

Abstract
Diabetic kidney disease (DKD) has a complex and prolonged pathogenesis involving many cell types in the kidney as well as extrarenal factors. It is clinically silent for many years after the onset of diabetes and usually progresses over decades. Given this complexity, a comprehensive and unbiased molecular approach is best suited to help identify the most critical mechanisms responsible for progression of DKD and those most suited for targeted intervention. Systems biological investigations provide such an approach since they examine the entire network of molecular changes that occur in a disease process in a comprehensive way instead of focusing on a single abnormal molecule or pathway. Systems biological studies can also start with analysis of the disease in humans, not in animal or cell culture models that often poorly reproduce the changes in human DKD. Indeed, in the last decade, systems biological approaches have led to the identification of critical molecular abnormalities in DKD and have directly led to development of new biomarkers and potential treatments for DKD.
Keywords: transcriptomics, proteomics, metabolomics, diabetes, epidermal growth factor
Diabetic kidney disease (DKD) is the major cause of chronic kidney disease (CKD) and end-stage renal disease (ESRD) in the United States and probably in the world.1 Despite some progress in reducing mortality and delaying kidney disease in the last few decades due to improved glycemic control, blood pressure lowering and the use of renin-angiotensin system blockade, the percentage of diabetic patients who develop kidney failure has not materially declined.2 Thus, millions of individual patients worldwide are in urgent need of new approaches to treatment that will actually prevent progression to ESRD. One reason for the slow progress in finding adequate therapies for DKD is the lack of comprehensive understanding of the underlying pathogenic mechanisms. Unfortunately, targeting single pathways and molecules based on hypothesis-driven research has resulted in no significant advances in treatment in the last 25 years. Unraveling the underlying mechanisms for DKD is complicated by the likelihood that a number of molecular processes interrelate over a number of years to cause the tissue damage that is the ultimate manifestation of the disease. A comprehensive and unbiased molecular approach is best suited to reveal this complex pathogenesis. Systems biological methods provide such an approach and have the additional important advantage that they can start with human disease samples to help ensure that the processes being studied are relevant to human DKD. This is important since animal and cell culture models fail to recapitulate many aspects of DKD found in humans.3 In this brief review, we will discuss how a systems biological approach to understand and treat DKD has advanced the field over the last decade or so.
Systems Biological Approaches to DKD
There are many definitions to systems biology and different definitions apply to different areas of research. For those interested in disease pathogenesis, system biology may be defined as an information-rich discovery process that analyzes biological systems and their behavior as regulatory networks. Studying these networks in multiple relevant tissues and associating them to clinically relevant data can identify the complex biological patterns that underlie disease onset and progression. To restate this in terms of DKD research, systems biology uses a set of experimental tools and computational approaches that comprehensively identify networks of molecular changes that occur in patients with DKD compared to normal individuals, and focuses when possible on changes in individual kidney cells or complexes of interrelated kidney cells. The analysis of these altered networks is used to identify associations among the many molecular changes that best predict and are likely to enhance disease progression or amelioration. Pragmatically, these studies are often restricted to one “scale” of molecular analysis (e.g., transcriptomics focusing on gene expression phenotypes) although they may combine several scales, so called “multiscalar” or “multi-omics” approaches (Figure 1), which often give the most powerful results. By their nature, systems biological studies are often hypothesis-generating, not hypothesis-testing. While not an issue for biomarker discovery, potential mechanisms of disease and therapeutic targets identified by systems biological approaches need to be validated by more conventional experimental methodologies. Examples of how systems biological approaches can be interwoven with more conventional experimental studies are noted in the sections on DKD biomarkers and targets for therapy, below.
Figure 1.
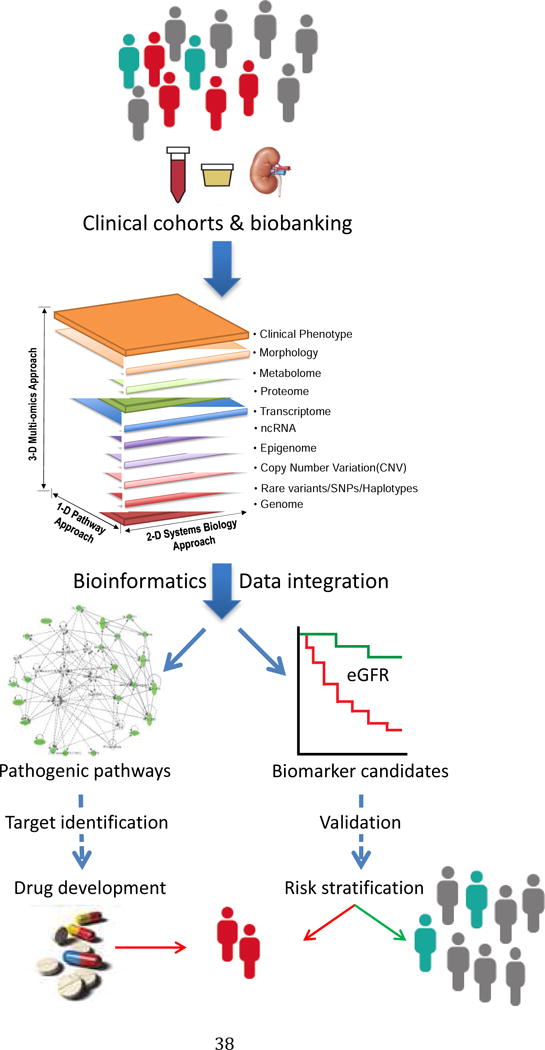
Schema for systems biological studies of diabetic kidney disease. Starting from clinical data and biosamples, including kidney biopsy tissue, systems-wide interrogation of the molecular phenotype of human participants with DKD and appropriate non-DKD controls are analyzed with a variety of bioinformatics approaches to identify pathways and networks of molecular interactions (left) that may be responsible for DKD progression. From these pathways and networks, key pathogenic molecules are identified that can be targeted through drug development to interrupt disease progression. In parallel, these studies identify molecular biomarkers (right) which provide early diagnosis of DKD or predict the progression of kidney failure in DKD.
A number of systems biological reports on human DKD have been published over the past decade or more. Many of these report transcriptomic analyses that examine the changes in gene expression in diabetic kidney tissues. A number of epigenomic, proteomic and metabolomic analyses have also been reported. In the sections below, examples of each of these approaches will be described. This review is restricted to studies that have used human tissues and that demonstrate how systems biological approaches can move the field forward. It is not a systematic review. Moreover, we have not reviewed DKD genetic studies, as these have been extensively reviewed relatively recently4 and no major advances have occurred in this area since that publication. The examples below have been chosen for illustrative purposes only, not because we believe they are definitive.
The fundamental role of bioinformatics in systems biological studies
As noted above, systems biology is inherently computational. The associations and connections in genome-wide datasets can only be made with computational tools and the better and more sophisticated the computational tools, the better and more sophisticated the systems biological analysis. As will be noted below, many of the initial systems-wide studies utilized quite simple analytic tools and therefore provided little new insight into disease processes, though were important as proof-of-principle studies. Later studies have utilized a panoply of computational tools and have often generated specific tools to lead to the most meaningful observations and discoveries. We have emphasized these computational aspects, below.
Clinical cohorts and biobanking: the critical underpinning for clinically pertinent DKD studies
Because animal and cell culture models have only partly replicated the molecular phenotype found in humans with DKD and because diabetic animals do not develop progressive DKD it has become imperative for meaningful DKD research to utilize human phenotypic data as a benchmark. This is especially true for systems biological studies which synthesize data from all molecular changes in the examined tissue or cell-type and reflect species-specific pathways which, if taken from models, do not always overlap with those in humans.5 Thus, a critical underpinning for systems biological studies of DKD has been the establishment and availability of long-standing cohorts of patients with chronic kidney diseases including DKD. Especially important for this work has been the availability of carefully obtained and preserved kidney biopsy, blood and urine samples paired with detailed longitudinal clinical information acquired after kidney biopsies from participants in the cohorts. These have allowed identification of molecular pathways or molecules that best predict progression of DKD or are likely to be therapeutic targets without having to follow patients for years in the future. There now exist multiple CKD registries accompanied by large well-curated biobanks with multiple types of samples (including plasma, urine and kidney tissue) that can be used for molecular analysis.
Systems biological approaches to develop new DKD biomarkers
Noninvasive molecular biomarkers with better sensitivity and specificity are urgently needed for the early diagnosis of DKD as well as identification of patients who are most at risk of disease progression. An ideal DKD biomarker would be easily detectable in body fluids such as plasma or urine, would represent diverse molecular mechanisms that underlie DKD pathogenesis and would be kidney-specific in order to eliminate effects of extrarenal sources. Most useful biomarkers are either proteins or metabolites, and unbiased proteomic and metabolomic approaches have helped discover a number of new potential biomarkers. Although none of these DKD biomarkers have been fully validated, several promising candidates have been discovered by these methods.
