

Abstract
Although there are changes in gene expression and alterations in neuronal density and afferent inputs in the forebrain of trisomic mouse models of Down syndrome (DS) and Alzheimer’s disease (AD), there is a lack of systematic assessments of gene expression and encoded proteins within individual vulnerable cell populations, precluding translational investigations at the molecular and cellular level. Further, no effective treatment exists to combat intellectual disability and basal forebrain cholinergic neurodegeneration seen in DS. To further our understanding of gene expression changes before and following cholinergic degeneration in a well-established mouse model of DS/AD, the Ts65Dn mouse, we assessed RNA expression levels from CA1 pyramidal neurons at two adult ages (~6 months of age and ~11 months of age) in both Ts65Dn and their normal disomic (2N) littermates. We further examined a therapeutic intervention, maternal choline supplementation (MCS), which has been previously shown to lessen dysfunction in spatial cognition and attention, and have protective effects on the survival of basal forebrain cholinergic neurons (BFCNs) in the Ts65Dn mouse model. Results indicate that MCS normalized expression of several genes in key gene ontology categories, including synaptic plasticity, calcium signaling, and AD-associated neurodegeneration related to amyloid-beta peptide (Aβ) clearance. Specifically, normalized expression levels were found for endothelin converting enzyme-2 (Ece2), insulin degrading enzyme (Ide), Dyrk1a, and calcium/calmodulin-dependent protein kinase II (Camk2a), among other relevant genes. Single population expression profiling of vulnerable CA1 pyramidal neurons indicates that MCS is a viable therapeutic for long-term reprogramming of key transcripts involved in neuronal signaling that are dysregulated in the trisomic mouse brain which have translational potential for DS and AD.
Keywords: hippocampus, laser capture microdissection, choline supplementation, microarray, trisomic
Introduction
Down syndrome (DS) is caused by the triplication of human chromosome 21 (HSA21) and results in systemic deficits (Esbensen, 2010). DS is also the primary genetic cause of intellectual disability (ID), with cognitive deficits in hippocampal-dependent learning and memory, attention, and language and communication skills (Chapman and Hesketh, 2000; Esbensen, 2010; Lott, 2012; Rachidi and Lopes, 2008, 2010). Overexpression of >550 genes and putative protein-encoding transcripts, including >160 known genes termed the ‘DS critical region’ is thought to cause the neurological and peripheral phenotype associated with DS (Sturgeon and Gardiner, 2011). Further complicating the DS phenotype, by the mid-third decade of life, DS individuals develop pathological changes associated with Alzheimer’s disease (AD), including amyloid-beta peptide (Aβ) containing senile plaques, neurofibrillary tangles (NFTs), degeneration of cholinergic basal forebrain (CBF) neurons, and early endosomal abnormalities (Cataldo et al., 2000; Hartley et al., 2015; Hartley et al., 2014; Lai and Williams, 1989; Leverenz and Raskind, 1998; Mann et al., 1986; Sendera et al., 2000; Wisniewski et al., 1985). In addition, degeneration of the CBF neuron projection system, which provides the major cholinergic innervation to the entire cortical mantle and the hippocampus (Mesulam et al., 1983; Mufson et al., 2008; Rye et al., 1984), found in DS, is similar to that observed in AD (Bierer et al., 1995; Leverenz and Raskind, 1998; Mann et al., 1986; Wisniewski et al., 1985).
Mouse models have been generated that recapitulate aspects of DS and AD for translational study. The well-established segmental trisomy Ts65Dn mouse model closely replicates human DS neuropathology (Cataldo et al., 2003; Davisson et al., 1993; Granholm et al., 2000; Holtzman et al., 1996; Salehi et al., 2006; Seo and Isacson, 2005). This model translocates a segment of mouse chromosome 16 (MMU16) and mouse chromosome 17 (MMU17) orthologous to HSA21 to <10% of the centromere of MMU17, creating a freely segregating chromosome (Akeson et al., 2001; Kahlem et al., 2004; Reeves et al., 1995) with a conservation of ~70% homology for >85 protein coding gene sequences, with over 250 genes and putative gene products triplicated (Davisson et al., 1993; Gardiner et al., 2003; Sturgeon and Gardiner, 2011). Ts65Dn mice mimic many key features of the human DS pathology, including hippocampal-dependent learning and memory deficits (Ash et al., 2014; Escorihuela et al., 1995; Hyde and Crnic, 2001; Reeves et al., 1995; Velazquez et al., 2013), attentional deficits (Moon et al., 2010; Powers et al., 2016, 2017), heightened emotionality (Driscoll et al., 2004; Moon et al., 2010), and hyperactivity (Cooper et al., 2001; Hunter et al., 2003; Reeves et al., 1995). Glutamatergic neurotransmission dysfunction, including impaired hippocampal long-term potentiation and enhanced long-term depression, has been observed in trisomic mice (Kaur et al., 2014; Kleschevnikov et al., 2004; Rueda et al., 2010; Siarey et al., 1999). Like their human DS counterparts, the Ts65Dn mice have intact basal forebrain cholinergic neurons (BFCNs) and septohippocampal organization at birth (Granholm et al., 2000; Holtzman et al., 1996; Hunter et al., 2003), but after 6 months of age (MO), Ts65Dn mice have significant BFCN and associated CA1 hippocampal degeneration, along with astrocytic hypertrophy, and microglial activation (Cooper et al., 2001; Granholm et al., 2000; Holtzman et al., 1996; Kelley et al., 2014b; Seo and Isacson, 2005). The septohippocampal circuit is vulnerable in both DS and AD, and Ts65Dn mice recapitulate aspects of this vulnerability through reduced hippocampal neurogenesis, synapse loss, and deficits in neuroplasticity (Belichenko et al., 2004, 2009; Granholm et al., 2003; Insausti et al., 1998; Kelley et al., 2014a, 2016; Kleschevnikov et al., 2012; Kurt et al., 2000).
Current therapeutics for DS and AD are largely ineffective and expensive, and none prevent the progression of the disease. A potential treatment modality originating from the study of mouse models of DS and AD is to supplement the maternal diet with additional choline during pregnancy and lactation (Strupp et al., 2016). Choline is an essential nutrient critical for several key developmental pathways in the brain, including the biosynthesis of acetylcholine, which regulates neuronal proliferation, differentiation, migration, plasticity and synapse formation (Strupp et al., 2016). In addition, choline is a key substrate of the phosphatidylethanolamine N-methyltransferase (PEMT) pathway, which generates phosphatidylcholine species, crucial elements in neuronal and non-neuronal lipid membranes (Lindblom and Oradd, 2009; Wang et al., 2016; Zeisel, 2010; Zeisel and Niculescu, 2006). Choline is also the primary dietary source of methyl groups, a principal driver of gene expression regulation through epigenetic programming (McGowan et al., 2008; Mehedint et al., 2010). Current dietary recommendations for choline, especially in regard to pregnant women, are likely to be inadequate for the high demand for choline during gestation (Caudill et al., 2017; Institute of Medicine, 1998; Strupp et al., 2016). Notably, maternal choline supplementation (MCS) has been demonstrated by our group to improve spatial cognition and attentional function, protect BFCNs, and normalize adult hippocampal neurogenesis in Ts65Dn offspring (Ash et al., 2014; Kelley et al., 2014a, 2016; Powers et al., 2016, 2017; Strupp et al., 2016; Velazquez et al., 2013). MCS treatment also increases choline acetyltransferase (ChAT) intensity in the hippocampus prior to BFCN degeneration in the Ts65Dn mouse model (Kelley et al., 2016). Further investigation of aging and MCS treatment shows an increase in ChAT intensity in 2N animals, but not in Ts65Dn mice when examined pre-BFCN degeneration (4-6 MO) and post-BFCN degeneration (14-18 MO), however this may be explained by the fact that unsupplemented Ts65Dn mice display septohippocampal degeneration during the aging process (Kelley et al., 2016). MCS has also been shown to protect the cholinergic system in a mouse model of amyloid-beta precursor protein (APP) overexpression (Mellott et al., 2017) and behavioral and morphologic benefits in several models of neurodevelopmental disorders (Bearer et al., 2015; Ross et al., 2016; Scremin et al., 2015; Stevens et al., 2008; Ward et al., 2008, 2009), indicating that the effects are broadly neuroprotective across several disease entities.
To understand mechanisms and signaling pathways underlying benefits of MCS at the molecular and cellular level within vulnerable neuronal populations in DS and AD, we examined gene expression changes in CA1 pyramidal neurons at two distinct timepoints, young (~6 MO; pre-BFCN degeneration) and middle-aged (~11 MO; post-BFCN degeneration) via single population expression profiling analysis. We postulate that cognitive improvement via MCS is reflected in select gene expression and septohippocampal signaling pathway changes in trisomic mice.
Materials and Methods
Mice and maternal dietary protocol
Animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the Nathan Kline Institute/NYU Langone Medical Center and were in full accordance with NIH guidelines. Breeder pairs (female Ts65Dn and male C57Bl/6J Eicher × C3H/HeSnJ F1 mice) were purchased from Jackson Laboratories (Bar Harbor, ME) and mated at Nathan Kline Institute, Orangeburg, NY. Upon arrival, breeder pairs were assigned to receive one of two choline-controlled experimental diets: i) control rodent diet containing 1.1 g/kg choline chloride (AIN-76A; Dyets Inc., Bethlehem, PA, or ii) choline-supplemented diet containing 5.0 g/kg choline chloride (AIN-76A; Dyets Inc.), as described previously (Kelley et al., 2014a; Powers et al., 2016, 2017). The choline-supplemented diet provides approximately 4.5 times the concentration of choline consumed by our control group, and is within the normal physiological range (Detopoulou et al., 2008). The control diet supplies an adequate level of choline, so the offspring are not choline deficient. Breeder pairs were provided ad libitum access to water and their assigned diets. Standard cages contained paper bedding and several objects for enrichment (e.g., plastic igloo, t-tube, and cotton square). Mice were maintained on a 12-hour light-dark cycle under temperature- and humidity-controlled conditions.
Tissue preparation
Pups born to choline supplemented (Ts65Dn+ and 2N+) or unsupplemented maternal choline (Ts65Dn and 2N) dams were weaned on postnatal day 21 and provided ad libitum access to water and the control diet. Tail clips were taken and genotyped as described by Duchon et al., (Duchon et al., 2011). Mice were aged to approximately 6 MO and 11 MO and sacrificed for brain tissue accession. Mice used in this study included: 2N+ n=11 (6 MO), n= 8 (11 MO); 2N n= 12 (6 MO), n= 7 (11 MO); Ts65Dn+ n= 16 (6 MO), n= 8 (11 MO); and Ts65Dn n=19 (6 MO), n= 6 (11 MO). The study used both male and female mice. Mice were given an overdose of ketamine and xylazine and perfused transcardially with ice-cold 4% paraformaldehyde buffered in 0.15 M phosphate buffer. Tissue blocks containing the dorsal hippocampus were paraffin embedded and 6 μm-thick tissue sections were cut in the coronal plane on a rotary microtome for immunocytochemistry as described previously (Alldred et al., 2012; Ginsberg, 2005a; Ginsberg, 2010). RNase-free precautions were employed, and solutions were made with 18.2 mega Ohm RNase-free water (Nanopure Diamond, Barnstead, Dubuque, IA). A second independent, mixed gender cohort of animals was perfused with 1M phosphate buffer and brains were prepared for CA1 region microdissections at both timepoints for qPCR validation. Flash frozen samples from 2N+, 2N, Ts65Dn+, and Ts65Dn (n=9) per timepoint were accrued for RNA extraction.
Single cell microaspiration and TC RNA amplification
Laser capture microdissection (LCM) and terminal continuation (TC) RNA amplification procedures have been described in detail previously by our group (Alldred et al., 2008, 2009, 2012; Che and Ginsberg, 2004; Ginsberg, 2008). Individual CA1 pyramidal neurons were microaspirated via LCM (Arcturus PixCell IIe, ThermoFisher, South San Francisco, CA). One hundred cells were captured per reaction for population cell analysis (Alldred et al., 2008; Ginsberg, 2010). Microarrays (containing 100 LCM-captured CA1 neurons each) were performed per mouse brain (range 2-5 times per mouse). The full TC RNA amplification protocol is available at http://cdr.rfmh.org/pages/ginsberglabpage.html. This method entails synthesizing first strand cDNA complementary to the RNA template, re-annealing the primers to the cDNA, and finally in vitro transcription using the synthesized cDNA as a template. Briefly, microaspirated CA1 neurons were homogenized in Trizol reagent (ThermoFisher, Carlsbad, CA), chloroform extracted, and precipitated (Alldred et al., 2009, 2012, 2015a, 2015b). RNAs were reverse transcribed and single-stranded cDNAs were then subjected to RNase H digestion and re-annealing of the primers to generate cDNAs with double-stranded regions at the primer interfaces. Single stranded cDNAs were digested and samples were purified by Vivaspin 500 columns (Sartorius Stedim Biotech, Goettingen, Germany). Hybridization probes were synthesized by in vitro transcription using 33P and radiolabeled TC RNA probes were hybridized to custom-designed cDNA arrays without further purification.
