

Abstract
Fabry disease, an X-linked inherited lysosomal storage disorder, is caused by mutations in the gene encoding α-galactosidase, GLA. In patients with Fabry disease, glycosphingolipids accumulate in various cell types, triggering a range of cellular and tissue responses that result in a wide spectrum of organ involvement. Although variable, gastrointestinal symptoms are among the most common and significant early clinical manifestations; they tend to persist into adulthood if left untreated. To further understand the effects of sustained enzyme replacement therapy (ERT) with agalsidase beta on gastrointestinal symptoms in heterozygotes, a data analysis of female patients enrolled in the Fabry Registry was conducted. To be included, females of any age must have received agalsidase beta (average dose 1.0 mg/kg every 2 weeks) for at least 2.5 years. Measured outcomes were self-reported gastrointestinal symptoms (abdominal pain, diarrhea). Outcomes at baseline and last follow-up, and their change from baseline to last follow-up, were assessed. Relevant data were available for 168 female patients. Mean age at the start of ERT was 43 years and mean treatment duration 5.7 years. Baseline pre-treatment abdominal pain was reported by 45% of females and diarrhea by 39%. At last follow-up, 31% reported abdominal pain (p < 0.01) and 27% diarrhea (p < 0.01). The results of this Fabry Registry analysis suggest that while on sustained treatment with agalsidase beta (1.0 mg/kg every 2 weeks), both abdominal pain and diarrhea improved in many female patients with Fabry disease.
Electronic supplementary material
The online version of this chapter (doi:10.1007/8904_2017_28) contains supplementary material, which is available to authorized users.
Keywords: Abdominal pain, Agalsidase beta, Diarrhea, Enzyme replacement therapy, Fabry disease, Fabry registry, Gastrointestinal symptoms
Introduction
Fabry disease (OMIM 301500) is an X-linked inherited lysosomal storage disorder caused by mutations in the GLA gene encoding the enzyme α-galactosidase (α-Gal) that lead to deficient or absent activity. This lack of activity results in the accumulation of globotriaosylceramide (GL-3) and other glycosphingolipids in the plasma and a variety of cell types including capillary endothelial, renal, cardiac, and nerve cells (Germain 2010). Glycolipid accumulation starts very early in life, is progressive, and triggers a range of cellular and tissue responses causing progressive damage to multiple organs. Life-threatening complications involving the kidneys, heart, and cerebrovascular system may start developing in the third decade of life (Wilcox et al. 2008; Germain 2010). The clinical presentation in female patients ranges from asymptomatic to, occasionally, a severe classic phenotype and depends in part on the mutation and the X-chromosome inactivation (Lyonization) profile (Germain 2010; Echevarria et al. 2016).
Gastrointestinal symptoms are among the most common and significant early clinical manifestations of Fabry disease, and may include abdominal pain, bloating, early satiety, intermittent/chronic diarrhea, constipation, recurrent nausea and vomiting, and poor weight gain (Ries et al. 2005; Ramaswami et al. 2006; Hoffmann et al. 2007; Hopkin et al. 2008; Buda et al. 2013; Politei et al. 2016). Studies among pediatric patients report abdominal pain as the most common gastrointestinal symptom (boys: 27–44%; girls: 17–27%) followed by diarrhea (boys: 19–33%; girls: 19–28%) (Ries et al. 2005; Ramaswami et al. 2006; Hopkin et al. 2008). Onset generally occurs earlier in boys than in girls (Ramaswami et al. 2006; Hopkin et al. 2008). These gastrointestinal symptoms tend to persist into adulthood if patients remain untreated (MacDermot et al. 2001; Hoffmann et al. 2007) and should therefore be assessed thoroughly and managed adequately to reduce their impact on quality of life.
Enzyme replacement therapies (ERTs) available for the treatment of patients with Fabry disease include agalsidase beta (Fabrazyme® [Sanofi Genzyme, Cambridge, MA, USA]; 1.0 mg/kg intravenously every 2 weeks) (Eng et al. 2001) and agalsidase alfa (Replagal® [Shire Human Genetic Therapies, Inc., Cambridge, MA, USA]; 0.2 mg/kg intravenously every 2 weeks) (Schiffmann et al. 2001). Both ERTs are available in the European Union and other countries worldwide, but only agalsidase beta is licensed in the USA.
