

Abstract
In an effort to elucidate and engineer interactions in type II nonribosomal peptide synthetases, we analyzed biomolecular recognition between the essential peptidyl carrier proteins and adenylation domains using NMR spectroscopy, molecular dynamics, and mutational studies. Three peptidyl carrier proteins, PigG, PltL, and RedO, in addition to their cognate adenylation domains, PigI, PltF, and RedM, were investigated for their cross species activity. Of the three peptidyl carrier proteins, only PigG, showed substantial cross-pathway activity. Characterization of the novel NMR solution structure of holo-PigG and molecular dynamic simulations of holo-PltL and holo-PigG revealed differences in structures and dynamics of these carrier proteins. NMR titration experiments revealed perturbations of the chemical shifts of the loop 1 residues of these peptidyl carrier proteins upon their interaction with adenylation domain. These experiments revealed a key region for the protein-protein interaction. Mutational studies supported the role of loop 1 in molecular recognition, as mutations to this region of the PCPs significantly modulated their activities.
Graphical Abstract
Authors are required to submit a graphic entry for the Table of Contents (TOC) that, in conjunction with the manuscript title, should give the reader a representative idea of one of the following: A key structure, reaction, equation, concept, or theorem, etc., that is discussed in the manuscript. Consult the journal’s Instructions for Authors for TOC graphic specifications.
Type II non-ribosomal peptide synthetases (NRPSs) participate in hybrid biosynthetic pathways with fatty acid synthases, polyketide synthases, and type I NRPSs,1–3 producing complex natural products that often demonstrate valuable antimicrobial (pyoluteorin4, 5, vancomycin6, and kutznerides7, 8) and antitumor (prodiginines9, actinomycin10–13, and C-102714) activities. Type II NRPSs typically modify amino acids through oxidations, hydroxylations, and chlorinations,15–19 and these precursors provide unique features and diversity to natural products. Given their makeup of primarily stand-alone proteins, type II NRPSs are ideal targets for metabolic engineering efforts and study by solution-state NMR methods. Common to all NRPS pathways are the peptidyl carrier protein (PCP) and adenylation (A) domains; their activities are essential for incorporation of amino acids into the biosynthetic machinery. We and others have recently shown that PCPs sequester substrates and intermediates,2, 20 a feature likely to protect against substrate degradation until downstream enzymes become available for further chemical processing. The A domain facilitates covalent attachment of an amino acid to holo-PCP at the thiol terminus of 4′-phosphopantetheine (Figure 1B), a cofactor post-translationally attached to a conserved serine on the PCP.21, 22 Adenylation, using adenosine triphosphate (ATP) as a second substrate, serves as the sole entry for amino acids into NRPS pathways. Loading of PltL and PigG occurs by a two-step process: formation of prolyl adenylate from proline and ATP followed by transfer of the prolyl moiety to the PCP via aminoacylation of terminal thiol of phosphopantetheine. Crucially, understanding the recognition surfaces of the PCP and A domain interactions may allow for control of substrate incorporation into products.
Figure 1.
