A figure of merit (FoM) has been defined as the ratio of the DC electrical conductivity (σ DC) to the optical conductivity (σ opt) to evaluate these transparent conductors under development [Eq. (1)]:34
| (1) |
in which R sheet is the sheet resistance and T(λ) is the transmittance at λ=550 nm. State‐of‐the‐art TCEs have achieved a FoM of well over 100, exceeding the industry standard for ITO: 90 % transmittance, 100 Ω/sq (FoM=35).35, 36, 37, 38 Certain light‐emitting devices, such as liquid crystal displays and other touchscreens, have less stringent requirements where a FoM of 10 is deemed sufficient.35 Recently, Fang and co‐workers deposited Ag nanowires onto transparent cellulose nanopaper that had a sheet resistance as low as 1.9 Ω/sq at a transmittance of 86 % (FoM=1270).39 However, the low oxidation potential of Ag overlaps with the potential range needed for the reversible switching of many electrochromic polymers (−0.5 to +0.8 V vs. Ag/Ag+). The conducting polymer suspension, poly(3,4‐ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) has recently been demonstrated as one of the highest performing transparent conductors (FoM=142) and is well suited for coating onto cellulose substrates as an aqueous ink.37, 40, 41
Considering the use of PEDOT:PSS in ECDs, it is necessary to balance the tradeoff between surface resistance and transmissivity to determine the optimal film thickness. Thick films will have a lower resistance but a deep blue color, whereas thin films will have a higher resistance but offer more transmissivity. Various strategies have been developed to enhance the conductivity of a PEDOT:PSS film. Without modification, PEDOT:PSS films exhibit a relatively low conductivity of 0.1–1 S cm−1 because of the large excess of inactive PSS− inhibiting charge transport between PEDOT+ crystallites. The conductivity of PEDOT:PSS has been enhanced by orders of magnitude to well over 1000 S cm−1 through the incorporation of co‐solvents or by post treatments with strong acids.42, 43, 44, 45 This enhancement is attributed to a higher degree of ordering and increased domain purity of the PEDOT+ crystallites combined with a partial removal of excess PSS−.
The best‐performing post treatments use a combination of heat (>100 °C) and concentrated acids (>10 m) or solvents that are incompatible with TW substrates. Recently, Kumar and co‐workers demonstrated fast‐switching ECDs composed of complimentary poly[3,4‐di‐(2‐ethylhexlyoxy(thiophene)‐co‐(3,4‐ethylenedioxythiophene)] and Prussian blue‐based electrochromes deposited on PEDOT:PSS electrodes treated with dilute 1 m para‐toluene sulfonic acid/dimethyl sulfoxide (pTSA/DMSO). These ECDs achieved a 40 % device contrast at switching times of less than 2 s.46 Not only were these conductivities suitable to support fast‐switching devices, but the post‐treatments also lowered the PEDOT:PSS dedoping onset potential from −0.57 V for untreated films to −0.72 V versus Ag/Ag+.47, 48 This allows the PEDOT:PSS electrodes to remain compatible with lower‐oxidation‐potential materials such poly(3,4‐dioxypyrroles) (PXDOPs),49 3,4‐ethylenedioxythiophene (EDOT) co‐polymers,50 and 3,4‐ethylenedioxyselenophene co‐polymers (EDOS).51
In this work, we demonstrate a simple and relatively benign process for generating highly conducting PEDOT:PSS/TW electrodes for ECDs. The TW substrates derived from birch wood incorporate 30 % cellulose fibers by volume and exhibit improved strength and toughness compared to neat PMMA or glass. A post‐treatment with 1 m pTSA dissolved in ethylene glycol (EG) at room temperature increased the conductivity to 1200 S cm−1 and provided a conductive potential window of −0.7 to 0.8 V versus Ag/Ag+. Electrochemical impedance analysis of the pTSA/EG‐treated PEDOT:PSS revealed that small changes in capacitance and ion diffusion occur at potentials less than −0.5 V versus Ag/Ag+, which results in faster coloration than bleaching for the magenta‐to‐clear ECP (ECP‐Magenta)films and ECDs. The low driving voltage (0.8 V) and high haze give rise to an exceptionally colorless bleached state). The facile processing of both the electrode and active materials combined with the low energy requirement (3 mWh m−2) and optical memory make these TW ECDs attractive options for energy‐saving smart windows and roofs.
Results and Discussion
Properties of TW substrates
TW composites were prepared as reported previously.1 Birch wood was delignified by immersion in a solution of sodium hypochlorite buffered to pH 4.6. The delignified wood was infiltrated with prepolymerized methyl methacrylate and 2,2′‐azobis(2‐methylpropionitrile) (AIBN) initiator followed by a baking step at 70 °C to form the PMMA matrix. The mechanical properties of TW were evaluated using a three‐point flexural bending test, and microstructural analysis was performed to understand the structural properties of the composite. Typical stress–strain curves of TW and glass from the three‐point bending test are presented in Figure 1 a, showing the increased strength and dramatically improved toughness of TW. The stress–strain curve for glass demonstrates its brittle nature after small strains (0.20±0.02 %), whereas the TW exhibits yielding behavior followed by fracture at (2.9±0.2) % strain.
Figure 1.

a) Stress–strain curves comparing glass and birch TW composite with the failure strain labeled. b) SEM image of the TW fracture surface, scale bar 5 μm. c) Visible transmittance of birch TW with and without the PEDOT:PSS coating. Inset images show the uncoated (left) and coated (right) substrates with their corresponding sheet resistances.
The stress at break of (140±10) MPa for TW is higher than that of the original birch wood (98±7 MPa), neat PMMA (72±6 MPa), and glass (116±13 MPa). The excellent mechanical performance of the TW is mainly attributable to the reinforcing wood skeleton in the composite.1 In addition, the wood−PMMA bond integrity appears to be favorable for load transfer between the fibers and the matrix in the composites. The scanning electron micrograph (SEM) presented in Figure 1 b shows the fracture surface of the TW where the PMMA matrix was fully infiltrated into the hollow wood cell. Strong interfacial bonds between the cell wall and PMMA are evident by the rough fracture surface. Low‐magnification SEM images of the fracture surface are shown in Figure S1 in the Supporting Information, where the macroscale surface roughness around the fibers is further indication of efficient load transfer between the fibers and the matrix. The stiff and strong cellulosic skeleton leads to an improved mechanical performance compared to neat PMMA. The high failure strain of the TW corresponds to a work of fracture of 3.2 MJ m−3. This is 30 times higher than the work of fracture for glass samples (0.10 MJ m−3), which have fracture strains of (0.20±0.02) %. This highlights the benefit of using tough TW over glass if brittle fracture and shattering are a safety concern.