To further understand the effects of sustained treatment with agalsidase beta treatment on gastrointestinal symptoms in female patients with Fabry disease, we analyzed data from the Fabry Registry.
Methods
The Fabry Registry (NCT00196742; sponsor: Sanofi Genzyme) is a multicenter, international, longitudinal, observational program designed to track the natural history and outcome of patients with Fabry disease (Eng et al. 2007; https://www.fabrycommunity.com/en/Healthcare/Registry.aspx). Patient and investigator participation is voluntary. To be included in the present Fabry Registry analysis, females of any age must have received agalsidase beta as their initial source of ERT given at the licensed dose of 1.0 mg/kg every 2 weeks (averaged dose; dose range: ≥0.9 to <1.1 mg/kg every 2 weeks), and have had a baseline (defined as time of first agalsidase beta infusion) and at least one additional on-treatment (i.e., follow-up after ≥2.5 years of agalsidase beta treatment) assessment of abdominal pain and/or diarrhea. The observation interval extended from the time each patient began treatment until one of the following occurred: (1) their most recently reported data to the Fabry Registry through April 3, 2015; (2) agalsidase beta treatment was discontinued; or (3) agalsidase beta treatment was switched to another treatment.
Data analyzed included age at first Fabry symptom, at diagnosis, and at first agalsidase beta infusion, and duration of agalsidase beta follow-up. Measured outcomes were self-reported gastrointestinal symptoms (abdominal pain, diarrhea) since the last clinical assessment using responses “yes” [present] or “no” [not present] to the questions “Has the patient had abdominal pain since the last clinical assessment?” and “Has the patient had diarrhea since the last clinical assessment?” Patients were grouped by age at first agalsidase beta infusion: <30 years, 30–49 years, ≥50 years, and all ages; descriptive statistics were used to characterize the individuals within these age groups. Patients responding “yes” [present] or “no” [not present] at baseline and at ≥2.5 years of treatment were compared and p values calculated using a Chi-square distribution and the McNemar test statistic. Analyses of possible risk factors for lack of ERT response and occurrence of new gastrointestinal symptoms included occurrence of severe clinical events (as defined elsewhere [Hopkin et al. 2016]) before or within the first 6 months of ERT, presence of estimated glomerular filtration rate (eGFR) values ≤60 mL/min/1.73 m2 before or within the first 3 months of ERT (both chi-square), time from first symptom to first ERT, and time on ERT (both ANOVA). A p value <0.05 was considered to represent statistical significance. Statistical analyses were performed using SAS statistical software V.9.2 (SAS Institute, Cary, NC, USA).
Results
As of April 3, 2015 (cutoff date for the analysis), 2,564 female patients with Fabry disease had been enrolled in the Fabry Registry, of whom 895 had received agalsidase beta treatment and 168 had the data of interest (assessment of abdominal pain and/or diarrhea) at baseline and at ≥2.5 years of agalsidase beta treatment. Patient characteristics are summarized in Table 1. Of the 168 patients, 56% (n = 94) had a GLA mutation classified as “classic” in the Fabry disease mutation database (http://fabry-database.org/mutants/old.cgi) and 6% (n = 10) as later-onset mutations (p.N215S, n = 5; p.G35R; p.A97V; p.R112H; p.R301Q; p.I317T). Other mutations were either reported in the mutation database but not classified (10% [n = 17]) or not reported in the mutation database (14% [n = 23]). GLA mutations were not available for 14% (n = 24) of the patients.
Table 1.
