

ABSTRACT
Porcine epidemic diarrhea virus (PEDV) causes acute and devastating enteric disease in suckling piglets and results in huge economic losses in the pig industry worldwide. To establish productive infection, viruses must first circumvent the host innate immune response. In this study, we found that PEDV infection stimulated epidermal growth factor receptor (EGFR) activation, which has been linked to not only anticancer therapeutics, but also antiviral signaling. Therefore, we determined whether EGFR activation affected PEDV infection by using an activator or overexpression assay. The data showed that EGFR activation enhanced virus replication in both cases. We also found that specific inhibition of EGFR by either inhibitors or small interfering RNA (siRNA) led to a decrease in virus yields. Further analysis revealed that inhibition of EGFR produced augmentation of type I interferon genes. We next observed that the EGFR downstream cascade STAT3 was also activated upon PEDV infection. Similar to the case of EGFR, specific inhibition of STAT3 by either inhibitor or siRNA increased the antiviral activity of interferon and resulted in decreased PEDV RNA levels, and vice versa. The data on STAT3 depletion in combination with EGFR activation suggest that the attenuation of antiviral activity by EGFR activation requires activation of the STAT3 signaling pathway. Taken together, these data demonstrate that PEDV-induced EGFR activation serves as a negative regulator of the type I interferon response and provides a novel therapeutic target for virus infection.
IMPORTANCE EGFR is a transmembrane tyrosine receptor that mediates various cellular events, as well as several types of human cancers. In this study, we investigated for the first time the role of EGFR in PEDV infection. We observed that PEDV infection induced EGFR activation. The role of EGFR activation is to impair the antiviral activity of type I interferon, which requires the involvement of the EGFR downstream signaling cascade STAT3. Our findings reveal a new mechanism evolved by PEDV to circumvent the host antiviral response, which might serve as a therapeutic target against virus infection.
KEYWORDS: EGFR, PEDV, interferons
INTRODUCTION
Porcine epidemic diarrhea virus (PEDV) is an enveloped, single-stranded, positive-sense RNA virus that belongs to the genus Alphacoronavirus in the family Coronaviridae, order Nidovirales (1). PEDV is the etiological agent of porcine epidemic diarrhea (PED), which is characterized by watery diarrhea, vomiting, and dehydration in infected swine (2, 3). Although reported initially in a few countries in the late 1970s, PED now occurs worldwide in most major swine-raising countries, causing huge economic losses, as well as public health concerns (4–9).
During viral infection, the most important pathological characteristics of PED are acute destruction of intestinal villous enterocytes and villous atrophy in the jejunum and ileum (10, 11). We and others have reported that the integrity of the intestinal epithelium is impaired, and the structural destruction and disorganization of tight-junction proteins in PEDV-infected cells are observed (12–14). Epidermal growth factor receptor (EGFR) is expressed in a wide spectrum of epithelial cells, and EGFR-dependent signaling is involved in the disassembly of epithelial tight junctions and epithelial barrier permeability alteration (15–19).
EGFR is a transmembrane glycoprotein that is activated by binding to its cognate ligands, such as epidermal growth factor (EGF), transforming growth factor α, amphiregulin, heparin-binding EGF, and betacellulin (20). EGFR is a receptor tyrosine kinase, serving as a homeostatic regulator of cell proliferation, differentiation, adhesion, and survival (21, 22). Since EGFR is overexpressed in the majority of solid tumors, much effort has been directed at developing anticancer agents that can interfere with EGFR activity (23–25). Most recently, emerging evidence indicated that EGFR and its downstream pathways play a critical role in the outcome of virus infection by modulating innate immunity (26, 27). For example, inhibition of EGFR function during hepatitis C virus (HCV) infection upregulates the expression of interferon-stimulated genes and boosts the antiviral efficacy of interferon (28). Respiratory virus infection-induced EGFR activation suppresses endogenous airway epithelial antiviral signaling (29). Venkataraman et al. have reported that EGFR signaling is a key regulator of severe acute respiratory syndrome coronavirus (SARS-CoV)-induced lung damage (30). However, the role of EGFR in PEDV infection has not been defined. In this study, we examined the relationship between EGFR signaling and PEDV infection. We observed that PEDV infection activates EGFR and its downstream pathways, which increase viral infection by negatively regulating type I interferon (IFN-I) signaling. In addition, functional inhibition of EGFR during PEDV infection augmented the host antiviral response and resulted in a reduction in virus titers. The work presented here advances our understanding of the role of EGFR in regulating virus infection.
RESULTS
PEDV induces EGFR activation.
To investigate whether PEDV infection is able to activate EGFR, we measured EGFR phosphorylation by Western blotting (31, 32) after virus infection in IPEC-J2 cells (a porcine intestinal epithelial cell line). Levels of total EGFR were comparable between PEDV-infected and mock-treated cells, whereas incubation with PEDV induced EGFR phosphorylation within 5 min and reached maximum at 15 min (Fig. 1A). We next examined whether the phosphorylation of EGFR induced by PEDV was limited to the specific cell type used. To this end, we repeated these experiments with HEK293 and Vero E6 cells (33, 34). Similarly, the levels of phosphorylated EGFR (phospho-EGFR) were increased in PEDV-infected HEK293 (Fig. 1B) and Vero E6 (Fig. 1C) cells. These results demonstrated that PEDV infection augments EGFR activation without affecting total EGFR expression.
FIG 1.

(A to C) PEDV infection induces EGFR phosphorylation. IPEC-J2, HEK293, and Vero E6 cells were incubated with PEDV at an MOI of 1 for 2 h at 4°C. Unbound viruses were removed with PBS, and the cells were then cultured at 37°C for different times as indicated. Mock-infected cells were used as a control. Detergent lysates collected from the cells were directly subjected to reducing SDS-PAGE and blotted with MAbs against phospho-EGFR (pEGFR), EGFR, and β-actin. (D) UV-PEDV incubation activates EGFR. IPEC-J2 and HEK293 cells were incubated with UV-PEDV at an MOI of 1, and the cells were then further cultured for the indicated times. The cell lysates were subjected to immunoblotting with the antibodies indicated. (E) PEDV S-Flag protein interacts with EGFR. HEK293T cells were cotransfected with PEDV S-Flag (S) and EGFR (E). At 36 h after transfection, immunoprecipitation (IP) and immunoblotting were performed as described in Materials and Methods to examine interactions between PEDV S-Flag and EGFR.
To characterize the molecular events initiated by PEDV infection, we used UV irradiation-inactivated PEDV (UV-PEDV) as a control. Of interest, we observed an increase in phosphorylated EGFR in UV-PEDV-infected cells (Fig. 1D), which is similar to the case with replication-competent PEDV, suggesting that PEDV replication is not required for the induction of EGFR activation in target cells. UV-PEDV inactivation was verified by plaque assay (data not shown). Since there was no viral protein synthesized in UV-PEDV-infected cells, we hypothesized that the early steps of the virus life cycle, such as attachment, entry, and uncoating, could result in the virion-induced phosphorylation of EGFR. It is known that coronavirus spike (S) protein attaches to the cellular receptor to mediate viral entry (35, 36); therefore, we examined the interaction between PEDV S protein and EGFR using a coimmunoprecipitation (co-IP) assay in HEK293 cells. As shown in Fig. 1E, the existence of EGFR was detected only in the presence of PEDV S protein and not in the presence of empty vector, suggesting that PEDV surface S protein can activate EGFR by direct interaction. Together, these findings demonstrate that PEDV induces EGFR activation at the early stage of viral infection.
EGFR activation augments PEDV infection.
To characterize the role of EGFR activation in PEDV infection, EGF, a physical stimulus, was added to the target cells. EGF binding to EGFR can result in receptor dimerization, autophosphorylation, and activation of the intrinsic protein tyrosine kinase activity (20, 37). However, it has been reported that prolonged stimulation with EGF can cause ligand-induced EGFR degradation (38, 39), so the influences of treatment time with EGF on EGFR phosphorylation and total EGFR expression were compared by Western blotting. As shown in Fig. 2A, phosphorylation of EGFR was induced at 5 min and remained stable until 15 min in IPEC-J2 and HEK293 cells, while the levels of total EGFR had no obvious change over time. Therefore, stimulation with EGF for 15 min was conducted to activate EGFR for subsequent experiments unless otherwise stated. Next, IPEC-J2 cells were pretreated with EGF for 15 min and then infected with PEDV for an additional 48 h. We observed that viral RNA levels were significantly increased in EGF-treated cells compared with control cells, and virus titers were also increased, as measured by the 50% tissue culture infective dose (TCID50) (Fig. 2B). Western blot analysis confirmed that EGFR was activated and total EGFR levels remained constant upon EGF treatment (Fig. 2C). To confirm the effect of EGFR activation on PEDV infection, we repeated these experiments with HEK293 cells. Consistent with the findings obtained with IPEC-J2 cells, induction of EGFR phosphorylation by EGF significantly enhanced virus replication in HEK293 cells (Fig. 2D and E).