In terms of optical properties, the TW samples used in this study provide a transmittance of 80 % at λ=550 nm with high haze (70 %; Figure 1 c). The transmittance is uniform across the visible spectrum, which results in a colorless substrate. The high haze of TW is a unique feature compared to glass. This haze enables the TW to offer indoor privacy while its high transmittance continues to offer diffuse lighting (Figure S2).1 In addition, TW has a lower thermal conductivity (0.23 W m−1 K−1) than glass (1.0 W m−1 K−1). The TW properties highlighted here demonstrate several advantages over glass to generate energy‐saving smart windows/roofs when high transparency is not required.
Electrochemical properties of PEDOT:PSS films
Prior to the fabrication of ECDs, the redox properties of PEDOT:PSS were evaluated to determine its suitability as an electrode material for ECP‐Magenta and the minimally color‐changing charge‐storage polymer (MCCP; Figure S3). To support the complete redox switching of these polymers, PEDOT:PSS must maintain a high conductivity over a potential window from −0.5 to 0.8 V versus Ag/Ag+.
Despite its broad electroactive window, PEDOT+ is dedoped to its neutral, insulating form at low potentials. To probe the dedoping of PEDOT:PSS, in situ conductance was measured as a function of the potential from 0.8 to −1.2 V versus Ag/Ag+. The corresponding conductivity values were calculated from the conductance using the film cross‐sectional area and footprint dimensions (Figure 2 a). A sharp decrease in conductivity is observed at −0.7 and −0.6 V for pTSA/EG‐treated and untreated films, respectively. At −1.0 V, acid‐treated PEDOT:PSS is rendered insulating, whereas this transition occurs at −0.8 V for the untreated films. The difference in this onset of dedoping is consistent with previous reports and is likely a result of the enhanced ordering of the PEDOT chains.46, 47 This ordering extends the effective conjugation length of the polymer and reduces the oxidation potential for a portion of the pTSA/EG‐treated material. The presence of these well‐ordered domains is also evident from the cyclic voltammograms (CVs) shown in Figure S4, in which the pTSA/EG treatment generates additional redox peaks at −0.78 and −0.92 V for oxidation and reduction, respectively. As many of the reported cathodically coloring ECPs switch to their colored state at potentials above −0.7 V versus Ag/Ag+ and switch to their colorless states below 0.8 V versus Ag/Ag+, these results demonstrate that PEDOT:PSS is suitable for use as a TCE for these polymer‐based ECDs.11
Figure 2.
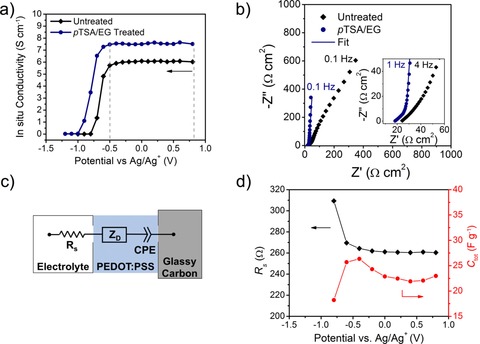
a) In situ conductivity of treated and untreated PEDOT:PSS from 0.8 to −1.2 V vs. Ag/Ag+ in 0.5 m tetrabutylammonium hexafluorophosphate/propylene carbonate (TBAPF6/PC) electrolyte. b) Nyquist plot in which the EIS spectra for treated and untreated PEDOT:PSS are compared (100 kHz to 0.1 Hz). Inset shows high‐frequency data points (100 kHz to 1 Hz) for treated and untreated PEDOT:PSS. c) Equivalent circuit used to fit the EIS data for treated PEDOT:PSS films. d) Fitted values for solution resistance and total capacitance versus potential for pTSA/EG‐treated PEDOT:PSS.
Electrochemical impedance spectroscopy (EIS) was used to further elucidate the physical changes occurring in the PEDOT:PSS layer over the potential window from −0.5 to 0.8 V and to understand how these changes affect the kinetics related to the various redox processes occurring during ECP switching. The Nyquist plot presented in Figure 2 b shows the EIS spectra for pTSA/EG‐treated PEDOT:PSS compared to untreated PEDOT:PSS measured at a DC potential of 0.0 V versus Ag/Ag+. The maximum impedance measured at f=0.1 Hz of the untreated films is |Z|=700 Ω cm2 compared to |Z|=340 Ω cm2 for the pTSA/EG‐treated films. This increased impedance for the untreated films is caused by the lower electronic conductivity of the film compared to the pTSA/EG‐treated PEDOT:PSS. The impedance measurements at high frequencies probe the ion diffusion in the film, which gives a sloped line at 45° in the Nyquist plot (Figure 2 b, inset). This behavior persists in the untreated films even at low frequencies, which is an indication for the presence of a slow diffusion process not seen for the pTSA/EG‐treated films. At low frequencies, the imaginary impedance increases with little change in the real impedance (vertical line) for the pTSA/EG‐treated PEDOT:PSS, indicating a purely capacitive response in the film. Both the reduction in total impedance and the emergence of this capacitive behavior in the pTSA/EG‐treated PEDOT:PSS relative to the untreated material highlight that the improved charge transport and electroactivity of the pTSA/EG‐treated PEDOT:PSS persists upon immersion in electrolyte.
In accordance with the in situ conductivity results, the onset of dedoping of the untreated PEDOT:PSS is apparent in the EIS spectra at −0.8 V at which a charge‐transfer semicircle appears in the high‐frequency region and the maximum impedance increases to |Z|=1200 Ω cm2 (Figure S5 a). The EIS spectra of pTSA/EG‐treated PEDOT:PSS show only a minimal change at this potential (Figure S5 b). This result gives further evidence that the untreated film becomes insulating below −0.8 V, whereas the pTSA/EG‐treated film remains highly electroactive. By fitting the EIS data for the pTSA/EG‐treated PEDOT:PSS with the equivalent circuit shown in Figure 2 c,52, 53, 54 the electrolyte resistance, diffusional time constant, and capacitance were extracted over the potential range from 0.8 to −0.8 V. The solution‐resistance (R s) element in the circuit takes into account the electrolyte resistance of the cell and can be impacted by a change in the resistance of the PEDOT:PSS electrode. The second element, a “finite‐length” Warburg diffusion element (Z D), accounts for the ion diffusion through the PEDOT:PSS film. Z D is a frequency‐dependent impedance defined in terms of the diffusional time constant (τ D) and the diffusional pseudocapacitance of the film (C D):55
| (2) |
in which j= , ω [rad s−1] is the angular frequency equal to 2 π f, and α is an exponent for which a value of 0.5 represents ideal diffusion described by Fick's second law.56 This element in the equivalent circuit reproduces both the diffusion‐limited behavior at high frequencies and the pseudocapacitive response at low frequencies seen in Figure 2 b. Previous studies have shown that PEDOT:PSS can form dense films with tortuous ion diffusion pathways that result in hindered “subdiffusion”.56 Ions in these films will diffuse more slowly than predicted by Fick's second law. This behavior was taken into account when fitting the EIS spectra for acid‐treated PEDOT:PSS in which a minor adjustment setting to α=0.48 improved the fits dramatically, giving rise to a goodness‐of‐fit value (χ 2) of 7.6×10−4. The constant phase element (CPE) is proportional to the PEDOT:PSS pseudocapacitance. It is defined as CPE=(1/jωC d) in which C d is the bulk electronic capacitance and p=1 (ideal) for all potentials between −0.6 and 0.4 V in which charging effects are absent. All EIS spectra for pTSA/EG‐treated and untreated films are shown in Figure S5b; the fit parameters are listed in Table S1.