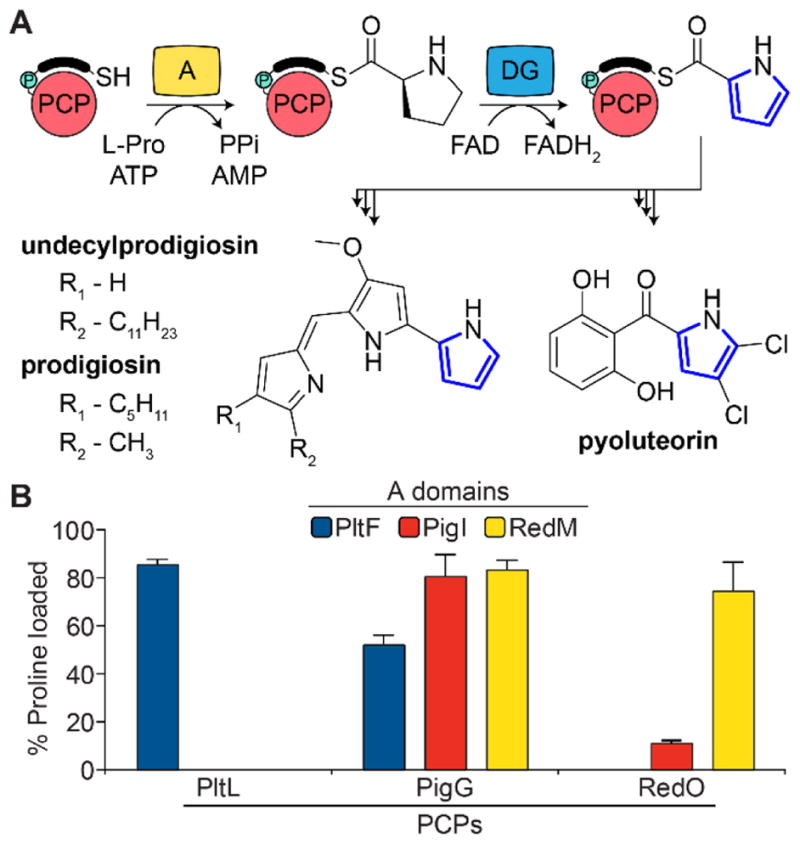
(A) Pyrrole formation in pyoluteorin and prodiginine biosynthesis. PCP, peptidyl carrier protein; A, adenylation domain; DG, dehydrogenase. The black bar and teal circle above the PCP denotes phosphopantetheine. (B) A domain activity with cognate and non-cognate PCPs.
Recent crystal structures of bound PCP•A domains have revealed the interfaces of these complexes,23–26 yet they cannot provide insights into the dynamic interactions that control the binding event. We have therefore chosen to compare the interactions of homologous type II PCP•A domains that catalyze an identical transformation: the incorporation of L-Pro in the first steps of prodigiosin, pyoluteorin, and undecylprodigiosin biosynthesis carried out by the PCP•A domain pairs PigG•PigI, PltL•PltF, and RedO•RedM, respectively (Figure 1A). Aminoacylation assays revealed different specificities between the homologous PCP•A domain partners. We utilize solution-phase NMR and MD simulations on the less homologous PltL and PigG PCPs to decipher the differences in their specificities. In addition, mutagenesis followed by activity assays were carried out on PltL and PigG in an attempt to modulate aminoacylation.
Previously, the homologous PCPs and A domains in the undecylprodigiosin and pyoluteorin pathways were shown to be specific for their cognate partner.15 We further examined the activity of PigG and PigI with these homologous pairs by monitoring the production of aminoacylated PCP product. Holo-PCP and A domain were incubated in the presence of proline and ATP for 5 min, then quenched with formic acid, and the substrate-loaded PCP species was observed by HPLC (Section C3 and Figure S1 of the Supporting Information). The assay confirmed that PltF and RedM loaded proline specifically onto their cognate PCPs, PltL and RedO, respectively (Figure 1B). Interestingly, RedO has slight recognition by PigI, while PigG was aminoacylated by all three A domains. The promiscuity of PigG is surprising, especially for PltF, considering the low seqgamuence identity (25.0%) between PigG and PltL. The identity between the prodiginine PCPs PigG and RedO is significantly higher (39.8%) (Figure S2 of the Supporting Information). The results reveal varying specificities between partners and provide a set of model systems to study the important interfaces for protein-protein recognition in type II NRPS systems. PigG’s observed promiscuity encouraged us to perform structural studies comparing PigG and the well-studied, non-prodiginine PCP PltL.