Baseline characteristics and ERT follow-up time for the 168 female Fabry patients who had abdominal pain (A) and/or diarrhea (B) status data available
| (A) Abdominal pain | |||||
|---|---|---|---|---|---|
| Age category for ERT initiation | Abdominal pain data availablea | Age at first FD symptom, mean (SD)b | Age at FD diagnosis, mean (SD)b | Age at first ERT, mean (SD) | ERT follow-up time, mean (SD) |
| <30 years | 32 | 11.5 (6.5) | 16.3 (7.5) | 20.8 (6.1) | 5.4 (1.9) |
| 30–49 years | 75 | 17 (11.8) | 34.1 (10.5) | 41.1 (5.3) | 5.7 (2.1) |
| ≥50 years | 59 | 31.7 (19.9) | 50.9 (13.2) | 58.2 (5.6) | 5.8 (2.3) |
| Overall | 166 | 20 (15.8) | 36.5 (16.7) | 43.3 (14.5) | 5.7 (2.1) |
| (B) Diarrhea | |||||
|---|---|---|---|---|---|
| Age category for ERT initiation | Diarrhea data availablea | Age at first FD symptom, mean (SD)b | Age at FD diagnosis, mean (SD)b | Age at first ERT, mean (SD) | ERT follow-up time, mean (SD) |
| <30 years | 32 | 11.5 (6.5) | 16.3 (7.5) | 20.8 (6.1) | 5.5 (1.8) |
| 30–49 years | 71 | 17.5 (11.9) | 34.6 (9.7) | 41.1 (5.3) | 5.8 (2.2) |
| ≥50 years | 57 | 31.2 (19.9) | 50.5 (13.3) | 58.2 (5.6) | 5.8 (2.2) |
| Overall | 160 | 19.9 (15.6) | 36.5 (16.5) | 43.1 (14.6) | 5.7 (2.1) |
ERT enzyme replacement therapy with agalsidase beta, FD Fabry disease, SD standard deviation
aData available at baseline and ≥2.5 years of ERT
bAge at first Fabry disease symptom and age at Fabry disease diagnosis were not known for all included patients
Baseline and treatment follow-up data for abdominal pain and diarrhea are shown in Figs. 1 and 2, respectively. Significantly fewer patients reported abdominal pain at last follow-up than at baseline (45% vs. 31%, p < 0.01). Likewise, significantly fewer patients reported diarrhea at last follow-up compared with baseline (39% vs. 27%, p < 0.01). Of patients reporting abdominal pain or diarrhea at baseline, 55% and 57%, respectively, reported its absence at last follow-up. Of those reporting having no abdominal pain or diarrhea at baseline, 20% and 16%, respectively, reported its presence at last follow-up.
Fig. 1.
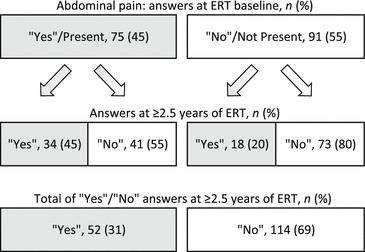
Abdominal pain responses (“yes” [present] or “no” [not present]) from 166 females with Fabry disease at treatment baseline and at ≥2.5 years of ERT. ERT enzyme replacement therapy with agalsidase beta
Fig. 2.
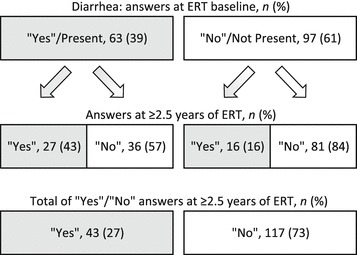
Diarrhea responses (“yes” [present] or “no” [not present]) from 160 females with Fabry disease at treatment baseline and at ≥2.5 years of ERT. ERT enzyme replacement therapy with agalsidase beta
The effect of age and severity on ERT response and occurrence of new gastrointestinal symptoms was analyzed and is presented in Supplementary Tables S1 and S2.
Discussion
This analysis represents the longest longitudinal follow-up of the effect of ERT on gastrointestinal symptoms in the largest cohort of female patients with Fabry disease (n = 168) reported to date. In this Fabry Registry cohort, 45% of the females had reported abdominal pain and 39% diarrhea at the start of agalsidase beta treatment, with improvements to 31% and 27%, respectively, after a mean duration of ERT with agalsidase beta of 5.7 years. As our study included only patients initiated on treatment following baseline assessments, these incidences cannot be directly compared with data from studies on the natural history of Fabry disease. Moreover, percentages of gastrointestinal symptoms in female Fabry patients vary throughout the literature (MacDermot et al. 2001; Hoffmann et al. 2007; Pensabene et al. 2016). In comparison, although scarce, epidemiological data on different abdominal symptoms in normal populations suggest that gastrointestinal symptoms are common. One large Danish population study found 1-year incidences of abdominal pain to range from 43 to 55% among females aged 30─60 years, with the vast majority reporting having pain weekly, monthly, or less frequently (Kay et al. 1994).