Examining the fitted parameters for the acid‐treated PEDOT:PSS, changes in resistance, diffusional time constant, and capacitance were monitored over 0.8 to −0.8 V (Table S2). The evolution of R s and total capacitance [C tot=(1/C D+1/C d)−1] as a function of potential are shown in Figure 2 d. The R s values remain relatively constant at 260 Ω between 0.8 and −0.6 V, consistent with the in situ conductance results. Similarly, the capacitance shows a slight increase from 22 F g−1 at 0.8 V to 27 F g−1 at −0.4 V coupled with an increase in the ion diffusion time constant from 0.086 to 0.15 s over the same potential range. This trend suggests that decreasing potentials cause morphological compaction of the PEDOT:PSS film, which hinders ion transport.57 Changes in the in situ conductance are not seen over this same potential range as ion diffusion cannot be perceived at low scan rates below 0.5 mV s−1. At −0.8 V, R s increases to 310 Ω and the total capacitance decreases from an average of 27 to 18 F g−1. These changes indicate that the PEDOT:PSS film transitions to an insulating state. These results from both the in situ conductance and the EIS measurements highlight that the PEDOT:PSS will remain charged and highly conductive in the potential window between −0.6 and 0.8 V versus Ag/Ag+ after treatment with pTSA/EG although minor changes in morphology occur in this range.
Properties of TW electrodes
To construct transparent electrodes using TW substrates, PEDOT:PSS was blade coated to achieve a thickness of roughly 140 nm and then treated with 1 m pTSA/EG at 40 °C. Above this thickness, the PEDOT:PSS will contribute a substantial blue tint to the electrochromic device and compromise the electrode transmittance and the optical contrast that can be achieved. This specific treatment method was chosen to avoid damage to the substrate while still providing a three‐order‐of‐magnitude increase in conductivity compared to untreated PEDOT:PSS (Table S2).
As a control experiment, treated PEDOT:PSS films were prepared on glass and achieved conductivities of 1200±100 S cm−1 (58±5 Ω/sq), which shows an improvement over EG (980±60 S cm−1) and 1 m pTSA/H2O (400±100 S cm−1) treatments. The improvement in conductivity using the pTSA/EG treatment is attributable to the combined effect of larger crystallite size and removal of PSSH.48, 58 The thickness of the PEDOT:PSS films is difficult to accurately measure on the TW substrates because of the relatively high surface roughness of 670±70 nm perpendicular to the wood fibers and (90±10) nm parallel to the fibers. Therefore, the PEDOT:PSS conductivity on TW is qualitatively compared by examining the transmittance versus sheet resistance curves (Figure S6). As the transmittance change varies in a similar fashion with the sheet resistance, the DC conductivity should be comparable.
The UV/Vis spectra comparing the transmittance of bare TW and PEDOT:PSS/TW electrode are shown in Figure 1 c. The TW electrodes provide 70 % transmittance at λ=550 nm with a 70 Ω/sq surface resistance (FoM=14). Ignoring the substrate, a FoM of 50 is achieved with the 1 m pTSA/EG treatment, which is on par with the commercial standard for ITO (FoM=35).
The color contribution from PEDOT:PSS can be quantified by using CIELAB color coordinates in which: L*, a*, and b* quantify the lightness (L*) and the hue/saturation of an object from green to red (a*) and from blue to yellow (b*). The color difference between two samples is given by Equation (3), defined as the Euclidian distance between each color coordinate.
| (3) |
At a color difference of ΔE*<2.4, two samples become indistinguishable to the human eye.59 Therefore, maintaining a color difference of less than 2.4 relative to standard white (L*=100, a*=0, b*=0) would be a perfectly color neutral electrode and would not impact the ECP contrast. The color difference between the PEDOT:PSS/TW electrodes (L*=87, a*=−1.2, b*=1.2) and bare TW (L*=90, a*=−1.8, b*=2.7) is only 3.8, highlighting the color neutrality of the PEDOT:PSS coating.
Next, the ECP‐Magenta films were evaluated by using cyclic voltammetry (CV) to compare their electrochemical response on pTSA/EG‐treated PEDOT:PSS/glassy carbon and on plain glassy carbon electrodes. The CV response for an ECP‐Magenta film is shown in Figure 3 where a similar current density profile is seen for both electrodes. This response highlights the ability to use PEDOT:PSS as an electrode to oxidize and reduce ECP‐Magenta. The anodic peak is shifted by 50 mV on the PEDOT:PSS electrodes compared to glassy carbon electrodes, while the cathodic peak remains at 0.10 V. The slight shift of the anodic peak indicates that there is a higher resistance to oxidize (bleach) the ECP when using PEDOT:PSS electrodes. An increase in current density over the entire potential range in Figure 3 is attributed to the pseudocapacitance of the PEDOT:PSS. This current contribution of the PEDOT:PSS can be determined by subtracting the ECP‐Magenta/glassy carbon current from that of the ECP‐Magenta/PEDOT:PSS/glassy carbon electrode (Figure S7 a). This subtracted current density matches the current density of PEDOT:PSS from 0.3 to 0.8 V at which ECP‐Magenta is oxidized, but is lower than the PEDOT:PSS current from −0.5 to 0.0 V at which the ECP‐Magenta film is charge neutral (Figure S7 b). The decrease in current seen at potentials less than 0 V indicates that neutral ECP‐Magenta blocks ion transport into the PEDOT:PSS electrode and, therefore, only a portion of the film is electrochemically accessed.
Figure 3.
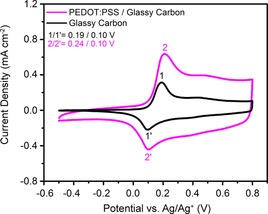
a) CVs of ECP‐Magenta on glassy carbon and on PEDOT:PSS/glassy carbon working electrodes at 20 mV s−1 in 0.5 m TBAPF6/PC. The anodic and cathodic redox peaks for ECP‐Magenta are labeled 1/1′ for the glassy carbon electrode and 2/2′ for the PEDOT:PSS/glassy carbon electrode.