We began by determining the NMR solution structure of holo-PigG to facilitate comparisons with our previous holo-PltL solution-state NMR structure (Figure 2) (Figures S3–S5 and Table S1 of the Supporting Information). Previously reported, NMR solution structures of PltL in holo- and pyrrolyl forms demonstrated that, like type II fatty acid and polyketide carrier proteins,2, 27, 28 type II PCPs have the capacity to sequester their substrates. Holo-PigG and holo-PltL have similar structural features. Both possess a unique interruption in helix III,2 and the N-terminal portion of helix II in both PCPs has slight positive potential that could form electrostatic interactions with an A domain, as this PCP region is proximal to the A domain in recent crystal structures PCP-A didomains (Figure 2C–D).23–26 In contrast, loop 1 of these PCPs varies significantly between the two structures (Figure 2C–D). The loop 1 N-terminal region of PigG (residues 15–34) has a strong negative potential, while the same region of loop 1 of PltL (residues 19–40) has a weak positive potential. Interestingly in enterobactin biosynthesis, loop 1 of the PCP EntB is in close proximity to the EntE Asub domain, a flexible C-terminal region of the A domain that is important for PCP recognition (Figure S6 of the Supporting Information).23
Figure 2.

The solution NMR structures and electrostatic potentials (ESPs) of PCPs. (A,B) Strucutre of holo-PltL and holo-PigG. Structure color maps represent the CSP in Figure 3. (C,D) ESPs of holo-PltL (C) and holo-PigG (D). (E,F) Backbone (heavy atom) root mean square deviations of holo-PltL (E) and holo-PigG (F). Data from 5 independent cMD simulations are colored uniquely.
To compare the structural dynamic differences between the two PCPs, conventional MD (cMD) and Gaussian accelerated MD (GaMD) simulations were performed. Analysis of simulation data sets revealed significantly larger backbone heavy root mean square deviations (RSMDs) in holo-PigG indicating that PigG is less well-ordered than PltL (Figure 2E–F and Figures S7–S8 of the Supporting Information). The greater flexibility of holo-PigG may allow it to sample conformations that can be recognized by non-cognate adenylation domains.
NMR titration experiments were next performed for each PCP with the A domains PltF and PigI to inform the interface residues required for protein-protein recognition. To capture the productive interactions between carrier protein and A domain in the presence of the struc-turally important29 amino acid adenylate species, we performed titrations in the presence of ATP and proline but with methyl protection of the holo-PCP thiol to prevent aminoacylation. Therefore, the A domain will catalyse adenylation (prolyl-AMP formation), but not thioesterifcation (prolyl-PltL) during the titration. The S-methylated holo-PCP (mholo-PCP) was generated by synthesizing a methylated coenzyme A (CoA) (Figure S9 of the Supporting Information), which was subsequently covalently ligated to apo-PCP by the action of Sfp.21, 22 1H-15N HSQC spectra were collected using mholo-15N-PltL or mholo-15N-PigG solutions with PCP•A domain molar ratios ranging from 0 to 1.6 and the chemical shift perturbations (CSPs) and peak intensities of the assigned backbone peaks were calculated (Figure 3 and Figures S10–S11 of the Supporting Information). For PltL titrations, significant linear peak perturbations and peak intensity loss were observed as PltF concentrations in-creased, while no significant perturbations and intensity loss were observed when PigI was introduced (Figure 2A and 3A–C), indicating a lack of interaction with the non-cognate A domain. For PigG titrations, significant peak perturbations and intensity loss were observed as both PigI and PltF concentrations were increased (Figure 2B and 3D–E), which agrees with the PigG promiscuity seen in product formation assays (Figure 1B). In fact, PigG NMR signal was effectively lost when PltF exceeded 0.4 molar equivalence, suggesting that PigG has a significantly greater affinity for PltF than PigI. The NMR titrations coincide with the aminoacylation assay which indicate PltL is recognized solely by PltF and PigG is recognized by both PltF and PigI (Figure 1B). Interestingly, when both PltL and PigG interacted with an A domain, significant peak perturbations and intensity loss were observed in residues of loop 1 (Figures 2A–B and 3B–E). Furthermore, the “diminished” residues of PigG at 0.4 equivalence PltF were located in loop 1 (residues 20–21, 28–29, 32–33, 35) of PigG (Figure 3E and Figure S11 of the Supporting Information), an indication of binding of this region to the significantly larger A domain. The dramatic loss of PigG signal in presence of PltF is consistent with the decreased product formation by the noncognate A domain compared to PigI (Figure 1B); the greater affinity of PigG toward PltF may delay product release and reduce overall turnover. Further inspection of the simulation data with respect to the loops of these PCPs suggest subtle differences in their flexibilities. The largest loop fluctuations in the PltL occur at the beginning of loop 1; whereas the end of loop 1 undergoes greater fluctuations in PigG (Figure S12 of the Supporting Information). Given this observation, we next performed mutational analysis on the PCPs to further assess the importance of the loop 1 region.