Microarray platforms and hybridization
Array platforms consist of 1 μg of linearized cDNA purified from plasmid preparations adhered to high-density nitrocellulose (Hybond XL, GE Healthcare, Piscataway, NJ) using an arrayer robot (VersArray, Bio-Rad, Hercules, CA) (Ginsberg, 2005b, 2008). Each cDNA and/or expressed sequence-tagged cDNA (EST) was verified by sequence analysis and restriction digestion. Mouse and human clones were employed on the custom-designed array. Approximately 576 cDNAs/ESTs were utilized for the younger cohort and approximately 649 genes were utilized for the older cohort, organized into 22 gene ontology (GO) groups (Table I). The older cohort of animals had additional genes newly available and/or implicated in DS/AD pathology. Additional genes were mainly added to 3 GO categories, protein phosphatases and kinases, stress response, and sirtuins (Table I). The majority of genes are represented by one transcript on the array platform, although the neurotrophin receptors TrkA, TrkB, and TrkC are represented by ESTs that contain the extracellular domain (ECD) as well as the tyrosine kinase domain (TK) (Ginsberg et al., 2006, 2010).
Table I.
Gene expression alterations by genotype and dietary treatment
| Gene Ontology Category | % altered* by Genotype** | % altered* by MCS*** | ||
|---|---|---|---|---|
| Age of mice | ~6 MO | ~11 MO | ~6 MO | ~11 MO |
| Autophagy and endosomal-lysosomal | 12.0 | 16.0 | 68.0 | 82.0 |
| Alzheimer’s disease | 8.6 | 26.8 | 51.4 | 80.5 |
| Cell death | 12.1 | 17.6 | 78.8 | 82.4 |
| Channels | 20.8 | 16.7 | 95.8 | 79.2 |
| Cytoskeletal elements | 14.3 | 5.6 | 68.6 | 75.0 |
| GABAergic neurotransmission | 4.5 | 9.1 | 72.7 | 72.7 |
| Glial-enriched markers | 27.3 | 9.1 | 63.6 | 72.7 |
| Glucose utilization | 7.7 | 15.4 | 84.6 | 69.2 |
| Glutamatergic neurotransmission | 11.8 | 23.5 | 61.8 | 85.3 |
| G-proteins | 18.2 | 24.2 | 93.9 | 75.8 |
| Immediate-early genes | 14.3 | 0.0 | 85.7 | 85.7 |
| Monoaminergic neurotransmission | 22.2 | 8.9 | 75.6 | 93.3 |
| Neurodevelopment | 18.2 | 26.1 | 81.8 | 78.3 |
| Neurotrophins | 21.1 | 5.0 | 52.6 | 75.0 |
| Peptides | 18.8 | 16.7 | 68.8 | 88.9 |
| Proteases | 23.5 | 14.7 | 70.6 | 58.8 |
| Protein phosphatases/kinases | 22.9 | 16.4 | 79.2 | 78.7 |
| Sirtuins | n.a. | 12.5 | n.a. | 100.0 |
| Steroid-related markers | 3.8 | 7.7 | 57.7 | 84.6 |
| Stress response genes | 14.3 | 18.2 | 42.9 | 90.9 |
| Synaptic-related markers | 31.3 | 9.4 | 81.3 | 78.1 |
| Transcription factors | 6.9 | 16.7 | 72.4 | 86.7 |
| Total # of Genes Changed | 93 | 99 | 417 | 526 |
| Total # of Genes Assayed | 576 | 649 | 576 | 649 |
| Total % Changed | 16.1 | 15.3 | 72.4 | 81.0 |
%(Genes Changed/Genes Assayed)
2N vs. Ts65Dn;
Choline Normal vs. MCS; n.a. = not assayed
Statistical procedures for custom-designed microarray analysis have been described in detail previously (Alldred et al., 2015a, 2015b; Ginsberg, 2014; Schafer et al., 2015). Gene expression differences due to genotype or dietary condition were assessed with respect to the hybridization signal intensity ratio of the total signal of all array genes. For each gene the signal intensity ratio was modeled as a function of mouse study group, using mixed effects models with random mouse effect to account for the correlation between repeated assays on the same mouse (McCulloch et al., 2011). Significance was judged at the level α=0.01, two-sided; false discovery rate (FDR) based on an empirical null distribution due to strong correlation between genes (Benjamini and Hochberg, 1995; Efron, 2007) was controlled at level α=0.1. Expression levels were graphed using a bioinformatics software package (GeneLinker Gold, Predictive Patterns, Kingston, ON).
qPCR
qPCR was performed on microdissected CA1 sections from an independent cohort of animals containing the hippocampal CA1 region from ~6 MO and ~11 MO Ts65Dn and 2N mice. Taqman qPCR primers (ThermoFisher, Waltham, MA) were utilized for qPCR (Table II). Samples were assayed on a real-time qPCR cycler (PikoReal, ThermoFisher) in 96-well optical plates with coverfilm as described previously (Alldred et al., 2008, 2012, 2015a, 2015b; Jiang et al., 2010). Standard curves and cycle threshold (Ct) were generated using standards obtained from total mouse brain RNA. The ddCT method was employed to determine relative gene level differences between study groups (ABI, 2004; Alldred et al., 2008; Ginsberg, 2010; Jiang et al., 2010). Succinate dehydrogenase A (Sdha) qPCR products were used as a control, as this probe demonstrated the least amount of variability of the 3 control primers tested (Gapdh, Hprt1, and Sdha; data not shown). Negative controls consisted of the reaction mixture without input RNA. The four study groups (Ts65Dn+, Ts65Dn, 2N+, and 2N) were compared with respect to the PCR product synthesis for each gene tested. For each gene, the PCR product synthesis was modeled as a function of mouse study group, using mixed effects models with random mouse effect to account for the correlation between repeated assays on the same mouse (Alldred et al., 2015a, 2015b; McCulloch et al., 2011). Significance was judged at the level α=0.05, two-sided.
Table II.
qPCR TaqMan gene probes utilized for CA1 region gene expression profiling
| Gene name | ID |
|---|---|
| App | Mm01344172_m1 |
| Apbb1 | Mm01274044_g1 |
| Bace1 | Mm00478671_m1 |
| Camk2a | Mm01258148_m1 |
| Casp8 | Mm00802247_m1 |
| Dyrk1a | Mm00432934_m1 |
| Gapdh | Mm99999915_g1 |
| Hsf1 | Mm01201402_m1 |
| Hprt1 | Mm01318747_g1 |
| Mapk13 | Mm00442493_m1 |
| p75NTR | Mm01309638_m1 |
| Prkaa2 | Mm01264790_m1 |
| Sdha | Mm01352360_m1 |
| Sesn2 | Mm004060686_m1 |
| Synj1 | Mm01215509_m1 |
Results
To determine beneficial gene expression changes associated with perinatal MCS treatment, we examined CA1 pyramidal neurons at two distinct timepoints, young mice at ~6 MO (prior to BFCN degeneration) and middle-aged mice at ~11 MO (following BFCN degeneration), postulating that in addition to known cognitive benefits (Strupp et al., 2016), MCS would delay septohippocampal degeneration seen in Ts65Dn offspring (Ash et al., 2014), and approximate the gene expression levels of 2N mice. We also evaluated the impact of MCS on 2N littermates to assess the effect of early maternal choline supplementation in normal offspring.
Microarray results show marked gene expression changes at both timepoints as a function of dietary treatment and genotype in each of the 22 GO categories assayed (Table I). Interestingly, results indicate a much higher percentage of gene changes occur due to maternal dietary treatment (choline normal versus MCS) than to genotype (Ts65Dn versus 2N) (Table I). Specifically, ~6 MO mice display significant expression changes in 72.4% of genes assayed (417 of 576 genes) due to dietary intervention (e.g., Ts65Dn versus Ts65Dn+ and 2N versus 2N+), whereas 16.1% of gene expression changes are due to genotype alone (93 of 576 genes; e.g., Ts65Dn versus 2N). Similar results are seen in the ~11 MO cohort with significant expression changes observed in 81% of the genes assayed due to maternal dietary choline intake level (526 of 649 genes), whereas 15.3% of gene expression changes are found by genotype (99 of 649 genes assayed). The remarkable alteration in expression levels by MCS treatment supports the notion that dietary methyl donor intake has a significant impact on overall gene expression patterns, likely through epigenetic programming.
Select aberrant gene expression profiles in ~6 MO Ts65Dn mice are attenuated by MCS
Results indicate gene expression changes in every GO category queried to date due to both maternal dietary treatment and genotype. For greatest mechanistic value, we sought to identify genes where genotype-driven expression level changes were impacted by MCS treatment (e.g., significant differences between Ts65Dn and 2N littermates that normalized in Ts65Dn+ mice). Accordingly, 11 genes were significantly upregulated in Ts65Dn mice that were normalized in Ts65Dn+ mice. Several of these genes are implicated in synaptic plasticity and AD-associated neurodegeneration GO categories, including the membrane-bound zinc-dependent metalloprotease endothelin converting enzyme-2 (Ece2), and another zinc metallopeptidase, insulin degrading enzyme (Ide) (Table I; Fig. 1, A–B). Interestingly, both Ece2 and Ide degrade Aβ peptides (Eckman et al., 2003; Pacheco-Quinto and Eckman, 2013; Qiu et al., 1998; Wang et al., 2010). Additional synaptic-related genes included transcriptional regulator paired box 6 (Pax6), synaptojanin 1 (Synj1), synaptopodin (Synpo), and pan-neurotrophin growth factor receptor (p75NTR) (Table I; Fig 1C–F). Attenuated transcripts also included four genes involved in calcium-signaling regulatory pathways, adenylate cyclase 1 (Adcy1), calcium/calmodulin-dependent protein kinase II (Camk2a), mitogen-activated protein kinase 13 (Mapk13), and neuropeptide Y receptor Y1 (Npy1r), suggesting that calcium regulation is important in the process of initiating BFCN degeneration (Table I; Fig. 2A–D).
Figure 1.
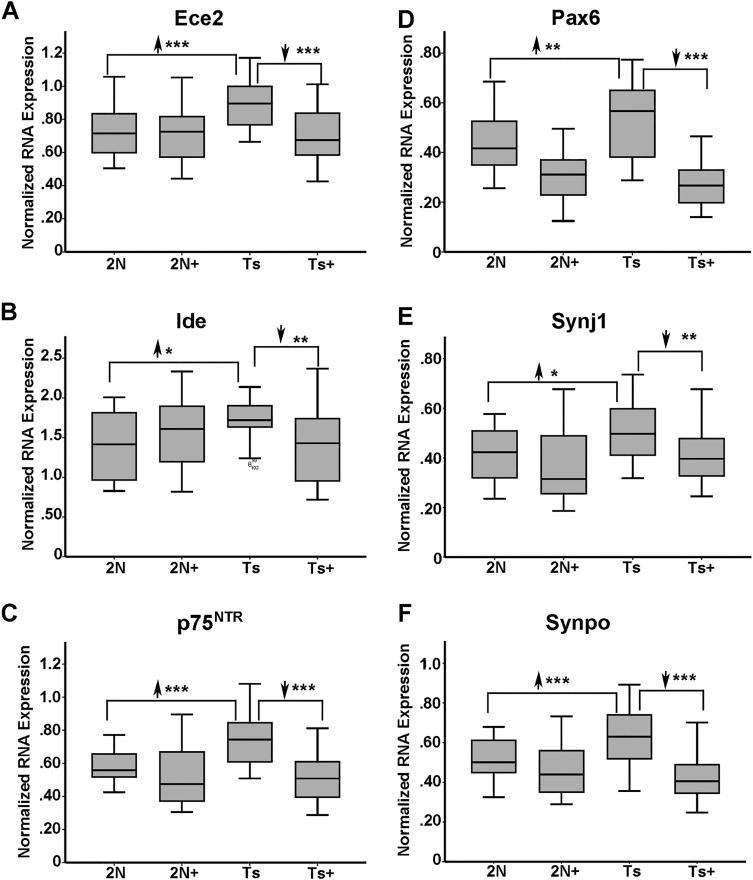
~6 MO MCS-treated trisomic offspring show upregulation and reversal of select genes in synaptic plasticity and neurodegeneration GO categories. A. Ece2 is upregulated 1.24 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. B. Ide is upregulated 1.22 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. C. p75NTR is upregulated 1.26 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. D. Pax6 is upregulated 1.23 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice relative to 2N and Ts levels by MCS treatment, while Pax6 levels in Ts+ are not significantly different from 2N+ levels. E. Synj1 is upregulated 1.22 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. F. Synpo is upregulated 1.24 fold (2N versus Ts) in Ts65Dn mice and is downregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. Note that 2N mice show no significant differences due to MCS treatment in the presented genes except Pax6. Key: *, p<0.01; **, p<0.005; ***, p<0.001. Upward arrowhead, significant upregulation; downward arrowhead, significant downregulation. For figure clarity, we used the abbreviation Ts (for Ts65Dn mice) and Ts+ (for Ts65Dn MCS treated mice).