ECP‐Magenta electrochromism on TW electrodes
Having established the ability of PEDOT:PSS to support the redox‐switching of ECP‐Magenta when deposited on a small‐area glassy carbon electrode (0.07 cm2), we proceeded to investigate the feasibility of using PEDOT:PSS as the sole conducting electrode on TW substrates (6 cm2). The CVs of ECP‐Magenta on PEDOT:PSS/TW (70±30 Ω/sq) and on an ITO/glass electrode with an equivalent sheet resistance (70 Ω/sq) are shown in Figure 4 a. The anodic oxidation peaks from ECP‐Magenta are found at the same potential on both the ITO and PEDOT:PSS/TW electrodes, whereas the reduction peak is shifted cathodically (−60 mV) on PEDOT:PSS/TW. This similarity in redox peaks between electrodes highlights that PEDOT:PSS supports sufficient lateral electron transport to oxidize/reduce the larger‐area ECP‐Magenta film on reasonable time scales. The higher current recorded on the PEDOT:PSS/TW electrodes is attributed to a contribution from the pseudocapacitance of PEDOT:PSS as observed previously in Figure 3 c. The charge stored in the ECP‐Magenta working electrode is 1.7 mC cm−2 for ITO and 3.3 mC cm−2 for PEDOT:PSS/TW. This enhancement in capacity for the PEDOT:PSS/TW electrode is even larger than that for a PEDOT:PSS/glass electrode (2.8 mC cm−2; Figure S8 a) because of the slightly larger surface area caused by the roughness on the TW substrate.
Figure 4.
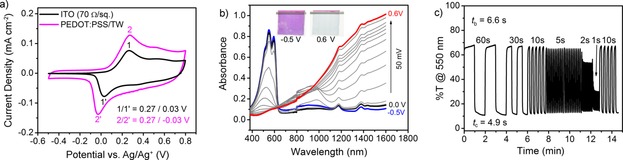
a) CVs of ECP‐Magenta films on PEDOT:PSS/TW and ITO/glass electrodes measured at 20 mV s−1. b) Stepwise spectral change upon the electrochemical oxidation of ECP‐Magenta films from −0.5 to 0.6 V. Inset photographs show the ECP‐Magenta film in its neutral (−0.5 V) and oxidized (0.6 V) state. c) Film switching kinetics for potential square‐wave pulse times ranging from 60 to 1 s. t b and t c to reach 95 % of full contrast are labeled.
Examining the spectral change upon oxidation of ECP‐Magenta on PEDOT:PSS/TW, a strong visible absorption from the neutral polymer is observed at potentials below 0 V. At higher potentials, the polymer is oxidized to form radical cation species at 0.1 V, which are converted to broadly absorbing dication charge carriers above 0.6 V versus Ag/Ag+ (Figure 4 b). This spectral change upon electrochemical oxidation is similar to the behavior seen on ITO electrodes (Figure S9 a) with matching contrasts of 68 Δ%T. For comparison, the ECP‐Magenta films on PEDOT:PSS/glass have a similar contrast of 66 Δ%T (Figure S8 b). These transitions to radical cation and dication charge‐carrier bands occur at the same potentials for PEDOT:PSS/TW compared to ITO and PEDOT:PSS/glass electrodes. Although PEDOT:PSS exhibits its own electrochromism in this potential range, the use of thin PEDOT:PSS films makes this change nearly imperceptible. The PEDOT:PSS absorbance change between −0.5 and 0.8 V is 0.06 (9 %T; Figure S10).
The switching kinetics of ECP‐Magenta were evaluated by applying potential square‐wave pulses between −0.5 and 0.6 V for durations ranging from 60 to 1 s (Figure 4 c). Switching times are quantified by calculating the time required to reach 95 % of the full contrast upon both bleaching (t b) and coloring (t c). The ECP‐Magenta film on PEDOT:PSS/TW electrodes bleaches in 6.6 s and recolors in 4.9 s. This switching speed is slightly slower than the switching speed on PEDOT:PSS/glass (t b=4.1 s, t c=1.9 s; Figure S8 c) and on ITO glass (t b=1.7 s, t c=3.7 s; Figure S9 b). For both glass and TW substrates, the PEDOT:PSS electrodes support faster coloration than bleaching, whereas the trend is opposite on ITO. This behavior can be explained considering the EIS results presented in Figure 2 d and Table S1. Unlike ITO, which is electrochemically inactive in the potential window required to switch the ECP, PEDOT:PSS exhibits small changes in capacitance and diffusional resistance. As the ECP‐Magenta is bleached, electrons removed from the film are injected into the PEDOT:PSS electrode. The injection of electrons temporarily reduces a portion of the PEDOT+, resulting in both the compaction of the film and hindered charge transport through the electrode as shown by the EIS results. In the opposite process in which the ECP is recolored, electrons are transferred from the PEDOT:PSS electrode into the ECP film, and the highly conducting, fast‐ion‐diffusion properties of the PEDOT:PSS are maintained.
TW electrochromic devices
Electrochromic devices (4×2.5 cm) were assembled by sandwiching gel electrolyte between two PEDOT:PSS/TW electrodes, one coated with ECP‐Magenta as the working electrode and the other coated with MCCP as the counter electrode. MCCP was used for the counter electrode of the device as a charge‐storage layer that remains colorless in both its neutral and oxidized states. When constructing polymer‐based ECDs, one of the electrodes is usually oxidized prior to ECD assembly, setting the opposite charged state to achieve full device contrast.60 For the ECDs based on PEDOT:PSS/TW electrodes, however, no preoxidation was required, which highlights an advantage of using PEDOT:PSS as an electrode material. This phenomenon is likely a combined result of the partial oxidation of MCCP to a radical cation species seen after spray coating on the PEDOT:PSS (Figure S11) with the charge‐storage capacity of the PEDOT:PSS able to compensate the charge and discharge of the electrochromic polymer.
TW ECDs provided a vibrant magenta‐to‐colorless switch (ΔE*=43.4) between −0.5 to 0.8 V. The spectra showing the transition from the magenta state at −0.5 V to the transmissive state at 0.8 V are shown in Figure 5 a. The device contrast is 38 Δ%T at 550 nm compared to 46 Δ%T for devices constructed with PEDOT:PSS/glass electrodes. The higher contrast of the glass‐based devices is attributed to the higher transmittance of the substrate and not to a more complete bleaching of the ECP. At 0.8 V, the TW ECD exhibits an even transmittance of 48 % from 420 to 600 nm with a minor decrease to 42 % from 650 to 780 nm. This constant transmittance over visible wavelengths makes the TW ECD exceptionally color neutral at 0.8 V (L*=75, a*=−0.8, b*=−0.2). Glass‐based ECDs required a higher driving voltage of 1 V to reach the transmissive state. At this higher voltage, the counter electrode becomes reduced and, therefore, contributes more of a blue hue to the device (L*=75, a*=−2.6, b*=−4.5). This color is evident from the spectra shown in Figure S12 a in which the transmittance at 1 V increases from 37 % at 780 nm to 60 % at 420 nm.
Figure 5.
