


Abstract
In DNP MAS NMR experiments at ~80–110 K, the structurally important −13CH3 and −15NH3+ signals in MAS spectra of biological samples disappear due to the interference of the molecular motions with the 1H decoupling. Here we investigate the effect of these dynamic processes on the NMR line shapes and signal intensities in several typical systems: (1) microcrystalline APG, (2) membrane protein bR, (3) amyloid fibrils PI3-SH3, (4) monomeric alanine-CD3, and (5) the protonated and deuterated dipeptide N-Ac-VL over 78–300 K. In APG, the three-site hopping of the Ala-Cβ peak disappears completely at 112 K, concomitant with the attenuation of CP signals from other 13C’s and 15N’s. Similarly, the 15N signal from Ala-NH3+ disappears at ~ 173 K, concurrent with the attenuation in CP experiments of other 15N’s as well as 13C’s. In bR and PI3-SH3, the methyl groups are attenuated at ~95 K, while all other 13C’s remain unaffected. However, both systems exhibit substantial losses of intensity at ~243 K. Finally, with spectra of Ala and N-Ac-VL, we show that it is possible to extract site specific dynamic data from the temperature dependence of the intensity losses. Furthermore, 2H labeling can assist with recovering the spectral intensity. Thus, our study provides insight into the dynamic behavior of biological systems over a wide range of temperatures, and serves as a guide to optimizing the sensitivity and resolution of structural data in low temperature DNP MAS NMR spectra.
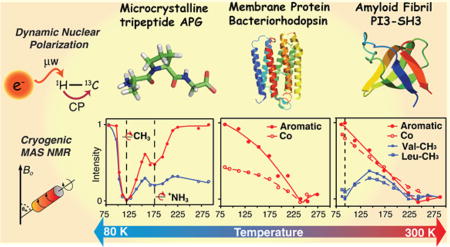
Graphical abstract
INTRODUCTION
Magic angle spinning (MAS) NMR spectroscopy is now established as a versatile and essential tool in structural biology.1–9 In particular, advances in sample preparation, methodology,10–19 and labeling strategies have dramatically improved the resolution of MAS spectra, thus making possible structural studies of large biomolecules not accessible with other techniques.20–24 Nevertheless, the primary limiting factor of MAS NMR is its inherently low sensitivity. An approach to circumvent this limitation is operation at cryogenic temperatures, since the Boltzmann population scales as 1/T.25,26 An even greater gain in sensitivity can be achieved by integrating dynamic nuclear polarization (DNP) into the MAS NMR experiments, where orders of magnitude enhancements of NMR signal intensities have been reported for peptide and protein samples.27–30 This permits experiments that are otherwise difficult or impossible to perform.31–35
To take advantage of this increased sensitivity, DNP/NMR experiments at 80–120 K are becoming widely accessible and heavily utilized. However, to date, the tremendous sensitivity gain overshadows the effects of molecular motions present in the ambient and low temperature spectra. For example, in many cases, side chain and backbone resonances of structural importance are absent for reasons that are glibly referred to as “dynamics” but are not clearly delineated or understood. Early 2H NMR studies on model compounds revealed 3-fold hopping (at 103–106 s−1) by −CD3 and −ND3+ groups at 130–200 and 200–310 K temperatures, respectively,36–39 and there have been a few investigations of these 3-fold processes and 2-fold flips of aromatic rings in peptides and proteins at low temperatures.40–47 However, to the best of our knowledge, none of them provide a detailed description of the effects of these processes on the accompanying loss in signal intensity in MAS spectra, especially in the 80–120 K regime. Furthermore, recent observation of heteronuclear polarization transfer under DNP has been attributed to methyl dynamics in proteins.48 Thus, the purpose of the experiments reported here is to provide an overview of the global and site-specific spectral intensity losses as a function of temperature. Thus, we combine data from 1H−13C/15N cross-polarization (CP), 13C Bloch decay MAS experiments recorded from a series of temperature-dependent MAS NMR spectra on five systems including (1) the microcrystalline tripeptide alanyl-prolyl-glycine (APG), (2) the membrane protein bacteriorhodopsin (bR), (3) amyloid fibrils of phosphatydinal-inositol-3-kinase SH3 domain (PI3-SH3), (4) monomeric −CH3 and −CD3 alanine, and (5) the protonated and deuterated dipeptide N-acetyl-valyl-leucine (N-Ac-VL) over the temperature range 78–300 K.
In APG, we find that the motion of the −NH3+ and −CH3 interferes with 1H decoupling and also compromises CP efficiencies, leading to specific and complete attenuation of spectral lines from these two groups at ~173 and ~112 K, respectively. In addition, these intensity losses propagate throughout the sample, causing a global loss of spectral intensity. At temperatures around 80 K, the interfering motions approach the rigid lattice limit and the signal intensity fully recovers. In this regime, the gain in signal intensity is purely due to the Boltzmann factor.
Two classes of larger protein systems commonly studied at low temperature and with DNP include membrane and amyloid proteins.49–52 Examples of these systems include bR in its native purple membrane and PI3-SH3. In both of these cases, the signal minimum due to the dynamics of −CH3 groups occurs at a lower temperature (95 K) and appears to be localized as opposed to the case of APG. In addition, other spectral regions such as the carbonyl and aromatic resonances are not affected and exhibit improved signals. Finally, we show that intensity losses due to the dynamic process can be partially and in some cases completely recovered by the introduction of 2H labeling. In particular, the introduction of −CD3 groups in Ala, N-Ac-VL, and the use of perdeuterated bR permit observation of these groups in cases where spectral lines from the −CH3 moiety are completely absent.
EXPERIMENTAL SECTION
Sample Preparation
Both uniformly 13C, 15N-labeled and 15N-labeled APG samples were diluted to 10% with the corresponding natural abundance APG to suppress any intermolecular couplings. This was accomplished by dissolving the mixture of labeled APG/natural abundance APG in a minimal amount of water (~45 mg/mL) followed by slow crystallization in a desiccator, and crystals forming in about a week. 40 mg of each APG sample was packed into a 4 mm Revolution NMR zirconia rotor.
Bacteriorhodopsin (bR), in its native purple membrane, was purified from Halobacterium salinarum grown in uniformly 13C, 15N-labeled peptone medium.53 Peptone was obtained from the anaerobic acid hydrolysis of Methylophilus methylotrophus cells grown on 13C-labeled methanol and 15N-labeled ammonium sulfate.54 The purple membranes were isolated using the method of Oesterhelt and Stoechenius.55 The sample was washed three times with 300 mM guanidine hydrochloride at pH 10.0. The sample was pelleted after every wash by centrifugation for 2 h at ~43,000g. The washed pellet was mixed with 5 mM AMUPol56 or 15 mM TOTAPOL57 in “DNP juice” consisting of d8-glycerol/D2O/H2O (60/30/10 volume ratio) and centrifuged once more.
The phosphatidyl-inositol-3-kinase SH3 domain PI3-SH3 fibril sample was uniformly 13C, 15N-labeled at the F, V, Y, and L residues. The fibrils were grown from a solution of monomeric protein by incubation at pH 2.0 and 25 °C for 14 days.58 “DNP juice” adjusted to pH 2.0 and supplemented with 15 mM TOTAPOL was added to the gel-like fibrils for cryoprotection and DNP experiments. Both bR and PI3-SH3 samples were packed into a 4 mm sapphire rotor for DNP/ NMR experiments.