Figure 2.
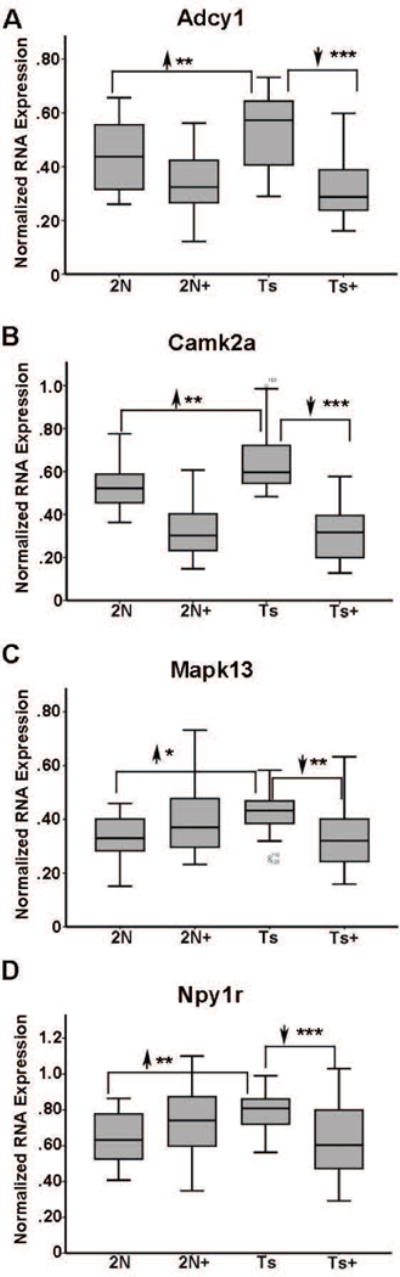
Select calcium signaling genes were normalized by MCS treatment in Ts65Dn mice at ~6 MO. A. Adcy1 is upregulated 1.21 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice relative to 2N and Ts levels by MCS treatment, while Adcy1 levels in Ts+ are not significantly different from 2N+ levels. B. Camk2a is upregulated 1.23 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice relative to 2N and Ts levels by MCS treatment, while Camk2a levels in Ts+ are not significantly different from 2N+ levels. C. Mapk13 is upregulated 1.28 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. D. Npy1r is upregulated 1.23 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. Significant differences due to MCS treatment in 2N mice were observed for Adcy1 and Camk2a. Key: *, p<0.01; **, p<0.005; ***, p<0.001. Upward arrowhead, significant upregulation; downward arrowhead, significant downregulation.
Select aberrant gene expression profiles in ~11 MO Ts65Dn mice are attenuated by MCS
In middle-aged Ts65Dn mice where BFCNs have degenerated, MCS treatment had profound effects in normalizing expression levels of several dysregulated genes in Ts65Dn+ mice. Specifically, significant normalization of 10 genes downregulated in Ts65Dn offspring occurred concomitantly with 12 genes being upregulated in Ts65Dn offspring that attained expression levels which did not differ significantly from 2N in the Ts65Dn+ offspring cohort. Attenuated genes belonged to GO-defined cellular pathways including cellular energy homeostasis, DS/AD response genes, aging and oxidative function, and excitation/inhibition of neuronal transmitters. DS/AD response genes attenuated by MCS included upregulated transcripts alpha-disintegrin and metalloproteinase domain-containing protein 10 (Adam10), dual specificity tyrosine phosphorylation regulated kinase 1A (Dyrk1a), which is triplicated in the Ts65Dn mouse, eukaryotic translation initiation factor 2 alpha kinase 2 (Eif2ak2; also termed Prkr), and Mapk13. DS/AD response genes normalized by MCS included downregulated transcripts amyloid-beta precursor protein binding family B member 1 (Apbb1; also known as Fe65), beta-site APP cleaving enzyme 1 (Bace1), caspase 8 (Casp8), and two truncated isoforms of tau, Mapt2N6D and Mapt2N6P (Table I; Fig 3A–I). Further, App is upregulated in Ts65Dn mice, and is partially attenuated by MCS treatment such that expression levels in Ts65Dn+ mice are significantly lower than Ts65Dn offspring, but still higher than 2N littermates (data not shown). Cellular energy homeostasis genes attenuated by MCS included upregulated transcripts dual specificity tyrosine phosphorylation regulated kinase 3 (Dyrk3), heat-shock transcription factor 1 (Hsf1) and heat-shock protein family A (Hsp70) member 1A (Hspa1a), protein kinase AMP-activated non-catalytic subunit alpha 2 (Prkaa2), protein kinase AMP-activated non-catalytic subunit gamma 1 (Prkag1), and sestrin 2 (Sesn2). Adenylate cyclase 9 (Adcy9) was the sole downregulated cellular homeostasis gene attenuated by MCS (Table I; Fig. 4 A–G). Further, genes involved in aging and oxidative function were normalized by MCS treatment, including corticotropin-releasing hormone (Crh, upregulated in Ts65Dn; Fig. 5A), potassium-voltage gated channel, subfamily B member 1 (Kcnb1, downregulated in Ts65Dn; Fig. 5B), potassium voltage-gated channel interacting protein 3 (Kcnip3, downregulated in Ts65Dn; Fig. 5C) and the transcription factor mutL homolog 1 (Mlh1, upregulated in Ts65Dn; Fig. 5D). Downregulated transcripts within the excitation/inhibition of neuronal transmitters pathway attenuated by MCS included the death domain-associated protein (Daxx, Fig. 6A) and gamma-aminobutyric acid (GABA) type A receptor beta-1 subunit (Gabrb1, Fig. 6B).
Figure 3.
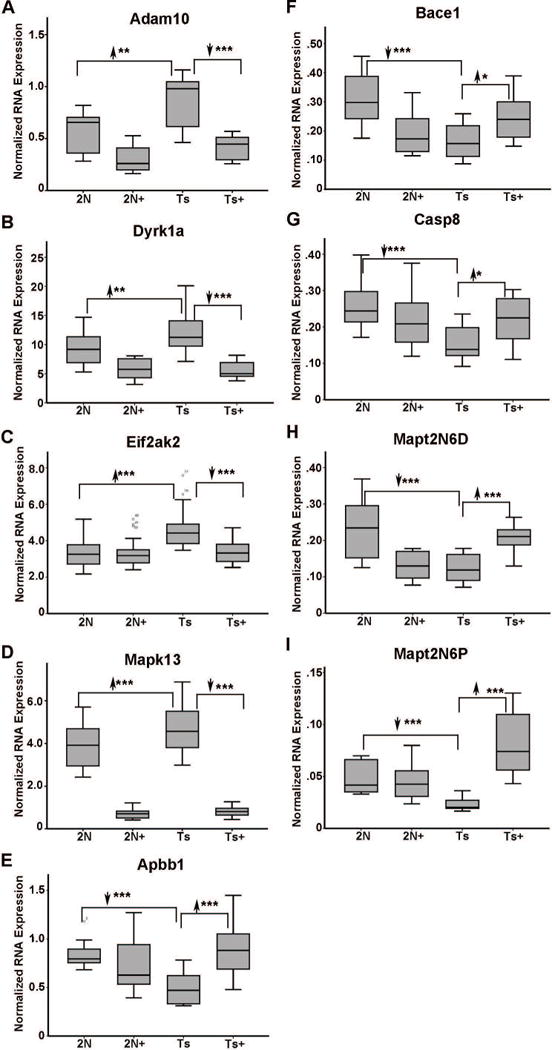
Expression levels impacted by perinatal MCS in ~11 MO MCS-treated offspring included select AD and DS response genes. A. Adam10 is upregulated 1.52 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. B. Dyrk1a is upregulated 1.30 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice relative to 2N and Ts levels by MCS treatment, while Dyrk1a levels in Ts+ are not significantly different from 2N+ levels. C. Eif2ak2 is upregulated 1.35 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. D. Mapk13 is upregulated 1.22 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice relative to 2N and Ts levels by MCS treatment, while Mapk13 levels in Ts+ are not significantly different from 2N+ levels. E. Apbb1 is downregulated 1.64 fold (2N versus Ts) in Ts65Dn mice and upregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. F. Bace1 is downregulated 1.92 fold (2N versus Ts) in Ts65Dn mice and upregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. G. Casp8 is downregulated 1.58 fold (2N versus Ts) in Ts65Dn mice and upregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. H. Mapt2N6D is downregulated 1.87 fold (2N versus Ts) in Ts65Dn mice and upregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. I. Mapt2N6P is downregulated 2.12 fold (2N versus Ts) in Ts65Dn mice and upregulated in Ts+ mice (Ts+ versus Ts) to significantly higher levels than Mapt2N6P expression in 2N and 2N+ mice. Key: *, p<0.01; **, p<0.005; ***, p<0.001. Upward arrowhead, significant upregulation; downward arrowhead, significant downregulation.
Figure 4.
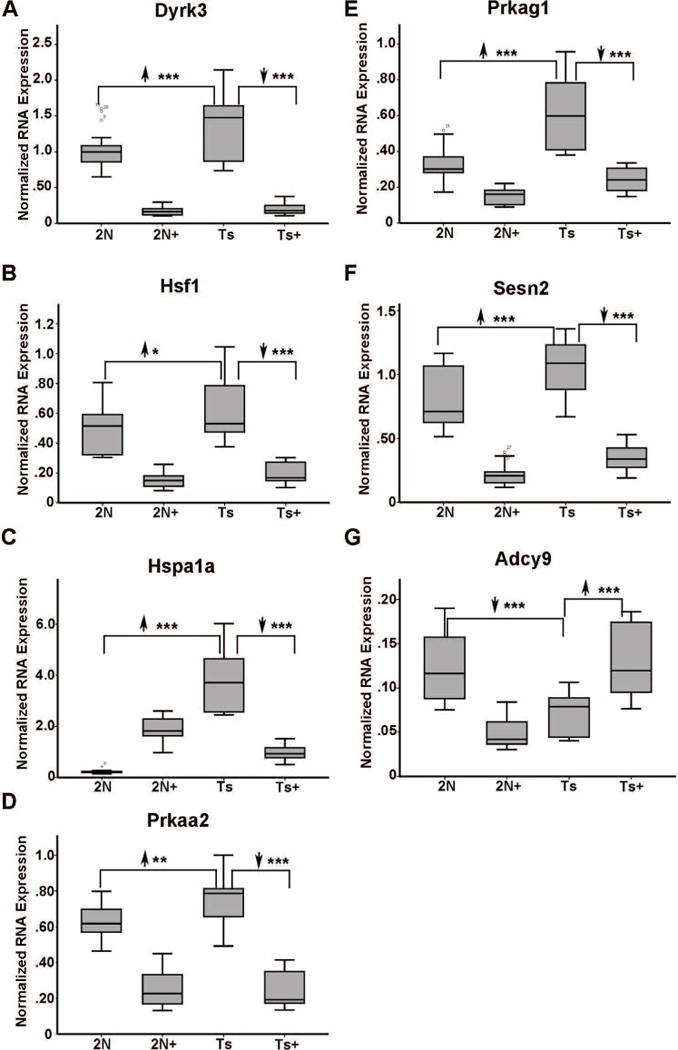
Expression levels impacted by perinatal MCS in ~11 MO MCS-treated offspring included select energy homeostasis genes. A. Dyrk3 is upregulated 1.32 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice relative to 2N and Ts levels by MCS treatment, while Dyrk3 levels in Ts+ are not significantly different from 2N+ levels. B. Hsf1 is upregulated 1.24 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice relative to 2N and Ts levels by MCS treatment, while Hsf1 levels in Ts+ are not significantly different from 2N+ levels. C. Hspa1a is upregulated 7.72 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice relative to 2N and Ts levels by MCS treatment, while Hspa1a levels in Ts+ are trend-level (p<0.02) lower than 2N+ levels but higher than 2N levels. D. Prkaa2 is upregulated 1.17 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice relative to 2N and Ts levels by MCS treatment, while Prkaa2 levels in Ts+ are not significantly different from 2N+ levels. E. Prkag1 is upregulated 1.83 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice relative to 2N and Ts levels by MCS treatment, while Prkag1 levels in Ts+ are not significantly different from 2N+ levels. F. Sesn2 is upregulated 1.32 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice relative to 2N and Ts levels by MCS treatment, while Sesn2 levels in Ts+ are not significantly different from 2N+ levels. G. Adcy9 is the sole downregulated gene in this GO category. Adcy9 is downregulated 1.73 fold in Ts65Dn mice and upregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. Key: *, p<0.01; **, p<0.005; ***, p<0.001. Upward arrowhead, significant upregulation; downward arrowhead, significant downregulation.
Figure 5.