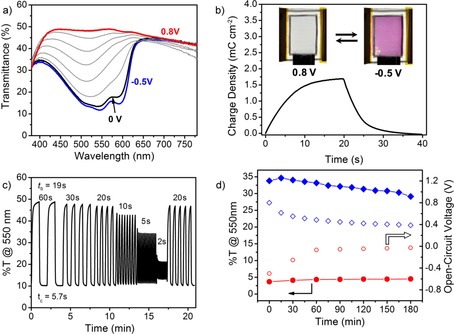
a) Stepwise spectral change of the magenta‐to‐clear TW ECD upon oxidation from −0.5 to 0.8 V. b) Charge density versus time for 20 s potential square‐wave pulses. Photographs of the device in its colorless (0.8 V) and colored (−0.5 V) states. c) Device transmittance at 550 nm for potential square‐wave pulses of 60 to 2 s. Coloration and beaching times are labeled. d) Optical memory for a 3 cm2 device constructed under an inert atmosphere showing the evolution of transmittance (filled points) and V OC (open points) in both the colored and bleached state.
We determined the charge required to switch the device by cycling the TW ECD between its extreme states at 0.8 and −0.5 V (Figure 5 b). As 0.8 V is applied to the device for 20 s, 1.7 mC cm−2 (0.47 μAh cm−2) is accumulated during the oxidation of the ECP‐Magenta working electrode and the device reaches its colorless form (inset image). The application of −0.5 V reduces the ECP‐Magenta working electrode and results in the recoloring of the device. This small charge required for device switching suggests that a standard 1500 mAh smartphone battery could power this 10 cm2 ECD for 300 000 cycles. The low energy requirement of 3 mWh m−2 and a maximum power of 2 Wm−2 further highlight the energy efficiency of these TW devices. The glass‐based ECDs are even more energy efficient for which a charge of 0.8 mC cm−2 (0.2 mAh m−2) is required for switching because of the lower amount of PEDOT:PSS on the smooth glass substrates (Figure S12 b).
The TW ECDs also show moderate switching speeds of t b=19 s and t c=5.7 s for 95 % of maximum contrast (Figure 5 c). These switching speeds match those of PEDOT:PSS/glass‐based ECDs (t b=21 s, t c=4.4 s; Figure S12 c). Although slower than state‐of‐the‐art electrochromic devices constructed on ITO/glass (<1 s, ≈2 cm2), these switching speeds are suitable for semistatic smart window/roof applications for which larger‐area devices (10–1000 cm2) currently exhibit switching times on the order of tens of seconds to several minutes.61
Polymer‐based ECDs also exhibit an optical memory where the bleached and colored states can be maintained without power consumption. This effect is attributed to the ability of conducting polymers to stabilize charge carriers in their oxidized, colorless state and their sufficiently high oxidation potentials that stabilize the colored state in air. To determine the optical memory, TW ECDs were constructed under an inert atmosphere and sealed to limit the amount of oxygen present in the electrolyte. The optical memory was tested by setting the device in its fully bleached or colored state and then removing the power input and measuring the change in transmittance as well as the open‐circuit voltage (V OC) over 3 h. The evolution of the transmittance at 550 nm with time for both optical states is shown in Figure 5 d. The transmittance decreases from 35 to 29 % for the bleached state and increases from 3.7 to 4.5 % for the colored state over 3 h. In comparison, the glass‐based ECD shows a comparable optical memory for both the bleached state (47–42 %) and the colored state (9.6–10.2 %) as shown in Figure S12 d.
The open data points in Figure 5 d show the change in V OC over the 3 h experiment. For both the bleached and colored state, the V OC relaxes rapidly to 0.5 and −0.1 V after 60 min but then changes only minimally over the final 2 h. The final V OC for the device was 0.39 V for the bleached state and −0.03 V for the colored state. This stability of the V OC allows the TW ECD to be reset to either the fully bleached or fully colored state with a minimal amount of charge. Rebleaching the device requires 0.08 mC cm−2 (0.02 mAh m−2), and recoloring the device requires 0.2 mC cm−2 (0.05 mAh m−2). A similar trend is seen for the glass‐based ECDs for which V OC decreases from 1 to 0.49 V in the bleached state and from −0.5 to 0.06 V in the colored state after 3 h. In agreement with the charge‐to‐switch values above, the glass‐based device requires slightly less charge to reset the bleached state (0.05 mC cm−2) and the colored state (0.03 mC cm−2).
Ultimately, the color change in the device over the 3 h open‐circuit hold is minimal for both the TW ECD and the glass‐based ECD (Figure S13). After 30 min, ΔE*=2.3 for the TW device and ΔE*=5.3 for the glass‐based device were obtained. This difference is owed to the initial color that is bluer for the glass‐based device as discussed above. After 3 h, a color change of ΔE*=7.8 was observed for the TW device compared to ΔE*=9.5 for the glass‐based device (Figure S13 a). Although there is a perceptible color change after 3 h, the bleached state of the device is largely maintained as shown in the photographs in Figure S13 c and d. The colored state of the devices has a better optical memory, and a very minimal color change is observed in the device over the 3 h test (ΔE*=4.9 for TW and ΔE*=1.7 for glass; Figure S13 e and f). Furthermore, the small amount of charge required to refresh each state (0.08 mC cm−2) means that the applying refresh pulses every 3 h for 21 h (7 pulses) would consume the same energy as one full switch between states. This optical memory further demonstrates the energy efficiency of these devices for smart window/roofing applications.
Conclusions
This work demonstrates the successful construction of electrochromic devices on transparent wood (TW) substrates. TW shows a great potential to replace glass for smart windows because of its high strength (140 MPa), excellent work of fracture (3.2 MJ m−3, 30 times higher than glass), low thermal conductivity (0.23 W mK−1), and diffuse transmittance (80 %T and haze of 70 %). Poly(3,4‐ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) treated with 1 m p‐toluene sulfonic acid/ethylene glycol demonstrated a conductivity of 1200 S cm−1 and a retention of this high conductivity and electroactivity over the potential window of interest: −0.6 to 0.8 V versus Ag/Ag+. Treated PEDOT:PSS is, therefore, a suitable electrode material for a vast range of cathodically coloring electrochromic polymers. A magenta‐to‐clear electrochromic polymer exhibits a nearly identical redox response on PEDOT:PSS/TW as on ITO (−0.5 to 0.8 V). Magenta‐to‐clear TW electrochromic devices demonstrated a contrast of 38 Δ%T (ΔE*=43.2) and a highly color neutral bleached state without the need for chemical or electrochemical preoxidation. The low driving voltage, optical memory, and the small charge required to switch the device between its colored and bleached states (1.7 mC cm−2, 0.02 μAh m−2) highlight the possibility to use these devices for energy‐saving, smart windows. Further advances in water‐processable electrochromic polymers will both expand the potential colors for TW electrochromic devices and allow more environmentally benign device fabrication and long‐term stability.