FIG 2.

EGFR activation enhances PEDV replication. (A) EGF treatment induces the phosphorylation of EGFR. IPEC-J2 and HEK293 cells were serum starved for 12 h and then incubated with 10 ng/ml EGF at 37°C for the indicated times. The cell lysates were blotted with antibodies against pEGFR, EGFR, and β-actin as indicated. (B and D) EGFR activation facilitates virus infection. IPEC-J2 (B) and HEK293 (D) cells were stimulated with 10 ng/ml EGF for 15 min. After washing, the cells were infected with mock control or PEDV for 48 h or 24 h, respectively. Total RNA was extracted from the cells, and PEDV RNA levels were assessed by quantitative RT-PCR using the primers listed in Table 1. Virus yields were determined by TCID50 assay. The results are representative of three independent experiments (means and SD). *, P < 0.05. The P value was calculated using Student's t test. (C and E) EGF treatment induces EGFR activation. Cells were treated with 10 ng/ml EGF at 37°C for 15 min, and the cell lysates were blotted with antibodies as indicated.
To further confirm that EGFR activation increases PEDV replication, we overexpressed EGFR in IPEC-J2 cells to specifically activate EGFR (40), and the cells were then infected with PEDV. As shown in Fig. 3A, EGFR overexpression enhanced the accumulation of viral N protein. Quantitative reverse transcription (RT)-PCR results showed that EGFR overexpression increased the levels of viral RNA, in contrast to the vector control (Fig. 3B). Similar to the results observed in IPEC-J2 cells, HEK293 cells treated with EGFR overexpression facilitated PEDV replication (Fig. 3C and D). In addition, results of TCID50 assays in both IPEC-J2 (Fig. 3E) and HEK293 (Fig. 3F) cells confirmed the positive effect of EGFR overexpression on the PEDV titers in comparison with the vector control. Taken together, these data indicate that virus-induced EGFR activation may facilitate PEDV infection.
FIG 3.
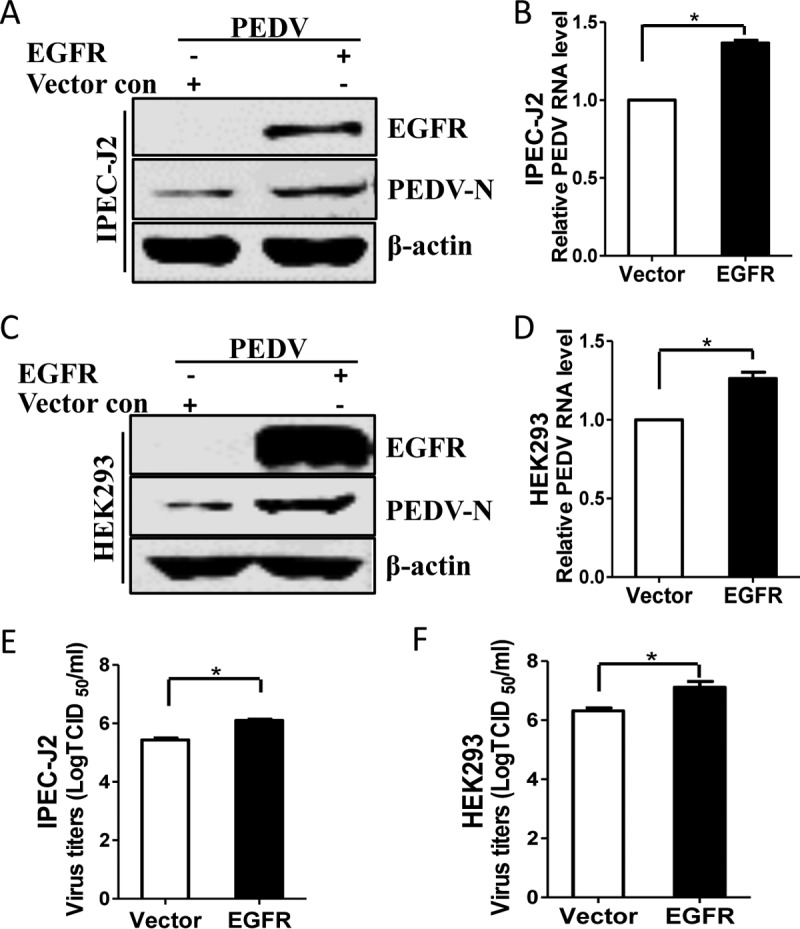
Overexpression of EGFR facilitates PEDV infection. (A and C) IPEC-J2 (A) and HEK293 (C) cells were transfected with pAAV/EGFR and pAAV vector control (con) for 24 h. The cells were then infected with PEDV for 48 h or 24 h. Detergent lysates were subjected to immunoblotting with the indicated antibodies. (B and D) EGFR overexpression enhances the levels of PEDV RNA. Total RNA was extracted from IPEC-J2 cells at 48 h postinfection (hpi) (B) or from HEK293 cells at 24 hpi (D) and analyzed by quantitative RT-PCR. (E and F) EGFR overexpression promotes PEDV titers. Virus samples were collected from IPEC-J2 cells at 48 hpi (E) or from HEK293 cells at 24 hpi (F) and measured by TCID50 assay. The results are representative of three independent experiments (means and SD). *, P < 0.05. The P value was calculated using Student's t test.
Pharmacological inhibition of EGFR decreases the yield of PEDV.
To further examine the effect of EGFR on PEDV infection, we next exposed cells to the inhibitors erlotinib and gefitinib, both of which can inhibit EGFR activation (41–43). The cytotoxic effects of erlotinib and gefitinib were examined with a Cell Counting Kit-8 (CCK-8) system (Dojindo Laboratory, Kumamoto, Japan) on IPEC-J2 and HEK293 cells. No significant toxicity of these two inhibitors was evident for the two types of cells at concentrations of <25 μM (Fig. 4A). The inhibitory effects of erlotinib and gefitinib on EGFR activation were also confirmed by Western blotting of cell lysates from EGF-treated cells in the presence or absence of 10 μM erlotinib or gefitinib (Fig. 4A, left). Thus, IPEC-J2 cells were pretreated with 10 μM erlotinib or gefitinib for 12 h and then infected with PEDV. As shown by immunofluorescence assay, the number of PEDV-positive cells was significantly lower in erlotinib- or gefitinib-treated IPEC-J2 cells than in a mock-treated control (Fig. 4B). Challenging cells with either erlotinib or gefitinib decreased viral protein synthesis (Fig. 4C) and viral RNA levels (Fig. 4D) in a concentration-dependent manner. The reduced titers of viruses in infected cell cultures containing erlotinib or gefitinib were further analyzed and confirmed by measuring the TCID50 (Fig. 4E). Consistent with the findings obtained with IPEC-J2 cells, the EGFR inhibitors erlotinib and gefitinib also had similar inhibitory effects on PEDV replication in HEK293 cells (Fig. 4), suggesting that virus-induced EGFR activation is involved in virus infection.
FIG 4.

EGFR inhibitors reduce PEDV infection. (A) Effects of inhibitors on EGFR function. IPEC-J2 and HEK293 cells were treated with the carrier control DMSO or EGFR-specific inhibitors, erlotinib (Erl) and gefitinib (Gef), at different concentrations for 72 h. Cell cytotoxicity was analyzed with the CCK-8 system as described in Materials and Methods. IPEC-J2 and HEK293 cells were also pretreated with an EGFR-specific inhibitor, Erl or Gef, at 10 μM or with DMSO for 12 h, followed by EGF stimulation (10 ng/ml) for 15 min. The levels of pEGFR and β-actin were analyzed by Western blotting. (B) EGFR inhibitors decrease the number of PEDV-positive cells. IPEC-J2 and HEK293 cells were pretreated with DMSO or EGFR inhibitors, Erl and Gef, at 10 μM for 12 h. After washing, the cells were infected with PEDV or mock control in the absence or presence of inhibitors. At 48 hpi or 24 hpi, the cell monolayers were fixed and examined for PEDV infection by IFA with an anti-PEDV spike protein MAb (3F12). The number of PEDV-positive cells was calculated. (C) Reduction of PEDV N protein by EGFR inhibitors is concentration dependent. Detergent lysates collected from IPEC-J2 and HEK293 cells were subjected to immunoblotting with antibodies as indicated. (D) EGFR inhibitors decreased PEDV RNA levels, as determined by quantitative RT-PCR. (E) Virus titers were reduced after inhibitor treatment, as detected by TCID50 assay. The results are representative of three independent experiments (means and SD). *, P < 0.05; ns, not significant. The P value was calculated using Student's t test.
Knockdown of endogenous EGFR expression reduces PEDV infection.
Our pharmacological data promoted further exploration of the relationship between EGFR activation and virus infection. Therefore, we used a specific small interfering RNA (siRNA) to knock down the endogenous expression of EGFR. When IPEC-J2 cells were transfected with EGFR-specific siRNA, the EGFR mRNA level was significantly decreased relative to that with control siRNA, and Western blot analysis of detergent lysates collected from cells transfected with EGFR siRNA revealed a clear reduction in the level of EGFR protein relative to that with control siRNA (Fig. 5A). At 24 h post-siRNA transfection, IPEC-J2 cells were inoculated with PEDV for an additional 48 h. We observed that the levels of viral protein were greatly decreased in the EGFR-specific siRNA transfection group compared with the control siRNA group (Fig. 5A). Knockdown of endogenous EGFR with siRNA also reduced virus loads as measured by quantitative RT-PCR and TCID50 assay (Fig. 5B). Consistent with the results in IPEC-J2 cells, we found that EGFR-specific siRNA significantly decreased the levels of virus titers in HEK293 cells, as well (Fig. 5C and D). Taken together, these data demonstrate that EGFR expression and function are relevant for PEDV infection and that EGFR is a host factor for PEDV infection.
FIG 5.
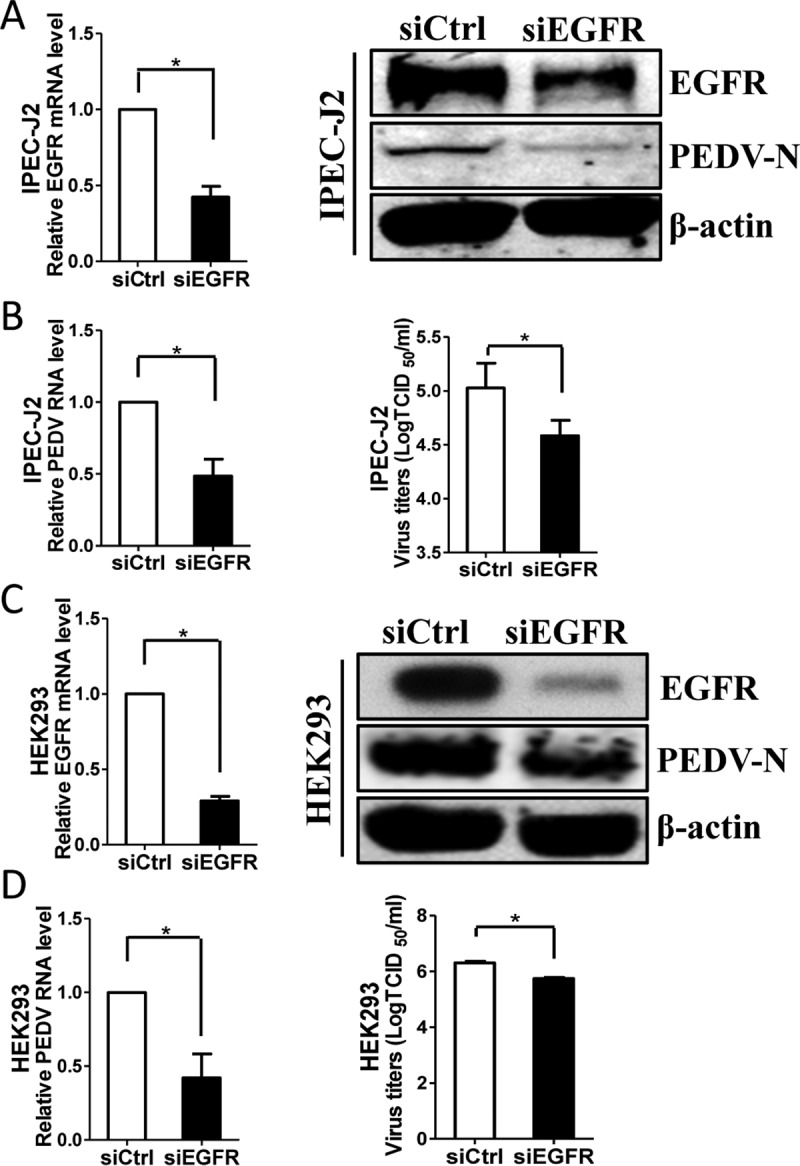
Knockdown of EGFR expression decreases PEDV infection. (A and C) Verification of EGFR knockdown efficiency. IPEC-J2 (A) and HEK293 (C) cells were transfected with EGFR-specific siRNA (siEGFR) or scrambled control siRNA (siCtrl) for 24 h, and the knockdown efficiency of EGFR was determined by quantitative RT-PCR and Western blotting. (B and D) Depletion of endogenous EGFR inhibited PEDV replication. After siRNA transfection for 24 h, cells were exposed to virus for 48 h (IPEC-J2) (B) or 24 h (HEK293) (D). The effect of EGFR knockdown on PEDV infection was determined by Western blotting (A and C, right), quantitative RT-PCR, and TCID50 assay. The results are representative of three independent experiments (means and SD). *, P < 0.05. The P value was calculated using Student's t test.
Depletion of endogenous EGFR expression enhances the antiviral response of type I interferon.
Given that inhibition of EGFR can decrease virus infection, it is worthwhile to further investigate the molecular mechanisms. IFN-I is the key innate immune cytokine produced by cells to trigger antiviral function (44, 45). Previous reports showed that EGFR-mediated signaling impairs the antiviral activity of IFN-I through cross talk with the interferon signaling molecules (28, 29, 36, 46). Therefore, we assessed the effect of EGFR on the signaling pathway of IFN-I by using specific siRNA targeting EGFR. Here, the mRNA levels of several interferon-stimulated genes (ISGs), including myxovirus resistance A (MxA), ISG15, and IFN-β, were analyzed by quantitative RT-PCR (47). The results showed that the mRNA levels of three ISGs, MxA, ISG15, and IFN-β, were significantly increased in EGFR siRNA-transfected IPEC-J2 (Fig. 6A) and HEK293 (Fig. 6B) cells, in contrast to the control siRNA treatments, suggesting that inhibition of EGFR activation restores cellular antiviral activity and thus decreases virus infection.
FIG 6.
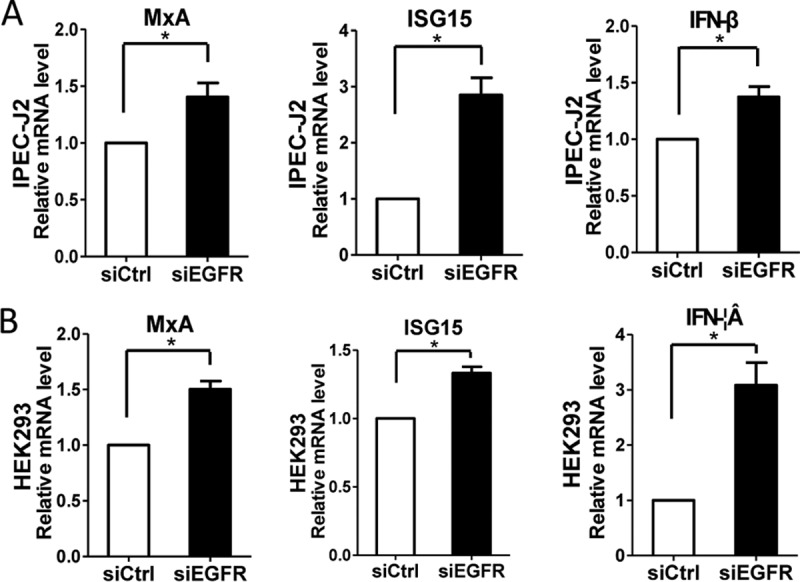
Depletion of EGFR expression enhanced the levels of several ISGs. IPEC-J2 (A) and HEK293 (B) cells were transfected with control siRNA or EGFR-specific siRNA for 24 h. Total RNA was extracted, and the mRNA levels of MxA, ISG15, and IFN-β were determined by quantitative RT-PCR. The results are representative of three independent experiments (means and SD). *, P < 0.05. The P value was calculated using Student's t test.
PEDV infection links to the activation of STAT3.
We next investigated the downstream mechanisms by which EGFR might regulate this cellular antiviral response. Studies in cancer models have suggested that EGFR physically interacts with signal transducers and activators of transcription 3 (STAT3), leading to the phosphorylation of STAT3 and subsequent gene regulation (48, 49). Therefore, we evaluated whether PEDV infection activates the EGFR downstream cascade STAT3 in target cells. Similar to the pattern of EGFR activation, the levels of phosphorylation of STAT3 were increased in the virus-infected IPEC-J2 (Fig. 7A) and HEK293 (Fig. 7B) cells. Previous reports have demonstrated that STAT3 negatively regulates the IFN-I-mediated antiviral response (50, 51), so we next determined the regulation effect of STAT3 on the IFN-I response. Here, S3I-201, an aminosalicylic compound, was added to the cell cultures to inhibit STAT3 phosphorylation and dimerization, thereby blocking its activation function (52). We observed that the mRNA levels of three ISGs, MxA, ISG15, and IFN-β, were significantly increased in IPEC-J2 (Fig. 7C) and HEK293 (Fig. 7D) cells in the presence of the STAT3 inhibitor S3I-201 at a concentration of 40 μM compared with a carrier control. Cells treated with the inhibitor S3I-201 at a concentration of <100 μM and untreated cells did not differ in cell morphology, viability, or numbers (Fig. 7E). These observations suggest that virus-induced STAT3 activation serves as a negative regulator of the IFN-I response.
FIG 7.

PEDV infection-induced STAT3 activation negatively regulates ISGs. (A and B) Infection with PEDV leads to STAT3 phosphorylation. IPEC-J2 (A) and HEK293 (B) cells were incubated with PEDV at an MOI of 1 for 2 h at 4°C. Unbound viruses were removed, and the cells were further incubated for different periods, as indicated. The cell lysates were blotted with the MAbs against phospho-STAT3 (pSTAT3), STAT3, and β-actin. The mock-infected cells were used as a control. (C and D) Inhibition of STAT3 function enhances antiviral responses. IPEC-J2 (C) and HEK293 (D) cells were treated with the STAT3-specific inhibitor S3I-201 (40 μM) for 24 h. The RNA levels of ISGs, MxA, ISG15, and IFN-β, were determined by quantitative RT-PCR. (E) Effect of STAT3 inhibitor on cell viability. IPEC-J2 and HEK293 cells were treated with S3I-201 at the indicated concentrations or with the carrier control DMSO for 72 h. Cell cytotoxicity was analyzed with the CCK-8 system as described in Materials and Methods. The results are representative of three independent experiments (means and SD). *, P < 0.05. The P value was calculated using Student's t test.
Role of STAT3 in PEDV infection.
To establish biological relevance, we investigated the consequences of interfering with STAT3 function for PEDV infection. IPEC-J2 and HEK293 cells were pretreated with the STAT3 inhibitor S3I-201, followed by virus inoculation. We found that S3I-201 treatment resulted in decreased expression of the viral N protein in a dose-dependent manner, as determined by Western blotting, and quantitative-RT-PCR analysis showed that S3I-201 treatment significantly decreased PEDV RNA levels in both IPEC-J2 and HEK293 cells in a concentration-dependent manner (Fig. 8A and B). To confirm the results with the inhibitor, a specific siRNA against STAT3 was introduced to knock down the endogenous expression of STAT3. A clear reduction of STAT3 mRNA and protein levels was observed in IPEC-J2 (Fig. 8C) and HEK293 (Fig. 8D) cells transfected with STAT3-specific siRNA, indicating that the STAT3 siRNA had worked properly. Under these conditions, the level of PEDV N protein was reduced in the STAT3 siRNA transfection group, and the reduced number of viruses in siRNA-transfected cells was confirmed by quantitative RT-PCR (Fig. 8C and D).
FIG 8.

STAT3 is involved in PEDV infection. (A and B) Reduction of virus loads by S3I-201 is concentration dependent. IPEC-J2 (A) and HEK293 (B) cells were treated with different concentrations of the STAT3 inhibitor S3I-201 for 24 h, followed by infection with PEDV. At 48 hpi (IPEC-J2) or 24 hpi (HEK293), virus infection was determined by Western blotting with antibodies to PEDV N protein and β-actin (loading control). Densitometric data for the PEDV N protein/actin ratio from three independent experiments are expressed as means and SD. Virus RNA levels were also assessed by quantitative RT-PCR. (C and D) Knockdown of STAT3 expression reduced PEDV infection. Cells were transfected with STAT3-specific siRNA for 24 h and then infected with PEDV for 48 h or 24 h. STAT3 knockdown efficiency was verified by quantitative RT-PCR and Western blotting. The effect of STAT3 knockdown on virus loads was determined by Western blotting and quantitative RT-PCR. (E and F) Overexpression of STAT3 increased PEDV infection. IPEC-J2 (E) and HEK293 (F) cells were transfected with pAAV/STAT3 or vector control. Twenty-four hours later, the cells were incubated with PEDV for an additional 48 h or 24 h. The levels of STAT3 protein and PEDV N protein were analyzed by Western blotting. Total RNA was also extracted from the cells, and the viral RNA levels were then assessed by quantitative RT-PCR. The results are representative of three independent experiments (means and SD). *, P < 0.05. The P value was calculated using Student's t test.
To further confirm the functional relevance of STAT3 to PEDV infection, STAT3 was overexpressed by transient-transfection assay. As indicated by Western blotting, the levels of viral protein were increased in STAT3-transfected IPEC-J2 and HEK293 cells compared with vector control-transfected cells (Fig. 8E and F). Furthermore, the viral RNA levels in both IPEC-J2 (Fig. 8E) and HEK293 (Fig. 8F) cells confirmed the positive effect of STAT3 overexpression on PEDV infection. Overall, these data indicate that virus-induced STAT3 activation impairs the host antiviral response against PEDV, which is similar to the results of the previous study of HCV infection (28).
PEDV-induced EGFR activation suppresses the host antiviral response via STAT3-mediated signaling.
A previous report demonstrated that EGFR activation can regulate the STAT3 signaling pathway through direct interaction (53), and we therefore examined the relationship between STAT3 and EGFR using EGF treatment. As shown in Fig. 9A, EGF incubation led to an increase of STAT3 phosphorylation in IPEC-J2 and HEK293 cells but no change in the levels of total STAT3 protein, suggesting that EGF induces rapid activation of STAT3. Under these conditions, we assessed the effect of the interaction between EGFR and STAT3 on PEDV infection. IPEC-J2 and HEK293 cells were first transfected with control siRNA or STAT3-specific siRNA for 24 h, and the cells were then cultured in the presence of EGF for 15 min. After washing, the cells were infected with PEDV for an additional 48 h or 24 h, and the cell lysates were collected for Western blot analysis. The results showed that EGF treatment increased viral protein synthesis and STAT3 siRNA decreased the levels of viral N protein, whereas the increase of viral N protein synthesis by activated EGFR was blocked by depletion of STAT3 expression (Fig. 9B and C). These findings indicate that virus-induced EGFR activation impairs the host antiviral response through the STAT3-mediated signaling pathway.
FIG 9.

PEDV-induced EGFR activation suppresses the host antiviral response via STAT3-mediated signaling. (A) EGF treatment induces phosphorylation of STAT3. Serum-starved IPEC-J2 and HEK293 cells were treated with 10 ng/ml EGF for 15 min. Then, the cells were lysed and subjected to Western blotting with antibodies against pSTAT3, STAT3, and β-actin protein. (B) Increase of viral N protein by EGF treatment is blocked by STAT3 siRNA in IPEC-J2 cells. Cells were transfected with control siRNA (siCtrl) or STAT3 siRNA (siST3) for 24 h, followed by stimulation with 10 ng/ml EGF for 15 min. The cells were then infected with PEDV for an additional 48 h. The cell lysates were blotted with antibodies as indicated. Densitometric data for the PEDV/actin ratio from three independent experiments are expressed as means and SD. (C) Increase of viral N protein by EGF treatment is blocked by STAT3 siRNA in HEK293 cells. The results are representative of three independent experiments (means and SD). *, P < 0.05. The P value was calculated using Student's t test.
DISCUSSION
EGFR is a transmembrane glycoprotein that was isolated over 30 years ago after the discovery of its ligand, EGF, in 1962 (54, 55). EGFR is best known for its classical function as a tyrosine kinase receptor and is widely distributed in epithelial cells. Activated EGFR recruits downstream signaling and plays pivotal roles in cell-cell communication. EGFR overexpression has been associated with a number of human cancers, and therefore, extensive efforts have been devoted to exploring anticancer therapeutics directed against EGFR (56). More recently, increasing evidence has shown that some viruses can induce EGFR activation and its subsequent signaling (27). In this study, we found that PEDV infection activated EGFR. This, in turn, suppressed the IFN-I signaling pathway and thus promoted viral infection, which was mediated by the EGFR downstream cascade STAT3. In addition, inhibition of EGFR during PEDV infection augmented IFN-I signaling, which resulted in a decrease in virus titers in target cells. These data suggest that virus-induced EGFR activation likely plays a role in the establishment of PEDV infection.
To infect its host, a virus must have some means of circumventing the IFN response, either by limiting IFN production or by blocking IFN functions (57). Like other viruses, PEDV must subvert the intrinsic antiviral defenses of target cells to produce progeny virions. We and others have reported that during PEDV infection virus can inhibit IFN production by encoding proteins that act as IFN antagonists or by restricting IFN functions through degrading IFN signaling molecules (33, 58–62). Here, we have found for the first time that PEDV infection induces activation of a receptor tyrosine kinase, EGFR, and its subsequent signaling. Several viruses, including rhinovirus, influenza virus, HCV, cytomegalovirus, and Epstein-Barr virus (EBV), have been shown to activate EGFR (46, 63–67). By using an activator or overexpression assay, we observed that activated EGFR led to an increase in the PEDV titer, whereas inhibition of EGFR activity by either inhibitors or siRNA resulted in a reduction of the PEDV titer. These findings indicate a positive regulation role of activated EGFR in virus infection. Further evidence from an siRNA knockdown assay showed that EGFR impaired the antiviral activity of type I interferon, suggesting the involvement of EGFR activation-mediated signaling pathways in virus infection. Our finding is similar to those described previously in studies of Pichinde virus, influenza A virus, EBV, and HCV (36, 65, 68, 69). This is another example of a coronavirus that directly impairs the host antiviral response. Several researchers have reported that inhibition of EGFR during viral infection resulted in decreased viral titers in vitro and in vivo (29, 70, 71), suggesting that inhibition of EGFR may result in decreased PEDV infection in pigs, as well. However, further work is needed to conclusively answer this question.
In the present study, by using the virions of UV-inactivated PEDV, we also found that virus replication is not required for EGFR activation. This suggests that binding of UV-inactivated virions to the cell surface may directly activate EGFR. A previous study revealed that UV-inactivated respiratory syncytial virus could induce mouse alveolar macrophages to secret interleukin-6 (IL-6) as efficiently as infectious virions (72). Riffault et al. have reported that UV-inactivated herpes simplex virus 1 triggers synthesis of IFN-I in mouse regional lymph nodes when delivered into the ear dermis (73). In addition, the cellular transcription factor NF-κB appears to be quickly upregulated after the binding of either live or UV-inactivated human cytomegalovirus to the cell surface (74). Since EGFR activation occurred in the target cells upon incubation with UV-inactivated PEDV, as well, a co-IP experiment was performed to address whether PEDV surface S protein interacted with EGFR. We observed that there was a direct interaction between EGFR and PEDV S protein, indicating that EGFR activation occurs at the early stage of viral infection.
Activated EGFR recruits different downstream signaling cascades, leading to the activation of several major pathways that are important for cell survival, proliferation, angiogenesis, and viral infection (27, 75, 76). The main downstream pathways of EGFR activation include those mediated by phosphatidylinositol 3-kinase (PI3K)-Akt-mTOR, Ras-Raf-MEK, and JAK2-STAT3 (49, 77, 78). Previous reports have demonstrated that STAT3, a downstream effector of EGFR signaling, suppresses the antiviral activity of type I interferon (48, 79). Considering that EGFR activation impairs the antiviral activity of type I interferon and facilitates PEDV replication, we assessed the involvement of STAT3 in virus infection. We first observed that the changes in STAT3 phosphorylation induced by virus infection were similar to the pattern of activated EGFR, suggesting that STAT3 is a downstream signaling cascade initiated by phosphorylated EGFR (28). Next, using the STAT3 inhibitor S3I-201, we found that inhibition of STAT3 activation resulted in the augmentation of type I interferon signaling, which is consistent with another study that showed that STAT3 negatively regulates the type I interferon-mediated response (50). Xue et al. have reported that inhibition of STAT3 phosphorylation by S3I-201 abrogates the antiviral ability of mature porcine IL-22 (mpIL-22) and the mpIL-22-induced expression of antimicrobial genes, such as BD-2, IL-18, survivin, and IFN-λ genes (80); however, they did not provide direct evidence of the involvement of STAT3 in PEDV infection. To address this controversial finding, we further analyzed the role of STAT3 in virus infection using genetic-modification methods. We observed that inhibition of STAT3 function resulted in the reduction of virus replication, whereas activated STAT3 facilitated virus replication. Sen et al. reported similar findings that STAT3 phosphorylation is required for efficient varicella-zoster virus replication and spread and that blocking STAT3 activation has antiviral activity against virus infection in vivo (51). In agreement with previous findings reported by Ho and Ivashkiv (81), our findings further support the idea that STAT3 expression and STAT3 function are relevant to PEDV infection.
Although previous data have shown that phosphorylated EGFR can mediate the activation of its downstream signaling cascade, STAT3, we needed direct evidence to confirm our hypothesis. We found that the augmentation effect of activated EGFR on virus infection was inhibited by STAT3 knockdown treatment. These data suggest that STAT3 is a downstream effector of the EGFR signaling pathway that negatively regulates the antiviral activity of type I interferon, as well. Our findings are similar to those in a previous report showing that EGFR-mediated STAT3 signaling impairs IFN-α production and facilitates HCV replication (28).
In summary, here, we have examined the relationship between EGFR activation and type I interferon pathways in the regulation of virus infection. Our findings have shown that EGFR positively regulates PEDV infection by activating the EGFR-STAT3 signaling pathway, which suppresses the antiviral activity of type I interferon. Although the signaling intermediates between PEDV and EGFR remain to be elucidated, we have demonstrated that PEDV S directly interacts with EGFR. In conclusion, we have uncovered a novel mechanism in which PEDV uses EGFR to suppress cellular antiviral defenses, which may present a potential therapeutic target against PEDV infection.
MATERIALS AND METHODS
Cells and viruses.
IPEC-J2 cells (porcine small intestine epithelial cell clone J2; ATCC) and HEK293 cells (human embryonic kidney epithelial cells; ATCC) were cultured in Dulbecco's minimum essential medium (DMEM) (Life Technologies, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (HyClone), 100 U/ml penicillin, 100 μg/ml streptomycin at 37°C in a 5% CO2 incubator (Thermo Scientific, USA). PEDV strain CV777 (GenBank accession number KT323979) was titrated in Vero E6 cells and stored at −80°C. To create replication-deficient virus, PEDV was UV irradiated using a CL-1000 cross-linker at 100 μJ/cm2 for 60 min on ice. Virus inactivation was verified by plaque assay.
Drug treatments and virus infection.
IPEC-J2 and HEK293 cells were serum starved overnight before the addition of EGF (Life Technologies, USA) at the indicated concentrations for the indicated times. After washing, some cells were infected with mock control or PEDV. For inhibitor treatments, IPEC-J2 and HEK293 cells were treated with various concentrations of the EGFR-specific inhibitors erlotinib (Selleck, USA) and gefitinib (Calbiochem, Germany), the STAT3-specific inhibitor S3I-201 (ApexBio, USA), or the carrier control dimethyl sulfoxide (DMSO) for 12 h or 24 h. Virus infection was then performed in the presence of these reagents. For the virus infection procedure, IPEC-J2 cells were infected with PEDV at a multiplicity of infection (MOI) of 1 for 48 h, and HEK293 cells were infected with virus at an MOI of 0.1 for 24 h. In this study, all virus infection experiments were conducted using the above-described procedure unless otherwise stated. The treated cells were then collected for subsequent analysis.
Overexpression and RNA interference.
The EGFR and STAT3 genes were amplified from porcine intestinal epithelial cell cDNA using the primers listed in Table 1 and then cloned into the pAAV vector (Stratagene) at the SalI and XhoI sites. The nucleotide sequences of the plasmids expressing EGFR and STAT3 were determined to ensure that the correct clones were used in the study. The vector carrying EGFR (pAAV/EGFR) or STAT3 (pAAV/STAT3) was transfected into cells. Transfection of plasmid DNA was performed with Lipofectamine 2000 (Invitrogen, USA) as recommended by the manufacturer. siRNA duplexes were designed specifically to knock down the endogenous expression of EGFR or STAT3 (Table 2). Cells were transfected with 100 nM siRNA duplexes using Lipofectamine RNAiMax reagent (Invitrogen, USA) according to the manufacturer's instructions. At 24 h posttransfection, the cells were infected with PEDV, followed by the indicated analysis.
TABLE 1.
PCR primers used in this study

a SalI sites are in boldface, and XhoI sites are underlined.
b Primer used for relative quantitative RT-PCR.
TABLE 2.
Sequences of sense strand of siRNA used to ablate EGFR and STAT3 protein expression in IPEC-J2 or HEK293 cells
| Target | Sense strand sequence (5′-3′) |
|---|---|
| Porcine EGFR-siRNA | CGCUGGAGGAGAAGAAAGUdTdT |
| Human EGFR-siRNA | GAGGAAAUAUGUACUACGAdTdT |
| Porcine STAT3-siRNA | GUCAGAUUGCUGGUCAAAUdTdT |
| Human STAT3-siRNA | CUGACUACACUGGCAGAGAdTdT |
| Control siRNA | UUCUCCGAACGUGUCACGUdTdT |
IFA.
Immunofluorescence assays (IFA) were performed as described previously with slight modification (13). Briefly, after EGFR inhibitor treatments, IPEC-J2 and HEK293 cells were infected with PEDV. The cells were fixed with 33.3% acetone for 30 min and then stained with mouse anti-PEDV spike protein monoclonal antibody (MAb) (3F12; Median Diagnostics, South Korea) for 1 h. After three washes with PBS, the cells were incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG for 45 min. After washing, the fluorescence was visualized with an Olympus inverted fluorescence microscope equipped with a camera.
Western blotting.
Western blot analysis was done as described previously (33) with slight modification. Treated cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (HaiGene, China) containing protease inhibitor cocktail and phosphatase inhibitors (Roche, Switzerland). After centrifugation, the lysate supernatants were fractionated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Merck Millipore, USA). The membranes were blocked and then incubated with the indicated primary antibodies. After hybridizing with horseradish peroxidase (HRP)-conjugated goat anti-rabbit or goat anti-mouse IgG, the membranes were visualized by addition of Super ECL Star (U.S. Everbright, China) and exposure to film, according to the manufacturer's instructions. The densitometric analysis was performed using ImageJ. The antibodies against phospho-EGFR (Tyr1068), EGFR, phospho-STAT3 (Tyr705), and STAT3 were obtained from Cell Signaling Technology, USA. Mouse anti-β-actin MAb was purchased from Santa Cruz Biotechnology. Anti-PEDV N protein MAb was stored in our laboratory.
Co-IP.
The PEDV S gene was amplified using the primers with Flag tags listed in Table 1 and then cloned into the pAAV vector (Stratagene). The plasmids carrying PEDV S protein (pAAV/PEDV S) in the presence or absence of EGFR (pAAV/EGFR) were transfected into HEK293T cells. At 36 h posttransfection, the treated cells were lysed in Triton lysis buffer (50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 150 mM NaCl, 5 mM MgCl2, 10% glycerol, 1% Triton X-100) plus protease inhibitor cocktail and phosphatase inhibitors (Roche, Switzerland). The supernatants were immunoprecipitated with anti-Flag affinity beads (Sigma-Aldrich, USA) overnight at 4°C. Immunoprecipitated samples were subjected to SDS-PAGE and transferred to PVDF membranes. The membranes were probed with either rabbit anti-EGFR MAb (CST, USA) or mouse anti-Flag MAb M2 (Sigma-Aldrich, USA).
Quantitative RT-PCR.
Quantitative RT-PCR analyses were carried out as described previously (82). After treatment, total RNA was extracted from cells and subjected to quantitative RT-PCR using specific primers listed in Table 1. The quantitative reactions were set up in triplicate using SYBR premixed Ex Taq (TaKaRa, Japan). Briefly, the relative quantification was calculated by the cycle threshold (ΔΔCT) method (83).
TCID50 assay.
The TCID50 assay was performed in Vero E6 cells according to the method of Reed and Muench as previously described (84). Briefly, cell monolayers were inoculated with serial dilutions of each virus stock and incubated for 4 days prior to observation of the presence of cytopathic effect.
Cell cytotoxicity assay.
Cell monolayers were incubated with each of three inhibitors (erlotinib, gefitinib, and S3I-201) at different concentrations or with the carrier control DMSO for 72 h. Cell viability was then measured with the CCK-8 system according to the manufacturer's instructions. Briefly, CCK-8 solution (10 μl per 100 μl of medium in each well) was added, the plates were incubated at 37°C for 1 h, and the absorbance was read at 450 nm.
Statistical analysis.
All statistical data were expressed as means and standard deviations (SD) of three independent experiments and analyzed using Student's t test. A P value of <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
The research was supported by grants from the National Natural Science Foundation of China (31572497) and the Natural Science Foundation of Heilongjiang Province (JC2016005 and LC201418).
REFERENCES
- 1.Fehr AR, Perlman S. 2015. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol 1282:1–23. doi: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kocherhans R, Bridgen A, Ackermann M, Tobler K. 2001. Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes 23:137–144. doi: 10.1023/A:1011831902219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song D, Park B. 2012. Porcine epidemic diarrhoea virus: a comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes 44:167–175. doi: 10.1007/s11262-012-0713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen J, Wang C, Shi H, Qiu H, Liu S, Chen X, Zhang Z, Feng L. 2010. Molecular epidemiology of porcine epidemic diarrhea virus in China. Arch Virol 155:1471–1476. doi: 10.1007/s00705-010-0720-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Q, Li G, Stasko J, Thomas JT, Stensland WR, Pillatzki AE, Gauger PC, Schwartz KJ, Madson D, Yoon K-J. 2014. Isolation and characterization of porcine epidemic diarrhea viruses associated with the 2013 disease outbreak among swine in the United States. J Clin Microbiol 52:234–243. doi: 10.1128/JCM.02820-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ducatelle R, Coussement W, Pensaert M, Debouck P, Hoorens J. 1981. In vivo morphogenesis of a new porcine enteric coronavirus, CV 777. Arch Virol 68:35–44. doi: 10.1007/BF01315165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang Y-W, Dickerman AW, Piñeyro P, Li L, Fang L, Kiehne R, Opriessnig T, Meng X-J. 2013. Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. mBio 4:e00737-13. doi: 10.1128/mBio.00737-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pensaert M, De Bouck P. 1978. A new coronavirus-like particle associated with diarrhea in swine. Arch Virol 58:243–247. doi: 10.1007/BF01317606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun R-Q, Cai R-J, Chen Y-Q, Liang P-S, Chen D-K, Song C-X. 2012. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg Infect Dis 18:161. doi: 10.3201/eid1801.111259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung K, Saif LJ. 2015. Porcine epidemic diarrhea virus infection: etiology, epidemiology, pathogenesis and immunoprophylaxis. Vet J 204:134–143. doi: 10.1016/j.tvjl.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jung K, Wang Q, Scheuer KA, Lu Z, Zhang Y, Saif LJ. 2014. Pathology of US porcine epidemic diarrhea virus strain PC21A in gnotobiotic pigs. Emerg Infect Dis 20:662. doi: 10.3201/eid2004.131685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung K, Annamalai T, Lu Z, Saif LJ. 2015. Comparative pathogenesis of US porcine epidemic diarrhea virus (PEDV) strain PC21A in conventional 9-day-old nursing piglets vs. 26-day-old weaned pigs. Vet Microbiol 178:31–40. doi: 10.1016/j.vetmic.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo X, Guo L, Zhang J, Xu Y, Gu W, Feng L, Wang Y. 2017. Tight junction protein occludin is a porcine epidemic diarrhea virus entry factor. J Virol 91:e00202-17. doi: 10.1128/JVI.00202-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao S, Gao J, Zhu L, Yang Q. 2014. Transmissible gastroenteritis virus and porcine epidemic diarrhoea virus infection induces dramatic changes in the tight junctions and microfilaments of polarized IPEC-J2 cells. Virus Res 192:34–45. doi: 10.1016/j.virusres.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Itallie CM, Balda MS, Anderson JM. 1995. Epidermal growth factor induces tyrosine phosphorylation and reorganization of the tight junction protein ZO-1 in A431 cells. J Cell Sci 108:1735–1742. [DOI] [PubMed] [Google Scholar]
- 16.Singh AB, Harris RC. 2004. Epidermal growth factor receptor activation differentially regulates claudin expression and enhances transepithelial resistance in Madin-Darby canine kidney cells. J Biol Chem 279:3543–3552. doi: 10.1074/jbc.M308682200. [DOI] [PubMed] [Google Scholar]
- 17.Basuroy S, Seth A, Elias B, Naren AP, Rao R. 2006. MAPK interacts with occludin and mediates EGF-induced prevention of tight junction disruption by hydrogen peroxide. Biochem J 393:69–77. doi: 10.1042/BJ20050959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raimondi F, Santoro P, Barone MV, Pappacoda S, Barretta ML, Nanayakkara M, Apicella C, Capasso L, Paludetto R. 2008. Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am J Physiology Gastrointest Liver Physiol 294:G906–G913. doi: 10.1152/ajpgi.00043.2007. [DOI] [PubMed] [Google Scholar]
- 19.Chen F, Hori T, Ohashi N, Baine AM, Eckman CB, Nguyen JH. 2011. Occludin is regulated by epidermal growth factor receptor activation in brain endothelial cells and brains of mice with acute liver failure. Hepatology 53:1294–1305. doi: 10.1002/hep.24161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herbst RS. 2004. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys 59:S21–S26. doi: 10.1016/j.ijrobp.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 21.Avraham R, Yarden Y. 2011. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat Rev Mol Cell Biol 12:104–117. doi: 10.1038/nrm3048. [DOI] [PubMed] [Google Scholar]
- 22.Lindsey S, Langhans SA. 2015. Epidermal growth factor signaling in transformed cells. Int Rev Cell Mol Biol 314:1–41. doi: 10.1016/bs.ircmb.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baselga J. 2001. The EGFR as a target for anticancer therapy—focus on cetuximab. Eur J Cancer 37:16–22. [DOI] [PubMed] [Google Scholar]
- 24.Han W, Lo H-W. 2012. Landscape of EGFR signaling network in human cancers: biology and therapeutic response in relation to receptor subcellular locations. Cancer Lett 318:124–134. doi: 10.1016/j.canlet.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herbst RS, Langer CJ. 2002. Epidermal growth factor receptors as a target for cancer treatment: the emerging role of IMC-C225 in the treatment of lung and head and neck cancers. Semin Oncol 29(Suppl 4):27–36. [DOI] [PubMed] [Google Scholar]
- 26.Yamashita M, Chattopadhyay S, Fensterl V, Saikia P, Wetzel JL, Sen GC. 2012. Epidermal growth factor receptor is essential for Toll-like receptor 3 signaling. Sci Signal 5:ra50. doi: 10.1126/scisignal.2002581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng K, Kitazato K, Wang Y. 2014. Viruses exploit the function of epidermal growth factor receptor. Rev Med Virol 24:274–286. doi: 10.1002/rmv.1796. [DOI] [PubMed] [Google Scholar]
- 28.Lupberger J, Duong FH, Fofana I, Zona L, Xiao F, Thumann C, Durand SC, Pessaux P, Zeisel MB, Heim MH. 2013. Epidermal growth factor receptor signaling impairs the antiviral activity of interferon-alpha. Hepatology 58:1225–1235. doi: 10.1002/hep.26404. [DOI] [PubMed] [Google Scholar]
- 29.Ueki IF, Min-Oo G, Kalinowski A, Ballon-Landa E, Lanier LL, Nadel JA, Koff JL. 2013. Respiratory virus-induced EGFR activation suppresses IRF1-dependent interferon λ and antiviral defense in airway epithelium. J Exp Med 210:1929–1936. doi: 10.1084/jem.20121401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Venkataraman T, Coleman CM, Frieman MB. 2017. Overactive epidermal growth factor receptor signaling leads to increased fibrosis after severe acute respiratory syndrome coronavirus infection. J Virol 91:e00182-17. doi: 10.1128/JVI.00182-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Downward J, Parker P, Waterfield M. 1984. Autophosphorylation sites on the epidermal growth factor receptor. Nature 311:483–485. doi: 10.1038/311483a0. [DOI] [PubMed] [Google Scholar]
- 32.Yarden Y, Schlessinger J. 1987. Epidermal growth factor induces rapid, reversible aggregation of the purified epidermal growth factor receptor. Biochemistry 26:1443–1451. doi: 10.1021/bi00379a035. [DOI] [PubMed] [Google Scholar]
- 33.Guo L, Luo X, Li R, Xu Y, Zhang J, Ge J, Bu Z, Feng L, Wang Y. 2016. Porcine epidemic diarrhea virus infection inhibits interferon signaling by targeted degradation of STAT1. J Virol 90:8281–8292. doi: 10.1128/JVI.01091-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J, Guo L, Xu Y, Yang L, Shi H, Feng L, Wang Y. 2017. Characterization of porcine epidemic diarrhea virus infectivity in human embryonic kidney cells. Arch Virol 162:2415–2419. doi: 10.1007/s00705-017-3369-2. [DOI] [PubMed] [Google Scholar]
- 35.Bosch BJ, van der Zee R, de Haan CA, Rottier PJ. 2003. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol 77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oshiumi H, Miyashita M, Okamoto M, Morioka Y, Okabe M, Matsumoto M, Seya T. 2015. DDX60 is involved in RIG-I-dependent and independent antiviral responses, and its function is attenuated by virus-induced EGFR activation. Cell Rep 11:1193–1207. doi: 10.1016/j.celrep.2015.04.047. [DOI] [PubMed] [Google Scholar]
- 37.Dawson JP, Berger MB, Lin C-C, Schlessinger J, Lemmon MA, Ferguson KM. 2005. Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Mol Cell Biol 25:7734–7742. doi: 10.1128/MCB.25.17.7734-7742.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlessinger J. 1986. Allosteric regulation of the epidermal growth factor receptor kinase. J Cell Biol 103:2067–2072. doi: 10.1083/jcb.103.6.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sorkin A. 2001. Internalization of the epidermal growth factor receptor: role in signalling. Biochem Soc Trans 29:480–484. doi: 10.1042/bst0290480. [DOI] [PubMed] [Google Scholar]
- 40.Lee H, Seo A, Kim E, Jang M, Kim Y, Kim J, Kim S, Ryu H, Park I, Im S. 2015. Prognostic and predictive values of EGFR overexpression and EGFR copy number alteration in HER2-positive breast cancer. Br J Cancer 112:103–111. doi: 10.1038/bjc.2014.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bean J, Brennan C, Shih J-Y, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S. 2007. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cataldo VD, Gibbons DL, Pérez-Soler R, Quintás-Cardama A. 2011. Treatment of non–small-cell lung cancer with erlotinib or gefitinib. N Engl J Med 364:947–955. doi: 10.1056/NEJMct0807960. [DOI] [PubMed] [Google Scholar]
- 43.Kim T, Murren J. 2002. erlotinib OSI/Roche/Genentech. Curr Opin Investig Drugs 3:1385–1395. [PubMed] [Google Scholar]
- 44.González-Navajas JM, Lee J, David M, Raz E. 2012. Immunomodulatory functions of type I interferons. Nat Rev Immunol 12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ivashkiv LB, Donlin LT. 2014. Regulation of type I interferon responses. Nat Rev Immunol 14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu K, Gualano RC, Hibbs ML, Anderson GP, Bozinovski S. 2008. Epidermal growth factor receptor signaling to Erk1/2 and STATs control the intensity of the epithelial inflammatory responses to rhinovirus infection. J Biol Chem 283:9977–9985. doi: 10.1074/jbc.M710257200. [DOI] [PubMed] [Google Scholar]
- 47.Schneider WM, Chevillotte MD, Rice CM. 2014. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lo H-W, Hsu S-C, Ali-Seyed M, Gunduz M, Xia W, Wei Y, Bartholomeusz G, Shih J-Y, Hung M-C. 2005. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 7:575–589. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 49.Park OK, Schaefer TS, Nathans D. 1996. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc Natl Acad Sci U S A 93:13704–13708. doi: 10.1073/pnas.93.24.13704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang W-B, Levy DE, Lee C-K. 2011. STAT3 negatively regulates type I IFN-mediated antiviral response. J Immunol 187:2578–2585. doi: 10.4049/jimmunol.1004128. [DOI] [PubMed] [Google Scholar]