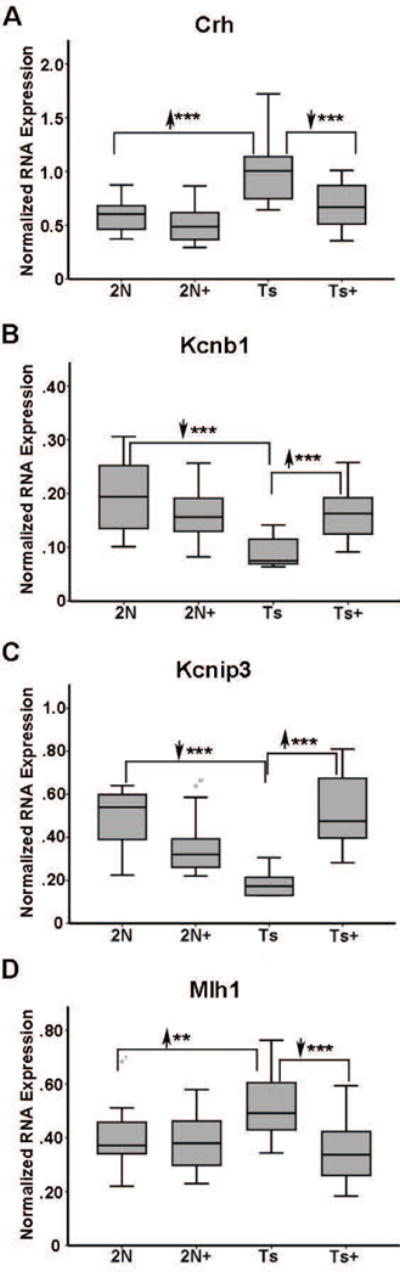
Select age-related and oxidative function genes were reversed by MCS in ~11 MO trisomic mice. A. Crh is upregulated 1.74 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. B. Kcnb1 is downregulated 2.19 fold (2N versus Ts) in Ts65Dn mice and upregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. C. Kcnip3 is downregulated 2.62 fold (2N versus Ts) in Ts65Dn mice and upregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. D. Mlh1 is upregulated 1.32 fold (2N versus Ts) in Ts65Dn mice and downregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. Key: **, p<0.005; ***, p<0.001. Upward arrowhead, significant upregulation; downward arrowhead, significant downregulation.
Figure 6.
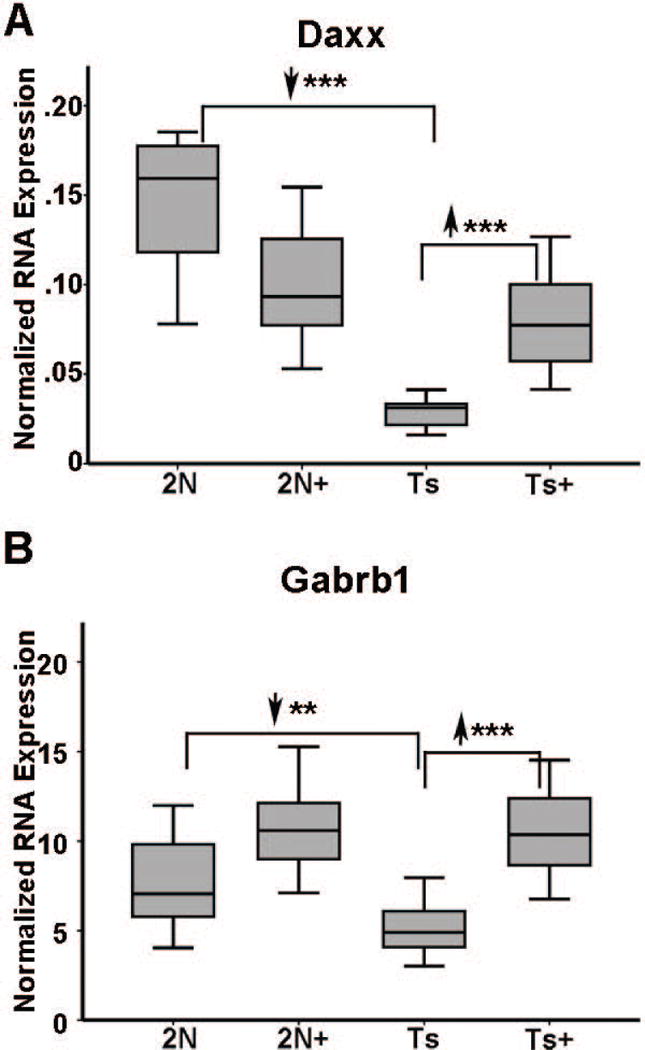
Downregulated neurotransmitter-related genes that were attenuated by MCS treatment included A Daxx is downregulated 5.17 fold (2N versus Ts) in Ts65Dn mice and upregulated in Ts+ mice (Ts+ versus Ts) to 2N+ levels by MCS, but not to 2N levels. B Gabrb1 is downregulated 1.43 fold (2N versus Ts) in Ts65Dn mice and upregulated in Ts+ mice (Ts+ versus Ts) to 2N levels by MCS. Key: **, p<0.005; ***, p<0.001. Upward arrowhead, significant upregulation; downward arrowhead, significant downregulation.
Assessment of select gene expression by qPCR
qPCR was performed on an independent cohort of ~6 MO trisomic and 2N mice using RNA extracted from CA1 hippocampal microdissections. Results indicated that Synj1 is significantly downregulated in Ts65Dn mice by dietary intervention (e.g., Ts65Dn+ versus Ts65Dn), although there was no genotype difference (e.g. Ts65Dn vs. 2N; Fig 7). Mapk13, Camk2a, and p75NTR did not show any significant differences in the admixed microdissected tissue (Fig 7).
Figure 7.
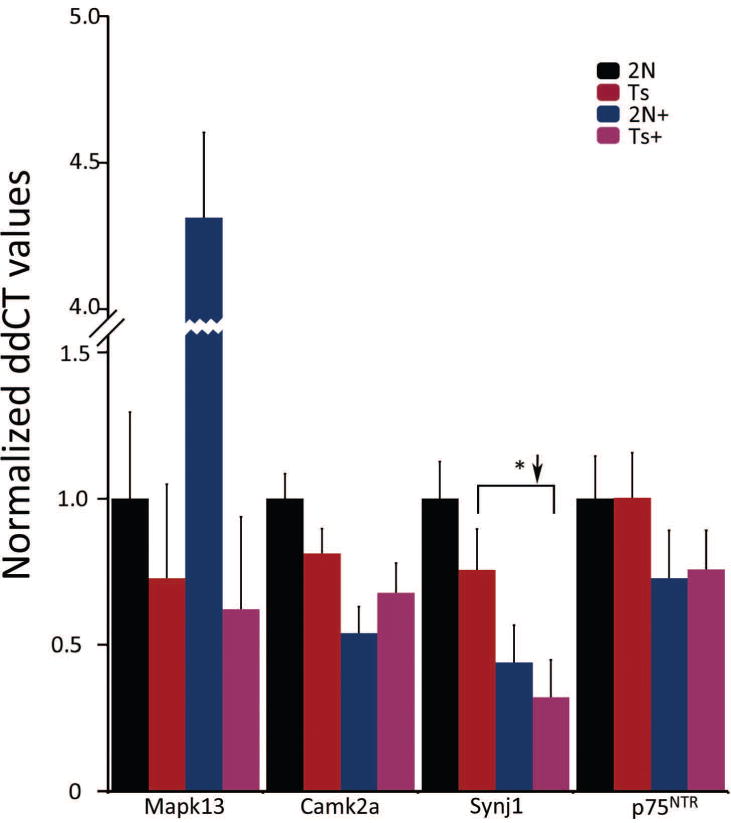
qPCR for select genes using subregional CA1 dissections of admixed cells in ~6 MO mice. Synj1 displays significant downregulation in MCS-treated Ts65Dn offspring, consistent with microarray studies. No genes had differential expression between genotypes, in contrast to microarray data from microdissected CA1 pyramidal neurons. Key: *, p<0.05.
Using an independent ~11 MO cohort, qPCR revealed that Apbb1, App, and Casp8 showed partial or full normalization of genotype-related changes by MCS. However, expression differences for Apbb1 and Casp8 in the admixed tissues assayed by qPCR were in the opposite direction of the microarray results (Fig. 8A), illustrating one of the caveats of comparing single-population expression profiles with admixed cell population transcript analyses. In contrast, Bace1 did not a show significant change in Ts65Dn mice nor attenuation due to MCS (Fig. 8A). Genes involved in stress response and cell homeostasis also showed either partial or fully normalized gene expression to 2N levels in Ts65Dn+ mice, including Dyrk1a, Hsf1, Mapk13, and Prkaa2, consistent with microarray results (Fig. 8B). In contrast, Sesn2 did not show significant differences by maternal diet or genotype (Fig. 8B).
Figure 8.
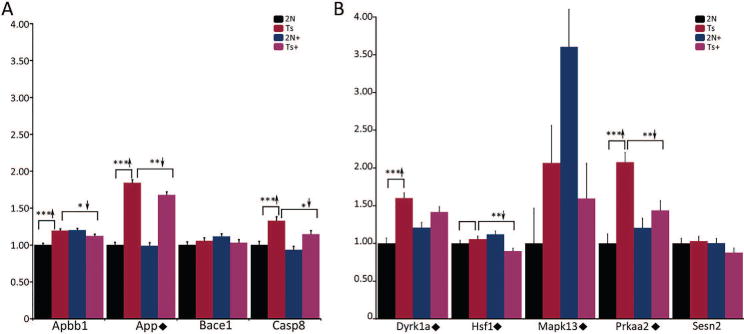
qPCR for select genes using subregional CA1 dissections of admixed cells in ~11 MO mice. A. Apbb1 and Casp8 were upregulated in Ts65Dn mice and displayed significant reduction in MCS treated trisomic offspring. App is upregulated in Ts65Dn mice, and partially recovers to 2N levels by MCS treatment. Bace1 showed no significant differences based on diet or genotype. B. Dyrk1a and Prkaa2 were upregulated in trisomic mice, and displayed normalization (Dyrk1a trend-level p<0.067) with MCS treatment in Ts65Dn offspring. Hsf1 and Mapk13 were downregulated due to MCS treatment in trisomic mice (Mapk13 trend-level p<0.11). Sesn2 showed no significant differences based on diet or genotype. Key: *, p<0.05; **, p<0.01; ***, p<0.001. Upward arrowhead, significant upregulation; downward arrowhead, significant downregulation. Diamond, qPCR results validate microarray findings.
Discussion
We report significant expression level changes in young and middle-aged Ts65Dn mice that were normalized by MCS, including select genes in GO categories that represent AD, cellular stress, synaptic plasticity, and cell death pathways, along with other changes that would indicate MCS treatment beneficially alters gene expression within vulnerable CA1 pyramidal neurons both prior to and following BFCN degeneration. While many additional genes showed expression level changes due to either genotype or MCS treatment, we focused on those gene expression changes in the DS model that were attenuated or reversed by MCS treatment to fully determine the mechanistic effects of the MCS treatment in an individual cell type vulnerable to degeneration, as seen in AD and DS individuals. There is greater attenuation of aberrant Ts65Dn gene expression by MCS in the ~11 MO cohort than in the ~6 MO cohort, indicating the long-term benefits of this early dietary intervention. Interestingly, young mice did not display normalization of downregulated genes, indicating that the gene expression changes that are upregulated in Ts65Dn mice may be the critical first response at the onset of BFCN degeneration. We highlight some of the key findings in relevant pathways.
Calcium signaling and metabolic dysregulation
In ~6 MO trisomic mice, MCS rescued aberrant expression involved in Ca2+ signaling and metabolic dysregulation. Consistent with our findings, Camk2a expression is upregulated in postmortem AD hippocampus and correlates with amyloid and NFT load (McKee et al., 1990; Wang et al., 2005). Downregulation of pathological Camk2a expression may be beneficial to homeostatic calcium signaling. Further support for normalization of calcium signaling comes from modification of the Camk2a target Npy1r (Lin et al., 2014), which independently has been shown to be regulated by BDNF as well as products of the choline PEMT pathway (Agrawal et al., 2014). Additionally, Adcy1 is a calcium-sensitive apoptosis gene, which catalyzes the formation of cyclic AMP, and upon activation, releases glutamate (Cheng and Yakel, 2015; Conti et al., 2009). Adcy1 is also required for maintenance of hippocampal-dependent remote contextual fear memory (Shan et al., 2008), indicating a need for proper gene expression regulation. Another gene involved in metabolic regulation is Mapk13, which when knocked out results in positive metabolic regulation by increasing glucose tolerance and enhancing insulin secretion (Sumara et al., 2009). Conversely, upregulation of Mapk13 results in increased immune system activation and tau phosphorylation (Cavallini et al., 2013; Feijoo et al., 2005; Risco et al., 2012). Mapk13 is the only gene we found that was aberrantly upregulated in both ~6 MO and ~11 MO Ts65Dn offspring of dams fed a standard choline diet, which may result in aberrant tau phosphorylation. These results indicate a need for homeostatic regulation of calcium signaling and metabolic regulation in the DS brain, especially at the onset of BFCN degeneration.