Experimental Section
Materials
Sodium chlorite (NaClO2) from Sigma–Aldrich was used for delignification to prepare TW. The wood used here was birch, bought from Glimakra of Sweden AB. Methyl methacrylate (MMA) was purchased from Sigma–Aldrich for the synthesis of infiltrated polymer using AIBN (Sigma–Aldrich) as the initiator. PEDOT:PSS ink was prepared by mixing Clevios PH1000 (Heraeus) with Triton‐X100 at 0.5 wt %. A solution of 1 m pTSA monohydrate (Alfa Aesar, 98 %) dissolved in EG (Sigma Aldrich, 99.8 %) was used as the conductivity enhancer. Tetrabutylammonium hexafluorophosphate (TBAPF6) was purchased from Acros Organics and recrystallized from hot ethanol. Acetonitrile (ACN) and propylene carbonate (PC) were purified using a solvent purification system from Vac atmospheres. Poly[2,2‐bis(2‐ethylhexyloxymethyl)‐propylene‐1,3‐dioxy)‐3,4‐thiophene‐2,5‐diyl] (ECP‐Magenta) was synthesized by direct (hetero)arylation polymerization (number‐average molecular weight M n=15 kDa, Dispersity (Đ)=1.5), and poly(3,4‐propylene dioxypyrrole‐C18) was prepared by dehalogenative polycondensation (M n=95.6 kDa, Đ=1.8; Figure S1) according to previously published procedures.62, 63 Toluene (99.5 %, Fischer Scientific) was used to dissolve the polymers at 5 mg mL−1. TBAPF6 dissolved in PC (0.5 m) was used as a liquid electrolyte for film measurements with the addition 6 wt % of PMMA (Sigma Aldrich; weight‐average molecular weight M w=996 kDa) dissolved in 0.5 m TBAPF6/PC for the device gel electrolyte. The Ag/Ag+ reference electrode was built using a 10 mm AgNO3/0.5 m TBAPF6/ACN (Acros Organics) solution in the body with a silver wire and calibrated at 73 mV versus Fc/Fc+ (Fc=ferrocene). VHB tape was purchased from 3m and used as a gasket material for the electrochromic devices.
Mechanical properties of TW substrates
TW was prepared according to previously reported methods.1 Birch wood with a thickness of 0.6 mm was first delignified using 1 wt % of NaClO2 at 80 °C in acetate buffer solution (pH 4.6) until the wood became white. The delignified samples were then washed with deionized water and dehydrated with ethanol and acetone sequentially. Finally, TW was obtained by vacuum‐infiltrating prepolymerized MMA and polymerization in an oven at 70 °C for 4 h. MMA prepolymerization was conducted at 75 °C for 15 min using 0.3 wt % AIBN as the initiator. Three‐point bending mechanical tests of the TW and glass were performed by using an Instron 5944 with a 500 N load cell. The tests were performed at a 10 % min−1 strain rate and a span of 30 mm. All samples were cut into strips (5 mm×60 mm) for testing.
Characterization of PEDOT:PSS films
To evaluate the redox properties of PEDOT:PSS, films of approximately 2 μm in thickness were prepared on glassy‐carbon button electrodes (0.07 cm2). The PH1000 ink (2 μL) was drop cast onto the glassy carbon and dried at 50 °C in a vacuum oven for 1 h to remove excess water. Films were treated by immersion into 1 m pTSA/EG (RT, 5 min) followed by three rinses with deionized water and subsequent drying at 60 °C. CV and EIS were performed in a three‐electrode cell (Ag/Ag+ reference electrode, platinum‐flag counter electrode) by using a Gamry Reference 3000 Potentiostat/Galvanostat controlled by the Gamry Framework software. EIS spectra were recorded at various DC potentials versus Ag/Ag+ from 100 kHz to 0.1 Hz with a 5 mV excitation signal and were fitted using the ZView software. The film thickness was estimated to be (590±70) nm measured by using a Bruker DektakXT profilometer. At least three samples for each treatment condition were prepared to ensure reproducibility.
In situ conductance measurements were performed by using an Au interdigitated microelectrode with 50 digits, 5 μm in width, and 5 μm separation as the working electrode in a four‐electrode cell with a Ag/Ag+ reference and a Pt flag counter electrode. PH1000 ink (0.5 μL) was dropcast onto the interdigitated electrode area and dried at 50 °C under vacuum for 1 h. For this measurement, a Pine Bipotentiostat controlled by the Aftermath software was used to hold one of the working electrodes at a given potential while cycling the second working electrode ±0.05 V at 0.5 mV s−1.46, 64 The slope of the resulting current density versus potential (I–V) curve was used to determine the film conductance and normalized to the film cross‐sectional area and width to calculate the in situ conductivity.
Solid‐state sheet resistance was measured by using a Keithley 2400 source meter and four‐point probe using a van der Pauw geometry. The PEDOT:PSS/TW electrode transmittance was measured by using a Cary 5000 UV/Vis‐NIR instrument with an integrating sphere attachment (DRA 2500). Transmittance measurements were made with the sample in a diffuse/normal geometry, regular component included (di:0°) as recommended by the International Commission on Illumination (CIE).65 Colorimetric values (L*,a*,b*) were calculated from recorded spectra using the standard observer functions with a D50 illuminant.
Electrochromic film measurements
PEDOT:PSS electrodes were prepared on TW substrates by blade coating of the PH1000 ink (0.5 wt % Triton‐X100) followed by film post treatment as described above and were contacted with Cu tape. The blade‐coating parameters were: 400 μm gap height, 8 mm s−1 coating speed, and 5 μL cm−2 drop volume at 40 °C. ECP‐Magenta films were spray cast onto the PEDOT:PSS/TW electrode using an airbrush spray gun pressurized with nitrogen. ECP‐Magenta films were characterized on PEDOT:PSS electrodes by using CV and UV/Vis‐NIR spectroscopy (di:0° geometry) while holding the film at defined potentials in a three‐electrode cell (Ag/Ag+ reference, Pt‐flag counter electrode). To evaluate the switching time, ECP‐Magenta films were characterized by using chronoabsorptometry in which the film transmittance was monitored at 550 nm (ECP‐Magenta λ max) while the film was switched repeatedly between 0.6 and −0.5 V versus Ag/Ag+ at various pulse lengths by using a EG&G PAR 273 potentiostat/galvanostat controlled by the Corrware software. Switching speeds were evaluated by considering the time to reach 95 % of the maximum contrast upon coloring (t c) or bleaching (t b). Photographs were taken in a light booth with calibrated D50 lighting by using a Nikon D90 Digital SLR camera and are reported without further manipulation beyond cropping.