- 51.Sen N, Che X, Rajamani J, Zerboni L, Sung P, Ptacek J, Arvin AM. 2012. Signal transducer and activator of transcription 3 (STAT3) and survivin induction by varicella-zoster virus promote replication and skin pathogenesis. Proc Natl Acad Sci U S A 109:600–605. doi: 10.1073/pnas.1114232109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip MR, Jove R, McLaughlin MM, Lawrence NJ. 2007. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A 104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang X-D, Baladandayuthapani V, Lin H, Mulligan G, Li B, Esseltine D-LW, Qi L, Xu J, Hunziker W, Barlogie B. 2016. Tight junction protein 1 modulates proteasome capacity and proteasome inhibitor sensitivity in multiple myeloma via EGFR/JAK1/STAT3 signaling. Cancer Cell 29:639–652. doi: 10.1016/j.ccell.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cohen S. 1962. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J Biol Chem 237:1555–1562. [PubMed] [Google Scholar]
- 55.Cohen S, Carpenter G, King L. 1980. Epidermal growth factor-receptor-protein kinase interactions. Co-purification of receptor and epidermal growth factor-enhanced phosphorylation activity. J Biol Chem 255:4834–4842. [PubMed] [Google Scholar]
- 56.Scaltriti M, Baselga J. 2006. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res 12:5268–5272. doi: 10.1158/1078-0432.CCR-05-1554. [DOI] [PubMed] [Google Scholar]
- 57.Finlay BB, McFadden G. 2006. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 124:767–782. doi: 10.1016/j.cell.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 58.Clementz MA, Chen Z, Banach BS, Wang Y, Sun L, Ratia K, Baez-Santos YM, Wang J, Takayama J, Ghosh AK. 2010. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J Virol 84:4619–4629. doi: 10.1128/JVI.02406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ding Z, Fang L, Jing H, Zeng S, Wang D, Liu L, Zhang H, Luo R, Chen H, Xiao S. 2014. Porcine epidemic diarrhea virus nucleocapsid protein antagonizes beta interferon production by sequestering the interaction between IRF3 and TBK1. J Virol 88:8936–8945. doi: 10.1128/JVI.00700-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang D, Fang L, Shi Y, Zhang H, Gao L, Peng G, Chen H, Li K, Xiao S. 2015. Porcine epidemic diarrhea virus 3C-like protease regulates its interferon antagonism by cleaving NEMO. J Virol 90:2090–2101. doi: 10.1128/JVI.02514-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Q, Shi K, Yoo D. 2016. Suppression of type I interferon production by porcine epidemic diarrhea virus and degradation of CREB-binding protein by nsp1. Virology 489:252–268. doi: 10.1016/j.virol.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu X, Wang D, Zhou J, Pan T, Chen J, Yang Y, Lv M, Ye X, Peng G, Fang L. 2017. Porcine deltacoronavirus nsp5 antagonizes type I interferon signaling by cleaving STAT2. J Virol 91:e00003-17. doi: 10.1128/JVI.00003-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barbier D, Garcia-Verdugo I, Pothlichet J, Khazen R, Descamps D, Rousseau K, Thornton D, Si-Tahar M, Touqui L, Chignard M. 2012. Influenza A induces the major secreted airway mucin MUC5AC in a protease–EGFR–extracellular regulated kinase–Sp1–dependent pathway. Am J Respir Cell Mol Biol 47:149–157. doi: 10.1165/rcmb.2011-0405OC. [DOI] [PubMed] [Google Scholar]
- 64.Diao J, Pantua H, Ngu H, Komuves L, Diehl L, Schaefer G, Kapadia SB. 2012. Hepatitis C virus induces epidermal growth factor receptor activation via CD81 binding for viral internalization and entry. J Virol 86:10935–10949. doi: 10.1128/JVI.00750-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kung C-P, Meckes DG, Raab-Traub N. 2011. Epstein-Barr virus LMP1 activates EGFR, STAT3, and ERK through effects on PKCδ. J Virol 85:4399–4408. doi: 10.1128/JVI.01703-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Soroceanu L, Akhavan A, Cobbs CS. 2008. Platelet-derived growth factor-α receptor activation is required for human cytomegalovirus infection. Nature 455:391–395. doi: 10.1038/nature07209. [DOI] [PubMed] [Google Scholar]
- 67.Zhu L, Lee P-K, Lee W-M, Zhao Y, Yu D, Chen Y. 2009. Rhinovirus-induced major airway mucin production involves a novel TLR3-EGFR–dependent pathway. Am J Respir Cell Mol Biol 40:610–619. doi: 10.1165/rcmb.2008-0223OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bowick GC, Fennewald SM, Scott EP, Zhang L, Elsom BL, Aronson JF, Spratt HM, Luxon BA, Gorenstein DG, Herzog NK. 2007. Identification of differentially activated cell-signaling networks associated with Pichinde virus pathogenesis by using systems kinomics. J Virol 81:1923–1933. doi: 10.1128/JVI.02199-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu Y, Shi Y, Yuan Q, Liu X, Yan B, Chen L, Tao Y, Cao Y. 2013. Epstein-Barr virus encoded LMP1 regulates cyclin D1 promoter activity by nuclear EGFR and STAT3 inCNE1 cells. J Exp Clin Cancer Res 32:90. doi: 10.1186/1756-9966-32-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kalinowski A, Ueki I, Min-Oo G, Ballon-Landa E, Knoff D, Galen B, Lanier LL, Nadel JA, Koff JL. 2014. EGFR activation suppresses respiratory virus-induced IRF1-dependent CXCL10 production. Am J Physiol Lung Cell Mol Physiol 307:L186–L196. doi: 10.1152/ajplung.00368.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset FL, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoel M, Raffelsberger W, Poch O, McKeating JA, Brino L, Baumert TF. 2011. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med 17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stadnyk AW, Gillan TL, Anderson R. 1997. Respiratory syncytial virus triggers synthesis of IL-6 in BALB/c mouse alveolar macrophages in the absence of virus replication. Cell Immunol 176:122–126. doi: 10.1006/cimm.1996.1075. [DOI] [PubMed] [Google Scholar]
- 73.Riffault S, Carrat C, Milon G, Charley B, Colle J. 2000. Transient IFN-γ synthesis in the lymph node draining a dermal site loaded with UV-irradiated herpes simplex virus type 1: an NK-and CD3-dependent process regulated by IL-12 but not by IFN-α/β. J Gen Virol 81:2365–2373. doi: 10.1099/0022-1317-81-10-2365. [DOI] [PubMed] [Google Scholar]
- 74.Yurochko AD, Hwang E, Rasmussen L, Keay S, Pereira L, Huang E. 1997. The human cytomegalovirus UL55 (gB) and UL75 (gH) glycoprotein ligands initiate the rapid activation of Sp1 and NF-kappaB during infection. J Virol 71:5051–5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lo H, Hung M. 2006. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer 94:184–188. doi: 10.1038/sj.bjc.6602941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yarden Y, Shilo B-Z. 2007. SnapShot: EGFR signaling pathway. Cell 131:1018.e1–1018.e2. doi: 10.1016/j.cell.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 77.Bowman T, Garcia R, Turkson J, Jove R. 2000. STATs in oncogenesis. Oncogene 19:2474. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 78.Navolanic PM, Steelman LS, McCubrey JA. 2003. EGFR family signaling and its association with breast cancer development and resistance to chemotherapy. Int J Oncol 22:237–252. [PubMed] [Google Scholar]
- 79.Schoggins JW. 2013. Regulating interferon antiviral activity: a role for epidermal growth factor receptor. Hepatology 58:1200–1202. doi: 10.1002/hep.26486. [DOI] [PubMed] [Google Scholar]
- 80.Xue M, Zhao J, Ying L, Fu F, Li L, Ma Y, Shi H, Zhang J, Feng L, Liu P. 2017. IL-22 suppresses the infection of porcine enteric coronaviruses and rotavirus by activating STAT3 signal pathway. Antiviral Res 142:68–75. doi: 10.1016/j.antiviral.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ho HH, Ivashkiv LB. 2006. Role of STAT3 in type I interferon responses: negative regulation of STAT1-dependent inflammatory gene activation. J Biol Chem 281:14111–14118. doi: 10.1074/jbc.M511797200. [DOI] [PubMed] [Google Scholar]
- 82.Guo L, Niu J, Yu H, Gu W, Li R, Luo X, Huang M, Tian Z, Feng L, Wang Y. 2014. Modulation of CD163 expression by metalloprotease ADAM17 regulates porcine reproductive and respiratory syndrome virus entry. J Virol 88:10448–10458. doi: 10.1128/JVI.01117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 84.Chua BH, Phuektes P, Sanders SA, Nicholls PK, McMinn PC. 2008. The molecular basis of mouse adaptation by human enterovirus 71. J Gen Virol 89:1622–1632. doi: 10.1099/vir.0.83676-0. [DOI] [PubMed] [Google Scholar]