Synaptic plasticity and AD-like neurodegeneration
Synj1 is triplicated in both the Ts65Dn mouse and human DS (Martin et al., 2014). We found that Synj1 expression levels are normalized by MCS. Upregulation of Synj1 has been linked to increases in Aβ and endosomal abnormalities in DS (Cossec et al., 2012; Martin et al., 2014; Zhu et al., 2013). Notably, genetic reduction of Synj1 leads to synaptic and behavioral rescue in an AD mouse model (McIntire et al., 2012). In contrast, Synpo is decreased in postmortem CA1 pyramidal neurons during the progression of dementia from mild cognitive impairment to AD, and this downregulation correlates with cognitive decline (Counts et al., 2014). Loss of Synpo has also been reported to coincide with loss of dendritic spines and spine apparatus, with concomitant deficits in learning and memory and synaptic plasticity (Deller et al., 2007). Therefore, a balance of Synpo expression is likely needed for proper synaptic activity and plasticity. Co-expression of p75NTR and App has been shown to result in apoptosis, and decrease of this pan-neurotrophin receptor has been linked to both prevention of the cognitive decline and reduction of amyloid burden (Fombonne et al., 2009; Yao et al., 2015). Further, p75NTR when bound to the precursor of NGF (proNGF) or the precursor to BDNF (proBDNF), can induce apoptosis (Mufson et al., 2012; Pedraza et al., 2005; Teng et al., 2005) indicating that upregulation of this receptor is potentially detrimental to neuronal health and survival. Ece2 has been identified as a regulator of Aβ clearance (Eckman et al., 2003; Pacheco-Quinto and Eckman, 2013). However, upregulation of Ece2 is found in postmortem AD patients with vascular pathology, indicating a need for balanced expression (Miners et al., 2010, 2014). Likewise, Ide is a thiol metallo-endopeptidase that acts as a protease regulating cerebral Aβ levels (Qiu et al., 1998; Wang et al., 2010). However, Ide-driven breakdown of Aβ competes with insulin as a substrate (Haque and Nazir, 2014), and the higher insulin levels seen in DS (Anwar et al., 1998; Bergholdt et al., 2006) would likely inhibit Ide-mediated clearance of Aβ. Collectively, these findings indicate that normalization of expression changes in CA1 pyramidal neurons in Ts65Dn offspring by MCS may be the result of decreased cellular stress and metabolic homeostasis. Furthermore, attenuated expression of genes involved in synaptic plasticity and AD-like neurodegeneration may underlie the amelioration of cognitive deficits, notably memory and attentional impairments seen in Ts65Dn+ offspring as previously shown by our group (Ash et al., 2014; Kelley et al., 2014a, 2016; Powers et al., 2016, 2017; Strupp et al., 2016; Velazquez et al., 2013).
Cellular homeostasis regulation
We hypothesize that maternal choline supplementation is neuroprotective to the offspring, especially as they age. Specifically, MCS treatment partially restores cellular homeostasis, resulting in Ts65Dn mice that do not aberrantly express genes involved in cellular energy regulation, effectively attenuating aberrant phenotypic effects on DS/AD response genes, aging and oxidative function and excitation and inhibition of neuronal transmitters. For example, we observed normalized expression of two catalytic subunits in the 5′ adenosine monophosphate-activated protein kinase (AMPK) complex signaling cascade that interact with each other, Prkaa2 and Prkag1 (Wong and Lodish, 2006). Consistent with this observation, Prkab2, the beta catalytic subunit of the AMPK complex, was upregulated in the postmortem human AD CA1 region by microarray assessment (Blalock et al., 2011). Related genes attenuated by MCS associated with the AMPK or downstream pathways include Hsf1, Dyrk3, and Sesn2. Hsf1 has been previously shown to be involved in protein homeostasis (Su and Dai, 2016), and is activated by synaptic depolarization (Hooper et al., 2016). Further, Hsf1 nuclear translocation and activation upregulates multiple synaptic genes including Bdnf and Psd95 (Hooper et al., 2016; Wang et al., 2017). Dyrk3 regulates stability of the mammalian target of rapamycin complex 1 (mTORC1) pathway during cellular stress, which is a downstream target of AMPK (Wippich et al., 2013). Dyrk3 has also been shown to be involved with dendritic branching (Slepak et al., 2012), and in conjunction with Dyrk1a, promotes cell survival through phosphorylation of sirtuin 1 (Sirt1) (Guo et al., 2010), indicating dysregulation may negatively impact multiple signaling pathways, including the AMPK cascade. Sesn2 is a critical factor mediating reactive oxygen species formation, in part by inhibiting AMPK pathway member mTORC1 (Ebnoether et al., 2017). Sesn2 is also induced by Aβ exposure in vitro and upregulated in serum of AD patients (Chen et al., 2014; Rai et al., 2016). Therefore, normalization of the AMPK cascade and/or downstream signaling pathway members, as seen with MCS, may be considered therapeutic targets in DS and AD.
AD-related genes
We demonstrate several key genes implicated in AD and DS pathobiology are MCS-responsive in Ts65Dn mice. For example, Adam10 is upregulated in Ts65Dn mice and attenuated by MCS. Studies have shown increased Adam10 levels in AD hippocampus, and two rare Adam10 mutations attenuate α-secretase activity (Gatta et al., 2002; Kim et al., 2009). Adam10 is present and active in synaptic vesicles, including those enriched in APP C-terminal fragments (CTFs), and colocalizes with Bace1 and synaptophysin, which may result in APP processing within synaptic vesicles (Lundgren et al., 2015). Dyrk1a is upregulated in DS and AD (Ferrer et al., 2005; Kimura et al., 2007; Lockstone et al., 2007), may dysregulate the 3-repeat tau to 4-repeat tau ratio (Wegiel et al., 2011), and is part of the triplicated ‘DS critical region’ that has been implicated in cognitive impairment in DS. Importantly, normalizing Dyrk1a gene dosage in trisomic mice improves spatial and reference memory and related AD-like phenotypes (Altafaj et al., 2013; Garcia-Cerro et al., 2014, 2017), highlighting the importance of attenuating Dyrk1a expression levels in DS as a potential therapeutic approach.
There are also several downregulated genes involved in AD/DS pathology normalized in Ts65Dn+ mice, including Bace1. Although associated with β-cleavage of APP, leading to greater Aβ and βCTF expression, attenuation of Bace1 levels by MCS may indicate a counterintuitive normalization of APP processing in vulnerable neurons that reflects the importance of steady-state Aβ and βCTF levels, underscoring the importance of evaluating βCTF expression in DS/AD models (Jiang et al., 2010, 2016). Alternatively, Bace1 deficiency may be considered detrimental, as a loss of Bace1 expression impairs myelination (Hu et al., 2015) and causes hippocampal synaptic deficits (Petrus and Lee, 2014; Wang et al., 2014; Zhu et al., 2016), illustrating the need for normative Bace1 levels. MCS also restores the normal expression of two truncated forms of tau (Mapt2N6D and Mapt2N6P). Little is known about the normal expression of these truncated forms of tau, especially in relation to the DS phenotype, but this may reflect a compensatory change by MCS to permit optimized tau processing, including truncation of the N- and C-terminal domains. Further, the App binding protein Fe65 (Apbb1) is upregulated to 2N levels by MCS in trisomic mice, which may be neuroprotective. Moreover, in addition to its role in processing APP (Chow et al., 2015), Apbb1 is a key protein in the cellular response to genotoxic stress (Ryu et al., 2015). Taken together, these varied expression levels changes reveal the need for greater understanding of Aβ processing and tau metabolism within vulnerable neurons in DS and AD models.
Limitations of the study include a caveat that expression-profiling studies, even those that employ single-population analysis, do not infer causality, but provide highly relevant targets for the study of mechanistic interactions for in vitro and in vivo experiments as well as identify potential targets for clinical trials. We recognize that the Ts65Dn mouse model does not recapitulate the full pathobiology of AD or DS, and is one of many models that could have been chosen for these studies. Discrepancies between human AD, DS transcriptomic profiles, and the trisomic model could be envisioned, as the Ts65Dn mouse has >90 triplicated genes that are not located on HSA21 (Sturgeon and Gardiner, 2011). Despite lacking total genetic overlap with DS and not displaying NFTs or Aβ deposition like AD, Ts65Dn mice are one of the few models available that mimics both behavioral and cognitive decline of AD and DS, including age-related cognitive deficits such as spatial learning, working memory, reference memory, and attention (Ash et al., 2014; Escorihuela et al., 1995; Hyde and Crnic, 2001; Reeves et al., 1995; Velazquez et al., 2013). Future studies may include studying the use of MCS in aged Ts65Dn mice as well as employing Dp16 DS models, APP overexpressing models, and tauopathy models in conjunction with the MCS paradigm. Additional studies comparing the expression profile of trisomic septohippocampal neurons with septohippocampal neurons following cholinotoxic delivery, such as either by ethylcholine mustard aziridinium ion (AF64A) (Fan and Hanin, 1999) or 192 IgG-saporin (Wiley et al., 1991) may also be considered.
In conclusion, results showed significant normalization of gene expression in Ts65Dn mice within several key GO categories as a result of MCS treatment. At the younger age point, critical upregulated genes were normalized by MCS, including those involved in calcium regulation and synaptic plasticity, both pathways critical for the normal homeostatic function at the onset of BFCN degeneration. Similarly, middle-aged trisomic mice saw significant improvement due to early MCS treatment that had lifelong beneficial consequences. These included positive regulation of genes involved in AD pathology and transcripts associated with ID. Additionally, normalization of genes involved in cellular metabolism and homeostasis may prove to be critical for normal function in the aging DS brain. In sum, MCS treatment may help prolong normal cellular function as well as reduce cognitive decline brought on by BFCN degeneration within the septohippocampal circuit in Ts65Dn mice.
Acknowledgments
This study was supported by grants AG014449, AG043375, AG055328 and AG107617 from the National Institutes of Health and the Alzheimer’s Association. We thank Arthur Saltzman, M.S. for expert technical assistance.