Of the females reporting abdominal pain or diarrhea at baseline in our analysis, more than half did not report these symptoms at last follow-up. A minority of patients reported no gastrointestinal symptoms at baseline, but did report abdominal pain (20%) and diarrhea (16%) at last follow-up. Whether the new symptoms are due to Fabry disease or unrelated causes is unknown. In a supplemental analysis for the effects of age and disease severity on response to ERT, diarrhea was more persistent in women aged ≥50 years with eGFR values ≤60 mL/min/1.73 m2 (p < 0.01, Supplementary Table S2). Abdominal pain appeared during treatment more frequently in females aged 30─49 years who had been on ERT longer (5.1 vs. 6.8 years, p = 0.04, Supplementary Table S1). However, given the small sample size, the clinical significance of these findings remains unclear.
Several studies have reported improvements in gastrointestinal symptoms with ERT in both children and adult patients with Fabry disease (Banikazemi et al. 2005; Hoffmann et al. 2007; Wraith et al. 2008; Ramaswami et al. 2012). However, only one study reported ERT follow-up data for adult female patients separately (Hoffmann et al. 2007). This Fabry Outcome Survey registry study found that 2 years of agalsidase alfa treatment in 25 female patients reduced the incidence of abdominal pain from 40 to 20%, while the incidence of diarrhea increased compared with baseline (12–16%).
Knowledge about the pathophysiology of gastrointestinal symptoms in Fabry disease and mechanisms of action of ERT on these symptoms remains incomplete. Deposition of GL-3 in the mesenteric vascular endothelium, autonomic ganglia cells of Meissner’s and Auerbach’s plexus, and smooth muscle cells is believed to contribute to Fabry disease pathology in the gastrointestinal tract, and lead to endothelial dysfunction, vasculopathy, neuropathy, and myopathy, and subsequently to delayed gastric emptying and intestinal dysmotility (MacDermot et al. 2001; Buda et al. 2013).
Although data are lacking, it can be hypothesized that initiation of ERT exerts an effect on gastrointestinal symptoms by (partially) clearing GL-3 from cells in the gastrointestinal system and preserving cellular function or ameliorating cellular dysfunction. Complete clearance of GL-3 from endothelial cells and other cell types, improvement in small nerve fiber function, and quality of life have been reported in studies on agalsidase beta (Eng et al. 2001; Hilz et al. 2004; Germain et al. 2007; Watt et al. 2010). Indirect evidence suggests that a higher ERT dose may be of clinical benefit in ameliorating gastrointestinal symptoms. A recent study showed that patients treated with agalsidase alfa experienced more gastrointestinal pain than those treated with agalsidase beta (at reduced or licensed dose) (Lenders et al. 2016).
One of the challenges regarding the study of gastrointestinal symptoms in real-world clinical practice is that the symptoms evolve and change (e.g., progress and remit), making it difficult to design a study which would inarguably demonstrate that the response, or lack thereof, is due to the therapy rather than the passage of time or other factors. This holds especially for the study of female patients, where gastrointestinal symptoms before and during menses could be a confounder in studying these symptoms due to Fabry disease (Bernstein et al. 2014).
If gastrointestinal symptoms are the first and only manifestations, the diagnosis of Fabry disease may be challenging particularly if there is no positive family history. Misdiagnoses include irritable bowel syndrome, chronic inflammatory bowel disease (IBD), appendicitis, autoimmune disorders, Whipple’s disease, dermatomyositis, Crohn’s disease, celiac disease, and colon cancer (Buda et al. 2013; Politei et al. 2016). However, these medical conditions may coincide with Fabry disease, as Fabry patients in the authors’ clinics have been correctly diagnosed with concomitant autoimmune IBD, lactose intolerance, gluten enteropathy, bacterial overgrowth, pancreatitis, neuroendocrine bowel tumor, and pheochromocytoma.