[U−13C, 15N] and [−CD3−U−13C, 15N] alanine were purchased from Cambridge Isotope Laboratories (CIL), dissolved in water, and then crystallized in a desiccator. [U−13C, 15N] N-acetyl-l-Val-l-Leu (N-Ac-VL) and [1,2-13C] acetic anhydride were purchased from CIL. N-Ac-VL was synthesized by New England Peptide (Gardner, MA), using standard solid-phase methods, and purified by HPLC. N-Ac-VL was crystallized from a 1:1 (v/v) H2O:acetone solution.
NMR Spectroscopy
MAS spectra were recorded using a custom-designed triple resonance (1H, 13C, 15N) cryogenic MAS probe equipped with a sample exchange system59 on a home-built NMR spectrometer operating at 380 MHz 1H frequency (courtesy of Dr. D. J. Ruben). Two types of 1D NMR experiments were conducted for APG: 1H−13C/ 15N cross-polarization (CP) and 13C Bloch decay. For CP experiments, a spin-lock field of 50 kHz was employed on the proton channel. Experimental parameters including recycle delay, CP Hartmann-Hahn matching conditions, CP duration, and TPPM decoupling have all been optimized for all temperature dependent experiments. 1H TPPM decoupling fields of ω1H/2π = 83 kHz or 100 kHz and ωr/2π = 4.83 kHz were used unless stated otherwise.
Liquid nitrogen boil-off gas was used for both the bearing and drive gas streams, and the spinning frequency was controlled by a Bruker MAS controller.59 Both bearing and drive streams were cooled using a custom-designed heat exchanger, and the temperature was subsequently controlled using heating elements inside vacuum jacketed transfer lines with two PID controllers (Lakeshore, Westerville, OH). The sample temperature was monitored using a fiber optic temperature sensor (Neoptix, Quebec, Canada) that extends to the inside of the MAS stator. The fiber optic thermometers were calibrated by immersion in liquid nitrogen at 77 K. Note, during MAS experiments, we have consistently recorded temperatures in the range 72–77 K presumably due to Joule-Thompson cooling on the expansion of the N2 gas from the jets of the drive cup and bearings of the stator.
DNP Experiments
In order to investigate methyl group dynamics at DNP temperatures, 1H, 13C, 15N CP and homonuclear experiments were performed using a home-built DNP gyrotron instrument operating at 250 GHz/380 MHz with ~14W of microwave power. 60,61 Enhancement factors (ε) were calculated by comparing the signals obtained with and without µw irradiation.
Simulations
The simulations of the Ala-CH3 group dynamics in APG were performed using GAMMA62 with 100 powder orientations chosen using the ZCW scheme.63 Details of the parameters used can be found in the Supporting Information. In order to simulate the three-site hopping mechanism, the simulations were carried out in a composite Liouvillian space that facilitates a mutual-exchange mechanism. The dimensions of the exchange matrices for the four-spin CH3 and CD3 spin systems are 256 × 256 and 2916 × 2916, respectively. The amount of time required to simulate the FID of one crystallite orientation using one CPU core is ~1 min and ~1.5 days for CH3 and CD3, respectively. All simulations took ~2 weeks to compute using the ETH Brutus cluster with 384 CPU cores. The parameters chosen for the simulations are ωr/2π = 4.651 kHz, TPPM decoupling ω1H/2π = 83 kHz with 6.9 µs pulses, and phases ±15 degree. The size of the quadrupole coupling used for 2H nuclei is 167 kHz.
RESULTS
The 1D 13C spectra in Figure 1 illustrate the spectral resolution of two microcrystalline APG samples with differing isotopic labeling schemes at 80 K. In both cases, the spectra exhibit a resolution comparable to that obtained at room temperature. Specifically, Figure 1a shows a 13C CP MAS NMR spectrum of [U−12C, 15N]-APG and in the absence of 13C−13C J-couplings; the line width of the Gly-Co is as narrow as 27 Hz (0.28 ppm). In comparison, the line widths in the spectrum of [U−13C, 15N]-APG (Figure 1b) increase by a factor of 2 or more primarily due to one-bond J-couplings and higher order cross-terms that arise from the denser 13C network. Nevertheless, resolved J-splittings (~50 Hz) can still be distinguished in two out of three carbonyls, each of which has only one J-coupled neighboring 13C. On the other hand, each 13C in the aliphatic region is J-coupled to multiple neighbors, which obscures the splitting. The aliphatic carbons also have stronger CH dipolar coupling than that of carbonyl carbons, and would require stronger decoupling and higher spinning frequencies to be averaged out effectively. The data confirm that the optimal resolution in MAS spectra can be obtained with sparsely labeled 13C samples.64 In addition, they demonstrate that high resolution in well-ordered materials can be obtained at cryogenic temperatures (~80 K).
Figure 1.

13C CP-MAS spectrum of [U−15N] APG (a) and [U−13C, 15N] APG (b) at 80 K. Both samples were diluted to 10% in unlabeled APG to minimize the intermolecular couplings. The line width of the Gly-Co is 27 Hz (0.28 ppm). In part b, the line width is broadened by 13C−13C J-coupling evident in the doublet splitting in the carbonyl region with the Gly-Co and Ala-Co showing one-bond Cα−Co J-couplings of ~50 Hz. The fact that these resonances are not resolved in the aliphatic region is likely due to the presence of multiple J-couplings.
Figure 2 illustrates the temperature dependence of the 1H−13C/15N CP-MAS spectra acquired in the range from 73 to 295 K, where we observe several interesting spectral changes. First, in the transition between regions I and II at 225 K, there is a doubling in all of the side chain signals that is especially obvious on the Ala-Cβ line (δ ≈ 18 ppm at 295 K), as well as Pro-Cβ and Pro-Cγ (30 and 35 ppm at 295 K, respectively). A recent calorimetric study of APG crystals revealed that the spectral changes at 225 K, although in the vicinity of the famous protein glass transition,65 are likely the result of a polymorphic phase transition.66 Similar spectral changes at ~200 K were observed for the peptide N-f-MLF-OH.32 Second, in region III, the intensity of Ala-Cβ exhibits a local minimum that coincides with the disappearance of the 15N signal from the −NH3+ group at ~173 K. Third, at lower temperatures, in region IV between 148 and 96 K, a more significant loss of Ala-13Cβ signal intensity occurs. The intensity decreases dramatically and the Ala-13Cβ line disappears into the baseline at ~112 K, and there is a concurrent loss of spectral intensity in the 15N spectra at this temperature. We attribute the two signal minima in regions III and IV at ~173 and ~112 K to the 3-fold hopping rates of the −NH3+ and −CH3 groups, respectively, matching the 1H decoupling frequency. Finally, at lower temperatures, the hopping rate enters the slow exchange limit and the −CH3 and −NH3+ lines reappear and narrow and the intensity recovers fully at 73 K.
Figure 2.

Temperature-dependent 1H−13C CP (left column) and 1H−15N CP (right column) spectra of [U−13C, 15N] APG. Several important spectral changes are observed. In region III, between 213 and 148 K, the intensity of the Ala-NH3+ peak is buried under the noise level at ~173 K, which couples with the partial attenuation of the 13C spectrum. Region IV, below 148 K, exhibits the disappearance of the Ala-CH3 signal at ~113 K, coincident with the signal dip in the 15N spectrum. The spectral changes are further analyzed in Figure 3. The spectra were acquired with ωr/2π = 4.83 kHz, ω1H/2π = 83 kHz for TPPM decoupling.