References
- ABI. Guide to Performing Relative Quantitation of Gene Expression Using Real-Time Quantitative PCR. Applied Biosystems Product Guide. 2004:1–60. [Google Scholar]
- Agrawal R, Tyagi E, Vergnes L, Reue K, Gomez-Pinilla F. Coupling energy homeostasis with a mechanism to support plasticity in brain trauma. Biochim Biophys Acta. 2014;1842:535–546. doi: 10.1016/j.bbadis.2013.12.004. [DOI] [PubMed] [Google Scholar]
- Akeson EC, Lambert JP, Narayanswami S, Gardiner K, Bechtel LJ, Davisson MT. Ts65Dn – localization of the translocation breakpoint and trisomic gene content in a mouse model for Down syndrome. Cytogenet Cell Genet. 2001;93:270–276. doi: 10.1159/000056997. [DOI] [PubMed] [Google Scholar]
- Alldred MJ, Che S, Ginsberg SD. Terminal continuation (TC) RNA amplification enables expression profiling using minute RNA input obtained from mouse brain. Int J Mol Sci. 2008;9:2091–2104. doi: 10.3390/ijms9112091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alldred MJ, Che S, Ginsberg SD. Terminal continuation (TC) RNA amplification without second strand synthesis. J Neurosci Methods. 2009;177:381–385. doi: 10.1016/j.jneumeth.2008.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alldred MJ, Duff KE, Ginsberg SD. Microarray analysis of CA1 pyramidal neurons in a mouse model of tauopathy reveals progressive synaptic dysfunction. Neurobiol Dis. 2012;45:751–762. doi: 10.1016/j.nbd.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alldred MJ, Lee SH, Petkova E, Ginsberg SD. Expression profile analysis of hippocampal CA1 pyramidal neurons in aged Ts65Dn mice, a model of Down syndrome (DS) and Alzheimer’s disease (AD) Brain Struct Funct. 2015a;220:2983–2996. doi: 10.1007/s00429-014-0839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alldred MJ, Lee SH, Petkova E, Ginsberg SD. Expression profile analysis of vulnerable CA1 pyramidal neurons in young-middle-aged Ts65Dn mice. J Comp Neurol. 2015b;523:61–74. doi: 10.1002/cne.23663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altafaj X, Martin ED, Ortiz-Abalia J, Valderrama A, Lao-Peregrin C, Dierssen M, Fillat C. Normalization of Dyrk1A expression by AAV2/1-shDyrk1A attenuates hippocampal-dependent defects in the Ts65Dn mouse model of Down syndrome. Neurobiol Dis. 2013;52:117–127. doi: 10.1016/j.nbd.2012.11.017. [DOI] [PubMed] [Google Scholar]
- Anwar AJ, Walker JD, Frier BM. Type 1 diabetes mellitus and Down’s syndrome: prevalence, management and diabetic complications. Diabet Med. 1998;15:160–163. doi: 10.1002/(SICI)1096-9136(199802)15:2<160::AID-DIA537>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Ash JA, Velazquez R, Kelley CM, Powers BE, Ginsberg SD, Mufson EJ, Strupp BJ. Maternal choline supplementation improves spatial mapping and increases basal forebrain cholinergic neuron number and size in aged Ts65Dn mice. Neurobiol Dis. 2014;70:32–42. doi: 10.1016/j.nbd.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearer CF, Wellmann KA, Tang N, He M, Mooney SM. Choline ameliorates deficits in balance caused by acute neonatal ethanol exposure. Cerebellum. 2015;14:413–420. doi: 10.1007/s12311-015-0691-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko NP, Belichenko PV, Kleschevnikov AM, Salehi A, Reeves RH, Mobley WC. The “Down syndrome critical region” is sufficient in the mouse model to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of Down syndrome. J Neurosci. 2009;29:5938–5948. doi: 10.1523/JNEUROSCI.1547-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko PV, Masliah E, Kleschevnikov AM, Villar AJ, Epstein CJ, Salehi A, Mobley WC. Synaptic structural abnormalities in the Ts65Dn mouse model of Down Syndrome. J Comp Neurol. 2004;480:281–298. doi: 10.1002/cne.20337. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300. [Google Scholar]
- Bergholdt R, Eising S, Nerup J, Pociot F. Increased prevalence of Down’s syndrome in individuals with type 1 diabetes in Denmark: A nationwide population-based study. Diabetologia. 2006;49:1179–1182. doi: 10.1007/s00125-006-0231-6. [DOI] [PubMed] [Google Scholar]
- Bierer LM, Haroutunian V, Gabriel S, Knott PJ, Carlin LS, Purohit DP, Perl DP, Schmeidler J, Kanof P, Davis KL. Neurochemical correlates of dementia severity in Alzheimer’s disease: relative importance of the cholinergic deficits. J Neurochem. 1995;64:749–760. doi: 10.1046/j.1471-4159.1995.64020749.x. [DOI] [PubMed] [Google Scholar]
- Blalock EM, Buechel HM, Popovic J, Geddes JW, Landfield PW. Microarray analyses of laser-captured hippocampus reveal distinct gray and white matter signatures associated with incipient Alzheimer’s disease. J Chem Neuroanat. 2011;42:118–126. doi: 10.1016/j.jchemneu.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Petanceska S, Peterhoff CM, Terio NB, Epstein CJ, Villar A, Carlson EJ, Staufenbiel M, Nixon RA. App gene dosage modulates endosomal abnormalities of Alzheimer’s disease in a segmental trisomy 16 mouse model of Down syndrome. J Neurosci. 2003;23:6788–6792. doi: 10.1523/JNEUROSCI.23-17-06788.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudill MA, Strupp BJ, Muscalu L, Nevins JEH, Canfield RL. Maternal choline supplementation during the third trimester of pregnancy improves infant information processing speed: a randomized, double-blind, controlled feeding study. FASEB J. 2017 Dec 7; doi: 10.1096/fj.201700692RR. 2017. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallini A, Brewerton S, Bell A, Sargent S, Glover S, Hardy C, Moore R, Calley J, Ramachandran D, Poidinger M, et al. An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with Alzheimer disease. J Biol Chem. 2013;288:23331–23347. doi: 10.1074/jbc.M113.463984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman RS, Hesketh LJ. Behavioral phenotype of individuals with Down syndrome. Ment Retard Dev Disabil Res Rev. 2000;6:84–95. doi: 10.1002/1098-2779(2000)6:2<84::AID-MRDD2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Che S, Ginsberg SD. Amplification of transcripts using terminal continuation. Lab Invest. 2004;84:131–137. doi: 10.1038/labinvest.3700005. [DOI] [PubMed] [Google Scholar]
- Chen YS, Chen SD, Wu CL, Huang SS, Yang DI. Induction of sestrin2 as an endogenous protective mechanism against amyloid beta-peptide neurotoxicity in primary cortical culture. Exp Neurol. 2014;253:63–71. doi: 10.1016/j.expneurol.2013.12.009. [DOI] [PubMed] [Google Scholar]
- Cheng Q, Yakel JL. Activation of alpha7 nicotinic acetylcholine receptors increases intracellular cAMP levels via activation of AC1 in hippocampal neurons. Neuropharmacology. 2015;95:405–414. doi: 10.1016/j.neuropharm.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow WN, Cheung HN, Li W, Lau KF. FE65: Roles beyond amyloid precursor protein processing. Cell Mol Biol Lett. 2015;20:66–87. doi: 10.1515/cmble-2015-0002. [DOI] [PubMed] [Google Scholar]
- Conti AC, Young C, Olney JW, Muglia LJ. Adenylyl cyclases types 1 and 8 promote pro-survival pathways after ethanol exposure in the neonatal brain. Neurobiol Dis. 2009;33:111–118. doi: 10.1016/j.nbd.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JD, Salehi A, Delcroix JD, Howe CL, Belichenko PV, Chua-Couzens J, Kilbridge JF, Carlson EJ, Epstein CJ, Mobley WC. Failed retrograde transport of NGF in a mouse model of Down’s syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proc Natl Acad Sci U S A. 2001;98:10439–10444. doi: 10.1073/pnas.181219298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossec JC, Lavaur J, Berman DE, Rivals I, Hoischen A, Stora S, Ripoll C, Mircher C, Grattau Y, Olivomarin JC, et al. Trisomy for synaptojanin1 in Down syndrome is functionally linked to the enlargement of early endosomes. Hum Mol Genet. 2012;21:3156–3172. doi: 10.1093/hmg/dds142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, Alldred MJ, Che S, Ginsberg SD, Mufson EJ. Synaptic gene dysregulation within hippocampal CA1 pyramidal neurons in mild cognitive impairment. Neuropharmacology. 2014;79:172–179. doi: 10.1016/j.neuropharm.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davisson MT, Schmidt C, Reeves RH, Irving NG, Akeson EC, Harris BS, Bronson RT. Segmental trisomy as a mouse model for Down syndrome. Prog Clin Biol Res. 1993;384:117–133. [PubMed] [Google Scholar]
- Deller T, Bas Orth C, Del Turco D, Vlachos A, Burbach GJ, Drakew A, Chabanis S, Korte M, Schwegler H, Haas CA, et al. A role for synaptopodin and the spine apparatus in hippocampal synaptic plasticity. Ann Anat. 2007;189:5–16. doi: 10.1016/j.aanat.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Detopoulou P, Panagiotakos DB, Antonopoulou S, Pitsavos C, Stefanadis C. Dietary choline and betaine intakes in relation to concentrations of inflammatory markers in healthy adults: the ATTICA study. Am J Clin Nutr. 2008;87:424–430. doi: 10.1093/ajcn/87.2.424. [DOI] [PubMed] [Google Scholar]
- Driscoll LL, Carroll JC, Moon J, Crnic LS, Levitsky DA, Strupp BJ. Impaired sustained attention and error-induced stereotypy in the aged Ts65Dn mouse: a mouse model of Down syndrome and Alzheimer’s disease. Behav Neurosci. 2004;118:1196–1205. doi: 10.1037/0735-7044.118.6.1196. [DOI] [PubMed] [Google Scholar]
- Duchon A, Raveau M, Chevalier C, Nalesso V, Sharp AJ, Herault Y. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling Down syndrome. Mamm Genome. 2011;22:674–684. doi: 10.1007/s00335-011-9356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebnoether E, Ramseier A, Cortada M, Bodmer D, Levano-Huaman S. Sesn2 gene ablation enhances susceptibility to gentamicin-induced hair cell death via modulation of AMPK/mTOR signaling. Cell Death Discov. 2017;3:17024. doi: 10.1038/cddiscovery.2017.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckman EA, Watson M, Marlow L, Sambamurti K, Eckman CB. Alzheimer’s disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J Biol Chem. 2003;278:2081–2084. doi: 10.1074/jbc.C200642200. [DOI] [PubMed] [Google Scholar]
- Efron B. Correlation and large-scale simultaneous significance testing. J Am Stat Assoc. 2007;102:93–103. [Google Scholar]
- Esbensen AJ. Health conditions associated with aging and end of life of adults with Down syndrome. Int Rev Res Ment Retard. 2010;39:107–126. doi: 10.1016/S0074-7750(10)39004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escorihuela RM, Fernandez-Teruel A, Vallina IF, Baamonde C, Lumbreras MA, Dierssen M, Tobena A, Florez J. A behavioral assessment of Ts65Dn mice: a putative Down syndrome model. Neurosci Lett. 1995;199:143–146. doi: 10.1016/0304-3940(95)12052-6. [DOI] [PubMed] [Google Scholar]
- Fan QI, Hanin I. Effects of AF64A on gene expression of choline acetyltransferase (ChAT) in the septo-hippocampal pathway and striatum in vivo. Neurochem Res. 1999;24:15–24. doi: 10.1023/a:1020967711189. [DOI] [PubMed] [Google Scholar]
- Feijoo C, Campbell DG, Jakes R, Goedert M, Cuenda A. Evidence that phosphorylation of the microtubule-associated protein Tau by SAPK4/p38delta at Thr50 promotes microtubule assembly. J Cell Sci. 2005;118:397–408. doi: 10.1242/jcs.01655. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Barrachina M, Puig B, Martinez de Lagran M, Marti E, Avila J, Dierssen M. Constitutive Dyrk1A is abnormally expressed in Alzheimer disease, Down syndrome, Pick disease, and related transgenic models. Neurobiol Dis. 2005;20:392–400. doi: 10.1016/j.nbd.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Fombonne J, Rabizadeh S, Banwait S, Mehlen P, Bredesen DE. Selective vulnerability in Alzheimer’s disease: amyloid precursor protein and p75(NTR) interaction. Ann Neurol. 2009;65:294–303. doi: 10.1002/ana.21578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cerro S, Martinez P, Vidal V, Corrales A, Florez J, Vidal R, Rueda N, Arbones ML, Martinez-Cue C. Overexpression of Dyrk1A is implicated in several cognitive, electrophysiological and neuromorphological alterations found in a mouse model of Down syndrome. PLoS One. 2014;9:e106572. doi: 10.1371/journal.pone.0106572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cerro S, Rueda N, Vidal V, Lantigua S, Martinez-Cue C. Normalizing the gene dosage of Dyrk1A in a mouse model of Down syndrome rescues several Alzheimer’s disease phenotypes. Neurobiol Dis. 2017;106:76–88. doi: 10.1016/j.nbd.2017.06.010. [DOI] [PubMed] [Google Scholar]
- Gardiner K, Fortna A, Bechtel L, Davisson MT. Mouse models of Down syndrome: how useful can they be? Comparison of the gene content of human chromosome 21 with orthologous mouse genomic regions. Gene. 2003;318:137–147. doi: 10.1016/s0378-1119(03)00769-8. [DOI] [PubMed] [Google Scholar]