Limitations associated with using data from the Fabry Registry must be considered in the present analysis. Not all female patients with Fabry disease have been diagnosed and enrolled; however, it is likely that the more severely affected patients are being treated with ERT and are enrolled and followed in the Registry. The Fabry Registry contains observational data of Fabry patients only. It is not a prospective clinical trial and data interpretation is limited by the lack of an appropriately matched control group. For example, untreated women have not been analyzed to investigate how their gastrointestinal problems change over time. Relatively crude measures of self-reported abdominal pain and diarrhea have been used in the analysis, because sufficient data on change in frequency and intensity of abdominal pain and change in frequency of diarrhea are not available. These measures may be biased by recall, patient (mis)perception, and lack of specific definitions for the symptoms of interest. Although the Fabry Registry provides a recommended schedule of assessments, patients and treating physicians determine which assessments the patients will receive and the time intervals at which they will be conducted. Patients may have incompletely reported medical histories, may be lost to follow-up, or may have used concomitant medications to alleviate gastrointestinal symptoms. Finally, the analysis does not assess non-Fabry-related causes of gastrointestinal symptoms: sufficient data on the use of concomitant medications were not available for analysis, nor were patient records for other causes of gastrointestinal symptoms.
Conclusions
This analysis represents the longest longitudinal follow-up of the effect of ERT on gastrointestinal symptoms in the largest cohort of female patients with Fabry disease reported to date. Commonly reported gastrointestinal symptoms prior to initiation of agalsidase beta treatment included abdominal pain and diarrhea. The results of this Fabry Registry analysis suggest that sustained treatment with agalsidase beta (1.0 mg/kg every 2 weeks for a mean of 5.7 years) improves both abdominal pain and diarrhea in many female patients. Women who do not respond or who develop new abdominal symptoms on ERT should be investigated for alternate causes. Further studies are needed to better understand the pathology underlying the gastrointestinal symptoms and the mechanisms of action of ERT in the treatment of these symptoms. Gastrointestinal symptoms should be assessed thoroughly and managed adequately using available adjunctive therapies to reduce their impact on the patient’s well-being.
Electronic Supplementary Material
Analysis of possible risk factors for lack of ERT response and occurrence of new abdominal pain (DOCX 39 kb)
Analyses of possible risk factors for lack of ERT response and occurrence of new diarrhea (DOCX 38 kb)
Acknowledgments
The authors would like to thank the many patients who have agreed to participate in the Fabry Registry as well as the physicians and research coordinators who have entered clinical data on behalf of these patients.
Synopsis
Enzyme replacement therapy with agalsidase beta alleviates both abdominal pain and diarrhea in a significant proportion of female patients with Fabry disease.
Compliance with Ethics Guidelines
Conflict of Interest
See the financial activities section below.
This research was funded by Sanofi Genzyme, the sponsor of the Fabry Registry.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committees on human experimentation (institutional and national) and with the Helsinki Declaration of 1975 (revised in 2000). Each independent Fabry Registry site is responsible for obtaining a patient’s informed written consent to submit their health information to the Registry, and to use and disclose this information in subsequent aggregate analyses.
Author Contributions
William R. Wilcox, Ulla Feldt-Rasmussen, Alberto Ortiz, Dominique P. Germain, Frank Weidemann, and Robert J. Hopkin contributed to the concept/design of the manuscript, data acquisition, data analysis/interpretation, drafting the manuscript, and critically revising it.
Ana Maria Martins, Ana Jovanovic, Carmen Varas, and Katherine Nicholls contributed to the data acquisition, data analysis/interpretation, drafting the manuscript, and critically revising it.
Roberta M. Lemay contributed to the concept/design of the manuscript, data analysis/interpretation, drafting the manuscript, and critically revising it.
Financial Relationships for the Work Under Consideration for Publication
The authors received medical writing/editing support in the preparation of this manuscript from Alessia Piazza and Tom Rouwette of Excerpta Medica, funded by Sanofi Genzyme. The authors also thank Hans Ebels for providing medical writing/editing services and Badari Gudivada for providing programming support on behalf of Sanofi Genzyme. A. Ortiz was supported by ISCIII intensificación de la actividad investigadora. This research was funded by Sanofi Genzyme, the sponsor of the Fabry Registry.