Figure 3.
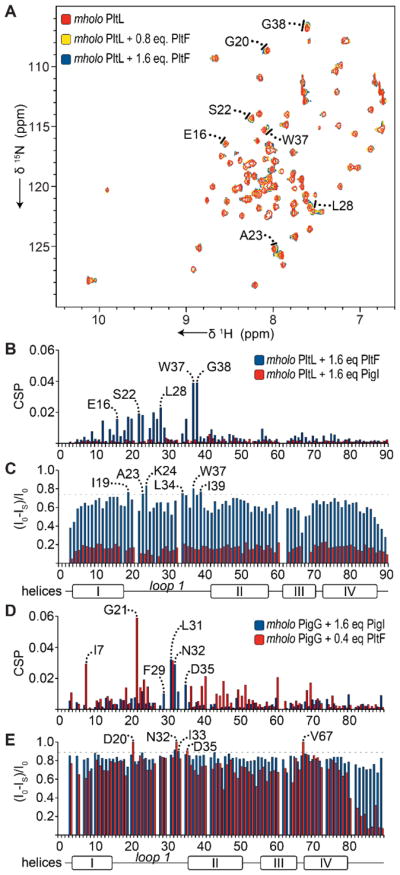
Perturbations of methylated holo- (mholo-) PigG and mholo-PltL due to inter-action with A domains. (A) 1H-15N HSQC overlays of mholo-15N-PltL with increasing PltF concentrations. (B,C) CSP plots (B) and intensity plots (C) of mholo-15N-PltL with PltF or PigI relative to mholo-15N-PltL alone. (D,E) CSP plots (D) and intensity plots (E) of mholo-15N-PigG with PigI or PltF relative to mholo-15N-PigG alone.
Based on the difference in dynamics of the PCPs and the activity in loop 1 in the presence of A domain, mutational studies were performed on the PCPs in three regions: residues on loop 1 (Figure 4, red), residues underneath loop 1 that interact with the region (Figure 4, blue), and residues on helix I and II that interact to hold the helices together via intramolecular interactions (Figure 4, orange). Focusing on polarity differences and length of the loop, residues of PigG were mutated to the corresponding residues on PltL in each region and vice-versa (Figure 4A and S13). Each mutant PCP species was analyzed in the aminoacylation assay with either PltF, PigI, or RedM to determine the influence of the different PCP regions. Mutant holo-PCP and A domain were incubated in the presence of proline and ATP for 30 min, then quenched with formic acid, and the substrate-loaded PCP species was observed by HPLC (Figure 4C and Figure S14 of the Supporting Information). The recognition of PigG mutant species 1 by all three A domains is significantly reduced compared to WT PigG and PigG mutant species 2 and 3. PltL mutant species did not significantly alter recognition with the A domains, although the PltL mutant species 1 did gain minimal function with RedM. The activities gained or lost for these mutants with the A domain illustrates the importance of loop 1 in the pyoluteorin and prodigiosin type II PCPs and highlights a region of carrier proteins that is critical to the specificities of molecular recognition in type II NRPSs.
Figure 4.

PCP loop 1 modification alters interactions with homologous A domains. Residues were mutated in loop 1 (red), underneath loop 1 (blue), and between helix I and II (orange). (A) Sequence alignment of PigG and PltL using MUSCLE.35 (B) holo-PigG NMR structure docked to model structure of PigI. Mutated residues are highlighted in red, blue, and orange and are explained in (C). (C) Aminoacylation assays with mutated PigG and either PigI or PltF. Astericks indicate PltL mutant 2 species was not analyzed due to instability.