- Gatta LB, Albertini A, Ravid R, Finazzi D. Levels of beta-secretase BACE and alpha-secretase ADAM10 mRNAs in Alzheimer hippocampus. Neuroreport. 2002;13:2031–2033. doi: 10.1097/00001756-200211150-00008. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD. Glutamatergic neurotransmission expression profiling in the mouse hippocampus after perforant-path transection. Am J Geriatr Psychiatry. 2005a;13:1052–1061. doi: 10.1176/appi.ajgp.13.12.1052. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD. RNA amplification strategies for small sample populations. Methods. 2005b;37:229–237. doi: 10.1016/j.ymeth.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD. Transcriptional profiling of small samples in the central nervous system. Methods Mol Biol. 2008;439:147–158. doi: 10.1007/978-1-59745-188-8_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD. Alterations in discrete glutamate receptor subunits in adult mouse dentate gyrus granule cells following perforant path transection. Anal Bioanal Chem. 2010;397:3349–3358. doi: 10.1007/s00216-010-3826-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD. Considerations in the use of microarrays for analysis of the CNS. Reference Module in Biomedical Research. 2014:1–7. [Google Scholar]
- Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y, Wuu J, Chao MV, Mufson EJ, Nixon RA, et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol Psychiatry. 2010;68:885–893. doi: 10.1016/j.biopsych.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J Neurochem. 2006;97:475–487. doi: 10.1111/j.1471-4159.2006.03764.x. [DOI] [PubMed] [Google Scholar]
- Granholm AC, Sanders L, Seo H, Lin L, Ford K, Isacson O. Estrogen alters amyloid precursor protein as well as dendritic and cholinergic markers in a mouse model of Down syndrome. Hippocampus. 2003;13:905–914. doi: 10.1002/hipo.10130. [DOI] [PubMed] [Google Scholar]
- Granholm AC, Sanders LA, Crnic LS. Loss of cholinergic phenotype in basal forebrain coincides with cognitive decline in a mouse model of Down’s syndrome. Exp Neurol. 2000;161:647–663. doi: 10.1006/exnr.1999.7289. [DOI] [PubMed] [Google Scholar]
- Guo X, Williams JG, Schug TT, Li X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J Biol Chem. 2010;285:13223–13232. doi: 10.1074/jbc.M110.102574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque R, Nazir A. Insulin-degrading enzyme: a link between Alzheimer’s and type 2 diabetes mellitus. CNS Neurol Disord Drug Targets. 2014;13:259–264. doi: 10.2174/18715273113126660139. [DOI] [PubMed] [Google Scholar]
- Hartley D, Blumenthal T, Carrillo M, DiPaolo G, Esralew L, Gardiner K, Granholm AC, Iqbal K, Krams M, Lemere C, et al. Down syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimers Dement. 2015;11:700–709. doi: 10.1016/j.jalz.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley SL, Handen BL, Devenny DA, Hardison R, Mihaila I, Price JC, Cohen AD, Klunk WE, Mailick MR, Johnson SC, et al. Cognitive functioning in relation to brain amyloid-beta in healthy adults with Down syndrome. Brain. 2014;137:2556–2563. doi: 10.1093/brain/awu173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Santucci D, Kilbridge J, Chua-Couzens J, Fontana DJ, Daniels SE, Johnson RM, Chen K, Sun Y, Carlson E, et al. Developmental abnormalities and age-related neurodegeneration in a mouse model of Down syndrome. Proc Natl Acad Sci USA. 1996;93:13333–13338. doi: 10.1073/pnas.93.23.13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper PL, Durham HD, Torok Z, Hooper PL, Crul T, Vigh L. The central role of heat shock factor 1 in synaptic fidelity and memory consolidation. Cell Stress Chaperones. 2016;21:745–753. doi: 10.1007/s12192-016-0709-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Hu J, Dai L, Trapp B, Yan R. Axonal and Schwann cell BACE1 is equally required for remyelination of peripheral nerves. J Neurosci. 2015;35:3806–3814. doi: 10.1523/JNEUROSCI.5207-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter CL, Bimonte HA, Granholm A-CE. Behavioral comparison of 4 and 6 month-old Ts65Dn mice: Age-related impairments in working and reference memory. Behav Brain Res. 2003;138:121–131. doi: 10.1016/s0166-4328(02)00275-9. [DOI] [PubMed] [Google Scholar]
- Hyde LA, Crnic LS. Age-related deficits in context discrimination learning in Ts65Dn mice that model Down syndrome and Alzheimer’s disease. Behav Neurosci. 2001;115:1239–1246. [PubMed] [Google Scholar]
- Insausti AM, Megias M, Crespo D, Cruz-Orive LM, Dierssen M, Vallina IF, Insausti R, Florez J, Vallina TF. Hippocampal volume and neuronal number in Ts65Dn mice: a murine model of Down syndrome. Neurosci Lett. 1998;253:175–178. doi: 10.1016/s0304-3940(98)00641-7. [DOI] [PubMed] [Google Scholar]
- Institute of Medicine NAoSU. Dietary reference intakes for thiamin, riboflavin, niacin, vitamin B6, folate, vitamin B12, pantothenic acid, biotin and choline. Washington, DC: National Academy Press; 1998. [PubMed] [Google Scholar]
- Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A, Ginsberg SD, Cataldo AM, Mathews PM, Nixon RA. Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci USA. 2010;107:1630–1635. doi: 10.1073/pnas.0908953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Rigoglioso A, Peterhoff CM, Pawlik M, Sato Y, Bleiwas C, Stavrides P, Smiley JF, Ginsberg SD, Mathews PM, et al. Partial BACE1 reduction in a Down syndrome mouse model blocks Alzheimer-related endosomal anomalies and cholinergic neurodegeneration: role of APP-CTF. Neurobiol Aging. 2016;39:90–98. doi: 10.1016/j.neurobiolaging.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlem P, Sultan M, Herwig R, Steinfath M, Balzereit D, Eppens B, Saran NG, Pletcher MT, South ST, Stetten G, et al. Transcript level alterations reflect gene dosage effects across multiple tissues in a mouse model of down syndrome. Genome Res. 2004;14:1258–1267. doi: 10.1101/gr.1951304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur G, Sharma A, Xu W, Gerum S, Alldred MJ, Subbanna S, Basavarajappa BS, Pawlik M, Ohno M, Ginsberg SD, et al. Glutamatergic transmission aberration: a major cause of behavioral deficits in a murine model of Down’s syndrome. J Neurosci. 2014;34:5099–5106. doi: 10.1523/JNEUROSCI.5338-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley CM, Ash JA, Powers BE, Velazquez R, Alldred MJ, Ikonomovic MD, Ginsberg SD, Strupp BJ, Mufson EJ. Effects of maternal choline supplementation on the septohippocampal cholinergic system in the Ts65Dn mouse model of Down syndrome. Curr Alzheimer Res. 2016;13:84–96. doi: 10.2174/1567205012666150921100515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley CM, Powers BE, Velazquez R, Ash JA, Ginsberg SD, Strupp BJ, Mufson EJ. Maternal choline supplementation differentially alters the basal forebrain cholinergic system of young-adult Ts65Dn and disomic mice. J Comp Neurol. 2014a;522:1390–1410. doi: 10.1002/cne.23492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley CM, Powers BE, Velazquez R, Ash JA, Ginsberg SD, Strupp BJ, Mufson EJ. Sex differences in the cholinergic basal forebrain in the Ts65Dn mouse model of Down syndrome and Alzheimer’s disease. Brain Pathol. 2014b;24:33–44. doi: 10.1111/bpa.12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Suh J, Romano D, Truong MH, Mullin K, Hooli B, Norton D, Tesco G, Elliott K, Wagner SL, et al. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate {alpha}-secretase activity. Hum Mol Genet. 2009;18:3987–3996. doi: 10.1093/hmg/ddp323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R, Kamino K, Yamamoto M, Nuripa A, Kida T, Kazui H, Hashimoto R, Tanaka T, Kudo T, Yamagata H, et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum Mol Genet. 2007;16:15–23. doi: 10.1093/hmg/ddl437. [DOI] [PubMed] [Google Scholar]
- Kleschevnikov AM, Belichenko PV, Faizi M, Jacobs LF, Htun K, Shamloo M, Mobley WC. Deficits in cognition and synaptic plasticity in a mouse model of Down syndrome ameliorated by GABAB receptor antagonists. J Neurosci. 2012;32:9217–9227. doi: 10.1523/JNEUROSCI.1673-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleschevnikov AM, Belichenko PV, Villar AJ, Epstein CJ, Malenka RC, Mobley WC. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J Neurosci. 2004;24:8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt MA, Davies DC, Kidd M, Dierssen M, Florez J. Synaptic deficit in the temporal cortex of partial trisomy 16 (Ts65Dn) mice. Brain Res. 2000;858:191–197. doi: 10.1016/s0006-8993(00)01984-3. [DOI] [PubMed] [Google Scholar]
- Lai F, Williams RS. A prospective study of Alzheimer disease in Down syndrome. Arch Neurol. 1989;46:849–853. doi: 10.1001/archneur.1989.00520440031017. [DOI] [PubMed] [Google Scholar]
- Leverenz JB, Raskind MA. Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: a regional quantitative analysis. Exp Neurol. 1998;150:296–304. doi: 10.1006/exnr.1997.6777. [DOI] [PubMed] [Google Scholar]
- Lin L, Hales CM, Garber K, Jin P. Fat mass and obesity-associated (FTO) protein interacts with CaMKII and modulates the activity of CREB signaling pathway. Hum Mol Genet. 2014;23:3299–3306. doi: 10.1093/hmg/ddu043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblom G, Oradd G. Lipid lateral diffusion and membrane heterogeneity. Biochim Biophys Acta. 2009;1788:234–244. doi: 10.1016/j.bbamem.2008.08.016. [DOI] [PubMed] [Google Scholar]
- Lockstone HE, Harris LW, Swatton JE, Wayland MT, Holland AJ, Bahn S. Gene expression profiling in the adult Down syndrome brain. Genomics. 2007;90:647–660. doi: 10.1016/j.ygeno.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Lott IT. Neurological phenotypes for Down syndrome across the life span. Prog Brain Res. 2012;197:101–121. doi: 10.1016/B978-0-444-54299-1.00006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren JL, Ahmed S, Schedin-Weiss S, Gouras GK, Winblad B, Tjernberg LO, Frykman S. ADAM10 and BACE1 are localized to synaptic vesicles. J Neurochem. 2015;135:606–615. doi: 10.1111/jnc.13287. [DOI] [PubMed] [Google Scholar]
- Mann DM, Yates PO, Marcyniuk B, Ravindra CR. The topography of plaques and tangles in Down’s syndrome patients of different ages. Neuropathol App Neurobiol. 1986;12:447–457. doi: 10.1111/j.1365-2990.1986.tb00053.x. [DOI] [PubMed] [Google Scholar]
- Martin SB, Dowling AL, Lianekhammy J, Lott IT, Doran E, Murphy MP, Beckett TL, Schmitt FA, Head E. Synaptophysin and synaptojanin-1 in Down syndrome are differentially affected by Alzheimer’s disease. J Alzheimers Dis. 2014;42:767–775. doi: 10.3233/JAD-140795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch CE, Searle SR, Neuhaus JM. Generalized, Linear, and Mixed Models. Second. New York: John Wiley & Sons; 2011. [Google Scholar]
- McGowan PO, Meaney MJ, Szyf M. Diet and the epigenetic (re)programming of phenotypic differences in behavior. Brain Res. 2008;1237:12–24. doi: 10.1016/j.brainres.2008.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntire LB, Berman DE, Myaeng J, Staniszewski A, Arancio O, Di Paolo G, Kim TW. Reduction of synaptojanin 1 ameliorates synaptic and behavioral impairments in a mouse model of Alzheimer’s disease. J Neurosci. 2012;32:15271–15276. doi: 10.1523/JNEUROSCI.2034-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Kosik KS, Kennedy MB, Kowall NW. Hippocampal neurons predisposed to neurofibrillary tangle formation are enriched in type II calcium/calmodulin-dependent protein kinase. J Neuropathol Exp Neurol. 1990;49:49–63. doi: 10.1097/00005072-199001000-00006. [DOI] [PubMed] [Google Scholar]
- Mehedint MG, Niculescu MD, Craciunescu CN, Zeisel SH. Choline deficiency alters global histone methylation and epigenetic marking at the Re1 site of the calbindin 1 gene. FASEB J. 2010;24:184–195. doi: 10.1096/fj.09-140145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellott TJ, Huleatt OM, Shade BN, Pender SM, Liu YB, Slack BE, Blusztajn JK. Perinatal choline supplementation reduces amyloidosis and increases choline acetyltransferase expression in the hippocampus of the APPswePS1dE9 Alzheimer’s disease model mice. PLoS One. 2017;12:e0170450. doi: 10.1371/journal.pone.0170450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Levey AI, Wainer BH. Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol. 1983;214:170–197. doi: 10.1002/cne.902140206. [DOI] [PubMed] [Google Scholar]
- Miners JS, Palmer JC, Tayler H, Palmer LE, Ashby E, Kehoe PG, Love S. Abeta degradation or cerebral perfusion? Divergent effects of multifunctional enzymes. Front Aging Neurosci. 2014;6:238. doi: 10.3389/fnagi.2014.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miners JS, van Helmond Z, Kehoe PG, Love S. Changes with age in the activities of beta-secretase and the abeta-degrading enzymes neprilysin, insulin-degrading enzyme and angiotensin-converting enzyme. Brain Pathol. 2010;20:794–802. doi: 10.1111/j.1750-3639.2010.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon J, Chen M, Gandhy SU, Strawderman M, Levitsky DA, Maclean KN, Strupp BJ. Perinatal choline supplementation improves cognitive functioning and emotion regulation in the Ts65Dn mouse model of Down syndrome. Behav Neurosci. 2010;124:346–361. doi: 10.1037/a0019590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Counts SE, Perez SE, Ginsberg SD. Cholinergic system during the progression of Alzheimer’s disease: therapeutic implications. Expert Rev Neurother. 2008;8:1703–1718. doi: 10.1586/14737175.8.11.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, He B, Nadeem M, Perez SE, Counts SE, Leurgans S, Fritz J, Lah J, Ginsberg SD, Wuu J, et al. Hippocampal proNGF signaling pathways and beta-amyloid levels in mild cognitive impairment and Alzheimer disease. J Neuropathol Exp Neurol. 2012;71:1018–1029. doi: 10.1097/NEN.0b013e318272caab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco-Quinto J, Eckman EA. Endothelin-converting enzymes degrade intracellular beta-amyloid produced within the endosomal/lysosomal pathway and autophagosomes. J Biol Chem. 2013;288:5606–5615. doi: 10.1074/jbc.M112.422964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedraza CE, Podlesniy P, Vidal N, Arevalo JC, Lee R, Hempstead B, Ferrer I, Iglesias M, Espinet C. Pro-NGF isolated from the human brain affected by Alzheimer’s disease induces neuronal apoptosis mediated by p75NTR. Am J Pathol. 2005;166:533–543. doi: 10.1016/S0002-9440(10)62275-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrus E, Lee HK. BACE1 is necessary for experience-dependent homeostatic synaptic plasticity in visual cortex. Neural Plast. 2014;2014:128631. doi: 10.1155/2014/128631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers BE, Kelley CM, Velazquez R, Ash JA, Strawderman MS, Alldred MJ, Ginsberg SD, Mufson EJ, Strupp BJ. Maternal choline supplementation in a mouse model of Down syndrome: Effects on attention and nucleus basalis/substantia innominata neuron morphology in adult offspring. Neuroscience. 2017;340:501–514. doi: 10.1016/j.neuroscience.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers BE, Velazquez R, Kelley CM, Ash JA, Strawderman MS, Alldred MJ, Ginsberg SD, Mufson EJ, Strupp BJ. Attentional function and basal forebrain cholinergic neuron morphology during aging in the Ts65Dn mouse model of Down syndrome. Brain Struct Funct. 2016;221:4337–4352. doi: 10.1007/s00429-015-1164-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem. 1998;273:32730–32738. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- Rachidi M, Lopes C. Mental retardation and associated neurological dysfunctions in Down syndrome: a consequence of dysregulation in critical chromosome 21 genes and associated molecular pathways. Eur J Paediatr Neurol. 2008;12:168–182. doi: 10.1016/j.ejpn.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Rachidi M, Lopes C. Molecular and cellular mechanisms elucidating neurocognitive basis of functional impairments associated with intellectual disability in Down syndrome. Am J Intellect Dev Disabil. 2010;115:83–112. doi: 10.1352/1944-7558-115.2.83. [DOI] [PubMed] [Google Scholar]
- Rai N, Kumar R, Desai GR, Venugopalan G, Shekhar S, Chatterjee P, Tripathi M, Upadhyay AD, Dwivedi S, Dey AB, et al. Relative alterations in blood-based levels of sestrin in Alzheimer’s disease and mild cognitive impairment patients. J Alzheimers Dis. 2016;54:1147–1155. doi: 10.3233/JAD-160479. [DOI] [PubMed] [Google Scholar]
- Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet. 1995;11:177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- Risco A, del Fresno C, Mambol A, Alsina-Beauchamp D, MacKenzie KF, Yang HT, Barber DF, Morcelle C, Arthur JS, Ley SC, et al. p38gamma and p38delta kinases regulate the Toll-like receptor 4 (TLR4)-induced cytokine production by controlling ERK1/2 protein kinase pathway activation. Proc Natl Acad Sci U S A. 2012;109:11200–11205. doi: 10.1073/pnas.1207290109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RG, Hunter SK, Hoffman MC, McCarthy L, Chambers BM, Law AJ, Leonard S, Zerbe GO, Freedman R. Perinatal phosphatidylcholine supplementation and early childhood behavior problems: evidence for CHRNA7 moderation. Am J Psychiatry. 2016;173:509–516. doi: 10.1176/appi.ajp.2015.15091188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda N, Llorens-Martin M, Florez J, Valdizan E, Banerjee P, Trejo JL, Martinez-Cue C. Memantine normalizes several phenotypic features in the Ts65Dn mouse model of Down syndrome. J Alzheimers Dis. 2010;21:277–290. doi: 10.3233/JAD-2010-100240. [DOI] [PubMed] [Google Scholar]
- Rye DB, Wainer BH, Mesulam MM, Mufson EJ, Saper CB. Cortical projections arising from the basal forebrain: a study of cholinergic and noncholinergic components employing combined retrograde tracing and immunohistochemical localization of choline acetyltransferase. Neuroscience. 1984;13:627–643. doi: 10.1016/0306-4522(84)90083-6. [DOI] [PubMed] [Google Scholar]
- Ryu S, Teles F, Minopoli G, Russo T, Rosenfeld MG, Suh Y. An epigenomic role of Fe65 in the cellular response to DNA damage. Mutat Res. 2015;776:40–47. doi: 10.1016/j.mrfmmm.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, Takimoto-Kimura R, Kleschevnikov AM, Sambamurti K, Chung PP, et al. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51:29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Schafer MJ, Dolgalev I, Alldred MJ, Heguy A, Ginsberg SD. Calorie restriction suppresses age-dependent hppocampal transcriptional signatures. PLoS One. 2015;10:e0133923. doi: 10.1371/journal.pone.0133923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scremin OU, Roch M, Norman KM, Djazayeri S, Liu YY. Brain acetylcholine and choline concentrations and dynamics in a murine model of the Fragile X syndrome: age, sex and region-specific changes. Neuroscience. 2015;301:520–528. doi: 10.1016/j.neuroscience.2015.06.036. [DOI] [PubMed] [Google Scholar]
- Sendera TJ, Ma SY, Jaffar S, Kozlowski PB, Kordower JH, Mawal Y, Saragovi HU, Mufson EJ. Reduction in TrkA-immunoreactive neurons is not associated with an overexpression of galaninergic fibers within the nucleus basalis in Down’s syndrome. J Neurochem. 2000;74:1185–1196. doi: 10.1046/j.1471-4159.2000.741185.x. [DOI] [PubMed] [Google Scholar]
- Seo H, Isacson O. Abnormal APP, cholinergic and cognitive function in Ts65Dn Down’s model mice. Exp Neurol. 2005;193:469–480. doi: 10.1016/j.expneurol.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Shan Q, Chan GC, Storm DR. Type 1 adenylyl cyclase is essential for maintenance of remote contextual fear memory. J Neurosci. 2008;28:12864–12867. doi: 10.1523/JNEUROSCI.2413-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siarey RJ, Carlson EJ, Epstein CJ, Balbo A, Rapoport SI, Galdzicki Z. Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in Down syndrome. Neuropharmacology. 1999;38:1917–1920. doi: 10.1016/s0028-3908(99)00083-0. [DOI] [PubMed] [Google Scholar]
- Slepak TI, Salay LD, Lemmon VP, Bixby JL. Dyrk kinases regulate phosphorylation of doublecortin, cytoskeletal organization, and neuronal morphology. Cytoskeleton (Hoboken) 2012;69:514–527. doi: 10.1002/cm.21021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens KE, Adams CE, Yonchek J, Hickel C, Danielson J, Kisley MA. Permanent improvement in deficient sensory inhibition in DBA/2 mice with increased perinatal choline. Psychopharmacology (Berl) 2008;198:413–420. doi: 10.1007/s00213-008-1170-3. [DOI] [PubMed] [Google Scholar]
- Strupp BJ, Powers BE, Velazquez R, Ash JA, Kelley CM, Alldred MJ, Strawderman M, Caudill MA, Mufson EJ, Ginsberg SD. Maternal choline supplementation: a potential prenatal treatment for Down syndrome and Alzheimer’s disease. Curr Alzheimer Res. 2016;13:97–106. doi: 10.2174/1567205012666150921100311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgeon X, Gardiner KJ. Transcript catalogs of human chromosome 21 and orthologous chimpanzee and mouse regions. Mamm Genome. 2011;22:261–271. doi: 10.1007/s00335-011-9321-y. [DOI] [PubMed] [Google Scholar]
- Su KH, Dai C. Metabolic control of the proteotoxic stress response: implications in diabetes mellitus and neurodegenerative disorders. Cell Mol Life Sci. 2016;73:4231–4248. doi: 10.1007/s00018-016-2291-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumara G, Formentini I, Collins S, Sumara I, Windak R, Bodenmiller B, Ramracheya R, Caille D, Jiang H, Platt KA, et al. Regulation of PKD by the MAPK p38delta in insulin secretion and glucose homeostasis. Cell. 2009;136:235–248. doi: 10.1016/j.cell.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD, Kermani P, Torkin R, Chen ZY, Lee FS, et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci. 2005;25:5455–5463. doi: 10.1523/JNEUROSCI.5123-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez R, Ash JA, Powers BE, Kelley CM, Strawderman M, Luscher ZI, Ginsberg SD, Mufson EJ, Strupp BJ. Maternal choline supplementation improves spatial learning and adult hippocampal neurogenesis in the Ts65Dn mouse model of Down syndrome. Neurobiol Dis. 2013;58:92–101. doi: 10.1016/j.nbd.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Liu Y, Huang L, Chen J, Li JJ, Wang R, Kim E, Chen Y, Justicia C, Sakata K, et al. A CNS-permeable Hsp90 inhibitor rescues synaptic dysfunction and memory loss in APP-overexpressing Alzheimer’s mouse model via an HSF1-mediated mechanism. Mol Psychiatry. 2017;22:990–1001. doi: 10.1038/mp.2016.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, de Jong DH, Ruhling A, Lesch V, Shimizu K, Wulff S, Heuer A, Glorius F, Galla HJ. Imidazolium-based lipid analogues and their interaction with phosphatidylcholine membranes. Langmuir. 2016;32:12579–12592. doi: 10.1021/acs.langmuir.6b02496. [DOI] [PubMed] [Google Scholar]
- Wang H, Megill A, Wong PC, Kirkwood A, Lee HK. Postsynaptic target specific synaptic dysfunctions in the CA3 area of BACE1 knockout mice. PLoS One. 2014;9:e92279. doi: 10.1371/journal.pone.0092279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wang R, Chen L, Bennett DA, Dickson DW, Wang DS. Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer’s brain. J Neurochem. 2010;115:47–57. doi: 10.1111/j.1471-4159.2010.06899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Chen GH, Hu XY, Lu YP, Zhou JN, Liu RY. The expression of calcium/calmodulin-dependent protein kinase II-alpha in the hippocampus of patients with Alzheimer’s disease and its links with AD-related pathology. Brain Res. 2005;1031:101–108. doi: 10.1016/j.brainres.2004.10.061. [DOI] [PubMed] [Google Scholar]
- Ward BC, Agarwal S, Wang K, Berger-Sweeney J, Kolodny NH. Longitudinal brain MRI study in a mouse model of Rett syndrome and the effects of choline. Neurobiol Dis. 2008;31:110–119. doi: 10.1016/j.nbd.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Ward BC, Kolodny NH, Nag N, Berger-Sweeney JE. Neurochemical changes in a mouse model of Rett syndrome: changes over time and in response to perinatal choline nutritional supplementation. J Neurochem. 2009;108:361–371. doi: 10.1111/j.1471-4159.2008.05768.x. [DOI] [PubMed] [Google Scholar]
- Wegiel J, Gong CX, Hwang YW. The role of DYRK1A in neurodegenerative diseases. FEBS J. 2011;278:236–245. doi: 10.1111/j.1742-4658.2010.07955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley RG, Oeltmann TN, Lappi DA. Immunolesioning: selective destruction of neurons using immunotoxin to rat NGF receptor. Brain Res. 1991;562:149–153. doi: 10.1016/0006-8993(91)91199-b. [DOI] [PubMed] [Google Scholar]
- Wippich F, Bodenmiller B, Trajkovska MG, Wanka S, Aebersold R, Pelkmans L. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell. 2013;152:791–805. doi: 10.1016/j.cell.2013.01.033. [DOI] [PubMed] [Google Scholar]
- Wisniewski KE, Dalton AJ, Crapper McLachlan DR, Wen GY, Wisniewski HM. Alzheimer’s disease in Down’s syndrome: clinicopathologic studies. Neurology. 1985;35:957–961. doi: 10.1212/wnl.35.7.957. [DOI] [PubMed] [Google Scholar]
- Wong KA, Lodish HF. A revised model for AMP-activated protein kinase structure: The alpha-subunit binds to both the beta- and gamma-subunits although there is no direct binding between the beta- and gamma-subunits. J Biol Chem. 2006;281:36434–36442. doi: 10.1074/jbc.M607410200. [DOI] [PubMed] [Google Scholar]
- Yao XQ, Jiao SS, Saadipour K, Zeng F, Wang QH, Zhu C, Shen LL, Zeng GH, Liang CR, Wang J, et al. p75NTR ectodomain is a physiological neuroprotective molecule against amyloid-beta toxicity in the brain of Alzheimer’s disease. Mol Psychiatry. 2015;20:1301–1310. doi: 10.1038/mp.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel SH. Choline: clinical nutrigenetic/nutrigenomic approaches for identification of functions and dietary requirements. J Nutrigenet Nutrigenomics. 2010;3:209–219. doi: 10.1159/000324357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel SH, Niculescu MD. Perinatal choline influences brain structure and function. Nutr Rev. 2006;64:197–203. doi: 10.1111/j.1753-4887.2006.tb00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K, Xiang X, Filser S, Marinkovic P, Dorostkar MM, Crux S, Neumann U, Shimshek DR, Rammes G, Haass C, et al. Beta-site amyloid precursor protein cleaving enzyme 1 inhibition impairs synaptic plasticity via seizure protein 6. Biol Psychiatry. 2016 Dec 26; doi: 10.1016/j.biopsych.2016.12.023. 2016. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Zhu L, Zhong M, Zhao J, Rhee H, Caesar I, Knight EM, Volpicelli-Daley L, Bustos V, Netzer W, Liu L, et al. Reduction of synaptojanin 1 accelerates Abeta clearance and attenuates cognitive deterioration in an Alzheimer mouse model. J Biol Chem. 2013;288:32050–32063. doi: 10.1074/jbc.M113.504365. [DOI] [PMC free article] [PubMed] [Google Scholar]