Relevant Financial Activities Outside the Submitted Work
William R. Wilcox consults for Sanofi Genzyme and is an investigator in clinical studies and trials sponsored by Sanofi Genzyme, Shire HGT, Amicus Therapeutics, and Protalix Corporation. These activities are monitored and are in compliance with the conflict of interest policies at Emory University School of Medicine.
Ulla Feldt-Rasmussen has received honoraria for lectures and participation in advisory boards from Sanofi Genzyme, Shire HGT, and Amicus Therapeutics, and has received research funds from Sanofi Genzyme and Shire HGT.
Ana Maria Martins is a member of the Fabry Registry Board of Advisors and has received research funds, travel support, and speaking fees from Sanofi Genzyme.
Alberto Ortiz is a consultant for Sanofi Genzyme and has received speaker fees from Shire HGT.
Roberta M. Lemay is an employee of Sanofi Genzyme.
Ana Jovanovic is a member of the Fabry Registry Board of Advisors and has received advisory board fees and honoraria for lectures from Sanofi Genzyme, Shire HGT, and Amicus Therapeutics.
Dominique P. Germain is a consultant for Sanofi Genzyme and Shire HGT and has received honoraria for lectures from Sanofi Genzyme, Shire HGT, and Amicus Therapeutics.
Carmen Varas is a member of the Fabry Registry Board of Advisors.
Katherine Nicholls has received research support and travel grants from Sanofi Genzyme, Amicus Therapeutics, and Shire HGT, and speaker honoraria from Sanofi Genzyme and Shire HGT.
Frank Weidemann has received speaker honoraria from Sanofi Genzyme and Shire HGT, is a member of the Fabry Registry European Board of Advisors, and has received travel assistance and speaker honoraria. Research grants were given to his institution by Sanofi Genzyme and Shire HGT.
Robert J. Hopkin consults with Sanofi Genzyme and Shire HGT and has been an investigator in clinical trials sponsored by Sanofi Genzyme, Shire HGT, and Amicus Therapeutics. These activities have been monitored and found to be in compliance with the conflict of interest policies at Cincinnati Children’s Hospital Medical Center.
Contributor Information
William R. Wilcox, Email: william.wilcox@emory.edu
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Banikazemi M, Ullman T, Desnick RJ. Gastrointestinal manifestations of Fabry disease: clinical response to enzyme replacement therapy. Mol Genet Metab. 2005;85:255–259. doi: 10.1016/j.ymgme.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Bernstein MT, Graff LA, Avery L, Palatnick C, Parnerowski K, Targownik LE. Gastrointestinal symptoms before and during menses in healthy women. BMC Womens Health. 2014;14:14. doi: 10.1186/1472-6874-14-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buda P, Książyk J, Tylki-Szymanska A. Gastroenterological complications of Anderson-Fabry disease. Curr Pharm Des. 2013;19:6009–6013. doi: 10.2174/13816128113199990347. [DOI] [PubMed] [Google Scholar]
- Echevarria L, Benistan K, Toussaint A, et al. X-chromosome inactivation in female patients with Fabry disease. Clin Genet. 2016;89:44–54. doi: 10.1111/cge.12613. [DOI] [PubMed] [Google Scholar]
- Eng CM, Fletcher J, Wilcox WR, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007;30:184–192. doi: 10.1007/s10545-007-0521-2. [DOI] [PubMed] [Google Scholar]
- Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase A – replacement therapy in Fabry’s disease. N Engl J Med. 2001;345:9–16. doi: 10.1056/NEJM200107053450102. [DOI] [PubMed] [Google Scholar]
- Fabry disease mutation database (H. Sakuraba). http://fabry-database.org/mutants/old.cgi. Accessed 7 Apr 2016
- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. doi: 10.1186/1750-1172-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain DP, Waldek S, Banikazemi M, et al. Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J Am Soc Nephrol. 2007;18:1547–1557. doi: 10.1681/ASN.2006080816. [DOI] [PubMed] [Google Scholar]
- Hilz MJ, Brys M, Marthol H, Stemper B, Dütsch M. Enzyme replacement therapy improves function of C-, Adelta-, and Abeta-nerve fibers in Fabry neuropathy. Neurology. 2004;62:1066–1072. doi: 10.1212/01.WNL.0000118207.84514.40. [DOI] [PubMed] [Google Scholar]
- Hoffmann B, Schwarz M, Mehta A, Keshav S. Gastrointestinal symptoms in 342 patients with Fabry disease: prevalence and response to enzyme replacement therapy. Clin Gastroenterol Hepatol. 2007;5:1447–1453. doi: 10.1016/j.cgh.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Hopkin RJ, Bissler J, Banikazemi M, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr Res. 2008;64:550–555. doi: 10.1203/PDR.0b013e318183f132. [DOI] [PubMed] [Google Scholar]
- Hopkin RJ, Cabrera G, Charrow J, et al. Risk factors for severe clinical events in male and female patients with Fabry disease treated with agalsidase beta enzyme replacement therapy: data from the Fabry Registry. Mol Genet Metab. 2016;119:151–159. doi: 10.1016/j.ymgme.2016.06.007. [DOI] [PubMed] [Google Scholar]
- Kay L, Jørgensen T, Jensen KH. Epidemiology of abdominal symptoms in a random population: prevalence, incidence, and natural history. Eur J Epidemiol. 1994;10:559–566. doi: 10.1007/BF01719573. [DOI] [PubMed] [Google Scholar]
- Lenders M, Canaan-Kühl S, Krämer J, et al. Patients with Fabry disease after enzyme replacement therapy dose reduction and switch-2-year follow-up. J Am Soc Nephrol. 2016;27:952–962. doi: 10.1681/ASN.2015030337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001;38:769–775. doi: 10.1136/jmg.38.11.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensabene L, Sestito S, Nicoletti A, Graziano F, Strisciuglio P, Concolino D. Gastrointestinal symptoms of patients with Fabry disease. Gastroenterol Res Pract. 2016;2016:9712831. doi: 10.1155/2016/9712831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politei J, Thurberg BL, Wallace E, Warnock D, Serebrinsky G, Durand C, Schenone AB. Gastrointestinal involvement in Fabry disease. So important, yet often neglected. Clin Genet. 2016;89:5–9. doi: 10.1111/cge.12673. [DOI] [PubMed] [Google Scholar]
- Ramaswami U, Whybra C, Parini R, et al. Clinical manifestations of Fabry disease in children: data from the Fabry Outcome Survey. Acta Paediatr. 2006;95:86–92. doi: 10.1080/08035250500275022. [DOI] [PubMed] [Google Scholar]
- Ramaswami U, Parini R, Pintos-Morell G, Kalkum G, Kampmann C, Beck M. Fabry disease in children and response to enzyme replacement therapy: results from the Fabry Outcome Survey. Clin Genet. 2012;81:485–490. doi: 10.1111/j.1399-0004.2011.01671.x. [DOI] [PubMed] [Google Scholar]
- Ries M, Gupta S, Moore DF, et al. Pediatric Fabry disease. Pediatrics. 2005;115:e344–e355. doi: 10.1542/peds.2004-1678. [DOI] [PubMed] [Google Scholar]
- Schiffmann R, Kopp JB, Austin HA, 3rd, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285:2743–2749. doi: 10.1001/jama.285.21.2743. [DOI] [PubMed] [Google Scholar]
- Watt T, Burlina AP, Cazzorla C, et al. Agalsidase beta treatment is associated with improved quality of life in patients with Fabry disease: findings from the Fabry Registry. Genet Med. 2010;12:703–712. doi: 10.1097/GIM.0b013e3181f13a4a. [DOI] [PubMed] [Google Scholar]
- Wilcox WR, Oliveira JP, Hopkin RJ, et al. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab. 2008;93:112–128. doi: 10.1016/j.ymgme.2007.09.013. [DOI] [PubMed] [Google Scholar]
- Wraith JE, Tylki-Szymanska A, Guffon N, et al. Safety and efficacy of enzyme replacement therapy with agalsidase beta: an international, open-label study in pediatric patients with Fabry disease. J Pediatr. 2008;152:563–570. doi: 10.1016/j.jpeds.2007.09.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analysis of possible risk factors for lack of ERT response and occurrence of new abdominal pain (DOCX 39 kb)
Analyses of possible risk factors for lack of ERT response and occurrence of new diarrhea (DOCX 38 kb)