The type II NRPS systems produce unique moieties in natural product biosynthesis, and their architecture is highly ammenable to the engineering of biosynthetic pathways. Here we demonstrated that type II PCPs in homologous pathways present different specificity profiles with regard to adenylation domain partners. Using NMR solution structures, MD simulations, and NMR titration experiments, loop 1 of these PCPs was identified as essential for recognition. These complement previous studies in the siderophore pathways,23, 30–32 demonstrating the significance of the loop 1 interface by altering A domain recognition in pyrrole biosynthesis. The PCP helix II surface is also considered to be a part of the PCP•A domain interface in other systems.23, 24 While our results indicate loop 1 is crucial for A domain recognition, smaller CSPs in helix II and higher mutation tolerance suggest helix II is less important than loop 1 in pyrrole PCP•A domain recognition. NRPS pathways rely on the fidelity of protein-protein interactions between each A domain and PCP cognate pair for the proper loading of starter units. A fundamental understanding of the interacttions between these partners should allow for engineering and eventual control over the identity of amino acids incorporated in NRPS pathways. The PCP loop 1 should also be considered when investigating other unique type II partners, including halogenases and cyclopropanases.33, 34 Control over PCP and partner enzyme recognition will contribute to the future engineering of carrier protein dependent pathways.
Supplementary Material
Acknowledgments
Funding was provided from NIH GM095970, GM31749, GM103426 and NIH/NCI T32 CA009523. We thank Drs. X. Huang and A. Mrse for NMR assistance, Dr. Y. Su for MS services, and the San Deigo Supercomputing Center (SDSC) for use of the Triton Shared Computing Cluster (TSCC).
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: XXXX
NMR structure was deposited to the PDB under 5JDX (holo-PigG)
References
- 1.Mantovani SM, Moore BS. J Am Chem Soc. 2013;135:18032–18035. doi: 10.1021/ja409520v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaremko MJ, Lee DJ, Opella SJ, Burkart MD. J Am Chem Soc. 2015;137:11546–11549. doi: 10.1021/jacs.5b04525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guenzi E, Galli G, Grgurina I, Gross DC, Grandi G. J Biol Chem. 1998;273:32857–32863. doi: 10.1074/jbc.273.49.32857. [DOI] [PubMed] [Google Scholar]
- 4.Hassan MN, Afghan S, Hafeez FY. Pest Manag Sci. 2011;67:1147–1154. doi: 10.1002/ps.2165. [DOI] [PubMed] [Google Scholar]
- 5.Howell C, Stipanovic R. Phytopathology. 1980;70:712–715. [Google Scholar]
- 6.Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, Kaplan SL, Karchmer AW, Levine DP, Murray BE, Rybak JM, Talan DA, Chambers HF. Clinical Infectious Diseases. 2011;52:285–292. doi: 10.1093/cid/cir034. [DOI] [PubMed] [Google Scholar]
- 7.Pohanka A, Menkis A, Levenfors J, Broberg A. J Nat Prod. 2006;69:1776–1781. doi: 10.1021/np0604331. [DOI] [PubMed] [Google Scholar]
- 8.Broberg A, Menkis A, Vasiliauskas R. J Nat Prod. 2006;69:97–102. doi: 10.1021/np050378g. [DOI] [PubMed] [Google Scholar]
- 9.Williamson NR, Fineran PC, Leeper FJ, Salmond GPC. Nature Rev Microbiol. 2006;4:887–899. doi: 10.1038/nrmicro1531. [DOI] [PubMed] [Google Scholar]
- 10.Khatua S, Nair CN, Ghosh K. J Pediatr Hematol/Oncol. 2004;26:777–779. doi: 10.1097/00043426-200411000-00020. [DOI] [PubMed] [Google Scholar]
- 11.Uberti EMH, Fajardo MdC, Ferreira SVVR, Pereira MV, Seger RC, Moreira MAR, Torres MD, de Nápoli G, Schmid H. Gynecol Oncol. 2009;115:476–481. doi: 10.1016/j.ygyno.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 12.Jaffe N, Paed D, Traggis D, Salian S, Cassady JR. Cancer. 1976;38:1925–1930. doi: 10.1002/1097-0142(197611)38:5<1925::aid-cncr2820380510>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 13.D’Angio GJ, Evans A, Breslow N, Beckwith B, Bishop H, Farewell V, Goodwin W, Leape L, Palmer N, Sinks L, Sutow W, Tefft M, Wolff J. Cancer. 1981;47:2302–2311. doi: 10.1002/1097-0142(19810501)47:9<2302::aid-cncr2820470933>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 14.Liu W, Christenson SD, Standage S, Shen B. Science. 2002;297:1170–1173. doi: 10.1126/science.1072110. [DOI] [PubMed] [Google Scholar]
- 15.Thomas MG, Burkart MD, Walsh CT. Chem Biol. 2002;9:171–184. doi: 10.1016/s1074-5521(02)00100-x. [DOI] [PubMed] [Google Scholar]
- 16.Galm U, Wendt-Pienkowski E, Wang L, George NP, Oh TJ, Yi F, Tao M, Coughlin JM, Shen B. Mol Biosyst. 2009;5:77–90. doi: 10.1039/b814075h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin S, Van Lanen SG, Shen B. J Am Chem Soc. 2008;130:6616–6623. doi: 10.1021/ja710601d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin S, Van Lanen SG, Shen B. J Am Chem Soc. 2007;129:12432–12438. doi: 10.1021/ja072311g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galonić DP, Vaillancourt FH, Walsh CT. J Am Chem Soc. 2006;128:3900–3901. doi: 10.1021/ja060151n. [DOI] [PubMed] [Google Scholar]
- 20.Goodrich AC, Frueh DP. Biochemistry. 2015;54:1154–1156. doi: 10.1021/bi501433r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mootz HD, Finking R, Marahiel MA. J Biol Chem. 2001;276:37289–37298. doi: 10.1074/jbc.M103556200. [DOI] [PubMed] [Google Scholar]
- 22.Beld J, Sonnenschein EC, Vickery CR, Noel JP, Burkart MD. Nat Prod Rep. 2014;31:61–108. doi: 10.1039/c3np70054b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sundlov Jesse A, Shi C, Wilson Daniel J, Aldrich Courtney C, Gulick Andrew M. Chem Biol. 2012;19:188–198. doi: 10.1016/j.chembiol.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitchell CA, Shi C, Aldrich CC, Gulick AM. Biochemistry. 2012;51:3252–3263. doi: 10.1021/bi300112e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reimer JM, Aloise MN, Harrison PM, Martin Schmeing T. Nature. 2016;529:239–242. doi: 10.1038/nature16503. [DOI] [PubMed] [Google Scholar]
- 26.Drake EJ, Miller BR, Shi C, Tarrasch JT, Sundlov JA, Leigh Allen C, Skiniotis G, Aldrich CC, Gulick AM. Nature. 2016;529:235–238. doi: 10.1038/nature16163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beld J, Cang H, Burkart MD. Angew Chem Int Ed. 2014;53:14456–14461. doi: 10.1002/anie.201408576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haushalter RW, Filipp FV, Ko KS, Yu R, Opella SJ, Burkart MD. ACS Chem Biol. 2011;6:413–418. doi: 10.1021/cb200004k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gulick AM. ACS Chem Biol. 2009;4:811–827. doi: 10.1021/cb900156h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marshall CG, Burkart MD, Meray RK, Walsh CT. Biochemistry. 2002;41:8429–8437. doi: 10.1021/bi0202575. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Z, Lai JR, Walsh CT. Proc Natl Acad Sci USA. 2007;104:11621–11626. doi: 10.1073/pnas.0705122104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goodrich AC, Harden BJ, Frueh DP. J Am Chem Soc. 2015;137:12100–12109. doi: 10.1021/jacs.5b07772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dorrestein PC, Yeh E, Garneau-Tsodikova S, Kelleher NL, Walsh CT. Proc Natl Acad Sci USA. 2005;102:13843–13848. doi: 10.1073/pnas.0506964102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vaillancourt FH, Yeh E, Vosburg DA, O’Connor SE, Walsh CT. Nature. 2005;436:1191–1194. doi: 10.1038/nature03797. [DOI] [PubMed] [Google Scholar]
- 35.Edgar RC. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.