Electrochromic device assembly and characterization
Magenta‐to‐clear ECDs were fabricated using PEDOT:PSS electrodes prepared as described using either TW or glass substrates. ECP‐Magenta was spray cast onto the working electrode to an absorbance of ≈0.8 whereas MCCP was spray cast onto the counter electrode to an absorbance of ≈0.4 for correct charge balancing. The ECP chemical structures are shown in Figure S3. Film thicknesses were monitored by airbrush spraying onto a glass slide adjacent to the electrode and measured at their maximum wavelength of absorption (550 nm for ECP‐Magenta and 310 nm for MCCP). The active area of the device was defined by using a VHB tape in a double gasket configuration. The ECP‐Magenta‐ and MCCP‐coated substrates were then sandwiched together with the gel electrolyte. To evaluate the device optical memory under ideal conditions, smaller 3 cm2 devices were prepared in a glovebox with an Ar atmosphere and sealed by using an ADCO HelioSeal PVS‐101a edge sealant. Potential square‐wave measurements were recorded by using a EG&G PAR 273 potentiostat (Corrware control). Spectral stepwise oxidation and device switching kinetics were evaluated as described above. The optical memory was evaluated by applying 0.8 or −0.5 V to the device for 30 s and holding the device at open circuit. Spectra were recorded every 15 min in the bleached state and every 30 min in the colored state. Colorimetric values and color difference were calculated as described above.
Conflict of interest
Electrochromic polymers used in this project are licensed by NXN Licensing.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support of this work is from the Renewable Bioproducts Institute at the Georgia Institute of Technology. The authors thank BASF for the synthesis of the electrochromic polymers used in this study. L.B. acknowledges funding from the European Research Council Advanced Grant No. 742733, Wood NanoTech.
A. W. Lang, Y. Li, M. De Keersmaecker, D. E. Shen, A. M. Österholm, L. Berglund, J. R. Reynolds, ChemSusChem 2018, 11, 854.
Contributor Information
Prof. Lars Berglund, Email: blund@kth.se.
Prof. John R. Reynolds, Email: reynolds@chemistry.gatech.edu.
References
- 1. Li Y., Fu Q., Yu S., Yan M., Berglund L., Biomacromolecules 2016, 17, 1358. [DOI] [PubMed] [Google Scholar]
- 2. Zhu M., Song J., Li T., Gong A., Wang Y., Dai J., Yao Y., Luo W., Henderson D., Hu L., Adv. Mater. 2016, 28, 5181. [DOI] [PubMed] [Google Scholar]
- 3. Li T., Zhu M., Yang Z., Song J., Dai J., Yao Y., Luo W., Pastel G., Yang B., Hu L., Adv. Energy Mater. 2016, 6, 1601122. [Google Scholar]
- 4. Li Y., Fu Q., Rojas R., Yan M., Lawoko M., Berglund L., ChemSusChem 2017, 17, 3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li Y., Yu S., Veinot J. G. C., Linnros J., Berglund L., Sychugov I., Adv. Opt. Mater. 2017, 5, 1600834. [Google Scholar]
- 6. Vasileva E., Li Y., Sychugov I., Mensi M., Berglund L., Popov S., Adv. Opt. Mater. 2017, 5, 1700057. [Google Scholar]
- 7. Yu Z., Yao Y., Yao J., Zhang L., Chen Z., Gao Y., Luo H., J. Mater. Chem. A 2017, 5, 6019. [Google Scholar]
- 8. Gan W., Xiao S., Gao L., Gao R., Li J., Zhan X., ACS Sustainable Chem. Eng. 2017, 5, 3855. [Google Scholar]
- 9. Wan J., Song J., Yang Z., Kirsch D., Jia C., Xu R., Dai J., Zhu M., Xu L., Chen C., Wang Y., Wang Y., Hitz E., Lacey S. D., Li Y., Yang B., Hu L., Adv. Mater. 2017, 29, 1703331. [DOI] [PubMed] [Google Scholar]
- 10. Chen C., Zhang Y., Li Y., Dai J., Song J., Yao Y., Gong Y., Kierzewski I., Xie J., Hu L., Energy Environ. Sci. 2017, 10, 538. [Google Scholar]
- 11. Granqvist C. G., Thin Solid Films 2014, 564, 1. [Google Scholar]
- 12. Cai G., Wang J., Lee P. S., Acc. Chem. Res. 2016, 49, 1469. [DOI] [PubMed] [Google Scholar]
- 13. Mortimer R. J., Annu. Rev. Mater. Res. 2011, 41, 241. [Google Scholar]
- 14. Groenendaal L., Zotti G., Aubert P. H., Waybright S. M., Reynolds J. R., Adv. Mater. 2003, 15, 855. [Google Scholar]
- 15. Franke E. B., Trimble C. L., Hale J. S., J. Appl. Phys. 2000, 88, 5777. [Google Scholar]
- 16. Dyer A. L., Grenier C. R. G., Reynolds J. R., Adv. Funct. Mater. 2007, 17, 1480. [Google Scholar]
- 17. Dyer A. L., Thompson E. J., Reynolds J. R., ACS Appl. Mater. Interfaces 2011, 3, 1787. [DOI] [PubMed] [Google Scholar]
- 18. Bulloch R. H., Kerszulis J. A., Dyer A. L., Reynolds J. R., ACS Appl. Mater. Interfaces 2015, 7, 1406. [DOI] [PubMed] [Google Scholar]
- 19. Österholm A. M., Shen D. E., Gottfried D. S., Reynolds J. R., Adv. Mater. Technol. 2016, 1, 1600063. [Google Scholar]
- 20. Argun A. A., Aubert P.-H., Thompson B. C., Schwendeman I., Gaupp C. L., Hwang J., Pinto N. J., Tanner D. B., MacDiarmid A. G., Reynolds J. R., Chem. Mater. 2004, 16, 4401. [Google Scholar]
- 21. J. F. Ponder, Jr. , Österholm A. M., Reynolds J. R., Chem. Mater. 2017, 29, 4385. [Google Scholar]
- 22. Bulloch R. H., Reynolds J. R., J. Mater. Chem. C 2016, 4, 603. [Google Scholar]
- 23. Shi P., Amb C. M., Dyer A. L., Reynolds J. R., ACS Appl. Mater. Interfaces 2012, 4, 6512. [DOI] [PubMed] [Google Scholar]
- 24. Llordés A., Wang Y., Fernandez-Martinez A., Xiao P., Lee T., Poulain A., Zandi O., Saez Cabezas C. A., Henkelman G., Milliron D. J., Nat. Mater. 2016, 15, 1267. [DOI] [PubMed] [Google Scholar]