Transcriptomic studies
A study from our group illustrated the feasibility of using transcriptomic data from human kidney biopsies to identify critical kidney-specific pathophysiologic molecules based on alterations in gene expression and then to test whether the protein products of these mRNAs could serve as urinary biomarkers to predict CKD progression.6 Following this strategy, urinary epidermal growth factor (uEGF) was identified as an independent predictor of eGFR slope and of the composite CKD progression endpoint of ESRD or 40% reduction of baseline eGFR after adjusting for age, sex, baseline eGFR and ACR. Moreover, uEGF added improved prognostic accuracy to eGFR and ACR in predicting progression in 3 separate CKD cohorts from different parts of the world. Although there was no specific DKD cohort included in this study, DKD patients were highly represented in one of the cohorts, and 135 CKD patients with diabetes, 70 of whom had biopsy-proven DKD diagnosis, contributed to the final results. A subgroup analysis of these diabetic patients also revealed a strong correlation between uEGF and eGFR or eGFR slope,6 supporting the prognostic value of uEGF in DKD. More recently, a urinary proteomics study in a mouse model by Betz and colleagues was used as a basis for human studies to also show that uEGF predicts CKD in diabetic patients without albuminuria.7 Thus, the combination of transcriptomics and targeted proteomics has identified a new kidney specific urinary biomarker that has been validated in multiple cohorts in 2 studies.
Proteomic studies
Proteomic studies have produced less rapid success in this area than was initially predicted a decade ago8 due to multiple challenges such as the complexity of proteomics technology; strict requirements for standardized specimen collection, processing and storage; difficulties in quantification of urinary markers with low abundances; and the large variation in urine protein excretion between individuals. Despite these difficulties, a urinary biomarker panel was derived from capillary electrophoresis-mass spectrometry (MS) analysis of urine samples of patients with CKD. This panel of 273 urinary peptides, the CKD273 classifier,9 has subsequently been validated in several CKD cohorts, including those with DKD. The CD273 classifier has been shown to predict onset of DKD in type 2 diabetic patients.10,11 In a meta-analysis of CKD patient data from the Human Urinary Proteome database in which over 75% of the patients had diabetes, the CKD273 classifier also better predicted a sustained decline in eGFR in patients with early DKD (eGFR 70–80 mL/min/1.73m2) than did baseline albuminuria.2,12 Finally, the classifier has now been used to help stratify patients for a randomized controlled trial of spironolactone in the treatment of early DKD.13 Results with the CKD273 classifier have been consistent enough that the US Food and Drug Administration has encouraged “the further development of CKD273…to be used in combination with current measures (i.e., albuminuria, serum creatinine) in early phase clinical trials in … DKD to identify patients with early stage disease who may be more likely to progress.”11
Metabolomic Studies
Metabolomic analyses can also identify useful biomarker panels14. Similar to proteomics, sample processing and handling are critical to the success of metabolomic studies. Additional confounding factors, in addition to those that beset proteomic biomarker approaches, may impact metabolomics studies. These confounding factors were recently and comprehensively reviewed by Hocher et al.,14 including genetic background, sex, age, body mass index, medication, lifestyle, circadian rhythms, hormonal status, and nutrition and fasting. These confounding factors, together with a lack of large prospective study cohorts and a dependency on sophisticated bioinformatics techniques for data interpretation, resulted in the slow progress of DKD metabolomic biomarker identification. However there have been promising examples such as the study carried out by Niewczas and colleagues.15 This nested case-control study used plasma samples of type 2 diabetic patients with normal or mildly impaired baseline renal function from the Joslin Kidney Study cohort, and aimed to identify metabolites that were associated with progression to ESRD. Metabolomic profiles of 40 patients who developed ESRD during 8–12 years of follow-up were compared to those of 40 control patients who were alive but did not progress to ESRD. Seventy eight metabolites previously reported to be elevated in ESRD (uremic solutes) were identified in this study, and 16 of them were elevated in the baseline plasma of cases years before ESRD developed. Essential amino acids and their derivatives were significantly depleted in the cases. These findings remained statistically significant after adjustment for albumin excretion rate, eGFR or HbA1c. Uremic solute differences were then confirmed by targeted quantitative metabolite measurements. Abnormal plasma concentrations of uremic solutes and essential amino acids were associated with progression to ESRD. The findings from this exploratory study will need to be replicated in independent and prospective study cohorts.
One concern about biomarker candidates derived from unbiased proteomic and metabolomics approach, especially if they are expressed in other tissues, is whether a biomarker candidate truly represents processes in the kidneys and whether it may have reduced specificity due to its production by extra-renal tissues. These concerns are obviated by selecting candidate biomarkers that appear to be derived solely from kidney cells.
Multiscalar studies
Multi-omic approaches to biomarker discovery that combine transcriptomics with either metablomics or proteomics have recently become easier as investigators can now take advantage of published DKD transcriptomic datasets. To facilitate access to such datasets, a web-based search and analytical platform, Nephroseq (www.nephroseq.org), was established. This user-friendly tool includes all published human DKD (and other CKD) transcriptomic datasets to allow for easy data mining and in-depth data analysis. As an example of the utility of such a tool, we identified from published literature a non-exhaustive list of putative protein biomarkers for kidney diseases and determined the association of the expression levels for the genes encoding each biomarker with kidney function in DKD patients from two Nephroseq datasets.6,16,17 In Table 1, we have displayed the association of gene expression with eGFR as well as the gene expression differences between DKD patients and normal controls for each biomarker. Using these data, investigators can assess whether the mRNA expression for the gene encoding a putative protein biomarker in kidney tissue correlates with disease pathophysiology. If it does, this increases the likelihood of the protein serving as a useful diagnostic or prognostic biomarker in DKD patients.
Table 1.
Association of intrarenal gene expression levels with kidney function for published DKD protein biomarker candidates.
| Gene Symbol/Alias | Glomerulus | Tubulointerstitium | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Dataset 1 | Dataset 2 | Dataset 1 | Dataset 2 | ||||||
|
| |||||||||
| Corr. eGFR | DKD vs control | Corr. eGFR | DKD vs control | Corr. eGFR | DKD vs control | Corr. eGFR | DKD vs control | ||
| ADIPOQ | −0.08 | 0.89* | 0.60 | 0.97 | 0.23 | 0.97 | 0.01 | 0.87* | 38–41 |
| AGT/SERPINA8 | 0.41 | 0.57* | 0.01 | 1.39 | 0.52* | 0.75 | 0.56 | 0.78 | 42–44 |
| AMBP/A1M | 0.34 | 0.86 | 0.58 | 1.15 | 0.18 | 0.93 | −0.67* | 0.98 | 42,45,46 |
| CCL2 | 0.01 | 3.11* | −0.40 | 1.65 | −0.39 | 1.56* | −0.09 | 3.47* | 41,47–53 |
| COL4A1 | −0.36 | 1.74* | −0.53 | 1.07 | −0.64* | 1.95* | −0.21 | 4.41* | 42,54,55 |
| CST3/Cystatin C | 0.09 | 1.30* | −0.47 | 1.72 | −0.78* | 1.05 | −0.07 | 1.32 | 42,56–59 |
| CTGF/CCN2 | 0.15 | 0.89 | 0.28 | 0.21* | −0.39 | 0.46* | 0.44 | 1.95 | 51,60–62 |
| CXCL10/IP10 | 0.46 | 2.01 | −0.01 | 0.97 | −0.51* | 1.13 | −0.34 | 2.13 | 53 |
| CXCL8/IL8 | −0.38 | 1.37 | 0.1 | 1.50 | −0.45 | 1.17 | −0.34 | 4.03* | 47,53 |
| EGF | 0.81* | 0.33* | 0.10 | 0.42* | 0.73* | 0.37* | 0.83* | 0.19* | 6,7 |
| FABP1/L-FABP | 0.52 | 0.24* | 0.37 | 1.02 | 0.51* | 0.55* | 0.45 | 0.51 | 42,63,64 |
| FGF23 | −0.43 | 0.88* | −0.29 | 1.04 | −0.19 | 0.92* | −0.46 | 0.92 | 65,66 |
| GH1 | −0.25 | 0.91* | 0.18 | 1.09 | −0.17 | 0.93* | −0.35 | 0.9 | 67–69 |
| GRN | −0.01 | 1.16* | −0.42 | 1.08 | −0.38 | 1.16* | −0.24 | 1.53* | 70 |
| HAVCR1/KIM1 | NA | NA | 0.05 | 1.25* | NA | NA | 0.08 | 1.22 | 65,71–77 |
| HIF1A | −0.43 | 1.09 | 0.11 | 1.26 | −0.44 | 1.25* | −0.03 | 1.55* | 78 |
| HP | −0.19 | 1.01 | 0.29 | 1.17 | −0.67* | 1.07 | −0.20 | 1.08 | 79,80 |
| IFNG | 0.02 | 1.01 | NA | NA | −0.25 | 1.00 | NA | NA | 81,82 |
| IGF1 | −0.15 | 0.29* | −0.11 | 0.69 | −0.62* | 0.82* | 0.30 | 0.98 | 78,83 |
| IL10 | −0.28 | 1.00 | 0.18 | 1.10 | −0.10 | 0.94 | 0.05 | 0.98 | 47,81 |
| IL6 | −0.04 | 1.05 | 0.46 | 0.98 | −0.36 | 0.90 | −0.41 | 1.01 | 47,81 |
| KL | 0.36 | 0.42* | −0.02 | 0.89 | 0.33 | 0.59* | 0.72* | 0.42 | 84–88 |
| LCN2/NGAL | −0.37 | 1.35 | 0.05 | 1.54* | −0.46 | 1.83* | −0.47 | 2.57* | 42,58,65,72–74,76,78,89–91 |
| MMP2 | −0.62* | 2.3* | −0.07 | 1.39* | −0.57* | 1.34* | −0.57 | 1.42 | 69 |
| MMP7 | −0.48 | 2.43* | −0.52 | 4.09* | −0.55* | 4.27* | −0.44 | 13.32* | 69,92 |
| NAGLU/NAG | 0.71* | 0.98 | 0.73* | 1.13 | 0.58* | 0.78* | 0.41 | 0.71 | 42,74,76,93,94 |
| NPHS1 | 0.54 | 0.43* | 0.32 | 0.12* | 0.32 | 0.46* | 0.27 | 0.66* | 42,95,96 |
| NPPB | −0.13 | 0.88* | 0.35 | 1.04 | −0.38 | 0.99 | −0.38 | 0.78* | 56,97 |
| RBP4 | 0.14 | 0.39* | 0.79* | 0.62 | 0.28 | 0.68 | −0.23 | 0.26* | 49,94,98–100 |
| TF | 0.12 | 0.91 | 0.48 | 1.20* | 0.00 | 1.00 | 0.34 | 0.93 | 101 |
| TNF | −0.21 | 1.42* | 0.65 | 1.15 | −0.15 | 1.12 | −0.62 | 0.90 | 75,81,102 |
| TNFRSF1A/TNFR1 | −0.18 | 1.07 | −0.35 | 1.39* | −0.70* | 0.94 | −0.23 | 1.31* | 72,75,103,104 |
| TNFRSF1B/TNFR2 | 0.07 | 2.42* | −0.18 | 1.52* | −0.52* | 1.29* | −0.64* | 1.57* | 75,103–105 |
| UMOD | 0.38 | 0.26* | −0.08 | 0.31* | 0.63* | 0.70 | 0.48 | 0.33* | 106,107 |
| VCAM1 | −0.31 | 1.67* | −0.48 | 1.37* | −0.59* | 2.00* | 0.12 | 2.64* | 47,108 |
| VEGFA/VEGF | 0.43 | 0.64* | 0.35 | 0.38* | 0.70* | 0.64* | 0.67* | 0.60 | 66,78,88 |
| VWF | −0.21 | 1.27 | −0.73* | 1.94* | −0.31 | 1.41* | −0.64* | 2.58* | 78,82 |
Gene expression data for both glomeruli and tubulointerstitium were abstracted from two independent published transcriptomic human DKD datasets (see identification below) for candidate biomarkers. Association of gene expression with eGFR was determined as was the relative biomarker expression in DKD vs. normal controls. Shaded areas depict biomarkers in which either (or both) eGFR correlation or gene expression differences were statistically significant in both datasets.
Corr. eGFR: correlation of gene expression with baseline eGFR. NA: data not available.
Identification of pathogenic pathways and therapeutic targets for DKD
Transcriptomic studies
Most comprehensive systems biological studies of human DKD have been transcriptomic analyses. These studies have depended largely on the availability of small amounts of mRNA from appropriately preserved human biopsy material that became largely available with biobanking efforts that started in Europe18 and spread rapidly to other continents. An early study of 2 diabetic and 2 cadaveric donor kidneys was published in 2004.19 This study served mainly as proof-of-principle for the use of transcriptomics, but had too few subjects for appropriate statistical analysis and depended on control samples (deceased donors) which subsequently were shown to have abnormal gene expression profiles likely due to inflammatory and other pathway activation in brain-dead donors.20 Nonetheless, this study did make several important observations that were confirmed in subsequent publications, including that glomerular gene expression profiles fall with progressive DKD, due in part to severe reduction of podocyte specific gene expression, such as VEGF and nephrin.
One of the first human transcriptomic analyses using sufficient numbers of patient samples, appropriate controls and unbiased mRNA expression screening found evidence of enhanced inflammatory stress response in the tubulointerstitium of patients with progressive DKD. Pathway mapping suggested that nuclear factor-kappa B (NF-kB) was a major upstream activator of many inflammatory genes active in progressive DKD when compared to early DKD with 54 of 138 NF-kB responsive genes being upregulated in progressive disease and only one being enhanced in early disease. In addition, this study identified a NF-kB promoter module that appeared to be specifically responsive in the inflammatory stress of progressive DKD.20 The unbiased assessment that pinpointed NF-kB activation in progressive human DKD helped cement the role of this pro-inflammatory transcription factor in kidney gene regulation and the pathogenesis of progressive DKD. Many additional studies in animal models and humans have now supported a role for NF-kB and downstream genes, such as CCL2 and CCL5, in the chronic inflammation that drives progressive DKD.
A second transcriptomic analysis was performed using glomeruli and tubulointerstitial tissues from patients with both early DKD and progressive DKD as well as those from normal kidneys (living donor kidney samples). In this analysis, one of the most prominent pathway changes detected was in the JAK-STAT signaling pathway. Virtually all JAK-STAT genes were expressed at substantially higher levels in the glomeruli from patients with early DKD compared to normal controls. Conversely, tubulointerstitial JAK-STAT gene expression was not elevated in early DKD but was high in the patients with more progressive DKD and the degree of elevation of JAK-STAT mRNA levels was tightly inversely correlated with the decline in eGFR (i.e., JAK-STAT gene expression increased as eGFR decreased).21 A similar pattern of progressive increase in JAK-STAT gene expression was not seen in glomerular samples as patients with progressive DKD had lower levels of glomerular JAK-STAT mRNAs than did those with early disease, though glomerular levels in progressive disease tended to still be higher than in normal controls. These data have been confirmed by transcriptomic studies in other DKD cohorts17 using Nephroseq. Moreover, unpublished gene expression analysis of STAT3-dependent genes in glomeruli in early DKD showed a highly significant increase in mRNA levels of those genes compared to normal controls (S. Eddy et al., unpublished data), implying increased activation as well as expression of JAK-STAT signaling in the glomerulus early DKD. A number of studies have suggested that JAK-STAT activation promotes progression of DKD and a recently published report found that human-like increases in JAK2 expression specifically in podocytes promoted much more severe DKD in a mouse model.22 Finally, a phase 2 study of a JAK1/2 inhibitor in participants with moderately severe DKD showed a significant reduction in albuminuria, as well as decreased inflammatory biomarkers, including plasma TNF receptor levels, in a phase 2 study that has been reported in preliminary form.23
Another comprehensive transcriptomic assessment was performed on glomeruli and tubulointerstitium from patients with advanced DKD (average eGFR for patients used for glomerular studies was 31 and the average eGFR for patients used for tubulointerstitial studies was 22).17 In this analysis, approximately 70% of differentially expressed glomerular genes were decreased in diabetic patients, whereas the opposite was true in the tubulointerstitium. This study demonstrated reduced expression of many podocyte specific genes in diabetic glomeruli, but an increase in inflammation and fibrosis related gene expression in both glomeruli and tubulointerstitium from DKD patients. A specific increase in a complement activation signal in the glomeruli of these patients was found suggesting that disease progression was stimulated by low level activation of the complement system in glomerular cells.17
Some general conclusions can be derived from these studies that apply to transcriptomic as well as other systems-wide analyses in DKD:
kidney biopsies from sufficient numbers of individual patients need to be analyzed to allow for appropriate statistical comparison to control groups;
false discovery rates need to be applied to each analysis when using unbiased discovery approaches to account for multiple comparisons;
appropriate control tissues (including those from normal healthy controls and controls with similar exposure to disease condition, such as diabetes, but without kidney complications) need to be utilized. At present the gold-standard normal control tissue is biopsy tissue taken from living donor kidneys at the time of transplantation, but equally important, though more difficult to obtain, is biopsy tissue taken from diabetic patients without any clinically or pathologically detectable DKD;
glomerular gene expression patterns appear to be quite different in early vs. late DKD so early biopsies (including protocol biopsies) should be performed to obtain needed early pathogenic information. Indeed, based on an analysis of early DKD gene expression changes, glomerular gene expression in general tends to be increased in early DKD, as it is in diabetic rodent glomeruli, and then reduced as DKD progresses,5 whereas tubulointerstitial expression tends to increase with DKD progression.17
transcription factor activity can be deduced from systematic assessment of downstream regulated gene expression, as was noted above for both NF-kB and STAT3 genes;
the central role of inflammation in progressive DKD in both glomeruli and tubulointerstitium has been confirmed by these transcriptomic studies. This suggests that targeting this chronic inflammatory state with agents that inhibit JAK/STAT, complement or cytokine/chemokine signaling could be effective in slowing progression of disease, as several small randomized controlled trials have suggested.23–25
Epigenomic studies
Epigenetic effects on gene transcription in response to environmental exposures are profound and the many environmental abnormalities in diabetes certainly participate in the pathogenic alterations resulting in altered gene expression in DKD. Non-genomic regulation of gene transcription is determined by multiple mechanisms including DNA methylation on cytosines, in CG dinucleotide sequences, and a complex set of post-translational modifications of histone proteins via methylation and acetylation. While a number of epigenetic studies have been conducted in both humans and models with DKD, only a very few have performed true genome-wide assessment of epigenetic alterations in human DKD.
An early genome-wide analysis of DKD patients examined DNA methylation predominately in CG Islands within proximal promoter regions of nearly 15,000 genes in blood from patients with type 1 diabetes, either with or without overt nephropathy as determined by albuminuria.26 This study found changes in methylation status that were correlated with DKD in 19 gene promoters. One of these promoters was in a gene that had been previously found to have a genetic variant associated with diabetic nephropathy, UNC13B, a protein expressed in the kidney cortical epithelia and upregulated by hyperglycemia.27 As with many initial studies using systems biological methods, this report served as proof-of-principle that further genome-wide epigenomic analyses could be performed, but given its limitations including comparison of groups that were unlikely to have been completely distinct (many of the “non-nephropathy” group likely had significant undetected disease), the use of blood samples instead of kidney samples, and the lack of bioinformatic pathway or network analysis, little new understanding of DKD pathogenesis emerged from this analysis.
In a somewhat newer and more powerful study, genome-wide cytosine DNA-methylation changes were assessed in tubular regions of normal parts of transplant and tumor nephrectomy specimens from patients with chronic kidney diseases, mostly DKD, and those without kidney disease.28 The investigators then identified differentially demethylated regions between the DKD patients and controls with 70% of these being in the DKD patients. Since demethylation permits transcription, this suggests that enhanced transcription in these regions occurred in the DKD tubules, which agrees in general with transcriptomic data that suggest a general pattern of increased gene expression. The demethylated regions were found to be mostly in non-promoter regulatory regions of the genome that contained consensus-binding motifs for kidney-specific transcription factors that were likely to coordinate the observed increases in gene transcription in 415 of the 1092 hypomethylated genes.28 Bioinformatic pathway analysis indicated that the deduced transcriptional changes in the DKD group were profibrotic or involved in renal development.
This second study shows the potential power of genome-wide epigenomic studies in DKD, and certainly future studies will likely shed more light on how the diabetic environment regulates gene transcription. Such studies will likely point to important targets for treatment either by altering the diabetic milieu in specific ways or by directly targeting the most important epigenetically regulated genes. Similar studies are emerging from specific analysis of non-coding RNAs (such as micro-RNAs and long non-coding RNAs) that also transduce environmental effects on gene transcription through a variety of mechanisms. Though formally transcriptomic in nature, these changes in non-coding RNAs result in multiple epigenetic downstream gene expression changes, similar to what occurs with DNA methylation and histone acetylation and methylation, and transduce environmental alterations to transcriptomic ones quite efficiently.
Proteomic studies
Similar to the situation with epigenomics, it has been difficult to perform truly systems-wide proteomic analysis of human DKD. As noted in the biomarker section, above, technical issues have slowed truly proteome-wide studies, but the field is moving rapidly and such analyses will likely become common in the near future. In addition, a newer focus on urinary peptidomics may also accelerate identification of important pathogenic pathways.
A relatively early non-targeted proteomics study was performed on urine from 36 participants: 12 healthy controls, 12 diabetic participants with no albuminuria or other evidence of kidney disease, and 12 participants with diabetes and albuminuria.29 The investigators identified proteins that were either exclusive to the diabetes + albuminuria group or were at least substantially higher in that group than in the other groups. They were able to successful detect over 150 intact protein signatures. Megalin and cubulin were among 2 of the most abundant urinary proteins found only in the diabetes + albuminuria group. These endocytic proteins are highly expressed in the proximal tubules and are responsible for uptake of most filtered proteins. This finding suggested to the authors that some proximal tubular pathology could be contributing to loss of these 2 proteins which in turn could augment proteinuria. That surmise, made in 2009, is in accord with the more recent focus on the proximal tubule as a direct contributor to albuminuria and to proximal tubular dysfunction playing an important role in the early pathogenesis of DKD.30 Importantly, they found a number of other proteins that were significantly augmented in the urine of diabetic participants with albuminuria. These findings and others were recently assessed by a systematic review of all published DKD urine proteomic studies that used a sophisticated bioinformatics approach to help identify candidates most likely to provide insights into DKD pathogenesis.31
This analytic review by Van et al. surveyed all urinary proteomic studies of DKD in the literature and included for its analyses only those that examined human samples, that used MS and that compared levels in diabetic patients and controls. The authors reviewed a total of 31 studies that met those criteria. In addition to their systematic review, the authors provided new bioinformatic analyses that were generally absent from the original studies. By this approach, they were able to identify important pathways and networks enriched in both early and progressive DKD. This analysis pointed to the critical role of extracellular matrix regulation and metabolism, cholesterol and lipid dysregulation, inflammation, regulation of the immune response, and cell adhesion as the critical processes that drove early disease. Similar to findings of the transcriptomic analyses noted above, these investigators found that many biological processes that were enriched in early DKD were not similarly enriched in progressive disease.
One protein noted in the analysis by Han, et al.31 that is highly elevated in the urine of DKD patients is retinol-binding protein 4 (RBP4). RBP4 is a protein synthesized primarily by liver cells and adipocytes that is generally freely filtered by glomerulus. It is then reabsorbed by proximal tubule proteins such as megalin and cubulin, as noted above. By some accounts RBP4 is the best biomarker of proximal tubular function and is particularly elevated in diseases of specific proximal tubular dysfunction such as causes of Fanconi Syndrome.32 Although it may function as a proximal tubular biomarker for DKD, it is also possible that urinary RBP4 could bind and deplete retinoic acid from the proximal tubule as it is found to bind retinoic acid avidly in the urine. Retinoic acid reduces macrophage-dependent injury and fibrosis and enhances repair in the kidney after acute kidney injury.33 If such protection is compromised more rapid progression of DKD might occur after acute injury events, and conversely, development of ways to deliver retinoic acid to the proximal tubule or reduce loss might be an important long-term strategy to prevent DKD progression. While this hypothesis has no evidence to support it at present, it is testable and provides an example of how urinary proteomics can generate new hypotheses for testing.
Metabolomic studies
Besides being helpful in identifying potential DKD biomarkers, as noted above, metabolomic studies have resulted in new insights about the progression of DKD. The most helpful studies have focused on metabolites in the urine of DKD patients. Urine samples provide a broader range of metabolites than do blood samples and have the additional advantage of providing a window into kidney metabolism, especially that of tubular cells.
An early urine metabolomic analysis was performed on type 2 diabetic patients without significant albuminuria who were divided into 2 groups based on eGFR.34 There was a clear separation in the pattern of urinary metabolite levels for the two groups, even though the kidney function differed only moderately between the groups. In the set of metabolites that best correlated with low eGFR, a number of them showed a stronger association with low eGFR than did conventional uremic toxins such as indoxyl sulfate. However, given the study design, none of these metabolites could be directly linked to progression of DKD.
An excellent example of a systematic urine metabolomic study in DKD patients was reported by Sharma et al. This study provided a comprehensive analysis of the urinary metabolome in patients with progressive DKD (mean eGFR of 36 ml/min/1.73m2) and included many important control groups and validation sets.35 In this analysis, 13 urinary metabolites were found to significantly associate with DKD + reduced eGFR in both screening and validation sets. Interestingly, all 13 metabolites had lower concentrations in progressive DKD patients than in healthy controls or in diabetic participants without kidney disease. This panel of urinary metabolites appeared to represent a signature of progressive DKD as opposed to that of diabetes, per se, or chronic kidney disease in general. On performing a protein interaction network analysis, the investigators found that 11 of these metabolites, which included tricarboxylic acid (TCA) cycle and amino acid metabolites, were linked by a metabolic network dominated by mitochondrial enzymes, suggesting a generalized defect in mitochondrial metabolism (Table 2). Exosomal mitochondrial DNA analysis and immunohistochemistry showed that mitochondrial content and therefore mitochondrial biogenesis was reduced in kidney tissue (apparently most notably in proximal tubules) of patients with progressive DKD. In summary, while not showing cause and effect in this cross-sectional study, the investigators used a metabolic signature in the urine of patients with progressive DKD to uncover a profound defect in mitochondrial function in the kidney cortex.
Table 2.
Metabolites that are significantly associated with DKD + reduced eGFR35
| 3-hydroxy isovalerate* |
| Aconitic acid* |
| Citric acid* |
| 2-ethyl 3-OH propionate 5* |
| Glycolic acid |
| Homovanillic acid |
| 3-hydroxy isobutyrate* |
| 2-methyl acetoacetate* |
| 3-methyl adipic acid* |
| 3-methyl crotonyl glycine* |
| 3-hydroxy propionate* |
| Tiglylglycine* |
| Uracil* |
connected by a protein-protein interaction network linking the enzymes involved in production of these metabolites
Another example, also from Sharma’s group, is an elegant multi-omic approach using metabolomics, proteomics and sophisticated bioinformatics to identify central proteins that organize pathobiological processes in DKD.36 Taking publically available global human metabolic pathways and combining these with a protein interaction network based on previously reported studies, the investigators developed a new bioinformatic resource called MetBridge. MetBridge can be used to identify all enzymes that interact directly with metabolites of interest as well as all proteins that directly interact with those enzymes. In this way MetBridge identifies proteins that directly interact with more than one metabolic pathway, which the investigators called “bridge proteins.” These bridge proteins are likely to be key regulatory proteins given their central role in physically organizing multiple metabolic pathways. With this toolkit in hand the investigators took the urinary metabolites identified in their previous study, described above,35 that were significantly associated with DKD + reduced eGFR and mapped those onto MetBridge. They found several bridge proteins that directly interacted with the metabolic pathways that generated the 13 urinary metabolites. At the top of the list was MDM2 (Mouse double minute 2 homolog, also known as E3 ubiquitin-protein ligase) whose expression was substantially decreased in both diabetic glomerulus and tubulointerstitium compared to normal controls, and which interacted with a large number of critical enzymes identified in the MetBridge analysis. In addition, 6 of the 13 urinary metabolites were directly associated with enzymes that bound to MDM2. The investigators then showed that reduction or inhibition of MDM2 in podocytes or in tubular epithelia in mice led to severe glomerular or tubular pathology, proving that decreased expression or activity of this bridge protein to diabetic levels resulted in severe kidney damage. Thus, by using a bioinformatics-driven, multi-omics approach, the investigators identified a key interacting protein that had not been previously suspected to be responsible for pathogenic metabolite changes in DKD. While the central role of MDM2 still needs to be proven in human DKD, other studies have begun to point to the importance of this protein in DKD.37
Conclusion
Systems biological approaches to research have contributed in multiple ways to our understanding of DKD pathogenesis, have identified new targets for therapy and have identified new important biomarkers to allow us to better diagnose, forecast and treat progressive DKD. Systems biological studies have emphasized the foundational roles of kidney inflammation and tubular mitochondrial dysfunction in the progression of DKD and have helped refocus research on human disease, not models. While a new breakthrough treatment has not yet emerged from these studies, several candidates have been identified and such treatments are likely on the horizon. That is truly the promise of systems biology in DKD.
Clinical Summary.
Systems biological approaches examine the network of all molecular changes that occur in a disease process instead of focusing on a single abnormal molecule (e.g., gene or protein) or pathway.
Diseases such as DKD that involve multiple cell types in the kidney; result from multiple metabolic, inflammatory and signaling changes; and evolve over years or decades are well suited for systems biological investigation.
A major advantage of systems biological investigation is that it can start with investigation of DKD in humans and does not initially rely on models that generally fail to fully represent the human disease process.
Multi-omic approaches that combine two different types of systems biological investigations (e.g., transcriptomics plus proteomics) can result in more precise identification of disease biomarkers and pathophysiologic molecular targets.
New biomarkers of progressive DKD and molecular targets for the treatment of DKD have been identified by systems biological approaches.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.2016 USRDS annual data report: Epidemiology of kidney disease in the United States National Institutes of Health. National Institute of Diabetes and Digestive and Kidney Disease; Bethesda, MD.: 2016. https://www.usrds.org/adr.aspx. [Google Scholar]
- 2.Afkarian M, Zelnick LR, Hall YN, et al. Clinical Manifestations of Kidney Disease Among US Adults With Diabetes, 1988–2014. JAMA. 2016;316(6):602–610. doi: 10.1001/jama.2016.10924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brosius FC, 3rd, Alpers CE, Bottinger EP, et al. Mouse models of diabetic nephropathy. Journal of the American Society of Nephrology: JASN. 2009;20(12):2503–2512. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Regele F, Jelencsics K, Shiffman D, et al. Genome-wide studies to identify risk factors for kidney disease with a focus on patients with diabetes. Nephrol Dial Transplant. 2015;30(Suppl 4):iv26–34. doi: 10.1093/ndt/gfv087. [DOI] [PubMed] [Google Scholar]
- 5.Hodgin JB, Nair V, Zhang H, et al. Identification of cross-species shared transcriptional networks of diabetic nephropathy in human and mouse glomeruli. Diabetes. 2013;62(1):299–308. doi: 10.2337/db11-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ju W, Nair V, Smith S, et al. Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker. Science translational medicine. 2015;7(316):316ra193. doi: 10.1126/scitranslmed.aac7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Betz BB, Jenks SJ, Cronshaw AD, et al. Urinary peptidomics in a rodent model of diabetic nephropathy highlights epidermal growth factor as a biomarker for renal deterioration in patients with type 2 diabetes. Kidney international. 2016;89(5):1125–1135. doi: 10.1016/j.kint.2016.01.015. [DOI] [PubMed] [Google Scholar]
- 8.Klein JB. Applying proteomics to detect early signs of chronic kidney disease: where has the magic gone? Expert review of proteomics. 2017;14(5):387–390. doi: 10.1080/14789450.2017.1315303. [DOI] [PubMed] [Google Scholar]
- 9.Good DM, Zurbig P, Argiles A, et al. Naturally occurring human urinary peptides for use in diagnosis of chronic kidney disease. Mol Cell Proteomics. 2010;9(11):2424–2437. doi: 10.1074/mcp.M110.001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindhardt M, Persson F, Zurbig P, et al. Urinary proteomics predict onset of microalbuminuria in normoalbuminuric type 2 diabetic patients, a sub-study of the DIRECT-Protect 2 study. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2016 doi: 10.1093/ndt/gfw292. [DOI] [PubMed] [Google Scholar]
- 11.Nkuipou-Kenfack E, Zurbig P, Mischak H. The long path towards implementation of clinical proteomics: Exemplified based on CKD273. Proteomics Clinical applications. 2017;11:5–6. doi: 10.1002/prca.201600104. [DOI] [PubMed] [Google Scholar]
- 12.Pontillo C, Jacobs L, Staessen JA, et al. A urinary proteome-based classifier for the early detection of decline in glomerular filtration. Nephrol Dial Transplant. 2016 doi: 10.1093/ndt/gfw239. [DOI] [PubMed] [Google Scholar]
- 13.Lindhardt M, Persson F, Currie G, et al. Proteomic prediction and Renin angiotensin aldosterone system Inhibition prevention Of early diabetic nephRopathy in TYpe 2 diabetic patients with normoalbuminuria (PRIORITY): essential study design and rationale of a randomised clinical multicentre trial. BMJ Open. 2016;6(3):e010310. doi: 10.1136/bmjopen-2015-010310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hocher B, Adamski J. Metabolomics for clinical use and research in chronic kidney disease. Nature reviews Nephrology. 2017;13(5):269–284. doi: 10.1038/nrneph.2017.30. [DOI] [PubMed] [Google Scholar]
- 15.Niewczas MA, Sirich TL, Mathew AV, et al. Uremic solutes and risk of end-stage renal disease in type 2 diabetes: metabolomic study. Kidney international. 2014;85(5):1214–1224. doi: 10.1038/ki.2013.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ju W, Greene CS, Eichinger F, et al. Defining cell-type specificity at the transcriptional level in human disease. Genome research. 2013;23(11):1862–1873. doi: 10.1101/gr.155697.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, Susztak K. Transcriptome analysis of human diabetic kidney disease. Diabetes. 2011;60(9):2354–2369. doi: 10.2337/db10-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen CD, Frach K, Schlondorff D, Kretzler M. Quantitative gene expression analysis in renal biopsies: a novel protocol for a high-throughput multicenter application. Kidney international. 2002;61(1):133–140. doi: 10.1046/j.1523-1755.2002.00113.x. [DOI] [PubMed] [Google Scholar]
- 19.Baelde HJ, Eikmans M, Doran PP, Lappin DW, de Heer E, Bruijn JA. Gene expression profiling in glomeruli from human kidneys with diabetic nephropathy. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2004;43(4):636–650. doi: 10.1053/j.ajkd.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 20.Schmid H, Boucherot A, Yasuda Y, et al. Modular activation of nuclear factor-kappaB transcriptional programs in human diabetic nephropathy. Diabetes. 2006;55(11):2993–3003. doi: 10.2337/db06-0477. [DOI] [PubMed] [Google Scholar]
- 21.Berthier CC, Zhang H, Schin M, et al. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes. 2009;58(2):469–477. doi: 10.2337/db08-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H, Nair V, Saha J, et al. Podocyte-specific JAK2 overexpression worsens diabetic kidney disease in mice. Kidney international. 2017 doi: 10.1016/j.kint.2017.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuttle KR. American Diabetes Association Meeting. Boston: 2015. Baricitinib in Diabetic Kidney Disease: Results from a Phase 2, Multicenter, Randomized, Double-Blind, Placebo-Controlled Study. [Google Scholar]
- 24.Menne J, Eulberg D, Beyer D, et al. C-C motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol Dial Transplant. 2017;32(2):307–315. doi: 10.1093/ndt/gfv459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Zeeuw D, Bekker P, Henkel E, et al. The effect of CCR2 inhibitor CCX140-B on residual albuminuria in patients with type 2 diabetes and nephropathy: a randomised trial. The lancet Diabetes & endocrinology. 2015;3(9):687–696. doi: 10.1016/S2213-8587(15)00261-2. [DOI] [PubMed] [Google Scholar]
- 26.Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genomics. 2010;3:33. doi: 10.1186/1755-8794-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mooyaart AL, Valk EJ, van Es LA, et al. Genetic associations in diabetic nephropathy: a meta-analysis. Diabetologia. 2011;54(3):544–553. doi: 10.1007/s00125-010-1996-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ko YA, Mohtat D, Suzuki M, et al. Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome biology. 2013;14(10):R108. doi: 10.1186/gb-2013-14-10-r108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thrailkill KM, Nimmo T, Bunn RC, et al. Microalbuminuria in type 1 diabetes is associated with enhanced excretion of the endocytic multiligand receptors megalin and cubilin. Diabetes care. 2009;32(7):1266–1268. doi: 10.2337/dc09-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilbert RE. Proximal Tubulopathy: Prime Mover and Key Therapeutic Target in Diabetic Kidney Disease. Diabetes. 2017;66(4):791–800. doi: 10.2337/db16-0796. [DOI] [PubMed] [Google Scholar]
- 31.Van JA, Scholey JW, Konvalinka A. Insights into Diabetic Kidney Disease Using Urinary Proteomics and Bioinformatics. Journal of the American Society of Nephrology: JASN. 2017;28(4):1050–1061. doi: 10.1681/ASN.2016091018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Norden AG, Lapsley M, Unwin RJ. Urine retinol-binding protein 4: a functional biomarker of the proximal renal tubule. Adv Clin Chem. 2014;63:85–122. doi: 10.1016/b978-0-12-800094-6.00003-0. [DOI] [PubMed] [Google Scholar]
- 33.Chiba T, Skrypnyk NI, Skvarca LB, et al. Retinoic Acid Signaling Coordinates Macrophage-Dependent Injury and Repair after AKI. Journal of the American Society of Nephrology: JASN. 2016;27(2):495–508. doi: 10.1681/ASN.2014111108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ng DP, Salim A, Liu Y, et al. A metabolomic study of low estimated GFR in non-proteinuric type 2 diabetes mellitus. Diabetologia. 2012;55(2):499–508. doi: 10.1007/s00125-011-2339-6. [DOI] [PubMed] [Google Scholar]
- 35.Sharma K, Karl B, Mathew AV, et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. Journal of the American Society of Nephrology: JASN. 2013;24(11):1901–1912. doi: 10.1681/ASN.2013020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saito R, Rocanin-Arjo A, You YH, et al. Systems biology analysis reveals role of MDM2 in diabetic nephropathy. JCI Insight. 2016;1(17):e87877. doi: 10.1172/jci.insight.87877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang H, Lei CT, Ye C, et al. MDM2 is implicated in high-glucose-induced podocyte mitotic catastrophe via Notch1 signalling. J Cell Mol Med. 2017 doi: 10.1111/jcmm.13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bjornstad P, Pyle L, Kinney GL, et al. Adiponectin is associated with early diabetic kidney disease in adults with type 1 diabetes: A Coronary Artery Calcification in Type 1 Diabetes (CACTI) Study. Journal of diabetes and its complications. 2017;31(2):369–374. doi: 10.1016/j.jdiacomp.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.von Scholten BJ, Reinhard H, Hansen TW, et al. Urinary biomarkers are associated with incident cardiovascular disease, all-cause mortality and deterioration of kidney function in type 2 diabetic patients with microalbuminuria. Diabetologia. 2016;59(7):1549–1557. doi: 10.1007/s00125-016-3937-0. [DOI] [PubMed] [Google Scholar]
- 40.Lenghel AR, Kacso IM, Bondor CI, Rusu C, Rahaian R, Gherman Caprioara M. Intercellular adhesion molecule, plasma adiponectin and albuminuria in type 2 diabetic patients. Diabetes research and clinical practice. 2012;95(1):55–61. doi: 10.1016/j.diabres.2011.08.028. [DOI] [PubMed] [Google Scholar]
- 41.Fujita H, Morii T, Koshimura J, et al. Possible relationship between adiponectin and renal tubular injury in diabetic nephropathy. Endocrine journal. 2006;53(6):745–752. doi: 10.1507/endocrj.k06-016. [DOI] [PubMed] [Google Scholar]
- 42.Zylka A, Gala-Bladzinska A, Rybak K, Dumnicka P, Drozdz R, Kusnierz-Cabala B. Role of new biomarkers for the diagnosis of nephropathy associated with diabetes type 2. Folia medica Cracoviensia. 2015;55(4):21–33. [PubMed] [Google Scholar]
- 43.Lee MJ, Kim SS, Kim IJ, et al. Changes in Urinary Angiotensinogen Associated with Deterioration of Kidney Function in Patients with Type 2 Diabetes Mellitus. Journal of Korean medical science. 2017;32(5):782–788. doi: 10.3346/jkms.2017.32.5.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mizushige T, Kobori H, Nishijima Y, et al. Urinary Angiotensinogen Could Be a Prognostic Marker of Renoprotective Effects of Alogliptin in Patients with Type 2 Diabetes. Journal of diabetes research. 2015;2015:517472. doi: 10.1155/2015/517472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hong CY, Hughes K, Chia KS, Ng V, Ling SL. Urinary alpha1-microglobulin as a marker of nephropathy in type 2 diabetic Asian subjects in Singapore. Diabetes care. 2003;26(2):338–342. doi: 10.2337/diacare.26.2.338. [DOI] [PubMed] [Google Scholar]
- 46.Shore N, Khurshid R, Saleem M. Alpha-1 microglobulin: a marker for early detection of tubular disorders in diabetic nephropathy. Journal of Ayub Medical College, Abbottabad: JAMC. 2010;22(4):53–55. [PubMed] [Google Scholar]
- 47.Hojs R, Ekart R, Bevc S, Hojs N. Markers of Inflammation and Oxidative Stress in the Development and Progression of Renal Disease in Diabetic Patients. Nephron. 2016;133(3):159–162. doi: 10.1159/000447434. [DOI] [PubMed] [Google Scholar]
- 48.Pena MJ, Heinzel A, Heinze G, et al. A panel of novel biomarkers representing different disease pathways improves prediction of renal function decline in type 2 diabetes. PloS one. 2015;10(5):e0120995. doi: 10.1371/journal.pone.0120995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Titan SM, Vieira JM, Jr, Dominguez WV, et al. Urinary MCP-1 and RBP: independent predictors of renal outcome in macroalbuminuric diabetic nephropathy. Journal of diabetes and its complications. 2012;26(6):546–553. doi: 10.1016/j.jdiacomp.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 50.Camilla R, Brachemi S, Pichette V, et al. Urinary monocyte chemotactic protein 1: marker of renal function decline in diabetic and nondiabetic proteinuric renal disease. Journal of nephrology. 2011;24(1):60–67. doi: 10.5301/jn.2010.1458. [DOI] [PubMed] [Google Scholar]
- 51.Tam FW, Riser BL, Meeran K, Rambow J, Pusey CD, Frankel AH. Urinary monocyte chemoattractant protein-1 (MCP-1) and connective tissue growth factor (CCN2) as prognostic markers for progression of diabetic nephropathy. Cytokine. 2009;47(1):37–42. doi: 10.1016/j.cyto.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 52.Tesch GH. MCP-1/CCL2: a new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am J Physiol Renal Physiol. 2008;294(4):F697–701. doi: 10.1152/ajprenal.00016.2008. [DOI] [PubMed] [Google Scholar]
- 53.Wolkow PP, Niewczas MA, Perkins B, et al. Association of urinary inflammatory markers and renal decline in microalbuminuric type 1 diabetics. Journal of the American Society of Nephrology: JASN. 2008;19(4):789–797. doi: 10.1681/ASN.2007050556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fiseha T. Urinary biomarkers for early diabetic nephropathy in type 2 diabetic patients. Biomarker research. 2015;3:16. doi: 10.1186/s40364-015-0042-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roscioni SS, de Zeeuw D, Hellemons ME, et al. A urinary peptide biomarker set predicts worsening of albuminuria in type 2 diabetes mellitus. Diabetologia. 2013;56(2):259–267. doi: 10.1007/s00125-012-2755-2. [DOI] [PubMed] [Google Scholar]
- 56.Langsford D, Tang M, Cheikh Hassan HI, Djurdjev O, Sood MM, Levin A. The Association between Biomarker Profiles, Etiology of Chronic Kidney Disease, and Mortality. American journal of nephrology. 2017;45(3):226–234. doi: 10.1159/000454991. [DOI] [PubMed] [Google Scholar]
- 57.Mussap M, Dalla Vestra M, Fioretto P, et al. Cystatin C is a more sensitive marker than creatinine for the estimation of GFR in type 2 diabetic patients. Kidney international. 2002;61(4):1453–1461. doi: 10.1046/j.1523-1755.2002.00253.x. [DOI] [PubMed] [Google Scholar]
- 58.Garg V, Kumar M, Mahapatra HS, Chitkara A, Gadpayle AK, Sekhar V. Novel urinary biomarkers in pre-diabetic nephropathy. Clinical and experimental nephrology. 2015;19(5):895–900. doi: 10.1007/s10157-015-1085-3. [DOI] [PubMed] [Google Scholar]
- 59.Pavkov ME, Knowler WC, Hanson RL, et al. Comparison of serum cystatin C, serum creatinine, measured GFR, and estimated GFR to assess the risk of kidney failure in American Indians with diabetic nephropathy. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2013;62(1):33–41. doi: 10.1053/j.ajkd.2012.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gerritsen KG, Leeuwis JW, Koeners MP, et al. Elevated Urinary Connective Tissue Growth Factor in Diabetic Nephropathy Is Caused by Local Production and Tubular Dysfunction. Journal of diabetes research. 2015;2015:539787. doi: 10.1155/2015/539787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brott DA, Furlong ST, Adler SH, et al. Characterization of renal biomarkers for use in clinical trials: effect of preanalytical processing and qualification using samples from subjects with diabetes. Drug design, development and therapy. 2015;9:3191–3198. doi: 10.2147/DDDT.S78792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nguyen TQ, Tarnow L, Jorsal A, et al. Plasma connective tissue growth factor is an independent predictor of end-stage renal disease and mortality in type 1 diabetic nephropathy. Diabetes care. 2008;31(6):1177–1182. doi: 10.2337/dc07-2469. [DOI] [PubMed] [Google Scholar]
- 63.Okazaki Y, Furuhashi M, Tanaka M, et al. Urinary excretion of fatty acid-binding protein 4 is associated with albuminuria and renal dysfunction. PloS one. 2014;9(12):e115429. doi: 10.1371/journal.pone.0115429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Choi HM, Park KT, Lee JW, et al. Urine neutrophil gelatinase-associated lipocalin predicts graft outcome up to 1 year after kidney transplantation. Transplantation proceedings. 2013;45(1):122–128. doi: 10.1016/j.transproceed.2012.05.080. [DOI] [PubMed] [Google Scholar]
- 65.Nielsen SE, Reinhard H, Zdunek D, et al. Tubular markers are associated with decline in kidney function in proteinuric type 2 diabetic patients. Diabetes research and clinical practice. 2012;97(1):71–76. doi: 10.1016/j.diabres.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 66.Agarwal R, Duffin KL, Laska DA, Voelker JR, Breyer MD, Mitchell PG. A prospective study of multiple protein biomarkers to predict progression in diabetic chronic kidney disease. Nephrol Dial Transplant. 2014;29(12):2293–2302. doi: 10.1093/ndt/gfu255. [DOI] [PubMed] [Google Scholar]
- 67.Turner G, Coates P, Warren S, Woodhead JS, Peters JR. Proximal tubular reabsorption of growth hormone and sodium/fluid in normo- and microalbuminuric insulin-dependent diabetes mellitus. Acta diabetologica. 1997;34(1):27–32. doi: 10.1007/s005920050061. [DOI] [PubMed] [Google Scholar]
- 68.Turner G, Coates P, Porter S, Peters JR, Woodhead JS. Urinary growth hormone measurements as a marker of renal tubular function in diabetes mellitus. Clinica chimica acta; international journal of clinical chemistry. 1993;220(1):19–30. doi: 10.1016/0009-8981(93)90003-m. [DOI] [PubMed] [Google Scholar]
- 69.Mayer G, Heerspink HJ, Aschauer C, et al. Systems Biology-Derived Biomarkers to Predict Progression of Renal Function Decline in Type 2 Diabetes. Diabetes care. 2017;40(3):391–397. doi: 10.2337/dc16-2202. [DOI] [PubMed] [Google Scholar]
- 70.Nicoletto BB, Krolikowski TC, Crispim D, Canani LH. Serum and Urinary Progranulin in Diabetic Kidney Disease. PloS one. 2016;11(10):e0165177. doi: 10.1371/journal.pone.0165177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.de Carvalho JA, Tatsch E, Hausen BS, et al. Urinary kidney injury molecule-1 and neutrophil gelatinase-associated lipocalin as indicators of tubular damage in normoalbuminuric patients with type 2 diabetes. Clinical biochemistry. 2016;49(3):232–236. doi: 10.1016/j.clinbiochem.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 72.Lin CH, Chang YC, Chuang LM. Early detection of diabetic kidney disease: Present limitations and future perspectives. World journal of diabetes. 2016;7(14):290–301. doi: 10.4239/wjd.v7.i14.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fu WJ, Li BL, Wang SB, et al. Changes of the tubular markers in type 2 diabetes mellitus with glomerular hyperfiltration. Diabetes research and clinical practice. 2012;95(1):105–109. doi: 10.1016/j.diabres.2011.09.031. [DOI] [PubMed] [Google Scholar]
- 74.Fufaa GD, Weil EJ, Nelson RG, et al. Association of urinary KIM-1, L-FABP, NAG and NGAL with incident end-stage renal disease and mortality in American Indians with type 2 diabetes mellitus. Diabetologia. 2015;58(1):188–198. doi: 10.1007/s00125-014-3389-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Coca SG, Nadkarni GN, Huang Y, et al. Plasma Biomarkers and Kidney Function Decline in Early and Established Diabetic Kidney Disease. Journal of the American Society of Nephrology: JASN. 2017 doi: 10.1681/ASN.2016101101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vaidya VS, Niewczas MA, Ficociello LH, et al. Regression of microalbuminuria in type 1 diabetes is associated with lower levels of urinary tubular injury biomarkers, kidney injury molecule-1, and N-acetyl-beta-D-glucosaminidase. Kidney international. 2011;79(4):464–470. doi: 10.1038/ki.2010.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nowak N, Skupien J, Niewczas MA, et al. Increased plasma kidney injury molecule-1 suggests early progressive renal decline in non-proteinuric patients with type 1 diabetes. Kidney international. 2016;89(2):459–467. doi: 10.1038/ki.2015.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shao Y, Lv C, Yuan Q, Wang Q. Levels of Serum 25(OH)VD3, HIF-1alpha, VEGF, vWf, and IGF-1 and Their Correlation in Type 2 Diabetes Patients with Different Urine Albumin Creatinine Ratio. Journal of diabetes research. 2016;2016:1925424. doi: 10.1155/2016/1925424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bhensdadia NM, Hunt KJ, Lopes-Virella MF, et al. Urine haptoglobin levels predict early renal functional decline in patients with type 2 diabetes. Kidney international. 2013;83(6):1136–1143. doi: 10.1038/ki.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang JK, Wang YY, Liu C, et al. Urine Proteome Specific for Eye Damage Can Predict Kidney Damage in Patients With Type 2 Diabetes: A Case-Control and a 5.3-Year Prospective Cohort Study. Diabetes care. 2017;40(2):253–260. doi: 10.2337/dc16-1529. [DOI] [PubMed] [Google Scholar]
- 81.Sangoi MB, de Carvalho JA, Tatsch E, et al. Urinary inflammatory cytokines as indicators of kidney damage in type 2 diabetic patients. Clinica chimica acta; international journal of clinical chemistry. 2016;460:178–183. doi: 10.1016/j.cca.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 82.Domingueti CP, Foscolo RB, Reis JS, et al. Association of Haemostatic and Inflammatory Biomarkers with Nephropathy in Type 1 Diabetes Mellitus. Journal of diabetes research. 2016;2016:2315260. doi: 10.1155/2016/2315260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Akturk M, Arslan M, Altinova A, et al. Association of serum levels of IGF-I and IGFBP-1 with renal function in patients with type 2 diabetes mellitus. Growth hormone & IGF research: official journal of the Growth Hormone Research Society and the International IGF Research Society. 2007;17(3):186–193. doi: 10.1016/j.ghir.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 84.Wu C, Wang Q, Lv C, et al. The changes of serum sKlotho and NGAL levels and their correlation in type 2 diabetes mellitus patients with different stages of urinary albumin. Diabetes research and clinical practice. 2014;106(2):343–350. doi: 10.1016/j.diabres.2014.08.026. [DOI] [PubMed] [Google Scholar]
- 85.Kim SS, Song SH, Kim IJ, et al. Decreased plasma alpha-Klotho predict progression of nephropathy with type 2 diabetic patients. Journal of diabetes and its complications. 2016;30(5):887–892. doi: 10.1016/j.jdiacomp.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 86.Lee EY, Kim SS, Lee JS, et al. Soluble alpha-klotho as a novel biomarker in the early stage of nephropathy in patients with type 2 diabetes. PloS one. 2014;9(8):e102984. doi: 10.1371/journal.pone.0102984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim HR, Nam BY, Kim DW, et al. Circulating alpha-klotho levels in CKD and relationship to progression. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2013;61(6):899–909. doi: 10.1053/j.ajkd.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 88.Kacso IM, Bondor CI, Kacso G. Soluble serum Klotho in diabetic nephropathy: relationship to VEGF-A. Clinical biochemistry. 2012;45(16-17):1415–1420. doi: 10.1016/j.clinbiochem.2012.07.098. [DOI] [PubMed] [Google Scholar]
- 89.Yuruk Yildirim Z, Nayir A, Yilmaz A, Gedikbasi A, Bundak R. Neutrophil Gelatinase-Associated Lipocalin as an Early Sign of Diabetic Kidney Injury in Children. Journal of clinical research in pediatric endocrinology. 2015;7(4):274–279. doi: 10.4274/jcrpe.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Papadopoulou-Marketou N, Skevaki C, Kosteria I, et al. NGAL and cystatin C: two possible early markers of diabetic nephropathy in young patients with type 1 diabetes mellitus: one year follow up. Hormones. 2015;14(2):232–240. doi: 10.14310/horm.2002.1520. [DOI] [PubMed] [Google Scholar]
- 91.Hafez MH, El-Mougy FA, Makar SH, Abd El Shaheed S. Detection of an earlier tubulopathy in diabetic nephropathy among children with normoalbuminuria. Iranian journal of kidney diseases. 2015;9(2):126–131. [PubMed] [Google Scholar]
- 92.Afkarian M, Zelnick LR, Ruzinski J, et al. Urine matrix metalloproteinase-7 and risk of kidney disease progression and mortality in type 2 diabetes. Journal of diabetes and its complications. 2015;29(8):1024–1031. doi: 10.1016/j.jdiacomp.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kern EF, Erhard P, Sun W, Genuth S, Weiss MF. Early urinary markers of diabetic kidney disease: a nested case-control study from the Diabetes Control and Complications Trial (DCCT) American journal of kidney diseases: the official journal of the National Kidney Foundation. 2010;55(5):824–834. doi: 10.1053/j.ajkd.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hong CY, Chia KS, Ling SL. Urinary protein excretion in Type 2 diabetes with complications. Journal of diabetes and its complications. 2000;14(5):259–265. doi: 10.1016/s1056-8727(00)00119-7. [DOI] [PubMed] [Google Scholar]
- 95.Fukuda A, Wickman LT, Venkatareddy MP, et al. Urine podocin:nephrin mRNA ratio (PNR) as a podocyte stress biomarker. Nephrol Dial Transplant. 2012;27(11):4079–4087. doi: 10.1093/ndt/gfs313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sato Y, Wharram BL, Lee SK, et al. Urine podocyte mRNAs mark progression of renal disease. Journal of the American Society of Nephrology: JASN. 2009;20(5):1041–1052. doi: 10.1681/ASN.2007121328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nielsen SE, Schjoedt KJ, Rossing K, et al. Levels of NT-proBNP, markers of low-grade inflammation, and endothelial dysfunction during spironolactone treatment in patients with diabetic kidney disease. Journal of the renin-angiotensin-aldosterone system: JRAAS. 2013;14(2):161–166. doi: 10.1177/1470320312460290. [DOI] [PubMed] [Google Scholar]
- 98.Ahn JM, Kim BG, Yu MH, Lee IK, Cho JY. Identification of diabetic nephropathy-selective proteins in human plasma by multi-lectin affinity chromatography and LC-MS/MS. Proteomics Clinical applications. 2010;4(6-7):644–653. doi: 10.1002/prca.200900196. [DOI] [PubMed] [Google Scholar]
- 99.Akbay E, Muslu N, Nayir E, Ozhan O, Kiykim A. Serum retinol binding protein 4 level is related with renal functions in Type 2 diabetes. Journal of endocrinological investigation. 2010;33(10):725–729. doi: 10.1007/BF03346678. [DOI] [PubMed] [Google Scholar]
- 100.Henze A, Frey SK, Raila J, et al. Evidence that kidney function but not type 2 diabetes determines retinol-binding protein 4 serum levels. Diabetes. 2008;57(12):3323–3326. doi: 10.2337/db08-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Howard RL, Buddington B, Alfrey AC. Urinary albumin, transferrin and iron excretion in diabetic patients. Kidney international. 1991;40(5):923–926. doi: 10.1038/ki.1991.295. [DOI] [PubMed] [Google Scholar]
- 102.Gupta S, Gambhir JK, Kalra O, et al. Association of biomarkers of inflammation and oxidative stress with the risk of chronic kidney disease in Type 2 diabetes mellitus in North Indian population. Journal of diabetes and its complications. 2013;27(6):548–552. doi: 10.1016/j.jdiacomp.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 103.Gohda T, Niewczas MA, Ficociello LH, et al. Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. Journal of the American Society of Nephrology: JASN. 2012;23(3):516–524. doi: 10.1681/ASN.2011060628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Niewczas MA, Gohda T, Skupien J, et al. Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. Journal of the American Society of Nephrology: JASN. 2012;23(3):507–515. doi: 10.1681/ASN.2011060627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Izumi Y, Yabe D, Taniguchi A, et al. Circulating TNF receptor 2 is associated with the development of chronic kidney disease in non-obese Japanese patients with type 2 diabetes. Diabetes research and clinical practice. 2013;99(2):145–150. doi: 10.1016/j.diabres.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 106.Lou NJ, Ni YH, Jia HY, et al. Urinary Microvesicle-Bound Uromodulin: A Potential Molecular Biomarker in Diabetic Kidney Disease. Journal of diabetes research. 2017;2017:3918681. doi: 10.1155/2017/3918681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chang CC, Chen CY, Huang CH, et al. Urinary glycated uromodulin in diabetic kidney disease. Clinical science. 2017 doi: 10.1042/CS20160978. [DOI] [PubMed] [Google Scholar]
- 108.Liu JJ, Yeoh LY, Sum CF, et al. Vascular cell adhesion molecule-1, but not intercellular adhesion molecule-1, is associated with diabetic kidney disease in Asians with type 2 diabetes. Journal of diabetes and its complications. 2015;29(5):707–712. doi: 10.1016/j.jdiacomp.2015.02.011. [DOI] [PubMed] [Google Scholar]