The temperature dependence of the signal intensity is a product of the Boltzmann factor, which has a T−1 dependence, and the intensity loss due to molecular motions, which varies with temperature. In order to isolate the effect of molecular dynamics, the contribution from the Boltzmann factor is removed by multiplying the integrated signal intensity by the corresponding temperature. Figure 3a shows the normalized signal intensity of Ala-Cβ (−CH3 group) with (red solid circles) and without (blue open circles) the correction for the Boltzmann factor. To correct for the Boltzmann factor, the normalized intensity was calculated as (I/T)/(I0/T0), where I0 is the intensity at the highest temperature T0, where experiments were performed and I is the intensity at temperature T. To facilitate data presentation, without the correction for the Boltzmann factor, the normalized intensity was calculated differently as (I/I0), where I0 is the intensity at the lowest temperature at which we perform experiments. Note that the integrated signal intensities were adjusted when necessary by their relative 1H or 13C T1’s measured at each temperature and incorporated into the intensity calculations. T1H and T13C values are plotted as a function of temperature in Figure S1 in the Supporting Information. The minima at ~112 and ~173 K in Figure 3 are visible even without this correction and are greatly amplified with the correction. Below 90 K and above 225 K, the adjusted signals in red are essentially the same, indicating that the signal intensities are unaffected by the effect of molecular dynamics in these regions. In particular, the signal intensity in these regions follows closely the T−1 dependence, resulting in a factor of 4 times higher intensity at 73 K compared to 295 K. Figure 3b illustrates that other sites such as Pro-Cα and Gly-Co also exhibit two signal minima in 1H−13C CP experiments and the entire spectrum is uniformly attenuated at 112 and 173 K. Figure 3c presents the data from the single-pulse 13C Bloch decay signals detected with 1H decoupling that show a minimum at 112 K, and a set of 13C Bloch decay spectra is included in Figure S2. Furthermore, at this temperature, the Ala-CH3 is also completely attenuated (Figure 3a), whereas other sites are nearly unaffected. In combination, the data in Figure 3b and c suggest that the signal minimum at ~112 K is associated with the 3-fold hopping rate of the Ala-CH3, which approximates to the Rabi frequency of the 1H RF fields during decoupling and/or CP. The minimum at ~173 K is attributed to the hopping of the Ala-NH3+ which manifests itself in the disappearance of its 1H−15N CP signal, as shown in Figure 3d. In comparison to the 1H−13C CP data (Figure 3b), the 1H−15N CP data also exhibit two minima but in reverse intensity order: nearly uniform signal attenuation at ~112 K and complete signal attenuation of Ala-NH3+ at ~173 K.
Figure 3.

Temperature dependence of the integrated peak intensities of [U−13C, 15N]-APG. The vertical dashed lines indicate the minima in the signal intensities at ~112 and ~173 K due to the hopping of −CH3 and −NH3+, respectively. (a) Ala-Cβ from 1H−13C CP with (red solid circles) and without (blue open circles) Boltzmann correction at each temperature. (b) Adjusted spectrum intensities of several peaks of APG plotted as a function of temperature from 1H−13C CP. Again, notice the two minima, ~112 K where the Ala-Cβ peak broadens beyond detection and ~173 K. (c) 13C Bloch decay experiments show the first minimum, indicating interference between methyl hopping and 1H decoupling. (d) Spectral intensities of 1H−15N CP alanine NH3+ as a function of temperature. The second minimum in part b is due to the hopping of the Ala-NH3+ group.
Motivated by the dramatic changes in signal intensities in the APG spectra, we extended our studies to the membrane protein bR, and to amyloid fibrils formed by PI3-SH3, to determine if a similar behavior is observed in these systems. Figure 4a illustrates that attenuation of methyl 13C resonances occurs in [U−13C, 15N]-bR. However, because of spectral overlap in this 248 amino acid, uniformly 13C/15N labeled protein, the extraction of accurate intensity data for these resonances is imprecise. Multidimensional experiments at higher fields are required to obtain better resolution. In contrast, the spectra in Figure 4c of [13C, 15N-FVYL]-PI3-SH3 are greatly simplified, which permits the extraction of the intensity of different functional groups, especially the Val-CH3 and Leu-CH3. In general, the spectra show that the overall intensity of carbonyl and aromatic 13C’s in both samples increases as temperature decreases except around 243 K (Figure 4b and d). In addition, the methyl groups exhibit another minimum at ~95 K in the case of [13C, 15N-FVYL]-PI3-SH3, but the intensities of the two −CH3 containing residues are partially recovered at 87 K and are expected to fully recover at lower temperatures. Incorporation of DNP into the experiments in this temperature regime will, in addition to the Boltzmann factor, boost the sensitivity. In Figure S5, we show 1D 1H−13C CP spectra of [13C, 15N-FVYL]-PI3-SH3 obtained with a signal enhancement of 35. A 2D RFDR spectrum of [13C, 15N-FVYL]-PI3-SH3 fibril with τmix = 1.6 ms was acquired in approximately 4 h. Without DNP, the same spectrum would require one month to achieve the same S/N.
Figure 4.

Temperature-dependent 1H−13C CP spectra of (a) [U−13C, 15N] bacteriorhodopsin and (c) [13C, 15N-FVYL]-PI3-SH3 amyloid fibrils without Boltzmann correction. In parts b and d, we show plots of normalized intensities for carbonyl, aromatic, and aliphatic regions in bR and PI3-SH3 fibrils as a function of temperature. Both the carbonyl and aromatic regions experience a minimum in intensity around 243 K followed by a steady increase as the temperature is decreased for both systems. The intensity of −CH3’s in the valines and leucines in PI3-SH3 also reveals a minimum at ~243 K, followed by a second minimum at ~95 K, where the intensity losses are again due to −CH3 hopping interfering with 1H decoupling and spin-lock fields. Both samples were cryoprotected in d8-glycerol/D2O/H2O (60/30/10 volume ratio). Full sets of spectra at different temperatures are provided in Figures S3 and S4. The spectra were acquired with ωr/2π = 4.83 kHz for bR and 7 kHz for PI3-SH3. In both cases, ω1H/2π = 83 kHz for TPPM decoupling.
In Figure 4b and d, the intensities are normalized to those obtained at 275 K for bR and 283 K for PI3-SH3. We note that the normalized intensities of the aromatic 13C’s at ~80–90 K can be larger than the contribution from the Boltzmann factor. This is due to the fact that at 300 K there is already a significant loss in signal intensity in aromatic ring spectra due to the 2-fold ring flips,67–70 which affects the polarization transfer during CP and interferes with decoupling. This phenomenon was observed in N-f-MLF-OH where the δ, δ′ and ε, ε′ signals from the Phe ring are absent in the 300 K 1D spectra. They start to reappear at 250–225 K and are fully developed at lower temperatures.32 In addition, in ZF-TEDOR spectra of PI3-SH3, the aromatic region is essentially empty at 300 K but is intense and well resolved at 90 K.71 Thus, the broad signal minimum around 243 K probably extends to the higher temperatures, ~280 K. Note also that protein samples used for DNP are cryoprotected by the glass-forming mixture of glycerol/water (60/40 volume ratio), as is the case for the bR and PI3-SH3 samples used in our study. Therefore, the signal minimum at 243 K coincides with the freezing of the glycerol/water mixture and a slowing of the 2-fold flips of the 24 aromatic rings in bR. We also note that this temperature is close to that of the protein glass transition, although it is generally centered at the somewhat lower temperature (~200 K).
It would clearly be desirable to recover the signal loss due to the interference between molecular dynamics and decoupling, and accordingly, we have examined the possibility of labeling methyl groups as −CD3’s.36,37 The rationale behind this approach is that the first-order 2H quadrupole coupling is inhomogeneous, the second-order 2H coupling is small at high fields, and 2H−2H dipole couplings, proportional to γI2, are a factor of 42 smaller than 1H−1H dipole couplings. Thus, MAS itself should average the 2H−2H and 2H−13C dipolar couplings and attenuate the intensity losses. Figure 5 shows temperature-dependent spectra obtained from the monomeric amino acid alanine containing either a −CH3 or −CD3 group. As expected, the 13C MAS spectra obtained from Ala-13CH3 exhibit dramatic intensity losses around 165 K. Note that the intensity also decreases uniformly across the spectrum and both the Cα and Co resonances are effectively suppressed, as was the case in APG (Figure 2). However, with the −13CD3 present, the Ala methyl line is not suppressed at 165 K and the intensity recovers at 81 K. Nevertheless, there is still considerable intensity loss at 165 K, and as discussed below, this is probably due to the −CD3 methyl hopping rate (~104–105 s−1) being similar to the magic angle spinning frequency. By interpolating the data from the Arrhenius plot reported by Beshah et al. for Ala-CD3, we obtain hopping rates of 1.4 × 105 at 165 K and 1.6 × 103 at 127 K,38 which confirms our hypothesis.
Figure 5.

Temperature dependent 13C MAS spectra of (a) [U−13C, 15N]-Ala with a 13CH3; (b) Ala-13CD3; (c) [U−1H, 13C, 15N] N-acetyl-l-Val-l-Leu; and (d) [U−2H, 13C, 15N] N-acetyl-l-Val-l-Leu. The spectra were acquired with ωr/2π = 6.2 kHz, ω1H/2π = 100 kHz for TPPM decoupling.
To explore the intensity losses in two other amino acids, namely, Leu and Val, we recorded spectra of protonated and −CD3 labeled N-Ac-VL. As shown in Figure 5c, the Val γ1 and γ2 lines in protonated N-Ac-VL exhibit significant intensity losses and are absent in the spectrum for T < 116 K. Note also that the line from Vγ2 disappears in the interval 116–173 K, whereas the Vγ1 line persists to ~116 K. This behavior is consistent with the fact that the 3-fold hopping rates, measured with 2H spectra, for the two −CH3 groups in Val differ by about an order of magnitude.38 Specifically, in N-Ac-dl-Val, they are 4.8 × 105 and 2.8 × 106 at 118 K, and therefore, the signal intensities of the methyl groups at 116 K (Figure 5d) are not severely attenuated and still detectable, as the hopping rates are ~1–2 orders of magnitude away from the interference regime (~104–105 s−1). In contrast, the Leu-CH3 line does not lose intensity even at 90 K and it can potentially be used for distance measurements at low temperatures. In deuterated N-Ac-VL, both groups are present at 90 K but with reduced intensity.
In DNP enhanced spectra of uniformly labeled bR, the intensity of methyl containing residues is also reduced. Figure 6a shows a 2D RFDR spectrum of 1H uniformly labeled bR at 190 K with cross-peaks corresponding to the 29 Ala and 18 Thr-CH3’s in bR that are not resolved at 380 MHz. In contrast, at 90 K (Figure 6b), the Ala Cβα cross-peaks are no longer observed in the H bR sample, while they are detected in the 2H uniformly labeled bR. Similarly, Thrγ2β cross-peaks are partially attenuated in the 1H spectrum compared to that at 190 K, but it is fully recovered in the spectrum of 2H bR This further verifies that an effective solution of methyl group attenuation could involve deuteration.
Figure 6.

2D DNP enhanced 13C−13C RFDR spectra of (a) U−1H, 13C, 15N bR doped with 5 mM AMUPol at 190 K, (b) 92 K and (c) 2D RFDR of U−2H, 13C, 15N bR containing 15 mM TOTAPOL at 92 K. Parts b and c were acquired with DNP microwave irradiation. Enhancements of 75 and 71 were obtained, respectively. All spectra were acquired with 2 ms mixing, 30 kHz 13C pulses, and 100 kHz 1H decoupling and spinning frequency ωr/2π = 7 kHz at ω0H/2π = 380 MHz. Both experiments took 7 h with 6 s recycle delay, 128 t1 increments, and 32 scans per increment.
Although the loss of signal intensity of the methyl groups at low temperature impedes many NMR experiments, including distance measurements, it encodes useful site-specific information about the dynamics of a group, for instance, the activation energy, Ea. The activation energy contains rich details about the local chemical and structural environment, and they can be extracted by first comparing experimental data at different temperatures with numerical simulations. Figure 7 shows the Arrhenius plot of the −CH3 group hopping rates of alanine extracted from simulations of the signal intensities in APG measured by observing 13C signals directly using a Bloch decay in the presence of 1H decoupling (data from Figure 3c). The least-squares fit yields an activation energy Ea of 7.2 ± 1 kJ/mol. This value is lower than the literature value of 20.0 kJ/mol39 obtained for the monomeric amino acid alanine. The activation energy of the −CH3 group in alanine is higher than that in the APG probably due to tighter crystal packing and thus more restricted rotation, hence the higher barrier for 3-fold hopping. However, this analysis is complicated by the fact that the methyl group is not strictly an isolated system, i.e., it is coupled to the nearby proton bath, especially the −NH3+ group via spin diffusion. A more detailed study of this effect on the activation energy can be performed by deuteration of the NH3+ group.
Figure 7.

Arrhenius plot of the three-site hopping rate of the Ala −CH3 group in APG. The least-squares fit (red line) yields an activation energy Ea of 7.2 ± 1 kJ/mol and a pre-exponential constant A of ~2 × 109 s−1.
DISCUSSION
It is well-known that the hopping rates of −CH3 and −NH3+ groups can be measured precisely by analyzing the 2H line shape of the deuterated analogues of these groups.36–39 Alternatively, Long et al. showed a strong correlation between the intensity of Bloch decay signals of 15N and the hopping rate in −NH3+.37 Using the same approach, we acquired 13C Bloch decay signals (Figure 3c) and assigned the signal minimum at ~112 K to the interference between the methyl group hopping and the 1H decoupling. Furthermore, the small signal attenuation (10%) of Pro-Cα and Gly-Co from Bloch decay experiments (Figure 3c) cannot account for the large signal loss (60%) of the same resonances from CP experiments (Figure 3b), suggesting that the methyl group hopping also causes inefficient CP at this temperature. This suggests that the hopping rate of the methyl group in APG at ~112 K is ~104–105 s−1, i.e., the same order of magnitude as the MAS frequency and/or 1H decoupling regime. The hopping rate can be extracted from the fitted Arrhenius plot (Figure 7) and we obtained a hopping rate of ~7 × 105 s−1, thus confirming our hypothesis.
In practice, the decoupling field and the spin-locking field are very close in strength. Thus, it is expected that interferences between these Rabi fields and the three-site hopping occur at the same temperature. Interference between the spin-locking field and the molecular dynamics is clearly the dominant mechanism responsible for the signal minimum at ~173 K in the 1H−13C CP data (Figure 3b), which does not appear in the 13C Bloch decay data (Figure 3c). At this matching condition, the spin-lock is inefficient for methyl 1H’s, causing a short 1H T1ρ. Furthermore, due to rapid 1H−1H spin diffusion and the combination of intra- and intermolecular contacts, the effect of a short T1ρ of the methyl 1H’s readily distributes itself throughout the molecule, resulting in a uniform signal loss at ~173 K in the 1H−13C CP data (Figure 3b). In contrast, the destructive interference effect on the decoupling appears to be more localized to the −CH3 and −NH3+. This is apparent in the signal minimum at ~112 K in Figure 3b and c as well as in the signal loss at ~173 K in the 1H−15N CP data (Figure 3d), which is due to interference of −NH3+ hopping with both the decoupling and spin-lock fields.
The diffusive hopping of −CH3 causes the first minimum at ~112 K, and the second at ~173 K is also caused by a similar phenomenon involving the −NH3+ group. The temperature or hopping rate at which the signal intensity is minimum is governed by both the Ea and the pre-exponential factor of the Arrhenius equation, A. However, if the values of A are comparable, then by comparing the previously published Ea values one can predict the temperature range at which the intensity is minimum. The higher activation energy of the −NH3+ group is due to its ability to form hydrogen bonds.37 For example, the Ea in Ala for −ND3+ is 40.5 kJ/mol,37 whereas that for the −CD3 group is 20.0 kJ/mol.39 Long has shown the Ea values of CD3 and CH3 groups are comparable. Similar to the case of −CH3, Long et al. shows that the hopping rate of −NH3+ in Ala reaches 5 × 104 s−1 at 243 K, which is significantly higher than 173 K implied by our data, pointing to a lower activation energy in APG, given that the values of A are comparable in both cases.
Our hypothesis on the correlation between molecular packing/flexibility and Ea is further supported by the data on larger systems including the membrane protein bR (Figure 4a and c) and amyloid fibrils of PI3-SH3 (Figure 4c and d). In contrast to the tripeptide APG, in bR and PI3-SH3, the hopping effect appears to be localized to the methyl groups. In [FVYL-13C, 15N] PI3-SH3, a sample in which only methyl groups of Val and Leu are labeled, we are able to observe the minimum in the intensity of the −CH3 groups at ~95 K, compared to ~112 K in APG, suggesting a lower Ea.
Our observations have a direct implication for the application of DNP to problems in structural biology. Carbonyl 13C’s, together with the aromatic side chains, are the least affected by the molecular dynamics and exhibit excellent sensitivity at low temperatures. This validates the approach using low temperature DNP to obtain long-range intermolecular distances involving aromatic side chains.71 Similarly, methyl groups are of proven importance in measuring long-range contacts in proteins due to their position at the termini of many amino acid side chains. Interestingly, as demonstrated by the data here, one cannot use DNP experiments in the 80–120 K temperature range to measure distances associated with certain protonated (−CH3) methyl groups. As hinted by the partial recovery of the methyl carbons at 87 K in Figures 3 and 4, the brute force solution to this problem is to perform experiments at even lower temperatures, which would then require cooling using liquid helium. A promising alternative approach is to fully or partially deuterate the methyl groups, and it was demonstrated some time ago that full deuteration largely prevents the intensity losses observed here.36 Thus, the benefit of this approach is 2-fold. First, it circumvents the detrimental intensity losses while maintaining sufficient CP from adjacent protons. Second, deuteration of proteins has been demonstrated to increase DNP enhancements by a factor of 3–4.31
We have initiated the investigation of such an approach. In particular, in Figure 5, we compared spectra of alanine with −CD3 and −CH3 methyl groups as well as between protonated and fully deuterated N-acetyl-l-Val-l-Leu. The methyl group −CH3 of alanine disappears completely at 165 K, while the alanine −CD3 signal experiences some level of attenuation but nevertheless survives. In N-Ac-VL, the Val Cγ1 and Cγ2 possess different activation energies, resulting in their disappearance at different temperatures, 90 and 116 K, respectively. This behavior is delayed in the perdeuterated N-Ac-VL spectra, and more importantly, valine Cγ1 and Cγ2 signals can still be observed at 90 K. These two carbons in N-Ac-Val were reported to have distinct Ea’s of 15.3 and 22.2 kJ/mol.38 Our results suggest that it is possible to maintain the signals from methyl groups at low temperature by deuteration. Further investigations including the incorporation of deuterium decoupling are underway and will be the topic of the subsequent studies.
It is worth noting that the signal loss at ~243 K can be as large as ~70% in the case of bR and occurs in the neighborhood of, but above, the temperature normally associated with the protein glass transition. It appears that two mechanisms could be responsible for this effect. First, this intensity loss could be due to the freezing of glycerol/water mixture. Thus, the signal attenuation is related to the hydration of the sample and protein–water interactions and is, therefore, ubiquitous in biological samples. Second, the loss could also be due to the two-site flipping of phenyl rings. bR contains 13 Phe’s and 11 Tyr’s in the amino acid sequence and preliminary 2H NMR data show intermediate exchange flipping rates at this temperature. We believe this is important, since protein samples are frequently studied at temperatures slightly lower than room temperature to slow some dynamic processes. Our results suggest that this approach may be suboptimal in terms of the overall sensitivity. It is therefore desirable to perform such experiments at even lower temperatures, but to date, these temperatures are not always achievable due to instrumental limitations.
Finally, the main focus of this Article is to address the effect of temperature on the line intensity of the −CH3 group, along with solutions to alleviate the line-broadening effect at certain temperatures due to interference between the hopping mechanism with MAS frequency and/or 1H decoupling and/ or CP. While such interference is problematic for some experiments, this temperature-dependent phenomena can be exploited and used to extract useful dynamic information about the system. Thus, we present here a new method to extract the activation energy of the −CH3 group (Figure 7) by monitoring the change of 13C signal intensities as a function of temperature. We are currently applying this novel approach to investigate the site-specific motions of other biological macromolecules like proteins, which could reveal important relations between the dynamics and functions of proteins.
CONCLUSION
In summary, we report effects of molecular motions on NMR signals of peptide and protein samples at cryogenic temperatures. In the microcrystalline tripeptide APG, a first-order polymorphic phase transition occurs at 225 K, which manifests itself in the line doubling in 13C NMR spectra. At lower temperatures, we observe a destructive interference effect from the 3-fold jump diffusion of the Ala-CH3 (at ~112 K) and of the Ala-NH3+ (at ~173 K) on the proton decoupling and/or the CP spin-lock fields. The effect on the decoupling appears to be localized, whereas the effect on the CP is readily transmitted throughout the molecule due to fast 1H−1H spin diffusion in a strongly coupled 1H bath.
We extend these experiments to larger biological systems consisting of the membrane protein bacteriorhodopsin and amyloid fibrils formed from PI3-SH3. At ~243 K, both samples exhibit significant signal loss, in the neighborhood of the protein glass transition. Similar to the case of APG, the methyl groups are attenuated at low temperatures. However, the effect does not propagate in an obvious way to other parts of the spectrum, which supports the approach of using low temperature DNP to obtain long-range distances involving aromatic side chains. Furthermore, the intensity minima occur at lower temperatures in these biological systems than in APG (95 K vs 112 K). Simulations of the intensity losses for the −CH3 group in APG suggest that it should be possible to use similar data to extract site-specific information on molecular dynamics. Finally, our study suggests deuteration of the methyl groups as −CD3 as another probe of long-range distance constraints using DNP at liquid nitrogen temperatures.
Supplementary Material
Acknowledgments
This work was supported by grants (EB-001960, EB-002804, EB-002026, and EB-001035) from the National Institute of Biomedical Imaging and Bioengineering. B.C. was partially funded by a DFG Research Fellowship CO802/1-1. K.O.T. is supported by Early Postdoc Mobility grant -165285 provided by the Swiss National Science Foundation (SNSF). We would like to thank Jeffrey Bryant, Ajay Thakkar, and Michael Mullins for their technical assistance and Drs. Sudheer Jawla and Richard Temkin for helping with DNP instrumentation. We would like to thank Prof. Matthias Ernst for useful discussions regarding the simulations. We are grateful to Dr. Marina Belenky and Angela Jacavone for the preparation of bR and PI3-SH3 samples, respectively.
ABBREVIATIONS
- MAS
magic angle spinning
- NMR
nuclear magnetic resonance
- ssNMR
solid state NMR
- CP
cross-polarization
- DNP
dynamic nuclear polarization
- RF
radio frequency
- RFDR
radio-frequency-driven recoupling
- S/N
signal-to-noise ratio
- APG
alanyl-prolyl-glycine
- bR
bacteriorhodopsin
- PI3-SH3
phosphatydinal-inositol-3-kinase SH3 domain
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.7b02066.
Spin—lattice relaxation T1 for 13C and 1H measured with saturation recovery, temperature dependence of 13C Bloch decay spectra of APG, full set of temperature-dependent 1H−13C CP spectra of [13C, 15N-FVYL]-PI3-SH3, full set of temperature-dependent 1H−13C CP spectra of bR, and 1D 1H−13C CP spectra and 2D RFDR DNP spectrum of [13C, 15N-FVYL]-PI3-SH3 fibril (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- 1.Renault M, Cukkemane A, Baldus M. Solid-State NMRS pectroscopy on Complex Biomolecules. Angew. Chem., Int. Ed. 2010;49:8346–8357. doi: 10.1002/anie.201002823. [DOI] [PubMed] [Google Scholar]
- 2.Lesage A. Recent Advances in Solid-State NMR Spectroscopy of Spin I = 1/2 Nuclei. Phys. Chem. Chem. Phys. 2009;11:6876–6891. doi: 10.1039/b907733m. [DOI] [PubMed] [Google Scholar]
- 3.Lange A, Becker S, Seidel K, Giller K, Pongs O, Baldus M. A Concept for Rapid Protein-Structure Determination by Solid-State NMR Spectroscopy. Angew. Chem., Int. Ed. 2005;44:2089–92. doi: 10.1002/anie.200462516. [DOI] [PubMed] [Google Scholar]
- 4.Cady SD, Schmidt-Rohr K, Wang J, Soto CS, DeGrado WF, Hong M. Structure of the Amantadine Binding Site of Influenza M2 Proton Channels in Lipid Bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castellani F, van Rossum BJ, Diehl A, Rehbein K, Oschkinat H. Determination of Solid-State NMR Structures of Proteins by Means of Three-Dimensional 15N-13C-13C Dipolar Correlation Spectroscopy and Chemical Shift Analysis. Biochemistry. 2003;42:11476–11483. doi: 10.1021/bi034903r. [DOI] [PubMed] [Google Scholar]
- 6.Brown SP, Spiess HW. Advanced Solid-State NMR Methods for the Elucidation of Structure and Dynamics of Molecular, Macromolecular, and Supramolecular Systems. Chem. Rev. 2001;101:4125–4155. doi: 10.1021/cr990132e. [DOI] [PubMed] [Google Scholar]
- 7.van der Wel PCA, Lewandowski JR, Griffin RG. Solid-State NMR Study of Amyloid Nanocrystals and Fibrils Formed by the Peptide GNNQQNY from Yeast Prion Protein Sup35p. J. Am. Chem. Soc. 2007;129:5117–30. doi: 10.1021/ja068633m. [DOI] [PubMed] [Google Scholar]
- 8.Andreas LB, Reese M, Eddy MT, Gelev V, Ni QZ, Miller EA, Emsley L, Pintacuda G, Chou JJ, Griffin RG. Structure and Mechanism of the Influenza A M2(18–60) Dimer of Dimers. J. Am. Chem. Soc. 2015;137:14877–14886. doi: 10.1021/jacs.5b04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Can TV, Sharma M, Hung I, Gor’kov PL, Brey WW, Cross TA. Magic Angle Spinning and Oriented Sample Solid-State NMR Structural Restraints Combine for Influenza A M2 Protein Functional Insights. J. Am. Chem. Soc. 2012;134:9022–9025. doi: 10.1021/ja3004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Paepe G. Dipolar Recoupling in Magic Angle Spinning Solid-State Nuclear Magnetic Resonance. Annu. Rev. Phys. Chem. 2012;63:661–684. doi: 10.1146/annurev-physchem-032511-143726. [DOI] [PubMed] [Google Scholar]
- 11.Rienstra CM. NMR Spectroscopy of Biological Solids. CRC Press; 2006. Magic-Angle Spinning Recoupling Techniques for Distance Determinations among Spin-1/2 Nuclei in Solid Peptides and Proteins; pp. 1–38. [Google Scholar]
- 12.Ernst M, Meier BH. Encyclopedia of Magnetic Resonance. John Wiley & Sons, Ltd; 2007. Adiabatic Polarization-Transfer Methods in MAS Spectroscopy. [Google Scholar]
- 13.Bjerring M, Rasmussen JT, Krogshave RS, Nielsen NC. Heteronuclear Coherence Transfer in Solid-State Nuclear Magnetic Resonance Using a Gamma-Encoded Transferred Echo Experiment. J. Chem. Phys. 2003;119:8916. [Google Scholar]
- 14.Ernst M. Heteronuclear Spin Decoupling in Solid-State NMR under Magic-Angle Sample Spinning. J. Magn. Reson. 2003;162:1–34. doi: 10.1016/s1090-7807(03)00074-0. [DOI] [PubMed] [Google Scholar]
- 15.Mithu VS, Pratihar S, Paul S, Madhu PK. Efficiency of Heteronuclear Dipolar Decoupling Schemes in Solid-State NMR: Investigation of Effective Transverse Relaxation Times. J. Magn. Reson. 2012;220:8–17. doi: 10.1016/j.jmr.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 16.Mithu VS, Paul S, Kurur ND, Madhu PK. Heteronuclear Dipolar Decoupling in Solid-State Nuclear Magnetic Resonance under Ultra-High Magic-Angle Spinning. J. Magn. Reson. 2011;209:359–363. doi: 10.1016/j.jmr.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 17.Van Beek JD, Carravetta M, Antonioli GC, Levitt MH. Spherical Tensor Analysis of Nuclear Magnetic Resonance Signals. J. Chem. Phys. 2005;122:244510. doi: 10.1063/1.1943947. [DOI] [PubMed] [Google Scholar]
- 18.Zhao X, Hoffbauer W, Schmedt auf der Gunne J, Levitt MH. Heteronuclear Polarization Transfer by Symmetry-Based Recou-pling Sequences in Solid-State NMR. Solid State Nucl. Magn. Reson. 2004;26:57. doi: 10.1016/j.ssnmr.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 19.Drobny GP, Long JR, Karlsson T, Shaw W, Popham J, Oyler N, Bower P, Stringer J, Gregory D, Mehta M, Stayton PS. Structural Studies of Biomaterials Using Double-Quantum Solid-State NMR Spectroscopy. Annu. Rev. Phys. Chem. 2003;54:531–571. doi: 10.1146/annurev.physchem.54.011002.103903. [DOI] [PubMed] [Google Scholar]
- 20.LeMaster DM, Kushlan DM. Dynamical Mapping of E. Coli Thioredoxin via 13C NMR Relaxation Analysis. J. Am. Chem. Soc. 1996;118:9255–9264. [Google Scholar]
- 21.Hong M. Determination of Multiple φ-Torsion Angles in Proteins by Selective and Extensive 13C Labeling and Two-Dimensional Solid-State NMR. J. Magn. Reson. 1999;139:389–401. doi: 10.1006/jmre.1999.1805. [DOI] [PubMed] [Google Scholar]
- 22.Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H. Structure of a Protein Determined by Solid-State Magic-Angle-Spinning NMR Spectroscopy. Nature. 2002;420:98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- 23.Jones DH, Cellitti SE, Hao X, Zhang Q, Jahnz M, Summerer D, Schultz PG, Uno T, Geierstanger BH. Site-Specific Labeling of Proteins with NMR-Active Unnatural Amino Acids. J. Biomol. NMR. 2010;46:89–100. doi: 10.1007/s10858-009-9365-4. [DOI] [PubMed] [Google Scholar]
- 24.Davis L, Chin JW. Designer Proteins: Applications of Genetic Code Expansion in Cell Biology. Nat. Rev. Mol. Cell Biol. 2012;13:168–182. doi: 10.1038/nrm3286. [DOI] [PubMed] [Google Scholar]
- 25.Concistre M, Johannessen OG, Carignani E, Geppi M, Levitt MH. Magic-Angle Spinning NMR of Cold Samples. Acc. Chem. Res. 2013;46:1914–1922. doi: 10.1021/ar300323c. [DOI] [PubMed] [Google Scholar]
- 26.Abragam A. The Principles of Nuclear Magnetism. Clarendon Press; Oxford, U.K.: 1961. p. 599. Vol. International series of monographs on physics. [Google Scholar]
- 27.Can TV, Ni QZ, Griffin RG. Mechanisms of Dynamic Nuclear Polarization in Insulating Solids. J. Magn. Reson. 2015;253:23–35. doi: 10.1016/j.jmr.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gelis I, Vitzthum V, Dhimole N, Caporini MA, Schedlbauer A, Carnevale D, Connell SR, Fucini P, Bodenhausen G. Solid-state NMR Enhanced by Dynamic Nuclear Polarization as a Novel Tool for Ribosome Structural Biology. J. Biomol. NMR. 2013;56:85–93. doi: 10.1007/s10858-013-9721-2. [DOI] [PubMed] [Google Scholar]
- 29.Barnes AB, De Paepe G, van der Wel PCA, Hu KN, Joo CG, Bajaj VS, Mak-Jurkauskas ML, Sirigiri JR, Herzfeld J, Temkin RJ, Griffin RG. High-Field Dynamic Nuclear Polarization for Solid and Solution Biological NMR. Appl. Magn. Reson. 2008;34:237–263. doi: 10.1007/s00723-008-0129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maly T, Debelouchina GT, Bajaj VS, Hu K-N, Joo C-G, Mak-Jurkauskas ML, Sirigiri JR, van der Wel PCA, Herzfeld J, Temkin RJ, Griffin RG. Dynamic Nuclear Polarization at High Magnetic Fields. J. Chem. Phys. 2008;128:052211. doi: 10.1063/1.2833582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akbey Ü, Franks WT, Linden A, Lange S, Griffin RG, Rossum B-Jv, Oschkinat H. Dynamic Nuclear Polarization of Deuterated Proteins. Angew. Chem., Int. Ed. 2010;49:7803–7806. doi: 10.1002/anie.201002044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bajaj VS, Mak-Jurkauskas ML, Belenky M, Herzfeld J, Griffin RG. Functional and Shunt States of Bacteriorhodopsin Resolved by 250 GHz Dynamic Nuclear Polarization-Enhanced Solid-State NMR. Proc. Natl. Acad. Sci. U. S. A. 2009;106:9244–9249. doi: 10.1073/pnas.0900908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mak-Jurkauskas ML, Bajaj VS, Hornstein MK, Belenky M, Griffin RG, Herzfeld J. Energy Transformations Early in The Bacteriorhodopsin Photocycle Revealed by DNP-Enhanced Solid-State NMR. Proc. Natl. Acad. Sci. U. S. A. 2008;105:883–888. doi: 10.1073/pnas.0706156105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Becker-Baldus J, Bamann C, Saxena K, Gustmann H, Brown LJ, Brown RCD, Reiter C, Bamberg E, Wachtveitl J, Schwalbe H, Glaubitz C. Enlightening the photoactive site of channelrhodopsin-2 by DNP-enhanced solid-state NMR spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2015;112:9896–9901. doi: 10.1073/pnas.1507713112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fricke P, Demers JP, Becker S, Lange A. Studies on the MxiH Protein in T3SS Needles Using DNP-Enhanced ssNMR Spectroscopy. ChemPhysChem. 2014;15:57–60. doi: 10.1002/cphc.201300994. [DOI] [PubMed] [Google Scholar]
- 36.Maus DC, Copie V, Sun B, Griffiths JM, Griffin RG, Luo S, Schrock RR, Liu AH, Seidel SW, Davis WM, Grohmann A. A Solid-State NMR Study of Tungsten Methyl Group Dynamics in [W(η5-C5Me5)Me4][PF6] J. Am. Chem. Soc. 1996;118:5665–5671. [Google Scholar]
- 37.Long JR, Sun BQ, Bowen A, Griffin RG. Molecular Dynamics and Magic Angle Spinning NMR. J. Am. Chem. Soc. 1994;116:11950–11956. [Google Scholar]
- 38.Beshah K, Griffin RG. Deuterium Quadrupole Echo NMR Study of Methyl Group Dynamics in N-acetyl-DL-(γ-d6)-Valine. J. Magn. Reson. 1989;84:268–74. [Google Scholar]
- 39.Beshah K, Olejniczak ET, Griffin RG. Deuterium NMR Study of Methyl Group Dynamics in L-alanine. J. Chem. Phys. 1987;86:4730–6. [Google Scholar]
- 40.Linser R, Sarkar R, Krushelnitzky A, Mainz A, Reif B. Dynamics in the Solid-State: Perspectives for the Investigation of Amyloid Aggregates, Membrane Proteins and Soluble Protein Complexes. J. Biomol. NMR. 2014;59:1–14. doi: 10.1007/s10858-014-9822-6. [DOI] [PubMed] [Google Scholar]
- 41.Vugmeyster L, Ostrovsky D, Ford JJ, Burton SD, Lipton AS, Hoatson GL, Vold RL. Probing the Dynamics of a Protein Hydrophobic Core by Deuteron Solid-State Nuclear Magnetic Resonance Spectroscopy. J. Am. Chem. Soc. 2009;131:13651–13658. doi: 10.1021/ja902977u. [DOI] [PubMed] [Google Scholar]
- 42.Vugmeyster L, Ostrovsky D, Khadjinova A, Ellden J, Hoatson GL, Vold RL. Slow Motions in the Hydrophobic Core of Chicken Villin Headpiece Subdomain and Their Contributions to Configurational Entropy and Heat Capacity from Solid-State Deuteron NMR Measurements. Biochemistry. 2011;50:10637–10646. doi: 10.1021/bi201515b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vugmeyster L, Ostrovsky D, Penland K, Hoatson GL, Vold RL. Glassy Dynamics of Protein Methyl Groups Revealed by Deuteron NMR. J. Phys. Chem. B. 2013;117:1051–1061. doi: 10.1021/jp311112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xue Y, Pavlova MS, Ryabov YE, Reif B, Skrynnikov NR. Methyl Rotation Barriers in Proteins from 2H Relaxation Data. Implications for Protein Structure. J. Am. Chem. Soc. 2007;129:6827–6838. doi: 10.1021/ja0702061. [DOI] [PubMed] [Google Scholar]
- 45.Latanowicz L. NMR Relaxation Study of Methyl Groups in Solids from Low to High Temperatures. Concepts Magn. Reson., Part A. 2005;27A:38–53. [Google Scholar]
- 46.Andrew ER, Hinshaw WS, Hutchins MG. Proton Magnetic-Relaxation in Crystalline Amino Acids. J. Magn. Reson. 1974;15:196–200. [Google Scholar]
- 47.Andrew ER, Hinshaw WS, Hutchins MG, Sjoblom ROI, Canepa PC. Proton Magnetic-Relaxation and Molecular-Motion in Polycrystalline Amino acids 2. Alanine, Isoleucine, Leucine, Methionine, Norleucine, Threonine and Valine. Mol. Phys. 1976;32:795–806. [Google Scholar]
- 48.Daube D, Aladin V, Heiliger J, Wittmann JJ, Barthelmes D, Bengs C, Schwalbe H, Corzilius B. Heteronuclear Cross-Relaxation under Solid-State Dynamic Nuclear Polarization. J. Am. Chem. Soc. 2016;138:16572–16575. doi: 10.1021/jacs.6b08683. [DOI] [PubMed] [Google Scholar]
- 49.Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, Donovan KJ, Michael B, Wall J, Linse S, et al. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc. 2016;138:9663–9674. doi: 10.1021/jacs.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lehnert E, Mao JF, Mehdipour AR, Hummers G, Abele R, Glaubitz C, Tampe R. Antigenic Peptide Recognition on the Human ABC Transporter TAP Resolved by DNP-Enhanced Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2016;138:13967–13974. doi: 10.1021/jacs.6b07426. [DOI] [PubMed] [Google Scholar]
- 51.Kaushik M, Bahrenberg T, Can TV, Caporini MA, Silvers R, Heiliger J, Smith AA, Schwalbe H, Griffin RG, Corzilius B. Gd(III) and Mn(II) complexes for dynamic nuclear polarization: small molecular chelate polarizing agents and applications with site-directed spin labeling of proteins. Phys. Chem. Chem. Phys. 2016;18:27205–27218. doi: 10.1039/c6cp04623a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fricke P, Mance D, Chevelkov V, Giller K, Becker S, Baldus M, Lange A. High resolution observed in 800 MHz DNP spectra of extremely rigid type III secretion needles. J. Biomol. NMR. 2016;65:121–126. doi: 10.1007/s10858-016-0044-y. [DOI] [PubMed] [Google Scholar]
- 53.Gochnauer MB, Kushner DJ. Growth and Nutrition of Extremely Halophilic Bacteria. Can. J. Microbiol. 1969;15:1157–63. doi: 10.1139/m69-211. [DOI] [PubMed] [Google Scholar]
- 54.Batey RT, Inada M, Kujawinski E, Puglisi JD, Williamson JR. Preparation of Isotopically Labeled Ribonucleotides for Multidimensional NMR-Spectroscopy of RNA. Nucleic Acids Res. 1992;20:4515–4523. doi: 10.1093/nar/20.17.4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oesterhelt D, Stoeckenius W. Isolation of the Cell Membrane of Halobacterium Halobium and Its Fractionation into Red and Purple Membrane. Methods Enzymol. 1974;31:667–78. doi: 10.1016/0076-6879(74)31072-5. [DOI] [PubMed] [Google Scholar]
- 56.Sauvee C, Rosay M, Casano G, Aussenac F, Weber RT, Ouari O, Tordo P. Highly Efficient, Water-Soluble Polarizing Agents for Dynamic Nuclear Polarization at High Frequency. Angew. Chem., Int. Ed. 2013;52:10858–10861. doi: 10.1002/anie.201304657. [DOI] [PubMed] [Google Scholar]
- 57.Song C, Hu K-N, Joo C-G, Swager TM, Griffin RG. TOTAPOL: A Biradical Polarizing Agent for Dynamic Nuclear Polarization Experiments in Aqueous Media. J. Am. Chem. Soc. 2006;128:11385–11390. doi: 10.1021/ja061284b. [DOI] [PubMed] [Google Scholar]
- 58.Guijarro JI, Sunde M, Jones JA, Campbell ID, Dobson CM. Amyloid Fibril Formation by an SH3 Domain. Proc. Natl. Acad. Sci. U. S. A. 1998;95:4224–4228. doi: 10.1073/pnas.95.8.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barnes AB, Mak-Jurkauskas ML, Matsuki Y, Bajaj VS, Wel PCAvd, DeRocher R, Bryant J, Sirigiri JR, Temkin RJ, Lugtenburg J, et al. Cryogenic sample exchange NMR probe for magic angle spinning dynamic nuclear polarization. J. Magn. Reson. 2009;198:261–270. doi: 10.1016/j.jmr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jawla S, Ni QZ, Barnes A, Guss W, Daviso E, Herzfeld J, Griffin R, Temkin R. Continuously Tunable 250 GHz Gyrotron with a Double Disk Window for DNP-NMR Spectroscopy. J. Infrared, Millimeter, Terahertz Waves. 2013;34:42–52. doi: 10.1007/s10762-012-9947-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barnes AB, Nanni EA, Herzfeld J, Griffin RG, Temkin RJ. A 250 GHz gyrotron with a 3 GHz tuning bandwidth for dynamic nuclear polarization. J. Magn. Reson. 2012;221:147–153. doi: 10.1016/j.jmr.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith SA, Levante TO, Meier BH, Ernst RR. Computer-Simulations in Magnetic-Resonance - An Object-Oriented Programming Approach. J. Magn. Reson., Ser. A. 1994;106:75–105. [Google Scholar]
- 63.Cheng VB, Suzukawa HH, Wolfsberg M. Investigations of a Nonrandom Numerical Method for Multimensional Integration. J. Chem. Phys. 1973;59:3992–3999. [Google Scholar]
- 64.Eddy MT, Belenky M, Sivertsen AC, Griffin RG, Herzfeld J. Selectively dispersed isotope labeling for protein structure determination by magic angle spinning NMR. J. Biomol. NMR. 2013;57:129–139. doi: 10.1007/s10858-013-9773-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee AL, Wand AJ. Microscopic Origins of Entropy, Heat Capacity and the Glass Transition in Proteins. Nature. 2001;411:501–504. doi: 10.1038/35078119. [DOI] [PubMed] [Google Scholar]
- 66.Markin AV, Markhasin E, Sologubov SS, Ni QZ, Smirnova NN, Griffin RG. Low-Temperature Polymorphic Phase Transition in a Crystalline Tripeptide l-Ala-l-Pro-Gly·H2O Revealed by Adiabatic Calorimetry. J. Phys. Chem. B. 2015;119:1787–92. doi: 10.1021/jp508710g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bajaj VS, van der Wel PCA, Griffin RG. Observation of a Low-Temperature, Dynamically Driven Structural Transition in a Polypeptide by Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2009;131:118–128. doi: 10.1021/ja8045926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rice DM, Wittebort RJ, Griffin RG, Meirovitch E, Stimson ER, Meinwald YC, Freed JH, Scheraga HA. Rotational Jumps of the Tyrosine Sidechain in Crystalline Enkephalin H2 NMR Lineshapes for Aromatic Rong Motion in Solids. J. Am. Chem. Soc. 1981;103:7707–7710. [Google Scholar]
- 69.Rice DM, Meinwald YC, Scheraga HA, Griffin RG. Tyrosyl Motion in Peptides H2 NMR Lineshapes and Spin Lattice Relaxation. J. Am. Chem. Soc. 1987;109:1636–1640. [Google Scholar]
- 70.Kamihira M, Naito A, Tuzi S, Saito H. Phenyl ring dynamics of enkephalin molecules and behavior of bound solvents in the crystalline states by H−2 NMR spectroscopy. J. Phys. Chem. A. 1999;103:3356–3363. [Google Scholar]
- 71.Bayro MJ, Debelouchina GT, Eddy MT, Birkett NR, MacPhee CE, Rosay M, Maas WE, Dobson CM, Griffin RG. Intermolecular Structure Determination of Amyloid Fibrils with Magic-Angle Spinning and Dynamic Nuclear Polarization NMR. J. Am. Chem. Soc. 2011;133:13967–13974. doi: 10.1021/ja203756x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.