- 25. Jensen J., Krebs F. C., Adv. Mater. 2014, 26, 7231. [DOI] [PubMed] [Google Scholar]
- 26. Ye S., Rathmell A. R., Chen Z., Stewart I. E., Wiley B. J., Adv. Mater. 2014, 26, 6670. [DOI] [PubMed] [Google Scholar]
- 27. Preston C., Fang Z., Murray J., Zhu H., Dai J., Munday J. N., Hu L., J. Mater. Chem. C 2014, 2, 1248. [Google Scholar]
- 28. Wu H., Kong D., Ruan Z., Hsu P.-C., Wang S., Yu Z., Carney T. J., Hu L., Fan S., Cui Y., Nat. Nanotechnol. 2013, 8, 421. [DOI] [PubMed] [Google Scholar]
- 29. Schneider J., Rohner P., Thureja D., Schmid M., Galliker P., Poulikakos D., Adv. Funct. Mater. 2016, 26, 833. [Google Scholar]
- 30. Wu Z., Chen Z., Du X., Logan J. M., Sippel J., Nikolou M., Kamaras K., Reynolds J. R., Tanner D. B., Hebard A. F., Rinzler A. G., Science 2004, 305, 1273. [DOI] [PubMed] [Google Scholar]
- 31. Gueye M. N., Carella A., Massonnet N., Yvenou E., Brenet S., Faure-Vincent J., Pouget S., Rieutord F., Okuno H., Benayad A., Demadrille R., Simonato J.-P., Chem. Mater. 2016, 28, 3462. [Google Scholar]
- 32. Tai Q., Chen B., Guo F., Xu S., Hu H., Sebo B., Zhao X.-Z., ACS Nano 2011, 5, 3795. [DOI] [PubMed] [Google Scholar]
- 33. Argun A. A., Cirpan A., Reynolds J. R., Adv. Mater. 2003, 15, 1338. [Google Scholar]
- 34. Haacke G., J. Appl. Phys. 1976, 47, 4086. [Google Scholar]
- 35. De S., Coleman J. N., ACS Nano 2010, 4, 2713. [DOI] [PubMed] [Google Scholar]
- 36. Worfolk B. J., Andrews S. C., Park S., Reinspach J., Liu N., Toney M. F., Mannsfeld S. C. B., Bao Z., Proc. Natl. Acad. Sci. USA 2015, 112, 14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Y., Zhu C., Pfattner R., Yan H., Jin L., Chen S., Molina-Lopez F., Lissel F., Liu J., Rabiah N. I., Chen Z., Chung J. W., Linder C., Toney M. F., Murmann B., Bao Z., Sci. Adv. 2017, 3, e1602076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen R., Das S. R., Jeong C., Khan M. R., Janes D. B., Alam M. A., Adv. Funct. Mater. 2013, 23, 5150. [Google Scholar]
- 39. Song Y., Jiang Y., Shi L., Cao S., Feng X., Miao M., Fang J., Nanoscale 2015, 7, 13694. [DOI] [PubMed] [Google Scholar]
- 40. Guo Y., Otley M. T., Li M., Zhang X., Sinha S. K., Treich G. M., Sotzing G. A., ACS Appl. Mater. Interfaces 2016, 8, 26998. [DOI] [PubMed] [Google Scholar]
- 41. Ryan J. D., Mengistie D. A., Gabrielsson R., Lund A., Müller C., ACS Appl. Mater. Interfaces 2017, 9, 9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nardes A. M., Janssen R. A. J., Kemerink M., Adv. Funct. Mater. 2008, 18, 865. [Google Scholar]
- 43. Rivnay J., Inal S., Collins B. A., Sessolo M., Stavrinidou E., Strakosas X., Tassone C., Delongchamp D. M., Malliaras G. G., Nat. Commun. 2016, 7, 11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yoon S., Sohn S., Kwon J., Park J. A., Jung S., Thin Solid Films 2016, 607, 55. [Google Scholar]
- 45. Kim G.-H., Nat. Mater. 2013, 12, 719. [DOI] [PubMed] [Google Scholar]
- 46. Singh R., Tharion J., Murugan S., Kumar A., ACS Appl. Mater. Interfaces 2017, 9, 19427. [DOI] [PubMed] [Google Scholar]
- 47. Park H.-S., Ko S.-J., Park J.-S., Kim J. Y., Song H.-K., Sci. Rep. 2013, 3, 2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mukherjee S., Singh R., Gopinathan S., Murugan S., Gawali S., Saha B., Biswas J., Lodha S., Kumar A., ACS Appl. Mater. Interfaces 2014, 6, 17792. [DOI] [PubMed] [Google Scholar]
- 49. Walczak R. M., Reynolds J. R., Adv. Mater. 2006, 18, 1121. [Google Scholar]
- 50. J. F. Ponder, Jr. , Österholm A. M., Reynolds J. R., Macromolecules 2016, 49, 2106. [Google Scholar]
- 51. J. F. Ponder, Jr. , Pittelli S. L., Reynolds J. R., ACS Macro Lett. 2016, 5, 714. [DOI] [PubMed] [Google Scholar]
- 52. Bobacka J., Lewenstam A., Ivaska A., J. Electroanal. Chem. 2000, 489, 17. [Google Scholar]
- 53. Chen C.-H., Kine A., Nelson R. D., LaRue J. C., Synth. Met. 2015, 206, 106. [Google Scholar]
- 54. Cui X., Martin D. C., Sens. Actuators B 2003, 89, 92. [Google Scholar]
- 55. Barsoukov E., Macdonald J. R., Impedance Spectroscopy: Theory, Experiment, And Applications, Wiley-Blackwell, Chichester, 2005. [Google Scholar]
- 56. Hernández-Labrado G. R., Contreras-Donayre R. E., Collazos-Castro J. E., Polo J. L., J. Electroanal. Chem. 2011, 659, 201. [Google Scholar]
- 57. Otero T. F., Boyano I., J. Phys. Chem. B 2003, 107, 4269. [Google Scholar]
- 58. Palumbiny C. M., Liu F., Russell T. P., Hexemer A., Wang C., Müller-Buschbaum P., Adv. Mater. 2015, 27, 3391. [DOI] [PubMed] [Google Scholar]
- 59. Schanda J., Colorimetry: Understanding the CIE System, Wiley, New York, 2007. [Google Scholar]
- 60. Shen D. E., Österholm A. M., Reynolds J. R., J. Mater. Chem. C 2015, 3, 9715. [Google Scholar]
- 61. Baetens R., Jelle B. P., Gustavsen A., Sol. Energy Mater. Sol. Cells 2010, 94, 87. [Google Scholar]
- 62. Estrada L. A., Deininger J. J., Kamenov G. D., Reynolds J. R., ACS Macro Lett. 2013, 2, 869. [DOI] [PubMed] [Google Scholar]
- 63. Knott E. P., Craig M. R., Liu D. Y., Babiarz J. E., Dyer A. L., Reynolds J. R., J. Mater. Chem. 2012, 22, 4953. [Google Scholar]
- 64. Morvant M. C., Reynolds J. R., Synth. Met. 1998, 92, 57. [Google Scholar]
- 65. CIE 15: Technical Report: Colorimetry Commission Internationale de L′eclairage, Vienna, Austria, 2004, https://archive.org/details/gov.law.cie.15.2004.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary